

Background: Interleukin (IL)-6 trans-signaling is inhibited by naturally occurring soluble gp130 (sgp130) variants.
Results: Alternative intronic polyadenylation in the gp130 gene creates the novel natural IL-6 trans-signaling inhibitor sgp130-E10.
Conclusion: Alternative mRNA polyadenylation contributes to regulation of IL-6 trans-signaling intensity.
Significance: This is the first report on a soluble isoform of the key cytokine co-receptor gp130 originating from alternative polyadenylation.
Keywords: Alternative Splicing, Interleukin, Polyadenylation, Signal Transduction, Protein-tyrosine Kinase (Tyrosine Kinase)
Abstract
Interleukin (IL)-6 signals via a receptor complex composed of the signal-transducing β-receptor gp130 and the non-signaling membrane-bound or soluble IL-6 receptor α (IL-6R, sIL-6R), which is referred to as classic and trans-signaling, respectively. IL-6 trans-signaling is functionally associated with the development of chronic inflammatory diseases and cancer. Soluble gp130 (sgp130) variants are natural inhibitors of trans-signaling. Differential splicing yields sgp130 isoforms. Here, we describe that alternative intronic polyadenylation in intron 10 of the gp130 transcript results in a novel mRNA coding for an sgp130 protein isoform (sgp130-E10) of 70–80 kDa. The sgp130-E10 protein was expressed in vivo in human peripheral blood mononuclear cells. To assess the biological activity of sgp130-E10, we expressed this variant as Fc-tagged fusion protein (sgp130-E10Fc). Recombinant sgp130-E10Fc binds to a complex of IL-6 and sIL-6R, but not to IL-6 alone, and specifically inhibits IL-6 trans-signaling. Thus, it might play an important role in the regulation of trans-signaling in vivo.
Introduction
The signaling complex of interleukin (IL)-6 is composed of the signal-transducing β-receptor gp130 3 and the non-signaling membrane-bound or soluble IL-6 receptor α (IL-6R, sIL-6R), referred to as classic and trans-signaling, respectively. IL-6 binds in a first step to the IL-6R, and this IL-6/sIL-6R complex recruits two gp130 molecules with high affinity, which initiates intracellular signal transduction. Soluble isoforms of gp130 (sgp130) compete with the membrane-bound gp130 for the binding to the complex of IL-6/sIL-6R, thereby specifically antagonizing IL-6 trans-signaling (1). Blocking IL-6 trans-signaling was shown to ameliorate chronic inflammatory diseases (2, 3). Levels of about 400 ng/ml sgp130 are found in normal human serum (4), but its cellular origin and the mechanism of its generation are still poorly understood. At least three soluble gp130 receptor isoforms of ∼110, 90, and 50 kDa have been detected in serum or urine (4–6). Only the protein corresponding to the differentially spliced mRNA of the smallest (50 kDa) isoform sgp130-RAPS was verified by Western blotting using specific antibodies directed against the novel C terminus of sgp130-RAPS (5, 7, 8). Two additional differentially spliced gp130 cDNAs were described, but have not been shown to be endogenously translated into sgp130 proteins (7, 8). The expected size of these variants would, if at all, correspond to the largest endogenous sgp130 variant of ∼110 kDa (4, 6). It is unknown whether sgp130 variants are also generated by ectodomain shedding of the transmembrane gp130 receptor.
Besides differential splicing, alternative intronic polyadenylation creates alternative protein isoforms (9). The use of an intronic polyadenylation site (PAS) leads to shortened mature mRNAs and thereby to truncated protein isoforms. If the C-terminal part of a full-length protein is important for its function, intronic polyadenylation might abolish or alter the function of the shortened protein isoform. Recently, soluble dominant-negative receptor tyrosine kinase isoforms were identified, which originated from alternative intronic polyadenylation (10).
In the present study, we have discovered a novel sgp130 mRNA, which results from alternative intronic polyadenylation in intron 10. The resulting isoform (sgp130-E10) consists of the four N-terminal domains of gp130 and ends in the loop region between domains 4 and 5. sgp130-E10 was found to be primarily expressed in peripheral blood mononuclear cells (PBMCs), resulting in a protein of 70–80 kDa. We expressed sgp130-E10 as Myc-His- or Fc-tagged variants and showed that sgp130-E10 specifically inhibits IL-6 trans-signaling, albeit with much lower efficiency than full-length sgp130.
EXPERIMENTAL PROCEDURES
Cells and Reagents
Hyper-IL-6 dependent Ba/F3-gp130 cells were created and selected as described previously (11). CHO-K1 cells and HEK-293 cells were purchased from the DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen; Braunschweig, Germany). Cells were cultured in standard DMEM growth medium consisting of DMEM high glucose medium (Life Technologies/Gibco; Darmstadt, Germany) supplemented with 10% FBS (Life Technologies/Gibco), penicillin (60 mg/liter, Life Technologies/Gibco), and streptomycin (100 mg/liter, Gibco) at 37 °C with 5% CO2 in a water-saturated atmosphere. For cultivation of Ba/F3-gp130 cells, the standard DMEM growth medium was additionally supplemented with 10 ng/ml Hyper-IL-6. Hyper-IL-6 is a fusion protein of IL-6 and the soluble IL-6 receptor that mimics IL-6 trans-signaling (12, 13). It was also used in an Fc-tagged form (Hyper-IL-6Fc). Hyper-IL-6 was expressed and purified as described previously (12). Antibodies against signal transducer and activator of transcription (STAT)-3 and its Tyr705-phosphorylated epitope were purchased from Cell Signaling Technology (Frankfurt am Main, Germany). The antibody directed against Myc tag was expressed and purified as described previously (14). The antibody directed against human Fc tag and secondary antibodies used for Western blot analyses were purchased from Pierce (Thermo Fisher Scientific). The recombinant protein sgp130Fc was expressed and purified as described previously (1, 15).
Generation of Hybridoma-producing Monoclonal gp130 E10 Antibodies
A peptide corresponding to amino acids 440–448 (CDFQGLYL) of the C-terminal region of sgp130-E10 was synthesized (Peptides&Elephants; Potsdam, Germany) and conjugated to BSA and KLH (Merck; Darmstadt, Germany) as carrier proteins as described previously (16) using sulfo-MBS (where MBS is m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester) as a cross-linker (Thermo Fisher Scientific/Perbio Science; Bonn, Germany). BALB/c mice were immunized intraperitoneally with 150 μg of peptide conjugated to KLH in 60 μl of phosphate-buffered saline (PBS; 137 mm NaCl, 2.7 mm KCl, 8.1 mm Na2HPO4, 1.5 mm KH2PO4) emulsified with 40 μl of GERBU adjuvant MM (GERBU Biotechnik; Heidelberg, Germany).
The mice were boosted intraperitoneally on days 14 and 21 with 100 μg of KLH-conjugated peptide emulsified with 20% of the adjuvant. The last two doses (100 μg) were performed on days 28 and 29 without adjuvant. The hybridoma fusion was performed on day 30; spleen cells from immunized animals were collected and fused with Ag8.653 myeloma cells using polyethylene glycol 1500 (Roche Diagnostics; Mannheim, Germany). The fused cells were cultured in selection medium (hypoxanthine-aminopterin-thymidine medium (HAT); Sigma-Aldrich; Taufkirchen, Germany) for 10 days. Hybridoma supernatants were screened on 96-well plates coated with the peptide of sgp130-E10 conjugated to BSA. Irrelevant peptide conjugated to BSA was used as a negative control.
