

Background: Quercetin inhibits prostate cancer growth, but its mechanisms are not fully elucidated.
Results: Quercetin binds to hnRNPA1 and hampers its return to the nucleus, causing accumulation of hnRNPA1 in the cytoplasm.
Conclusion: hnRNPA1 is a target of quercetin, driving cells toward apoptosis through expression regulation of cIAP1.
Significance: Understanding of the novel mechanism of actions on quercetin is crucial for cancer prevention and treatment in prostate cancer.
Keywords: Natural Product, Prostate Cancer, Protein Targeting, Proteomics, RNA-binding Protein, Quercetin, hnRNPA1, Chemical Proteomics, Molecular Target
Abstract
Quercetin, a flavonoid abundantly present in plants, is widely used as a phytotherapy in prostatitis and prostate cancer. Although quercetin has been reported to have a number of therapeutic effects, the cellular target(s) responsible for its anti-cancer action has not yet been clearly elucidated. Here, employing affinity chromatography and mass spectrometry, we identified heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) as a direct target of quercetin. A specific interaction between quercetin and hnRNPA1 was validated by immunoblotting and in vitro binding experiments. We found that quercetin bound the C-terminal region of hnRNPA1, impairing the ability of hnRNPA1 to shuttle between the nucleus and cytoplasm and ultimately resulting in its cytoplasmic retention. In addition, hnRNPA1 was recruited to stress granules after treatment of cells with quercetin for up to 48 h, and the levels of cIAP1 (cellular inhibitor of apoptosis), an internal ribosome entry site translation-dependent protein, were reduced by hnRNPA1 regulation. This is the first report that anti-cancer effects of quercetin are mediated, in part, by impairing functions of hnRNPA1, insights that were obtained using a chemical proteomics strategy.
Introduction
Quercetin is a naturally occurring dietary flavonol compound, abundant in a broad range of fruits, vegetables, wine, and tea, that exhibits several potentially beneficial properties, including anti-oxidant, anti-inflammatory, and anti-cancer effects, with almost no human toxicity (1). Quercetin has been used therapeutically to treat patients with prostatitis or prostate cancer (2). It has been shown to induce apoptosis, inhibit cell growth, and decrease expression of matrix metalloproteases in PC-3 cells; it also exhibits enhanced anti-cancer efficacy when combined with other dietary phytoestrogens (1, 3, 4). In addition, it appears to selectively inhibit the proliferation of tumor cells relative to normal cells (5). Quercetin exerts chemopreventive actions through induction of cell cycle arrest at G1 phase and/or G2/M phase through various mechanisms and by promoting increases in p53, p21, and p27 expression and decreases in Bcl-xL and cyclin D1 expression (1, 3, 5). Numerous cellular effects of quercetin have been recognized, and some proteins that interact directly with quercetin have been identified. However, a fuller understanding of the array of quercetin intracellular targets is crucial for establishing the molecular mechanisms of the anti-cancer effects of quercetin.
Among the previously identified proteins that can directly bind quercetin are MEK/Raf1 kinases of the MAPK cascade and PI3K. Binding of quercetin to MEK/Raf1 (in rat liver epithelial cells) or PI3K (in mouse JB6 P+ skin epidermal cells) suppresses the activity of the corresponding kinase induced by arsenite and tissue plasminogen activator, respectively (6, 7). Quercetin has also been reported to bind casein kinase 2, one of the regulators that modulate global caspase signaling, and inhibit its activity (8, 9). More recently, the heat shock proteins HSP70 and HSP90, RuvB-like 2 ATPases, and eIF-3 have also been identified as quercetin-binding proteins. All of these proteins have been considered potential targets in anti-cancer therapy; thus, there are numerous candidate mediators of the anti-cancer actions of quercetin (8, 9).
Chemical proteomics is an emerging strategy for comprehensive, proteome-wide drug target identification and an unbiased method for finding promiscuous protein targets interacting with drugs that mimic in vivo cellular interactions. This approach combines an immobilized drug affinity pull-down step with MS-based proteomics for protein identification (10). Several successful examples of the application of chemical proteomics have previously been reported. For example, utilizing this strategy, Ermakova et al. (11) identified vimentin as a molecular target of epigallocatechin gallate, revealing a possible mechanism for the anti-cancer action of this agent. Specifically, these authors showed that epigallocatechin gallate binding inhibited phosphorylation of vimentin at Ser-50 and Ser-55 by Cdk2 (cyclin-dependent kinase 2) and protein kinase A (PKA), preventing assembly of vimentin filament structures and thereby impairing the cellular space integration function of vimentin. Chemical proteomics also identified Nampt (nicotinamide phosphoribosyltransferase), which is involved in NAD biosynthesis and is frequently elevated in cancers with high energy requirements, as a target of CB30865, a folic acid analog that inhibits thymidylate synthase (12).
There has been growing recent interest in exploiting the activities of phytotherapeutic reagents to regulate tumor progression, especially in prostate disease. Among available nutraceuticals, quercetin is particularly promising in this context because of its effectiveness (13). Here, we employed a chemical proteomics strategy to identify the cellular targets of quercetin underlying its medicinal value in prostate diseases. In the first step, we used affinity chromatography to isolate quercetin-binding proteins from the PC-3 human prostate cancer cell line, applying cell lysates to quercetin-conjugated Sepharose beads. From the chromatography profile and the results of multiple additional analyses, including MS, we successfully identified a novel quercetin-binding protein. On the basis of our results, we propose a novel mechanism for quercetin suppression of cell growth in prostate tumors in which quercetin acts by binding hnRNPA12 to impair following cascade of protein expression (i.e. cIAP1 in this study). Our study highlights the potential contribution of hnRNPA1 in cIAP1 IRES-mediated translation control can be a common feature of quercetin's action in attenuating the progression of prostate cancers.
EXPERIMENTAL PROCEDURES
Materials
All media were obtained from Invitrogen; FBS was from Hyclone (Thermo Fisher Scientific). Quercetin was from Sigma-Aldrich, and CNBr-Sepharose 4B was obtained from GE Healthcare. [3H]Quercetin (1 mCi/ml in ethanol) was sourced from American Radiolabeled Chemicals Inc. The mouse anti-hnRNPA1 (4B10 clone) mAb, rabbit anti-PABP polyclonal antibody, anti-lamin antibody, and mouse β-actin mAb were purchased from Santa Cruz Biotechnology, Inc. The rabbit anti-vinculin polyclonal antibody, anti-α-tubulin, and rabbit anti-EF-1α polyclonal antibody were from GeneTex Inc. The rabbit anti-nucleolin polyclonal antibody was obtained from Abnova. The mouse anti-cIAP1 mAb was from R&D Systems Inc. The mouse anti-transportin 1 (Tnpo1) mAb (D45 clone) was purchased from Millipore. All restriction enzymes, ligases, and endonucleases used for cloning were from New England Biolabs.
Clones and Constructs
His-tagged, full-length hnRNPA1 (amino acids (aa) 1–320) was cloned from hnRNPA1 cDNA (accession number BC002355), inserted into the pET28a vector, and transformed into the Rosetta Escherichia coli strain (Merck). Truncated versions of hnRNPA1, including the N-terminal RNA binding domain (aa 1–196), the middle region (aa 182–268), and the C-terminal region (aa 268–320), were subcloned from the full-length plasmid, respectively. Recombinant full-length hnRNPA1 or truncated versions of hnRNPA1 were then expressed in E. coli strain BL21 (DE3, Merck) and purified by nickel affinity column chromatography. Recombinant full-length wild-type hnRNPA1 (WT hnRNPA1), used for transfection in PC-3 cells, was amplified by PCR, cloned into the p3XFLAG-CMV-14 plasmid (Sigma) to yield p3XFLAG-CMV-14 WT hnRNPA1, and transformed into JM109 E. coli (Promega). F2 mutant hnRNPA1 (p3XFLAG-CMV-14-F2mt- hnRNPA1) was constructed using the p3XFLAG-CMV-14 WT hnRNPA1 plasmid as a template and a forward primer containing mutations that converted S codons to A codons and a reverse primer that right-flanked the forward primer for self-ligation. PCR products were then purified and treated with DpnI endonuclease to digest templates, followed by self-ligation and transformation. For monitoring cellular localization of F2mt-hnRNPA1 by confocal microscopy, another F2mt-hnRNPA1 in pGFP-C1 vectors was generated using p3XFLAG-CMV-14-mt hnRNPA1 as a DNA source. For knockdown experiments, all shRNAs were provided from the Taiwan National RNAi core facility.
Cell Culture and Transient Transfection
PC-3 cells were routinely maintained in F12K medium (Invitrogen) supplemented with 10% FBS, 0.37% sodium bicarbonate, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. PC-3 cells were transiently transfected with various DNA constructs, including FLAG WT hnRNPA1 or FLAG F2mt-hnRNPA1, using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Transfected cells were then cultured in fresh medium containing 10% FBS for 16–18 h before treatment. In confocal microscopy experiments, cells were preseeded onto fibronectin-coated coverslips and transfected (FuGene 6; Promega) with FLAG WT hnRNPA1 and pGFP-C1-F2mt-hnRNPA1 plasmids 6 h postseeding. For knockdown experiments, lentiviral vector-mediated transductions of hnRNPA1- and cIAP1-specific shRNA were used. Lentiviruses expressing shRNAs against hnRNPA1, cIAP1, or control shRNA (shCtrl) were produced from HEK293FT cells. Medium containing viruses of hnRNPA1 shRNA, cIAP1 shRNA, or shCtrl was added to PC-3 cells. Experiments were conducted 3–5 days after transduction of shRNA.
Cell Treatment and Sample Preparation
The concentration of DMSO, used as a vehicle, was set at 0.4%. Before treating cells, the medium was replaced with fresh F-12K medium and supplemented with 10% FBS after incubation with quercetin or vehicle for 1 h. In immunoprecipitation (IP) experiments, 100 μm quercetin was added 24 h post-transfection, and cells were harvested after an additional 24 h. In confocal microscopy experiments, cells treated with 0.6 m d-sorbitol (Sigma) for 2.5 h were tested in parallel as a positive control for osmotic stress. Cells were washed and fixed after the indicated times, followed by immunofluorescence analysis.
Quercetin Uptake Assays
Cells were seeded at 5 × 105 cells/ml in 96-well plates 24 h before the experiment. Cells were washed and refed with incubation buffer (20 mm Tris-HCl, pH 7.4, 144 mm NaCl, 3 mm K2HPO4, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm d-glucose). Unlabeled quercetin and vehicle were added to a final DMSO concentration of 0.4% and incubated at 37 °C for 20 min. 3H-Labeled quercetin (30 nm) was subsequently added to each well, and cells were washed at designated times with incubation buffer. Nonspecific binding was defined as cells incubated in the presence of excess unlabeled ligands (100 μm). Radioactivity was measured using a scintillation counter (Microbeta, PerkinElmer Life Sciences).
Cell Viability Assays
Cell viability was measured using a CellTiter-Glo kit (Promega) following the manufacturer's protocol. Cells seeded at 3 × 104 cells/ml in a 96-well plate were incubated with quercetin at the indicated concentrations. After 24, 48, and 72 h, luciferin and Ultra-Glo recombinant luciferase were then added to each well, and the plate was incubated at room temperature for 10 min with constant shaking. Luminescence intensity was read using a microplate reader (Safire, Tecan), taking the luminescence reading of vehicle control as the 100% value.
Cell Apoptosis Assays
Cells were seeded on a 6-well plate of 1.2 × 105 cells/ml and incubated with quercetin for the indicated times. After harvesting, cells were stained with fluorescently labeled annexin V in combination with the dead cell marker, 7-aminoactinomycin D (annexin V assay) or with a DNA-binding dye and DEVD substrate together with 7-aminoactinomycin D (caspase-3/7 activity), as described by the manufacturer of the kits (Millipore). Cells were then analyzed using a Muse cell analyzer (Millipore). Apoptosis percentages in the respective assays were defined as annexin V-positive and caspase-3/7-positive groups.
Affinity Chromatography
Quercetin was first coupled to CNBr-activated Sepharose 4B matrix, according to the manufacturer's instructions (GE Healthcare). PC-3 cells were lysed and sonicated and then centrifuged at 14,500 rpm for 30 min, after which the supernatant was applied to the quercetin-Sepharose 4B column overnight at 4 °C. The mobile phase was T buffer (50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10 mm 6-aminohexanoic acid, 1 mm PMSF, 1 mm benzamidine hydrochloride, and 1 mm EDTA) running at a flow rate of 0.5 ml/min. Bound proteins were then eluted with T1 buffer (4 m urea, 1 m NaCl in T buffer). Proteins in eluted fractions were quantified using a 660-nm protein assay (Thermo Fisher Scientific) and separated by SDS-PAGE.
In-gel Digestion, MS Analysis, and Protein Identification
After silver staining, protein bands were excised from the SDS-PAGE and digested overnight with trypsin (Promega) at 37 °C. The tryptic peptide mixture was then eluted from the gel using 0.1% TFA in 60% acetonitrile and lyophilized. Tryptic peptides were resuspended in 0.1% TFA and analyzed by liquid chromatography-tandem MS (LC-MS/MS) using an LTQ-Orbitrap Velos hybrid mass spectrometer (Thermo Fisher Scientific). Peptide separations were performed online with MS by nanoflow LC (Dionex Ultimate 3000; Dionex Corp.). Samples were injected in a 10-μl volume of starting mobile phase (2% acetonitrile in 0.1% formic acid) at a flow rate of 750 nl/min onto a nanoACQUITY LC system with a 75 μm × 15-cm C18 column. The peptides were then separated by gradient elution at 300 nl/min in 90 min and injected onto a Thermo Scientific Velos Orbitrap electrospray mass spectrometer running at a resolution of 30,000 for precursors and 15,000 for fragment ions. Data-dependent MS/MS analysis was carried out using Xcalibur MS acquisition software (Xcalibur version 2.1; Thermo Fisher Scientific). Each scan cycle consisted of a full-scan MS acquired in profile mode at 60,000 resolution by the Orbitrap analyzer over the mass range 400–1800 m/z, followed by up to five data-dependent MS/MS scans of the five most intense peaks. Proteins were identified by automated searching (Mascot software) against the NCBI protein database.
Immunoblotting
Equal amounts of protein extract were separated by SDS-PAGE and transferred to a PVDF membrane, which was then blocked for 1 h. The membrane was further incubated overnight at 4 °C with primary antibodies. After six washing steps, the membranes were incubated for 1 h at 25 °C with secondary antibodies. Then, after an additional six washing steps, immunoreactive proteins were detected using ECL detection reagents (Millipore). Densitometry was performed using ImageQuant version 5.2 software from GE Healthcare.