Construction of Expression Plasmids
To create expression plasmids for sgp130-E10, we amplified the corresponding cDNA with specific primers from the plasmid pcDNA-DEST40_gp130-EYFP (17). The novel C terminus of sgp130-E10 was introduced via the reverse primer. The PCR product was phosphorylated with T4 polynucleotide kinase (Thermo Fisher Scientific Biosciences; St. Leon-Rot, Germany) and subcloned into HincII-digested pCR-Script vector. In the next step, the sgp130-E10 cDNA was subcloned via HindIII and NotI into a pcDNA3.1 vector (Life Technologies) containing either a Myc-His or an Fc tag.
Transfection and Selection of Stably Transfected CHO-K1 Cells
1 × 106 CHO-K1 cells were transiently transfected using TurboFectTM according to the manufacturer's instructions (Thermo Fisher Scientific Biosciences). The transfection efficiency was typically ∼70%, which was estimated by microscopic analysis (Keyence, BZ-9000) of green fluorescent protein expression 24 h after transfection. Transfected cells were cultured in standard DMEM growth medium; from 48 h after transfection onwards, transfected cells were selected in 1 mg/ml G-418 sulfate (Genaxxon Biosciences; Ulm, Germany) for at least 1 week. After 1 week of antibiotic selection, the remaining cells were detached with 0.05% (w/v) trypsin and 0.02% (w/v) EDTA in PBS and transferred to a new 10-cm cell culture dish. Cells were then cultured in standard DMEM growth medium with 1 mg/ml G-418 sulfate for another 4 days. Subsequently, cells were detached with trypsin/EDTA as described above and transferred to a 96-well plate at a theoretical concentration of 0.1 cells/well. After seeding, cells were cultured in standard DMEM growth medium with 1 mg/ml G-418 sulfate for another 9 days. To identify wells with single cell clones, all wells were initially and repeatedly checked by microscopy. Supernatants of each single-clone well were analyzed for sgp130-E10Fc or sgp130-E10Myc-His secretion by ELISA (DuoSet, DY228; R&D Systems; Wiesbaden, Germany). The cell clone with the highest concentration of sgp130-E10Fc or sgp130-E10Myc-His, respectively, in the supernatant was chosen for further cell expansion and protein production.
Proliferation Assay
Ba/F3-gp130 cells were washed three times with sterile PBS and suspended in standard DMEM growth medium without Hyper-IL-6 at 5 × 103 cells/well of a 96-well plate. The cells were cultured for 3 days in a final volume of 100 μl with additional 10 ng/ml Hyper-IL-6 and sgp130 variants as indicated under “Results.” The CellTiter-Blue cell viability assay (Promega; Mannheim, Germany) was used to determine the amount of viable cells following the manufacturer's instructions and measured on a Tecan Infinite 200pro fluorometer (excitation wavelength, 560 nm/9 nm; emission wavelength, 590 nm/20 nm; gain, 90; software, Tecan i-control, TECAN, Crailsheim, Germany). All values were measured in triplicate.
Coomassie Blue Staining
SDS-PAGE gels were incubated for 1 h in Coomassie Blue stain (water supplemented with 0.1% Coomassie R250, 10% acetic acid, and 40% methanol) under gentle agitation. Coomassie Blue stain was replaced by destaining solution (water supplemented with 20% methanol and 10% acetic acid) and kept under gentle agitation. Destaining solution was exchanged several times, until the background of the gel stain was minimized.
Western Blotting
For Western blotting, proteins separated by SDS-PAGE were transferred onto PVDF membranes (Roti®-PVDF; Carl Roth; Karlsruhe, Germany) by semidry electroblotting. Membranes were blocked in a solution of Tris-buffered saline (TBS; 10 mm Tris-HCl, pH 7.6 and 150 mm NaCl) supplemented with 0.05% Tween 20 and 5% (w/v) skimmed milk powder, and probed overnight with the indicated antibodies at 4 °C followed by incubation at room temperature for 1 h with horseradish peroxidase-conjugated secondary antibody (Thermo Fisher Scientific/Perbio Science). Immunoreactive proteins were detected using a chemiluminescence kit (Amersham BiosciencesTM ECLTM Prime Western blotting detection reagent; GE Healthcare; Freiburg, Germany) following the manufacturer's instructions.
Purification of Recombinant sgp130-E10Fc
2 × 106 cells of the selected CHO-K1 cell clone secreting sgp130-E10Fc into the cell culture supernatant were seeded into a 225-cm2 cell culture flask containing 110 ml of standard DMEM growth medium. After 6 days, the supernatant was harvested and centrifuged at 3,200 × g and 4 °C for 100 min. The resulting supernatant was filtered (bottle top filter, 0.22-μm pore diameter; Nalgene; Rochester, NY) and stored at −80 °C. Before chromatography, the supernatant was thawed, and its pH was adjusted to 6.6. The supernatant was loaded on a protein-A column (HiTrap protein A HP; GE Healthcare) at a flow rate of 3 ml/min. The column was washed with 30 column volumes of PBS. sgp130-E10Fc protein was eluted with a 3:7 mixture of 50 mm citric acid buffers adjusted to pH 5.5 and 3.25, respectively, at a flow rate of 1 ml/min. Fractions of 1 ml were collected. Fractions containing the protein peak were pooled, and the pH was adjusted to pH 7 with 1 m Tris. The protein was further purified by size exclusion chromatography (see below).
Purification of Recombinant sgp130-E10Myc-His
Cell culture supernatant containing sgp130-E10Myc-His was produced as described for sgp130-E10Fc. The pH of the supernatant was adjusted to 8, and the protein was purified by affinity chromatography using a HisTrapTM Excel column (GE Healthcare) according to the manufacturer's instructions. The protein was further purified by size exclusion chromatography (see below).
Size Exclusion Chromatography
The sgp130-E10Fc protein and the sgp130-E10Myc-His protein were further purified on a SuperdexTM 200 10/300 GL column (GE Healthcare) using PBS as the mobile phase with a constant flow rate of 1 ml/min. Fractions of 1 ml were collected and analyzed by SDS-PAGE or ELISA. sgp130-E10 protein-containing fractions were pooled, concentrated, and analyzed by SDS-PAGE, Western blotting, and ELISA.
Protein Concentration Measurements
The concentration of sgp130-E10 proteins was determined by ELISA (Human sgp130 DuoSet, R&D Systems) following the manufacturer's instructions.
Immunoprecipitation
Purified sgp130-E10Fc protein or conditioned HEK-293 cell medium containing sgp130Fc, sgp130-E10Fc, or sgp130-E10Myc-His after transient transfection was incubated with Hyper-IL-6- or Hyper-IL-6Fc-containing medium overnight at 4 °C followed by the addition of 50 μl of protein A-Sepharose (Pierce® protein A-Agarose, 50% slurry; Thermo Fisher Scientific) for at least 4 h at 4 °C. Immunoprecipitates were washed five times with washing buffer (20 mm Tris-HCl, pH 7.6, 150 mm NaCl, 0.5 mm EDTA, plus one tablet of cOmplete EDTA-free protease inhibitor (Roche Applied Science; Mannheim, Germany) in 50 ml of buffer). 100 μl of Laemmli sample buffer (5× Laemmli buffer stock solution: 500 mm Tris-HCl, pH 6.8, 2% (w/v) SDS, 2% (w/v) 2-mercaptoethanol, 20% (v/v) glycerol and 0.03% bromphenol blue) were added to the Sepharose and incubated at 95 °C for 10 min.
Binding Assay
Microtiter plates (F96 MaxiSorp NUNC-Immuno plate; Thermo Fisher Scientific) were coated with Hyper-IL-6 (5 μg/ml) in PBS or sgp130-E10Fc (5 μg/ml) in PBS overnight at room temperature. After blocking with 1% (w/v) BSA in PBS for 2 h, 50-μl aliquots of purified sgp130-E10Fc, sgp130-E10Myc-His, or sgp130Fc (50 pm to 100 nm) or IL-6 (0–1 μg/ml) were added. The plate was incubated for 1 h at room temperature. sgp130 variants bound to the plate were detected by the biotinylated detection antibody of the gp130 ELISA used for determination of gp130 concentration (see above) following the manufacturer's instructions. IL-6 bound to the plate was detected by the biotinylated IL-6 antibody of the human interleukin-6 ELISA kit (ImmunoTools; Friesoythe, Germany) following the manufacturer's instructions.
Competition Assay
Microtiter plates (F96 MaxiSorp NUNC-Immuno plate, Thermo Fisher Scientific) were coated with sgp130Fc (5 μg/ml) in PBS and incubated overnight at room temperature. After blocking with 1% (w/v) BSA in PBS for 2 h, 50-μl aliquots of Hyper-IL-6 (1 μg/ml) with purified sgp130-E10Fc or sgp130Fc (50 pm to 100 nm) were added. The plate was incubated for 1 h at room temperature. Hyper-IL-6 bound to the plate was detected by the biotinylated IL-6 antibody of the human interleukin-6 ELISA kit (ImmunoTools) following the manufacturer's instructions.
Stability Assay
For investigating the stability of sgp130-E10Fc in comparison with sgp130Fc, both proteins were added to standard DMEM growth medium consisting of DMEM high glucose medium supplemented with 10% FBS, penicillin (60 mg/liter), and streptomycin (100 mg/liter) with or without 10 ng/ml Hyper-IL-6 and incubated at 37 °C for 72 h in tubes. Every 24 h, a sample of 200 μl was taken and stored at −80 °C. Finally, all samples were analyzed by gp130 ELISA following the manufacturer's instructions (see above). The concentrations at 0 h were set to 100%, and the relative concentrations for later time points were calculated.
ELISA
To detect sgp130 serum levels, we used the human sgp130 DuoSet (R&D Systems) according to the manufacturer's instructions. To specifically detect sgp130-E10, we used E10/1 as capture antibody (final concentration 4 μg/ml in PBS) and performed all other steps according to the manufacturer's instructions for the human sgp130 DuoSet. Serum from healthy humans was obtained as described previously (18). Ethic approval for sgp130 determination in the serum of healthy volunteers for this study was obtained from the Institutional Review Board of the Heinrich-Heine-University.
Phospho-STAT3 Assay
For detection of phospho-STAT3, Ba/F3-gp130 cells were washed three times with sterile PBS and starved for 4 h in serum-free DMEM before adding 10 ng/ml Hyper-IL-6 and/or sgp130 variants for 15 min at 37 °C as indicated under “Results.” Hyper-IL-6 was preincubated with sgp130 variants for 30 min at 37 °C before adding to the cells. Subsequently, cells were centrifuged, and the cell pellet was directly heated in 100 μl of Laemmli sample buffer at 95 °C for 10 min followed by Western blotting as described above.
Sequence Alignment
Alignment of the intron 10 sequences of gp130 from different animal species was performed using the Clustal Omega software.
Isolation of PBMCs
For the isolation of RNA, PBMCs were isolated from total animal blood samples or from the buffy coat of nine healthy blood donors using LSM 1077 lymphocyte separation medium (PAA Laboratories; Cölbe, Germany) following the manufacturer's instructions. Cells were immediately frozen at −80 °C until RNA isolation.
For the detection of sgp130 protein forms in PBMCs, 35 ml of anti-coagulated blood were loaded onto 15 ml of Histopaque 1077 (Sigma-Aldrich). After centrifugation at 400 × g for 30 min at room temperature without braking, the layer containing the PBMCs was isolated and washed 4–5 times with PBS supplemented with 2 mm EDTA. For immunodetection of different forms of sgp130, 2.5 × 106 cells were lysed in lysis buffer (50 mm Tris/HCl, pH 7.5, 150 mm NaCl, 2 mm EDTA, 1% NP-40, 1% Triton X-100, cOmplete protease inhibitor mixture (Roche Applied Science)). After separation of proteins via 10% SDS-PAGE, sgp130 variants were detected using the B-R3 gp130 antibody (Abcam; Cambridge, UK) and the antibody E10/1 (see above; hybridoma supernatant diluted 1:200) for sgp130-E10 followed by mouse-POD (Thermo Fisher Scientific) as a secondary antibody. Protein bands were visualized by SuperSignal® West Femto maximum sensitivity substrate (Thermo Fisher Scientific).
RNA, cDNA, and RT-PCR
RNA was extracted from the cells using the peqGOLD total RNA kit (S-Line) (PEQLAB Biotechnologie; Erlangen, Germany) following the manufacturer's instructions, and 5 μg of RNA were used for cDNA synthesis using RevertAid reverse transcriptase (Thermo Fisher Scientific) following the manufacturer's instructions and using the oligo(dT) method. PCR reactions on cDNA templates were performed using DreamTaq DNA polymerase (Thermo Fisher Scientific) following the manufacturer's instructions and using adapted PCR protocols and primers. For amplifying sgp130-E10 cDNA from human, dog, and pig samples, we used primers annealing to exon 9 and intron 10. As a control, primers annealing to exon 9 and 11 were used to amplify all gp130 transcripts including also any sgp130 transcripts except for sgp130-RAPS and sgp130-E10. For mouse cDNA, primers annealing to exon 8 and intron 10 were used to amplify sgp130-E10, and primers annealing to exon 8 and exon 10 were used to amplify total gp130 as a control.
Oligonucleotides
Primers for the human sgp130-E10 isoform (5′-TACAGAACTGTACAACTCGTGTGG-3′ and (5′-CAAGAGTCAGGCTCCATCTCACAG-3′) and all human gp130 transcripts except for sgp130-RAPS (5′-TACAGAACTGTACAACTCGTGTGG-3′ and 5′-CTGGTCCATCAGCATATACTGGAG-3′) were used in PCR analysis. The sequences of the oligonucleotides used for amplification of gp130 transcripts of other mammals will be provided upon request.
Quantitative RT-PCR (qPCR)
Using SYBR®Green PCR master mix (Life Technologies/Applied Biosystems), the qPCR reaction was set up in triplicates using 25 ng of cDNA as a template. The relative expression levels of sgp130-E10 were calculated for each cell line and condition and also for primary cells after normalization to the respective gp130 levels. Student's t test was used for statistical analysis of the difference in expression between gp130 and sgp130-E10.
Surface Plasmon Resonance Spectroscopy
Experiments were performed on a BiacoreT200 Instrument (GE Healthcare) using PBS supplemented with 0.05% Tween 20 as running buffer. A human Fc capture antibody was immobilized to the surface of a CM5 sensor chip according to the manufacturer's instructions. Binding experiments were performed at 25 °C and a flow rate of 30 μl/min. For getting comparable binding signals, sgp130Fc and sgp130-E10Fc were diluted to give capture levels of 72 RU and 144 RU, respectively. Hyper-IL-6 samples were injected at concentrations from 0.594 to 151 nm using a 2-fold series dilution. The contact time of the association phase was 180 s followed by dissociation for 600 s. The high affinity component was evaluated by globally fitting the dissociation rate in the 400–600-s time window of the dissociation phase. The association rate was then determined for the two lowest concentrations. The binding curves simulated for the simple monovalent binding of the high affinity component were laid over the measured binding curve to illustrate where deviation occurs.
RESULTS
Identification of a Soluble gp130 Receptor mRNA Originating from Alternative Polyadenylation
Initially, we searched for alternative PAS in the introns of human gp130. A potential alternative PAS was identified in intron 10, which was accompanied by a premature stop codon in the potential sgp130-E10 transcript (Fig. 1A). Subsequently, we verified the transcription of sgp130-E10 by RT-PCR with primers spanning exon 9 to intron 10 and thereby excluding amplification of genomic DNA. A PCR product with the expected size of 271 bp was observed in all human cell lines tested (Fig. 1B). Amplification of gp130 using primers binding in exon 9 and exon 11 with an expected amplicon size of 485 bp was used as internal control (Fig. 1B).