Kd Measurement by Surface Plasmon Resonance
Recombinant full-length hnRNPA1 (aa 1–320) and truncated versions of hnRNPA1, including the N-terminal RNA binding domain (aa 1–196), the middle region (aa 182–268), and the C-terminal region (aa 268–320), were individually covalently coupled to a dextran matrix (CM5 chip) using a Biacore T200 system (GE Healthcare), following the manufacturer's protocols. Immobilized proteins were activated with N-hydroxysuccinimide and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide to form a carbodiamide linkage between carboxyl moieties on the dextran and primary amide groups on the protein. hnRNPA1 variants were dissolved in 10 mm acetate buffer (full-length, pH 4; N-terminal RNA binding domain and the C-terminal region, pH 4.5; middle region, pH 5) to a concentration of 20 μg/ml and flowed over the activated surface for 5 min with PBS. Unreacted groups were blocked with ethanolamine. PBS containing 0.05% (v/v) P20 (surfactant) and 5% DMSO was used for priming and conditioning the surface of the chips. Quercetin was serially diluted (from 200 to 3 μm) in PBS buffer and maintained in DMSO (5% (v/v)) (final concentrations from 10 to 0.15 μm). The baseline response was determined for 120 s (30 μl/min) before perfusing the immobilized proteins with the compounds to allow association to occur. Dissociation was then monitored over a period of an additional 120 s. The immobilized proteins were regenerated with 50 mm NaOH. Solvent correction was performed by subtracting DMSO-alone signals and responses of quercetin perfused over the dextran matrix from those of quercetin perfused over immobilized ligands. Data were analyzed using Biacore T200 evaluation software and fitted using GraphPad Prism software.
Immunofluorescence Analysis
Cells seeded on fibronectin (10 μg/ml)-coated coverslips were treated with quercetin (100 μm) for the indicated times. Cells were fixed with 3.7% paraformaldehyde for 15 min and permeabilized for 50 min with 0.1% Triton X-100 at room temperature. Cells were extensively washed with 1× PBS after each procedure. Cells were blocked with 5% BSA prior to adding mouse anti-hnRNPA1 (1:400) or rabbit anti-PABP (1:200) antibody. Subsequently, FITC-conjugated (1:400) or rhodamine-conjugated (1:200) anti-mouse or anti-rabbit secondary antibodies (Santa Cruz Biotechnology) and DAPI were used as appropriate. Cell images were captured using a TCS SP5 confocal microscope (Leica).
Immunoprecipitation
The binding of Tnpo1 to hnRNPA1 was examined by treating PC-3 cells with 100 μm quercetin for 24 and 48 h and lysing cells with RIPA buffer. Extracts containing 10 μg of protein were precleared with magnetic protein G beads (Millipore) at 4 °C for 1 h and then incubated with hnRNPA1 antibody-conjugated (2 μg of anti-hnRNPA1) protein G beads at 4 °C overnight. The precipitated immunocomplexes were harvested by extensive washing with ice-cold RIPA buffer. The immunoprecipitates were resuspended in sample buffer and subjected to electrophoresis and immunoblotting as described above.
Subcellular Fractionation
Briefly, cells were scraped, resuspended, and lysed in buffer A (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.5 mm DTT) containing protease inhibitors using a Dounce homogenizer. The lysed cells were then centrifuged at 3,000 rpm for 10 min, and the supernatant (cytoplasmic extract) was cleared by a further centrifugation at 14,800 rpm for 15 min. The pellet (nuclear extract) was washed and centrifuged at 5,100 rpm for 10 min, resuspended in S1 buffer (0.25 m sucrose and 10 mm MgCl2), and subsequently purified using a gradient of S3 buffer (0.88 m sucrose and 0.5 mm MgCl2).
RNA-binding Protein Immunoprecipitation (RIP) and Quantitative RT-PCR (RT-qPCR) Analysis
Cells were lysed in Dautry buffer (10 mm Tris-HCl, 140 mm NaCl, 1.5 mm MgCl2, 10 mm EDTA, and 0.5% Nonidet P-40 containing protease inhibitors, DNase (Ambion), and RNase Out (Invitrogen)). The cytoplasmic fraction was obtained by centrifugation at 3,000 rpm at 4 °C for 5 min. RNA·protein complexes were immunoprecipitated with anti-hnRNPA1 beads (ChIP protein A/G beads; Millipore) or mouse IgG-conjugated beads (control) for 2 h at 4 °C with constant shaking. After IP, RNA was isolated using TRIzol reagent (Invitrogen), according to the manufacturer's protocol. cDNA was generated from isolated RNA using an oligo(dT) primer and Superscript II kit (Invitrogen). The synthesized cDNA was used as the template for RT-qPCR using the SYBR Green PCR kit (Applied Biosystems) and analyzed on an ABI StepOne Plus system. Target mRNA levels were normalized to those of β-actin.
Data Analysis
All experiments were performed in duplicate at least three times, and representative results are presented. Statistical analyses of cell viability, apoptosis assays, densitometric quantifications and RT-qPCR data, presented as mean values ± S.E., were based on three independent experiments and were performed using GraphPad Prism software. The significance of differences of experimental means relative to corresponding controls was evaluated using Student's t test.
RESULTS
Workflow for Identification and Characterization of Quercetin-binding Proteins
Our aim was to establish a platform for screening and identifying quercetin-binding proteins. As shown in Fig. 1, quercetin-specific binding proteins were captured by quercetin-Sepharose beads, and eluted fractions were resolved by SDS-PAGE. Distinct proteins in gel-eluted bands were identified using MS and validated by immunoblotting analysis and in vitro binding assays. Specific targets were further characterized using confocal microscopy, IP, in vitro binding assays, RT-qPCR, and immunoblotting analysis.
FIGURE 1.
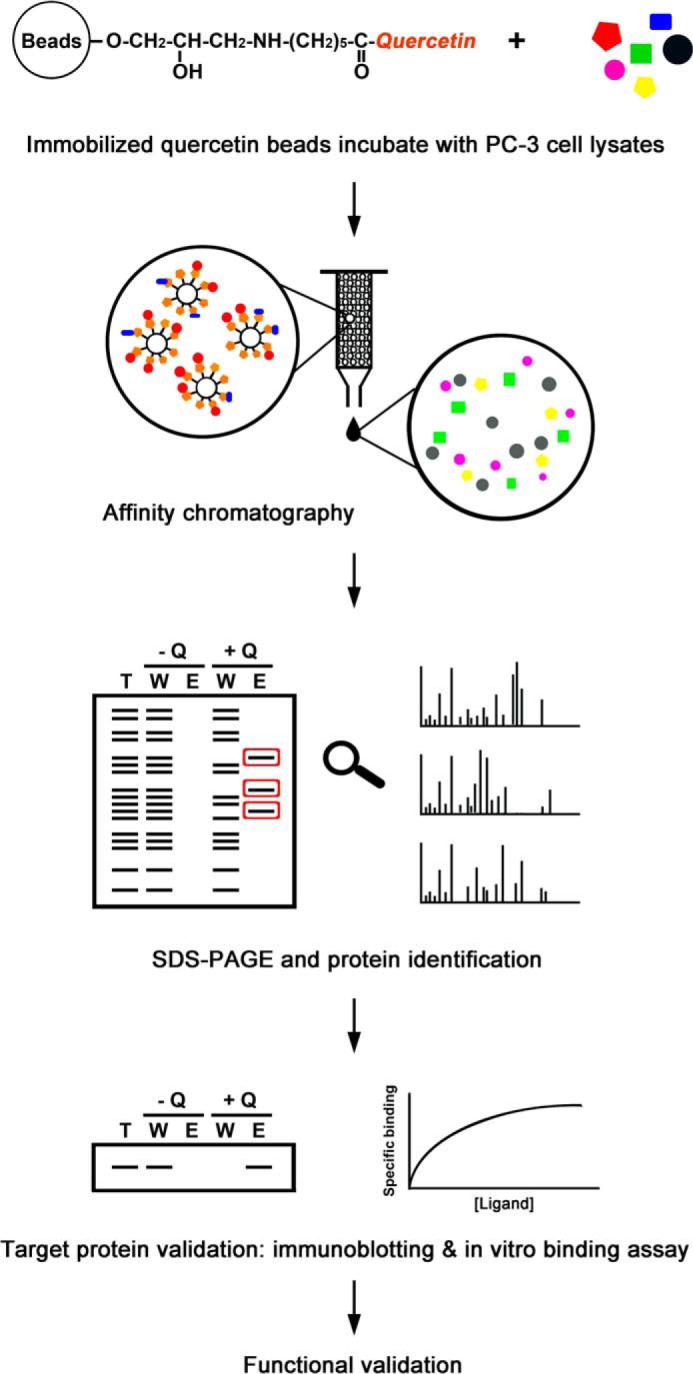
Schematic depiction of the workflow used to identify and characterize quercetin-binding proteins. Quercetin-specific binding proteins were captured by quercetin-Sepharose beads, and eluted fractions were resolved by SDS-PAGE. Distinct proteins in gel-eluted bands were identified using MS and validated by immunoblotting analyses and surface plasmon resonance binding assays. Specific targets were further characterized using a series of approaches, including confocal microscopy, IP, RIP, RT-qPCR, and immunoblotting analysis. Q, quercetin; T, total cell lysates; W, proteins that did not bind quercetin; E, bound proteins eluted.
Time Course of Quercetin Uptake, Cell Viability, and Apoptosis in Quercetin-treated PC-3 Cells
To evaluate the effects of quercetin, we measured the uptake of quercetin in PC-3 cells. Fig. 2A shows quercetin uptake by PC-3 cells after different incubation durations. In cells treated with 30 nm 3H-labeled quercetin, specific [3H]quercetin uptake increased after 5 min of incubation, reached a plateau after 20 min, and remained stable for up to 60 min. Radioactive intensity was significantly lower in the presence of excess unlabeled ligands (100 μm) than in vehicle-treated cells, indicating that PC-3 cells can effectively take up quercetin. As an initial step in identifying the target of quercetin in prostate cancer, we evaluated the cell growth-inhibitory activity of quercetin in PC-3 cells. As shown in Fig. 2B, quercetin induced cell death in a time- and concentration-dependent manner. The IC50 of quercetin for PC-3 cells was about 100 μm after a 24-h incubation. The reduction in cell viability resulted from apoptosis, as evidenced by staining for annexin V and activated caspase-3/7 (Fig. 2C). Collectively, these data demonstrate that the specific target of quercetin interaction in prostate cells is involved in inducing apoptosis.
FIGURE 2.

Quercetin uptake, cell viability, and apoptosis assay in quercetin-treated PC-3 cells. A, quercetin uptake was determined by quantifying the intracellular radioactivity of 3H-labeled quercetin at designated time points. B, cells were treated with the indicated concentrations of quercetin for 24, 48, and 72 h, and cell viability was analyzed by quantifying intracellular ATP level using a CellTiter-Glo kit. C, cells were stained with annexin V and caspase-3/7 together with 7-aminoactinomycin D for staining apoptotic cells and dead cells, respectively, after treatment with control or 100 μm quercetin for 24 and 48 h. *, p < 0.05; **, p < 0.01; ***, p < 0.001 with respect to control; Student's t test. QCT, quercetin; Ctrl, vehicle control. Error bars, S.E.
Analysis of Quercetin-binding Proteins
To identify direct targets of quercetin, we employed an affinity-based chemical proteomics strategy. We first coupled quercetin to CNBr-activated Sepharose beads and then incubated PC-3 cell lysates with quercetin-Sepharose beads, as described under “Experimental Procedures.” After eluting columns, fractions containing quercetin-bound proteins were analyzed by SDS-PAGE. Chromatography profiles revealed no nonspecific binding in the eluted fractions of Sepharose beads but revealed a number of different specifically bound proteins of quercetin-conjugated beads. We then performed in-gel digestions and LC-MS/MS to identify proteins that directly bound to quercetin. These analyses identified vinculin, nucleolin, EF-1α, and hnRNPA1 as quercetin-binding proteins (Fig. 3A and Table 1). To validate the MS results, we examined the interactions of these proteins with quercetin using immunoblotting analysis. As shown in Fig. 3B, vinculin, nucleolin, EF-1α, and hnRNPA1 were potentially direct, specific targets of quercetin; among these proteins, EF-1α is known to directly interact with quercetin (14). Given that hnRNPA1 was relatively abundant in the eluted fraction of quercetin beads and has not been previously reported to bind quercetin, we chose this protein for further investigation.
FIGURE 3.

Identification and validation of quercetin binding targets using immunoblotting and surface plasmon resonance binding assays. A, silver-stained gel showing proteins bound to the affinity column in the presence or absence of quercetin. T, total cell lysates; Un, unbound fraction; W, proteins that did not bind quercetin; E, bound proteins eluted. The indicated protein bands were excised from the gel and digested with trypsin, and the peptides were analyzed by MS. The identified quercetin-binding proteins are listed in Table 1. B, both unbound and bound proteins were separated by SDS-PAGE on 10% gels and immunoblotted with antibodies against vinculin, nucleolin, EF-1α, and hnRNPA1. C and E, recombinant full-length hnRNPA1 (aa 1–320) protein and the C-terminal region of hnRNPA1 (aa 268–320) were individually covalently coupled to a Biacore CM5 chip following the manufacturer's instructions. Serial dilutions of quercetin (final concentrations, 10 to 0.15 μm) were perfused over the immobilized proteins to allow association to occur, and dissociation was then monitored. Solvent correction was performed by subtracting vehicle-alone signals and those of quercetin perfused over uncoupled dextran matrix. D and F, saturation curve fitting of quercetin-full-length hnRNPA1 and quercetin-C-terminal region hnRNPA1 (aa 268–320) was plotted using GraphPad Prism software. The Kd values were calculated to be about 8.9 μm for full-length hnRNPA1 and about 1.7 μm for the C-terminal region of hnRNPA1. QCT, quercetin. RU, response units. Error bars, S.E.
TABLE 1.
Identification of quercetin-binding proteins by LC-MS/MS
An individual ion score of >43 indicates identity or extensive homology (p < 0.05). The protein score is derived from ion scores as a non-probabilistic basis for ranking protein hits.
| Band no. | Protein name | Protein mass/pI | NCBI accession number | Protein score | Matched peptides | Protein coverage | Description/Physiological function |
|---|---|---|---|---|---|---|---|
| Da/pI | % | ||||||
| 1 | Vinculin isoform VCL | 116,649/5.83 | gi|4507877 | 327 | 8 | 25% | Cytoskeleton. A membrane-cytoskeletal protein involved in linking integrin adhesion molecules to the actin cytoskeleton. |
| 2 | Nucleolin | 76,298/4.59 | gi|189306 | 259 | 3 | 7% | Protein biosynthesis. Organization of nucleolar chromatin, packaging of pre-RNA, rDNA transcription, and ribosome assembly. |
| Elongation factor-1α | 50,109/9.10 | gi|4503471 | 155 | 2 | 5% | Protein biosynthesis. An important GTP-binding protein that utilizes GTP to mediate the elongation step in protein synthesis by directing the aminoacyl-tRNA to ribosomes. | |
| 3 | Heterogeneous nuclear ribonucleoprotein A1 | 38,723/9.17 | gi|133254 | 167 | 10 | 29% | Protein biosynthesis. mRNA-binding protein regulating IRES-dependent translation. |
Kinetic Analysis of Quercetin-hnRNPA1 Interaction
To examine a possible direct interaction between hnRNPA1 and quercetin, we generated recombinant, full-length hnRNPA1 and expressed and purified it for analysis of quercetin binding by surface plasmon resonance. Fig. 3C shows the association and dissociation binding profile of full-length hnRNPA1 following titration of different concentrations of quercetin. To further identify the region of hnRNPA1 required for interaction with quercetin, we produced a series of different truncated versions of hnRNPA1 and evaluated their ability to interact with quercetin. The C-terminal region of hnRNPA1 was capable of binding to quercetin in a dose-dependent manner (Fig. 3E); the N-terminal RNA binding domain bound nonspecifically (data not shown); and the middle region of hnRNPA1 was unable to bind quercetin (data not shown). Saturation curves were further generated by plotting specific binding versus quercetin concentration. The Kd value of quercetin for binding to full-length hnRNPA1 was calculated to be about 8.9 μm, and that for the C-terminal region of hnRNPA1 was about 1.7 μm (Fig. 3, D and F). The Kd values obtained were similar to those obtained with in vitro radioactivity binding assays using 3H-labeled quercetin (data not shown). Thus, the C-terminal region of hnRNPA1 is required for interaction with quercetin.