FIGURE 1.

sgp130-E10 is conserved in many mammals and most abundantly expressed in blood cells. A, schematic overview of gp130 and the novel sgp130-E10 variant in comparison with the three transcripts formed by alternative polyadenylation. Exons are indicated by E-1 to E-17, intron 10 and the remaining part of intron 14 are indicated by I-10 and I-14, respectively. The stop codon used is highlighted by an asterisk. 5′-untranslated region (5′UTR), signal peptide (S), extracellular domains (D) D1–D6, transmembrane domain (TM), intracellular domain (ICD), and 3′-untranslated region (3′UTR) are also indicated. Polyadenylation (poly(A)) is indicated after E-17 for gp130 and in I-10 for sgp130-E10. sgp130-RAPS lacks exon 9, leading to a novel stop codon in exon 10. This results in a shortened soluble isoform containing domains 1 to 3. In sgp130-Diamant (8) a part of intron 14 remains in the mature mRNA leading to a novel stop codon in exon 15 just before the transmembrane domain starts. This results in a shortened soluble isoform containing every extracellular domain. In sgp130-Sharkey (7) exon 15 is spliced, leading to a novel stop codon in exon 16. This results in a shortened soluble isoform containing every extracellular domain. B, expression of sgp130-E10 and total gp130 (as internal control) in different human cell lines. A minimum of two independent experiments has been performed for each cell line. A representative experiment is shown. C, PBMCs were analyzed for the expression of sgp130-E10 performing a primer walk for the reverse primer. RNA from PBMCs of a healthy blood donor was extracted and reverse-transcribed using oligo(dT) primers. The resulting cDNA was used for PCRs amplifying sgp130-E10 using primers annealing to exon 9 and intron 10. The primer annealing to exon 9 was kept at the same position, whereas the reverse primer for intron 10 was moved downstream stepwise. The calculated distance of the annealing site of the primer to the alternative polyadenylation site is indicated. A representative experiment out of two independent experiments is shown. D, quantitative PCR using RNA from cell lines. Each cell line was analyzed in 2–4 independent experiments. Gp130 amplification was set to “mRNA fold 1,” and sgp130-E10 expression was calculated relative to the gp130 expression. Standard errors were calculated and indicated. Minimum of three independent cDNA samples are included for each cell line. E, quantitative PCR using RNA of PBMCs or mammary carcinoma cell lines (pooled data). Gp130 amplification was set to mRNA fold 1 and sgp130-E10 expression was calculated relative to gp130 expression. Standard errors were calculated and indicated. Mammary carcinoma cell lines comprise five different and independent cell lines, and PBMCs comprise PBMCs from seven different healthy blood donors. qPCR reactions have been performed for individual cDNAs, results have been pooled. F, alignment of the sequences of intron 10, which contains the alternative polyadenylation site, from different mammals. Exon 10 (E-10), intron 10 (I-10), and exon 11 (E-11) are indicated. Asterisks highlight the alternative stop codon, and arrows mark alternative polyadenylation sites (poly(A)). G, RT-PCR of RNA samples from PBMCs from dogs and pigs as well as spleen cells from mice to investigate expression of sgp130-E10.
By performing a PCR primer walk in intron 10, we indirectly showed that the alternative PAS was used for alternative polyadenylation. All primers for the distances −890 to −12 resulted in the expected PCR product sizes, in contrast to the primer that annealed 3′ of the potential alternative PAS (Fig. 1C), strongly indicating that the predicted alternative polyadenylation site is used for the transcript. qPCR revealed that about 2% of the total gp130 transcripts in the liver carcinoma cell lines HepG2 and Hep3B as well as in the human embryonic kidney cell line HEK-293 represented sgp130-E10. Amplification of total gp130 transcripts included membrane-bound and soluble gp130 transcripts generated by differential splicing without sgp130-RAPS and sgp130-E10. In the MCF-7 mammary carcinoma and Jurkat T cell lymphoma cell lines, the percentage of sgp130-E10 was about 7 and 60% of the total gp130 transcripts, respectively (Fig. 1D). This indicates that the expression of sgp130-E10 is higher in cells in which IL-6 trans-signaling is important for maintenance of the inflammatory state. Thus, sgp130-E10 might be a natural inhibitor of trans-signaling produced directly by pro-inflammatory cells at the site of inflammation to limit their own activation. To verify this finding, we investigated the expression of sgp130-E10 in five other mammary carcinoma cell lines (SKBR3, BT20, T47D, NCF12A, and Cal85-1) as well as in peripheral blood mononuclear cells (PBMCs). All mammary carcinoma cell lines had a similar expression of sgp130-E10 of about 4% of the total gp130 transcripts (Fig. 1E). In contrast, the percentage of sgp130-E10 was ∼21% of all gp130 transcripts in PBMCs (Fig. 1E).
Expression of the sgp130 variant identified by Diamant et al. (8) was very low because the qPCR signals were close to the negative control. We have not analyzed the expression of sgp130-RAPS.
An alignment of gp130-intron 10 sequences from different species revealed that the alternative PAS is located at the same position in humans, dogs, monkeys, and cats. Horses and pigs, however, feature potential polyadenylation sites at different locations in intron 10, whereas mice and rats do not have a canonical alternative PAS in intron 10. Moreover, transcripts from all examined species would feature a premature stop codon in the sgp130-E10 transcript with the exception of rats (Fig. 1F). To exemplarily examine mRNA expression of sgp130-E10 in other species, we isolated total RNA from PBMCs of dogs and pigs or spleen cells of mice. As predicted, RT-PCR detected sgp130-E10 transcripts in the dog and pig samples, but not in the murine samples (Fig. 1G). Again, amplification of sgp130-E10 was performed using primers for exon 9 and intron 11. PCR with primers for exon 8 and exon 10 was used as an internal gp130 amplification control (Fig. 1G). In conclusion, we identified a novel sgp130 transcript variant generated by alternative intronic polyadenylation that shows tissue-specific expression patterns.
sgp130-E10 protein Binds to IL-6/Soluble IL-6R (Hyper-IL-6), but Not to IL-6 Alone
Full-length gp130 consists of six extracellular domains (D1–D6), a transmembrane domain, and an intracellular domain (8). For sgp130-E10, exons 1 and 2 encode the 5′-untranslated region, exon 3 encodes the signal peptide, exons 4–8 encode the extracellular domains D1 to D3, and exons 9 and 10 encode D4 followed by four additional amino acids (GLYL) and the premature stop codon encoded by intron 10.
Binding of IL-6/sIL-6R to sgp130 is mediated by D1 (Ig-like domain) and D2/D3 (cytokine-binding module, CBM). We modeled sgp130-E10 in complex with IL-6/sIL-6R and concluded that sgp130-E10 should be able to inhibit IL-6 trans-signaling (Fig. 2A).
FIGURE 2.