Cytoplasmic Accumulation of hnRNPA1 and Reduced Tnpo1 Binding after Quercetin Treatment
hnRNPA1 is known to predominantly locate to the nucleus, where it mediates splicing of pre-mRNAs and transport of mRNAs from the nucleus to the cytoplasm (15). As shown in Fig. 4A, treatment with 100 μm quercetin for 24 h induced primarily a cytoplasmic pattern of hnRNPA1 accumulation. Previous studies have indicated that, during stresses, hnRNPA1 is phosphorylated and retained in the cytoplasm due to phosphorylation within the F-peptide adjacent to the nuclear localization signal region of the M9 domain, thereby hindering Tnpo1 binding to hnRNPA1 and tipping the balance toward apoptosis (16). To confirm that the change in localization was caused by impaired binding of Tnpo1 protein, we performed co-IP experiments. The results demonstrated a time-dependent reduction in Tnpo1 binding to hnRNPA1 after quercetin treatment (Fig. 4B).
FIGURE 4.
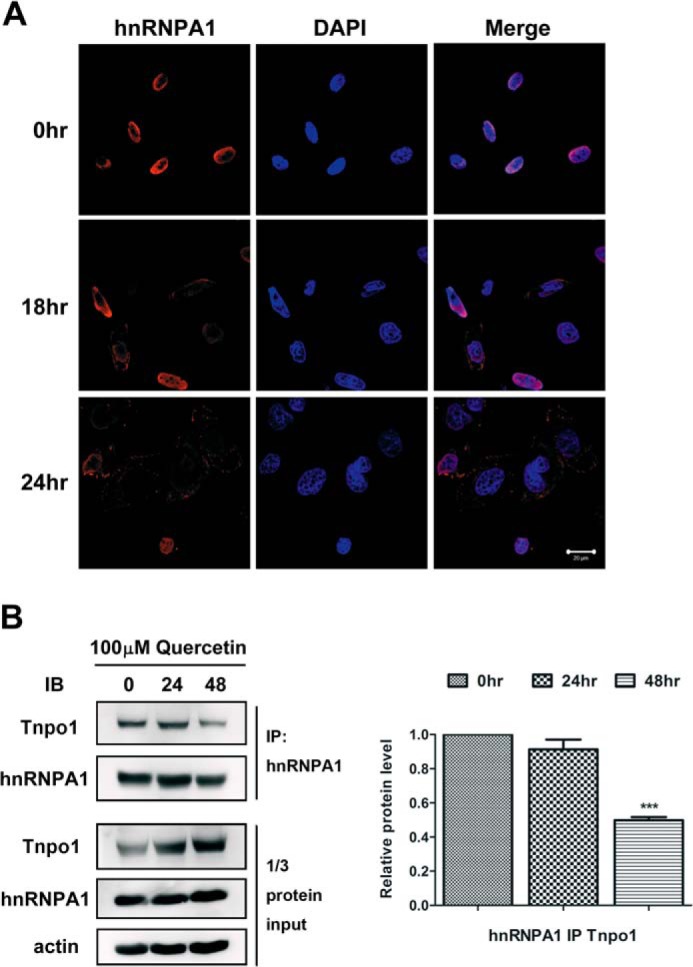
Cytoplasmic accumulation of hnRNPA1 and reduced Tnpo1 binding after quercetin treatment. A, PC-3 cells were treated with 100 μm quercetin for 18 and 24 h. Immunofluorescence analyses showed the localization of hnRNPA1 (red), and DAPI was used to stain nuclei. Merge, merged images. Scale bar, 20 μm. B, left, PC-3 cells were treated with 100 μm quercetin for 24 and 48 h. After treatment, cell lysates were immunoprecipitated with anti-hnRNPA1 antibodies, and proteins in immunoprecipitates were resolved by SDS-PAGE on 10% gels and probed with anti-Tnpo1 and anti-hnRNPA1 antibodies. In the bottom panel, one-third of the total protein lysate was loaded. Right, quantification of three independent immunoblotting analyses. The intensity of Tnpo1 was quantified by densitometry and normalized to actin. ***, p < 0.001 with respect to control. IB, immunoblot. Error bars, S.E.
The Mechanism Underlying the Quercetin-induced Change in hnRNPA1 Localization in PC-3 Cells Is Independent of F-peptide Phosphorylation
We next sought to determine whether the reduction in Tnpo1 binding induced by quercetin treatment was caused by phosphorylation within the F-peptide of hnRNPA1. To this end, we collected the cytoplasmic fraction of PC-3 cells after treatment with DMSO or 100 μm quercetin and determined the phosphorylation status of F-peptide by LC-MS/MS. However, these experiments revealed no change in the phosphorylation status of the F-peptide following quercetin treatment (Fig. 5A). To further verify that cytoplasmic relocalization was not due to F-peptide phosphorylation, we constructed F2mt-hnRNPA1, replacing four serine residues of the F-peptide with alanines (Fig. 5B). We again performed quercetin affinity chromatography, utilizing lysates from cells transfected with WT hnRNPA1 or F2mt-hnRNPA1 to determine whether F2mt-hnRNPA1 retained specific interactions with quercetin. The results demonstrated that modification of phosphorylation sites did not alter the specific binding of quercetin and hnRNPA1 (Fig. 5C). We also transiently co-transfected PC-3 cells with FLAG-tagged WT hnRNPA1 or GFP-tagged F2mt-hnRNPA1 and exposed cells to osmotic stress or quercetin treatment. As expected, cells expressing WT hnRNPA1 showed cytoplasmic accumulation following treatment with quercetin or exposure to osmotic stress. Surprisingly, although the F2mt-hnRNPA1 lacking F-peptide phosphorylation sites exhibited a change in localization from nucleus to cytoplasm in response to quercetin treatment, it was restricted to the nucleus following exposure to osmotic stress (Fig. 5D). Thus, quercetin binding specificity was retained for F2mt-hnRNPA1 (Fig. 5C), which, like WT hnRNPA1, became mislocalized to the cytoplasm following quercetin treatment (Fig. 5D). These results demonstrate that the change in hnRNPA1 localization following treatment with quercetin is mediated by a mechanism distinct from phosphorylation of the F-peptide of hnRNPA1.
FIGURE 5.

Localization of WT hnRNPA1 and F2mt-hnRNPA1 in transiently transfected PC-3 cells following quercetin treatment. A, mass spectra of F-peptides of cytoplasmic hnRNPA1 treated with vehicle (data not shown) or 100 μm quercetin for 24 h. B, schematic representation of the FLAG WT hnRNPA1 and FLAG F2mt-hnRNPA1 used in this work. The mutations of serines (positions 309–312) to alanines in the F-peptide are shown. C, PC-3 cells were transfected with FLAG WT hnRNPA1 or FLAG F2mt-hnRNPA1, and pull-down fractions were separated by SDS-PAGE, immunoblotted, and probed with anti-FLAG antibody. D, PC-3 cells co-transfected with FLAG WT hnRNPA1 or GFP-F2mt-hnRNPA1 were treated with quercetin for 24 h and then immunostained with antibodies against FLAG (red) and imaged. Nuclei were visualized by counterstaining with DAPI (blue). Merge, merged images. Scale bar, 20 μm. QCT, quercetin; OSM, osmotic stress; T, total cell lysates; W, proteins that did not bind quercetin; E, bound proteins eluted. Error bars, S.E.