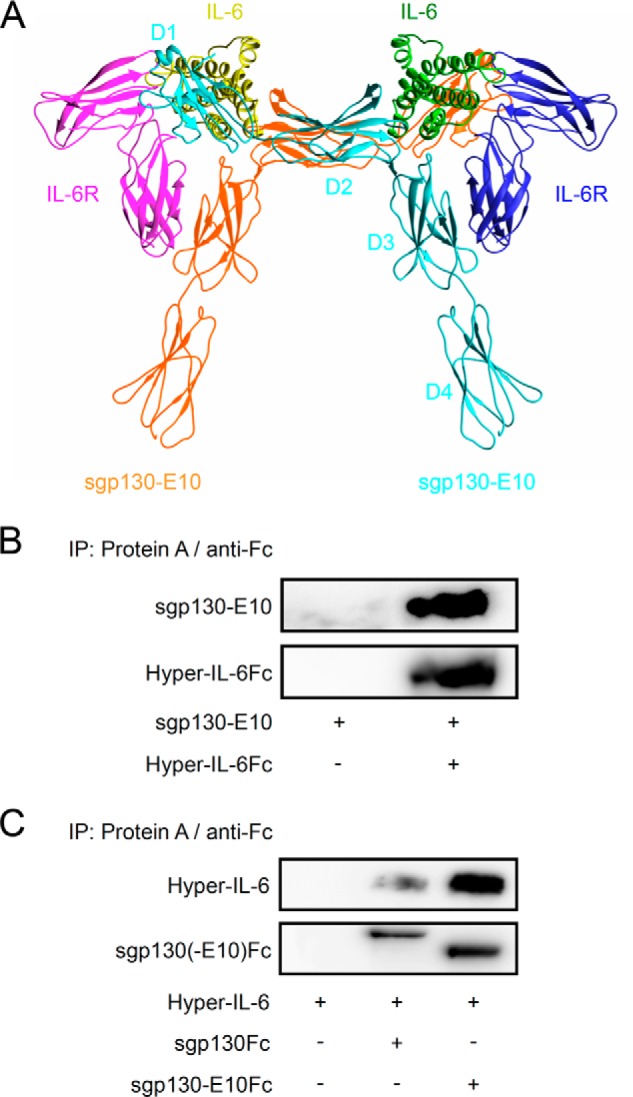
Structure of sgp130-E10 and its interaction with Hyper-IL-6. A, schematic overview of sgp130-E10 in complex with IL-6/IL-6R. B, immunoprecipitation with conditioned medium from HEK-293 cell cultures transiently transfected with plasmids coding for sgp130-E10Myc-His or Hyper-IL-6Fc. Supernatants were harvested 2 days after transfection followed by immunoprecipitations (IP) using protein A-agarose beads and Western blot analysis using primary antibodies directed against the Myc (precipitated antibody from cell culture supernatant in unknown concentration) or human Fc tags (0.4 μg/ml). Three independent experiments were performed. A representative experiment is shown. C, immunoprecipitation with conditioned medium from HEK-293 cell cultures transiently transfected with plasmids coding for sgp130-E10Fc or sgp130Fc. Supernatants were harvested 2 days after transfection followed by immunoprecipitations using protein A-agarose beads and Western blot analysis using primary antibodies directed against the human Fc tag (0.4 μg/ml) or IL-6 (2.5 μg/ml). A representative experiment out of three independent experiments is shown.
The sgp130-E10 cDNA was subcloned into a eukaryotic expression plasmid with either a C-terminal Myc and His tag or an Fc tag from a human IgG1 antibody. cDNAs coding for sgp130-E10Fc or sgp130Fc were transiently transfected into HEK-293 cells. sgp130Fc consists of the extracellular domains D1–D6 of gp130 fused to the Fc-part of a human IgG1 antibody. The Fc fusion results in a constant dimerization of sgp130 and, thus, in a 10–100-fold increase in binding efficiency to IL-6/sIL-6R as compared with monomeric sgp130 (1).
The resulting monomeric protein sgp130-E10Myc-His had an apparent molecular mass of 80–90 kDa (Fig. 2B). sgp130-E10Myc-His was precipitated from conditioned HEK-293 cell culture supernatants by Hyper-IL-6Fc (Fc-tagged Hyper-IL-6, a fusion protein of IL-6 and sIL-6R) in combination with protein A-agarose beads (Fig. 2B). In addition, conditioned HEK-293 cell culture supernatants containing sgp130-E10Fc or sgp130Fc were used to precipitate Hyper-IL-6 via protein A-agarose beads (Fig. 2C). Our results showed that sgp130-E10 in both a monomeric and a dimeric form is able to bind to the IL-6/sIL-6R complex represented by Hyper-IL-6.
Subsequently, stable CHO-K1 cell clones expressing sgp130-E10Myc-His or sgp130-E10Fc were generated and selected for high level expression by ELISA. Recombinant protein expression was determined by ELISA to be about 300 μg/liter supernatant for sgp130-E10Myc-His and 800 μg/liter for sgp130-E10Fc.
The final yield of pure protein was, however, much lower, namely about 1 μg/liter for sgp130-E10Myc-His and ∼40 μg/liter for sgp130-E10Fc, which was due to instability of the recombinant proteins and loss of recombinant proteins during the purification via His tag or Fc tag. For this reason, purification of sufficient amounts of sgp130-E10Myc-His for further experiments was not possible.
sgp130-E10Fc was purified by affinity chromatography and size exclusion chromatography. The purity and identity of sgp130-E10Fc was verified by Coomassie Blue staining of SDS-PAGE gels (Fig. 3A) and Western blotting (Fig. 3B). Just like sgp130Fc, purified sgp130-E10Fc was able to precipitate Hyper-IL-6 in combination with protein A-agarose beads, indicating that the purified sgp130-E10Fc was biologically active (Fig. 3C).
FIGURE 3.
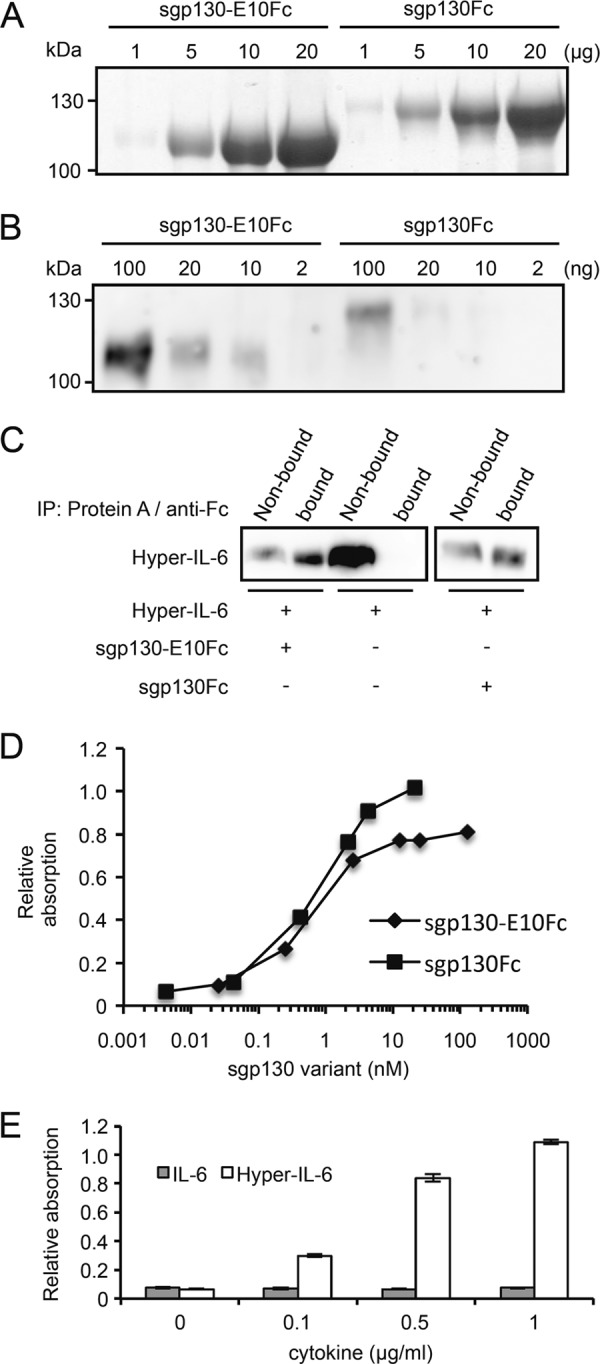
sgp130-E10Fc interacts with Hyper-IL-6, but not with IL-6 alone. A, Coomassie Blue stains of sgp130-E10Fc and sgp130Fc. Different amounts from 1 to 20 μg of sgp130-E10Fc and sgp130Fc were separated by SDS-PAGE and visualized by Coomassie Blue stain. B, Western blot analysis of purified sgp130-E10Fc and sgp130Fc. Different amounts from 2 to 100 ng of sgp130-E10Fc and sgp130Fc were separated by SDS-PAGE and detected by human Fc antibody. C, immunoprecipitation (IP) of Hyper-IL-6 by sgp130-E10Fc or sgp130Fc conjugated to protein A-agarose beads and detection by IL-6 antibody. A representative experiment out of two independent experiments is shown. D, concentration-dependent binding of sgp130 variants to immobilized Hyper-IL-6. A representative experiment out of two independent experiments is shown. E, like sgp130Fc, sgp130-E10Fc only binds the IL-6/sIL-6R complex represented by Hyper-IL-6, but not IL-6 alone. Standard errors were calculated and indicated.