Cytoplasmic Accumulation of hnRNPA1 after Quercetin Treatment Results in Greater IRES-dependent mRNA Binding but Diminished Protein Expression
It is known that cytoplasmic accumulation of hnRNPA1 leads to a reduction in IRES (internal ribosome entry site)-mediated translation of antiapoptotic mRNAs (16). To gain a better understanding of hnRNPA1 function following quercetin treatment, we used RIP RT-qPCR to isolate and identify mRNAs that interact with hnRNPA1. As shown in Fig. 6A, a greater amount of cIAP1 mRNA was retained in the cytoplasm after treatment with quercetin for 24 and 48 h compared with controls. A significantly greater enrichment of cIAP1 mRNA was observed in the hnRNPA1 RIP experiment (Fig. 6B), suggesting that hnRNPA1 modulates the stability of these mRNAs and prevents their degradation in the context of quercetin treatment. To determine the protein expression pattern of hnRNPA1 and cIAP1, we performed subcellular fractionation and immunoblotting accordingly. We observed that cytoplasmic relocation of hnRNPA1 triggered by quercetin causes a significant reduction in cIAP1 protein level (Fig. 6C). On the basis of these findings, we further performed a knockdown experiment to verify whether cIAP1 protein expression is regulated via hnRNPA1 in response to quercetin treatment. We observed that exposure of cells to quercetin reduced cIAP1 expression, and this reduction in cIAP1 protein levels can be abrogated by transient knockdown of hnRNPA1, indicating that the reduction of cIAP1 expression in response to quercetin is dependent on hnRNPA1 (Fig. 6D). Moreover, to confirm that quercetin-induced apoptosis was regulated through hnRNPA1 and cIAP1, we evaluated the apoptotic activity of cells treated with quercetin and cells transfected with shRNA targeting hnRNPA1, cIAP1, or a combination of them. As shown in Fig. 6E, individual knockdown of hnRNPA1 or cIAP1 had moderate effects on apoptosis, and the combination knockdown of cIAP1 and hnRNPA1 resulted in stronger induction of apoptosis, similar to quercetin treatment. In addition, we compared the cell viability of the shRNA hnRNPA1- or shRNA cIAP1-transfected cells relative to the respective controls; a reduced viability of the knockdown cells was cohesively close to quercetin treatment on controls. More importantly, when the hnRNPA1- and cIAP1- knockdown cells were treated with quercetin, a lesser reduction (13 and 14% respectively) than corresponding controls (39 and 35%) was found, indicating that the quercetin-induced cell death was partially diminished when hnRNPA1 or cIAP1 was eliminated (Fig. 6F). From the results above, we conclude hnRNPA1 and cIAP1 play important roles in regulating quercetin-induced cell death of PC-3 cells. Collectively, these results indicate that quercetin treatment increases mRNA-hnRNPA1 binding levels but decreases protein expression, assuming that mRNA·hnRNPA1 complexes are stored in temporary vesicles instead of executing translation functions at ribosomes. Thus, an immunofluorescence study was subsequently carried out. As shown in Fig. 6G, cytoplasmic hnRNPA1 and PABP, a protein marker of SGs, were appreciably co-localized in SGs, but not in P-bodies (processing bodies; data not shown), indicating that translation functions had stalled. IAPs (inhibitors of apoptosis protein) are a family of caspase inhibitors that specifically bind to caspase-3, -7, and -9 and thereby inhibit apoptosis. It has been reported that a reduction in the level of cIAP1 prompts death in susceptible cells (17–19). In this study, we found that less cIAP1 mRNA was released from hnRNPA1 (Fig. 6, A and B), and mRNA·hnRNPA1 complexes were present in SGs after quercetin treatment, suggesting that quercetin inhibits IRES-mediated antiapoptotic protein translation by trapping hnRNPA1 in the cytoplasm, ultimately leading to cell death through a reduction in cIAP1 levels as one of the mechanisms.
FIGURE 6.

Quercetin induces hnRNPA1 localization to SGs and reduces cIAP1 protein expression level. A, the mRNA level of cIAP1 in cytosolic fractions after treatment with 100 μm quercetin was analyzed. by RT-qPCR. B, cytoplasmic mRNA obtained from cytosolic extracts of cell lysates was immunoprecipitated with hnRNPA1 antibodies, purified, and quantified by RT-qPCR. C, PC-3 cells were treated with 100 μm quercetin for 24 and 48 h. Subcellular fractionation and lysates were separated by SDS-PAGE and analyzed by immunoblotting using the indicated antibodies. Respective cytoplasmic and nuclear fractions were loaded and analyzed by immunoblotting as indicated. α-Tubulin and lamin were used as cytosol and nucleus markers, respectively. Quantification of three independent analyses of cIAP1 protein is shown below the blot. The intensity was quantified by densitometry and normalized to that of actin. D, PC-3 cells transfected with shRNAs targeting hnRNPA1 and a non-targeting shRNA (shCtrl) were incubated with quercetin for 48 h. The cells were lysed and analyzed by immunoblotting accordingly with the indicated antibodies. E, PC-3 cells transfected with indicated shRNAs were analyzed. Cells were incubated with vehicle or quercetin (PC-3 control cells and shCtrl) for 48 h. The cells were stained with annexin V and caspase-3/7 together with 7-aminoactinomycin D for staining apoptotic cells and dead cells, respectively. F, PC-3 cells transfected with shRNAs targeting hnRNPA1, cIAP1, and shCtrl were incubated with quercetin for 48 h. Cell viability was analyzed by quantifying intracellular ATP level using a CellTiter-Glo kit. G, cells were fixed and double immunostained with antibodies against hnRNPA1 (green) or PABP (red). Nuclei were visualized by counterstaining with DAPI (blue). Merge, merged images. Scale bar, 20 μm. Arrows, formation of SGs. *, p < 0.05; **, p < 0.01 with respect to control. Error bars, S.E.
DISCUSSION
hnRNPA1 normally carries mRNAs from the nucleus to the cytosol and then rapidly shuttles back to the nucleus upon Tnpo1 binding (Fig. 7A). In this study, we demonstrated that this cycle is disrupted by treatment of cells with quercetin, resulting in cytoplasmic accumulation of hnRNPA1 and subsequent recruitment to SGs, ultimately putting cells on the path to apoptosis (Fig. 7B). This is the first study to identify hnRNPA1 as a direct target of quercetin and to reveal that the anti-cancer effects of quercetin in prostate cells are mediated, at least in part, by a loss of hnRNPA1 functions.
FIGURE 7.
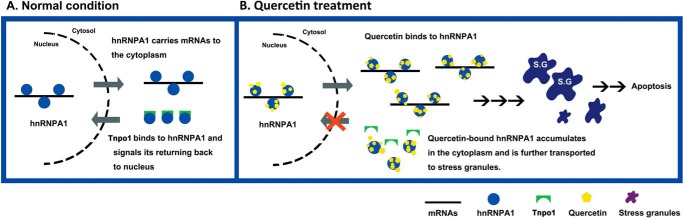
Model of quercetin binds to hnRNPA1 and induces apoptotic mechanism in PC-3 cells. A, in normal conditions, hnRNPA1 is released from mRNAs in the cytoplasm through binding Tnpo1 and returns back to the nucleus. B, upon quercetin treatment, quercetin binds to hnRNPA1 and hampers its returning back to the nucleus, resulting in accumulation of hnRNPA1 in the cytoplasm.
Among the proteins identified in this study was EF-1α, which is a documented quercetin binding partner. It has previously been shown that quercetin binding to EF-1α blocks formation of the EF-1α·GTP·tRNA ternary complex and further inhibits the ability of the complex to bind to ribosomes (14). Furthermore, overexpression of EF-1α is correlated with increased cell proliferation and oncogenic transformation in breast cancers (20) and is positively associated with prostate cancer progression (21). In addition to EF-1α, we identified three novel interacting proteins that collectively explain the cell growth-inhibitory impact of quercetin on PC-3 cells. Vinculin acts as a membrane-cytoskeletal protein that associates with focal adhesion plagues. A strong association between vinculin gene amplification and advanced prostate cancers has been documented, and knockdown of vinculin expression has been shown to reduce the growth of PC-3 cells, demonstrating that vinculin is a tumor-promoting protein (22). It is also worth noting that nucleolin, which has been implicated in ribosome assembly and nucleocytoplasmic transport, was identified in our study as a quercetin-interacting protein. Nucleolin has an RNA binding domain in the central region of its structure and has been reported to stabilize Bcl-2 mRNA in an AU-rich element-dependent manner in HL-60 cells (23, 24). In addition, nucleolin expressed at the cell surface binds a variety of ligands that have been implicated in tumorigenesis and angiogenesis (25, 26).
In this study, we mainly focused on hnRNPA1. The hnRNP family comprises more than 20 members whose primary functions are nascent transcript packaging, alternative splicing, and translational regulation. hnRNPs generally share the common property of nucleocytoplasmic shuttling to carry spliced mRNA out of the nucleus (15). In addition to these functions, hnRNPA1 has a regulatory role in IRES-dependent translation, modulating several proteins involved in the cell cycle, apoptosis, and differentiation. In a previous study (16), it was shown that hnRNPA1 represses translation of Bcl-xL mRNA during hypertonic stress in S/S cells. Osmotic stress was also shown to cause hnRNPA1 to accumulate in the cytoplasm, thus inhibiting IRES-mediated translation of XIAP mRNA (27). We found quercetin mediated cytoplasmic accumulation of hnRNPA1, resulting in decreased cIAP1 protein levels. This diminishment in cIAP1 levels can be reversed by knockdown of hnRNPA1, confirming that the suppression of cIAP1 expression following quercetin is dependent on hnRNPA1. We suggest that hnRNPA1 triggers apoptosis by inhibiting IRES-mediated translation of a group of antiapoptotic mRNAs. The importance of hnRNPA1 in tumorigenesis was first revealed by the discovery that hnRNPA1/A2 expression levels were increased in a variety of human cancers relative to that in normal tissues (28). Accumulating recent evidence indicates a significant role for hnRNPA1 as a biomarker of many types of cancers, including prostate cancer, colorectal cancer, breast cancer, liver cancer, and others (29, 30).
It has been widely reported that hnRNPA1 undergoes subcellular relocalization through phosphorylation within its F-peptide under stress conditions and accumulates in cytoplasmic granules, which have evolved in mammalian cells to facilitate cellular recovery from environmental stresses (31). Of particular interest, we found that hnRNPA1 was not phosphorylated at F-peptide sites in the context of quercetin treatment but rather bound via its C-terminal region to quercetin and proceeded toward SGs. Quercetin is known to inhibit nucleotide-dependent kinases through competition with ATP and GTP. The single ring backbone of quercetin is considered a critical structure for accessing the kinase binding site, which is positioned at or near the ATP fold of protein kinases (32). The structure of PI3K and quercetin revealed that quercetin binding sites are partially overlapping and coplanar with the space of the adenine moiety of ATP (33). Given this, we proposed that the interaction between quercetin and the F-peptide is analogous to ATP binding at the F-peptide and interferes with Tnpo1 interaction, resulting in cytoplasmic accumulation of hnRNPA1. However, structural studies will be required to provide definitive insight into these interactions.
Drug target identification and mechanism-of-action studies have important roles in drug discovery efforts. Many bioactive compounds have been found to be more promiscuous than originally anticipated, exerting desirable drug efficacy and undesirable toxic responses in parallel (8, 32). The identification of protein-binding profiles of an active compound will provide invaluable information for deciphering both beneficial and cytotoxic interactions of drugs. In contrast to traditional global protein profiling, chemical proteomics is a useful, affordable tool for conducting drug interacting profiles depending on the purpose of the study; relative protein quantification is also achievable by combining this approach with quantitative MS (34, 35). This approach reduces the time needed to explore a drug's targets and is usually used as an initial screening platform to prioritize a list of potential protein targets for verification. Our results demonstrated that hnRNPA1 is a promising therapeutic target in prostate cancer, and quercetin can be used as a chemical scaffold for further structural improvements designed to enhance its potency and selectivity. It is anticipated that chemical proteomics-based drug development will identify direct targets of drugs and serve as a starting point for delineating signaling networks and facilitating downstream structure modifications to eliminate off-target impacts in the clinic.
Acknowledgment
shRNA reagents were obtained from the National RNAi Core Facility Platform located at the Institute of Molecular Biology/Genomic Research Center, Academia Sinica.
This study was supported by National Science Council Grant NSC 101-2325-B-002-065, the Liver Disease Prevention and Treatment Research Foundation, and the Ministry of Education, Taiwan.
- hnRNP
- heterogeneous nuclear ribonucleoprotein
- IRES
- internal ribosome entry site
- cIAP1
- cellular inhibitor of apoptosis protein-1
- PABP
- poly(A)-binding protein
- Tnpo1
- transportin 1
- SGs
- stress granules
- IP
- immunoprecipitation
- aa
- amino acids
- ShCtrl
- control shRNA
- RIP
- RNA-binding protein immunoprecipitation
- RT-qPCR
- quantitative RT-PCR.