We performed an ELISA binding assay with Hyper-IL-6-coated plates and free sgp130Fc and sgp130-E10Fc to characterize binding of the sgp130 variants to Hyper-IL-6. In this assay, sgp130Fc and sgp130-E10Fc showed similar binding curves for Hyper-IL-6, suggesting comparable affinities (Fig. 3D). In addition, incubating sgp130-E10Fc-coated plates with increasing concentrations of IL-6 or Hyper-IL-6 showed that sgp130-E10Fc, like sgp130Fc (1), does not bind to IL-6 in the absence of sIL-6R (Fig. 3E).
The binding of sgp130Fc and sgp130-E10Fc to Hyper-IL-6 was studied by surface plasmon resonance spectroscopy. sgp130Fc (77 RU) and sgp130-E10Fc (144 RU) were captured through their Fc fusion partner using a human Fc monoclonal antibody followed by injection of Hyper-IL-6. As expected from the complex binding mechanism, stoichiometry, and the crystal structure, the binding curves exhibited more than one component. A low affinity component is clearly visible at high Hyper-IL-6 concentration, whereas at low Hyper-IL-6 concentrations, the high affinity component predominates (Fig. 4). For comparison, this high affinity component was analyzed using the simple 1:1 binding model, and the association and dissociation rate constants and the resulting equilibrium dissociation constant were determined (Table 1). The results show that Hyper-IL-6 binds 2.5 times more slowly to (1.3 × 106 m−1 s−1 versus 3.1 × 106 m−1 s−1) and dissociates three times more rapidly from (9.0 × 10−5 s−1 versus 3.1 × 10−5 s−1) sgp130-E10Fc than from sgp130Fc. This results in overall apparent affinities of KD = 9 pm for sgp130Fc and of KD = 71 pm for sgp130-E10Fc. The KD of sgp130Fc to Hyper-IL-6 was in the range of previously described values (15).
FIGURE 4.
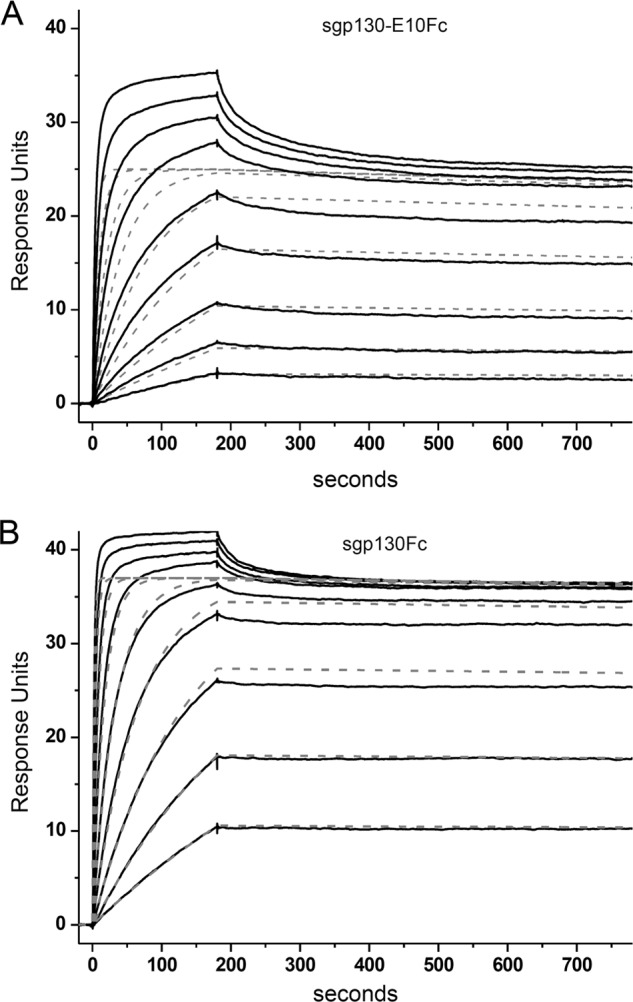
Analysis of the binding of Hyper-IL-6 to captured sgp130Fc and gp130-E10Fc by surface plasmon resonance spectroscopy. A, sgp130-E10Fc and B, sgp130Fc were captured to immobilized human Fc antibody, and Hyper-IL-6 was injected for 180 s at concentrations from 0.59 to 151 nm. The dissociation was followed for 600 s. The sensorgrams show double referenced curves. Dotted gray lines represent simulated curves for the proposed high affinity component of the observed complex binding curves (black lines).
TABLE 1.
Kinetic constants and affinities of Hyper-IL-6 to sgp130-E10Fc and sgp130Fc
| Ligand | Rcapture | Rmax | kon | koff | KD |
|---|---|---|---|---|---|
| RU | RU | m−1 × s−1 | s−1 | pm | |
| sgp130Fc | 72 | 37 | 3.1 × 106 | 3.1 × 10−5 | 9 |
| sgp130-E10Fc | 144 | 25 | 1.3 × 106 | 9.0 × 10−5 | 71 |
Generation of an sgp130-E10-specific Antibody and Detection of sgp130-E10 in PBMCs
Using the synthetic peptide CDFQGLYL deduced from the unique C terminus of sgp130-E10, we generated a monoclonal antibody in mice specifically detecting this sequence. One resulting mAb, E10/1, was tested in Western blots for specific detection of sgp130-E10 using recombinant sgp130-E10Fc, and sgp130Fc served as negative control. E10/1 was able to detect sgp130-E10Fc with a detection limit of ∼10 ng and without cross-reactivity to sgp130Fc (Fig. 5A).
FIGURE 5.
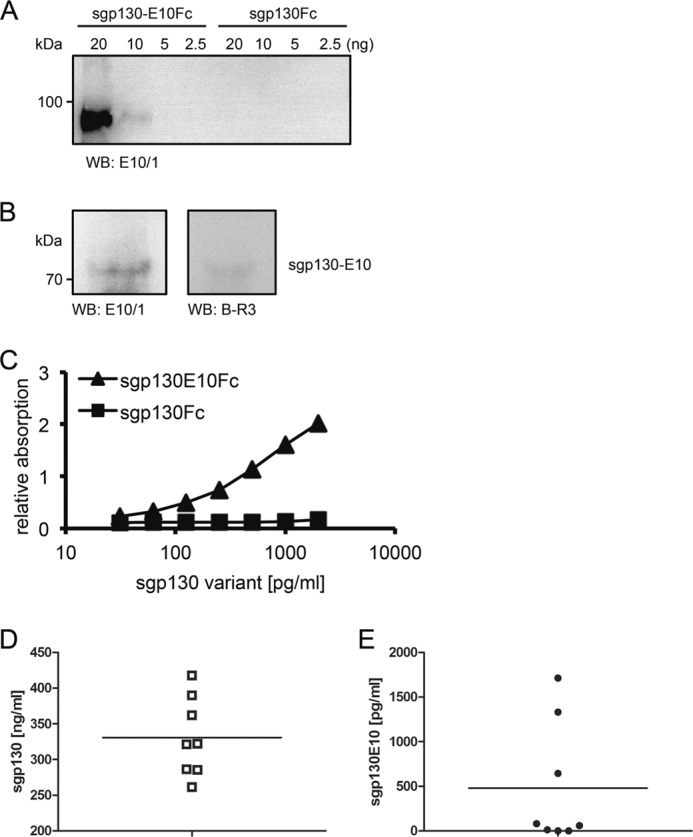
Specific detection of sgp130-E10 by the monoclonal sgp130-E10 antibody E10/1 in PBMCs and serum. A, Western blot (WB) analysis of sgp130-E10Fc and sgp130Fc in different concentrations. The monoclonal sgp130-E10 antibody E10/1 specifically detected recombinant sgp130-E10Fc with a detection limit of ∼10 ng; E10/1 did not detect sgp130Fc. B, Western blot (WB) analysis of the sgp130-E10 isoform in human PBMCs lysates using the gp130 antibody B-R3 and the novel sgp130-E10 antibody E10/1. sgp130-E10 was specifically detected at the expected apparent molecular mass of 70–80 kDa. C, ELISA with the sgp130-E10 antibody E10/1 as capture antibody, demonstrating the specific detection of sgp130-E10Fc, but not sgp130Fc. D and E, sgp130 (D) or sgp130-E10 (E) in the sera of eight healthy volunteers was quantified by specific ELISA.
We observed the highest mRNA level of sgp130-E10 in PBMCs. Therefore, PBMCs were analyzed for sgp130-E10 expression using the E10/1 mAb. As depicted in Fig. 5B, E10/1 detected a protein with an apparent molecular mass of 70–80 kDa in PBMCs, which corresponded to the expected size of sgp130-E10. The same protein was detected using the commercial gp130 mAb BR-3, albeit with a much lower efficacy. BR-3 specifically detects the extracellular domain D2 of gp130. Next, we used this novel antibody to establish an ELISA that specifically detects sgp130-E10, but not other sgp130 variants. As shown in Fig. 5C, E10/1 was able to capture sgp130-E10Fc in a dose-dependent manner, but not sgp130Fc. sgp130Fc is found in high concentrations in human serum of about 200–400 ng/ml (Fig. 5D), and we therefore asked whether sgp130-E10 contributes to these levels. Indeed, we were able to detect sgp130-E10 in the range of 1 ng/ml (1,713, 1,331, and 644 pg/ml) in 3 out of 8 serum samples from healthy donors (Fig. 5E). In conclusion, our data indicate that sgp130-E10 is translated into a protein in PBMCs, is the dominant sgp130 variant in these cells, and contributes to the sgp130 serum levels in healthy humans.
Biological Activity and Stability of sgp130-E10Fc
To judge the potential of sgp130-E10Fc as a drug candidate as compared with the “gold standard” sgp130Fc, we performed a competitive ELISA to compare the binding properties of sgp130Fc and sgp130-E10Fc to Hyper-IL-6. sgp130Fc was immobilized on a 96-well plate and incubated with saturating concentrations of Hyper-IL-6. Hyper-IL-6 was detected using labeled IL-6 mAbs. As expected, free sgp130Fc or sgp130-E10Fc reduced the amount of Hyper-IL-6 that was bound to immobilized sgp130Fc in a concentration-dependent manner. As compared with sgp130Fc, the concentration of sgp130-E10Fc needed to completely reduce the Hyper-IL-6 detection signal to baseline was 5–10-times higher (Fig. 6A). We conclude from these results that sgp130-E10Fc specifically binds to IL-6/sIL-6R, albeit with a 5–10-fold lower efficacy than sgp130Fc.
FIGURE 6.
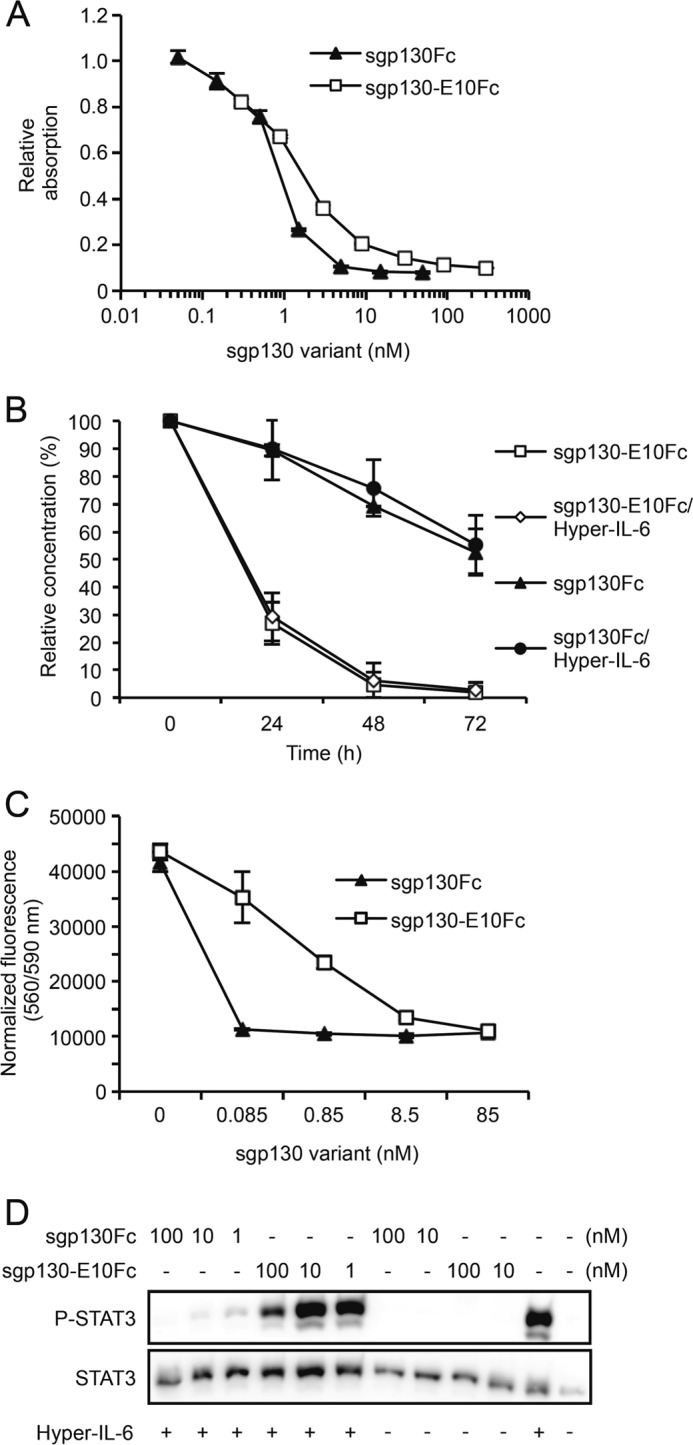
Purified sgp130-E10Fc is biologically active, but inferior to sgp130Fc in terms of binding to Hyper-IL-6, activity and stability. A, competitive ELISA with coated sgp130Fc and competition with either sgp130Fc or sgp130-E10Fc for Hyper-IL-6 binding. Standard errors were calculated and indicated. A representative experiment out of two independent experiments is shown. B, stability assay for sgp130Fc and sgp130-E10Fc. After incubation at 37 °C in cell culture medium for the indicated times, samples were analyzed using gp130 ELISA. The starting concentration was arbitrarily set to 100%, and the remaining protein was calculated and plotted. Standard errors were calculated and indicated. Two independent experiments were performed. Mean values of the two experiments were been calculated and are indicated. C, Ba/F3-gp130 cell proliferation assay comparing the bioactivities of sgp130-E10Fc and sgp130Fc. Standard errors were calculated and indicated. D, comparison of the inhibitory effect of sgp130-E10Fc or sgp130Fc on phosphorylation of STAT3 (P-STAT3) in Ba/F3-gp130 cells. A representative experiment out of three independent experiments is shown.