REFERENCES
- 1. Gupta S. C., Kim J. H., Prasad S., Aggarwal B. B. (2010) Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev. 29, 405–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murphy A. B., Macejko A., Taylor A., Nadler R. B. (2009) Chronic prostatitis: management strategies. Drugs 69, 71–84 [DOI] [PubMed] [Google Scholar]
- 3. Vijayababu M. R., Kanagaraj P., Arunkumar A., Ilangovan R., Aruldhas M. M., Arunakaran J. (2005) Quercetin-induced growth inhibition and cell death in prostatic carcinoma cells (PC-3) are associated with increase in p21 and hypophosphorylated retinoblastoma proteins expression. J. Cancer Res. Clin. Oncol. 131, 765–771 [DOI] [PubMed] [Google Scholar]
- 4. Kumar R., Verma V., Jain A., Jain R. K., Maikhuri J. P., Gupta G. (2011) Synergistic chemoprotective mechanisms of dietary phytoestrogens in a select combination against prostate cancer. J. Nutr. Biochem. 22, 723–731 [DOI] [PubMed] [Google Scholar]
- 5. Lamson D. W., Brignall M. S. (2000) Antioxidants and cancer, part 3: quercetin. Altern. Med. Rev. 5, 196–208 [PubMed] [Google Scholar]
- 6. Lee K. W., Kang N. J., Heo Y. S., Rogozin E. A., Pugliese A., Hwang M. K., Bowden G. T., Bode A. M., Lee H. J., Dong Z. (2008) Raf and MEK protein kinases are direct molecular targets for the chemopreventive effect of quercetin, a major flavonol in red wine. Cancer Res. 68, 946–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee K. M., Hwang M. K., Lee D. E., Lee K. W., Lee H. J. (2010) Protective effect of quercetin against arsenite-induced COX-2 expression by targeting PI3K in rat liver epithelial cells. J. Agric. Food Chem. 58, 5815–5820 [DOI] [PubMed] [Google Scholar]
- 8. Wang R. E., Hunt C. R., Chen J., Taylor J. S. (2011) Biotinylated quercetin as an intrinsic photoaffinity proteomics probe for the identification of quercetin target proteins. Bioorg. Med. Chem. 19, 4710–4720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Duncan J. S., Turowec J. P., Duncan K. E., Vilk G., Wu C., Lüscher B., Li S. S., Gloor G. B., Litchfield D. W. (2011) A peptide-based target screen implicates the protein kinase CK2 in the global regulation of caspase signaling. Sci. Signal. 4, ra30. [DOI] [PubMed] [Google Scholar]
- 10. Piersma S. R., Labots M., Verheul H. M., Jiménez C. R. (2010) Strategies for kinome profiling in cancer and potential clinical applications: chemical proteomics and array-based methods. Anal. Bioanal. Chem. 397, 3163–3171 [DOI] [PubMed] [Google Scholar]
- 11. Ermakova S., Choi B. Y., Choi H. S., Kang B. S., Bode A. M., Dong Z. (2005) The intermediate filament protein vimentin is a new target for epigallocatechin gallate. J. Biol. Chem. 280, 16882–16890 [DOI] [PubMed] [Google Scholar]
- 12. Fleischer T. C., Murphy B. R., Flick J. S., Terry-Lorenzo R. T., Gao Z. H., Davis T., McKinnon R., Ostanin K., Willardsen J. A., Boniface J. J. (2010) Chemical proteomics identifies Nampt as the target of CB30865, an orphan cytotoxic compound. Chem. Biol. 17, 659–664 [DOI] [PubMed] [Google Scholar]
- 13. Curtis Nickel J., Shoskes D., Roehrborn C. G., Moyad M. (2008) Nutraceuticals in prostate disease: the urologist's role. Rev. Urol. 10, 192–206 [PMC free article] [PubMed] [Google Scholar]
- 14. Marcinkiewicz C., Gałasiński W., Gindzieński A. (1995) EF-1 α is a target site for an inhibitory effect of quercetin in the peptide elongation process. Acta Biochim. Pol. 42, 347–350 [PubMed] [Google Scholar]
- 15. Han S. P., Tang Y. H., Smith R. (2010) Functional diversity of the hnRNPs: past, present and perspectives. Biochem. J. 430, 379–392 [DOI] [PubMed] [Google Scholar]
- 16. Bevilacqua E., Wang X., Majumder M., Gaccioli F., Yuan C. L., Wang C., Zhu X., Jordan L. E., Scheuner D., Kaufman R. J., Koromilas A. E., Snider M. D., Holcik M., Hatzoglou M. (2010) eIF2α phosphorylation tips the balance to apoptosis during osmotic stress. J. Biol. Chem. 285, 17098–17111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schimmer A. D. (2004) Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res. 64, 7183–7190 [DOI] [PubMed] [Google Scholar]
- 18. Gill C., Dowling C., O'Neill A. J., Watson R. W. (2009) Effects of cIAP-1, cIAP-2 and XIAP triple knockdown on prostate cancer cell susceptibility to apoptosis, cell survival and proliferation. Mol. Cancer 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qi Y., Xia P. (2012) Cellular inhibitor of apoptosis protein-1 (cIAP1) plays a critical role in beta-cell survival under endoplasmic reticulum stress: promoting ubiquitination and degradation of C/EBP homologous protein (CHOP). J. Biol. Chem. 287, 32236–32245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Al-Maghrebi M., Anim J. T., Olalu A. A. (2005) Up-regulation of eukaryotic elongation factor-1 subunits in breast carcinoma. Anticancer Res. 25, 2573–2577 [PubMed] [Google Scholar]
- 21. Liu H., Ding J., Chen F., Fan B., Gao N., Yang Z., Qi L. (2010) Increased expression of elongation factor-1α is significantly correlated with poor prognosis of human prostate cancer. Scand. J. Urol. Nephrol. 44, 277–283 [DOI] [PubMed] [Google Scholar]
- 22. Ruiz C., Holz D. R., Oeggerli M., Schneider S., Gonzales I. M., Kiefer J. M., Zellweger T., Bachmann A., Koivisto P. A., Helin H. J., Mousses S., Barrett M. T., Azorsa D. O., Bubendorf L. (2011) Amplification and overexpression of vinculin are associated with increased tumour cell proliferation and progression in advanced prostate cancer. J. Pathol. 223, 543–552 [DOI] [PubMed] [Google Scholar]
- 23. Ginisty H., Sicard H., Roger B., Bouvet P. (1999) Structure and functions of nucleolin. J. Cell Sci. 112, 761–772 [DOI] [PubMed] [Google Scholar]
- 24. Sengupta T. K., Bandyopadhyay S., Fernandes D. J., Spicer E. K. (2004) Identification of nucleolin as an AU-rich element binding protein involved in bcl-2 mRNA stabilization. J. Biol. Chem. 279, 10855–10863 [DOI] [PubMed] [Google Scholar]
- 25. Hovanessian A. G., Soundaramourty C., El Khoury D., Nondier I., Svab J., Krust B. (2010) Surface expressed nucleolin is constantly induced in tumor cells to mediate calcium-dependent ligand internalization. PLoS One 5, e15787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Christian S., Pilch J., Akerman M. E., Porkka K., Laakkonen P., Ruoslahti E. (2003) Nucleolin expressed at the cell surface is a marker of endothelial cells in angiogenic blood vessels. J. Cell Biol. 163, 871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lewis S. M., Veyrier A., Hosszu Ungureanu N., Bonnal S., Vagner S., Holcik M. (2007) Subcellular relocalization of a trans-acting factor regulates XIAP IRES-dependent translation. Mol. Biol. Cell 18, 1302–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patry C., Bouchard L., Labrecque P., Gendron D., Lemieux B., Toutant J., Lapointe E., Wellinger R., Chabot B. (2003) Small interfering RNA-mediated reduction in heterogeneous nuclear ribonucleoparticule A1/A2 proteins induces apoptosis in human cancer cells but not in normal mortal cell lines. Cancer Res. 63, 7679–7688 [PubMed] [Google Scholar]
- 29. Pino I., Pío R., Toledo G., Zabalegui N., Vicent S., Rey N., Lozano M. D., Torre W., García-Foncillas J., Montuenga L. M. (2003) Altered patterns of expression of members of the heterogeneous nuclear ribonucleoprotein (hnRNP) family in lung cancer. Lung Cancer 41, 131–143 [DOI] [PubMed] [Google Scholar]
- 30. Ma Y. L., Peng J. Y., Zhang P., Huang L., Liu W. J., Shen T. Y., Chen H. Q., Zhou Y. K., Zhang M., Chu Z. X., Qin H. L. (2009) Heterogeneous nuclear ribonucleoprotein A1 is identified as a potential biomarker for colorectal cancer based on differential proteomics technology. J. Proteome Res. 8, 4525–4535 [DOI] [PubMed] [Google Scholar]
- 31. Guil S., Long J. C., Cáceres J. F. (2006) hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell Biol. 26, 5744–5758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Russo M., Spagnuolo C., Tedesco I., Bilotto S., Russo G. L. (2012) The flavonoid quercetin in disease prevention and therapy: facts and fancies. Biochem. Pharmacol. 83, 6–15 [DOI] [PubMed] [Google Scholar]
- 33. Walker E. H., Pacold M. E., Perisic O., Stephens L., Hawkins P. T., Wymann M. P., Williams R. L. (2000) Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 6, 909–919 [DOI] [PubMed] [Google Scholar]
- 34. Schirle M., Bantscheff M., Kuster B. (2012) Mass spectrometry-based proteomics in preclinical drug discovery. Chem. Biol. 19, 72–84 [DOI] [PubMed] [Google Scholar]
- 35. Bantscheff M., Drewes G. (2012) Chemoproteomic approaches to drug target identification and drug profiling. Bioorg. Med. Chem. 20, 1973–1978 [DOI] [PubMed] [Google Scholar]