During protein purification, we observed a reduced stability of sgp130-E10Myc-His and sgp130-E10Fc as compared with sgp130Fc. Therefore, we analyzed the time-dependent stability of the Fc-compounds by ELISA. sgp130-E10Fc and sgp130Fc were incubated with or without Hyper-IL-6 in standard DMEM growth medium at 37 °C. The half-life of sgp130-E10Fc (12 h) was strongly reduced as compared with the half-life of sgp130Fc (72 h). Stability of both sgp130 variants was independent of the presence of Hyper-IL-6 (Fig. 6B).
Subsequently, we investigated whether sgp130-E10Fc was able to inhibit IL-6/sIL-6R-induced cellular proliferation and STAT3 phosphorylation. We used Ba/F3 cells stably transduced with human gp130, which proliferate in the presence of Hyper-IL-6 in a phospho-STAT3-dependent manner (11), as a model system. sgp130-E10Fc and sgp130Fc inhibited Ba/F3-gp130 cell proliferation in a dose-dependent manner. From the data of these experiments, we calculated an IC50 of 67 nm for sgp130-E10Fc (Fig. 6C) and concluded that, although sgp130-E10Fc does inhibit IL-6 trans-signaling, it is about 1,000-fold less active than sgp130Fc with an IC50 of 0.067 nm in this system (19). These results were supported by analyses of the Hyper-IL-6-induced STAT3 phosphorylation in Ba/F3-gp130 cells in the presence of increasing amounts sgp130-E10Fc or sgp130Fc. Although sgp130Fc inhibited STAT3 phosphorylation at concentrations of ∼1 nm, even 100 times more sgp130-E10Fc only reduced, but did not completely inhibit, STAT3 phosphorylation (Fig. 6D).
DISCUSSION
Hyper-activation of gp130 by IL-6 trans-signaling is due to elevated levels of IL-6 and sIL-6R and was observed in many inflammatory disorders, e.g. in rheumatoid arthritis (20–22). IL-6 trans-signaling is crucial for the development and maintenance of rheumatoid arthritis (23, 24). Global blockade of IL-6 classic and trans-signaling by the IL-6R-neutralizing antibody tocilizumab is approved in the United States, European Union, and Japan for the treatment of rheumatoid arthritis and other diseases. Blockade of IL-6 trans-signaling by soluble variants of gp130 might offer an attractive alternative treatment option by leaving IL-6 classic signaling intact (3, 19). An optimized variant of the trans-signaling inhibitor sgp130Fc is currently in clinical trials.
For sgp130Fc, a 2–5-fold molar excess is sufficient to inhibit IL-6/sIL-6R-mediated Ba/F3-gp130 cell proliferation. In sgp130Fc, the complete extracellular part of gp130 (domains D1–D6) was fused to a dimerization domain (Fc part of a human IgG antibody). Therefore, sgp130Fc mimics the receptor signaling complex on the cell membrane, which also exists as a preformed dimer (25). Interestingly, the preformed sgp130Fc dimer has a 10–100-fold higher efficacy in inhibiting IL-6/sIL-6R-mediated responses than the naturally occurring monomeric sgp130 isoform (1). Three soluble isoforms of gp130 have previously been found in human serum and urine, with apparent molecular masses of about 50, 90, and 110 kDa, respectively. So far, only the 50-kDa isoform sgp130-RAPS was assigned to a differentially spliced mRNA (5, 7, 8).
Recently, generation of soluble dominant-negative receptor tyrosine kinase isoforms by alternative intronic PAS has been described (10). Many of these receptors are hyperactivated in tumors, and blockade of the receptors is recognized as a drug target (26, 27). In this study, we demonstrated that intronic PAS also resulted in the generation of a novel gp130 mRNA isoform termed sgp130-E10, resulting in an sgp130 isoform with an apparent molecular mass of 70–80 kDa. We also showed that the designer protein sgp130-E10Fc inhibits IL-6 trans-signaling, albeit with a much lower efficacy than its counterpart sgp130Fc (1), which was proved by surface plasmon resonance spectroscopy. To our knowledge, this is the first report on the generation of soluble isoforms of receptors with associated Janus kinases by alternative polyadenylation. Furthermore, the sgp130-E10 mRNA isoform is not only produced in humans, but also in other mammals, suggesting a conserved alternative polyadenylation event with a function in vivo.
Inhibition of the small nucleolar ribonuclear protein U1, which is essential for splicing, has recently been shown to increase alternative intronic polyadenylation and generation of soluble receptor tyrosine kinase isoforms (10). It remains to be elucidated whether the same mechanism controls the mRNA transcription of sgp130-E10. Inhibition of small nucleolar ribonuclear protein U1 will not only induce soluble dominant-negative isoforms of receptor tyrosine kinases, but also reduce the level of membrane-bound receptors and thereby decrease signaling. Inhibition of small nucleolar ribonuclear protein U1 might, therefore, represent a beneficial future treatment option for IL-6-related diseases with hyperactive gp130 signaling.
Interestingly, Jurkat T cells and human PBMCs produced the highest levels of sgp130-E10 mRNA as compared with other cell types. Moreover, sgp130-E10 was detectable in PBMCs and in human serum. PBMCs are involved in inflammatory disorders (24). Production of the isoform sgp130-E10 by these cells might directly bring the IL-6 trans-signaling inhibitor into the center of inflammation, where it can act in an autocrine as well as paracrine way. The short half-life of sgp130-E10 could, however, guarantee that IL-6 trans-signaling is only transiently inhibited.
Acknowledgments
We acknowledge Prof. Niederacher and Ellen Honisch from the Brustklinik Düsseldorf for the RNA samples of mammary carcinoma cell lines and primary blood cells. We thank Dr. Engelhardt from the animal facility of the University of Düsseldorf for providing us with blood samples of different animals.
This work was funded by a grant from the Deutsche Forschungsgemeinschaft (SCHE 907/2-1, Bonn, Germany) (to J. S.).
- gp130
- glycoprotein 130 kDa
- sgp130
- soluble glycoprotein 130 kDa
- sgp130-E10
- soluble glycoprotein 130 kDa-exon 10 variant
- sgp130-RAPS
- soluble glycoprotein 130 kDa of the rheumatoid arthritis antigenic peptide-bearing soluble form
- Ba/F3-gp130 cells
- murine pro B cells stably transduced with human glycoprotein 130
- IL-6R
- interleukin-6 receptor
- sIL-6R
- soluble IL-6R
- Hyper-IL-6
- Hyper-interleukin-6, a fusion protein of human interleukin-6 and its soluble receptor
- PAS
- polyadenylation site
- KLH
- keyhole limpet hemocyanin
- PBMC
- peripheral blood mononuclear cell
- qPCR
- quantitative RT-PCR
- RU
- response units.
REFERENCES
- 1. Jostock T., Müllberg J., Ozbek S., Atreya R., Blinn G., Voltz N., Fischer M., Neurath M. F., Rose-John S. (2001) Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 268, 160–167 [DOI] [PubMed] [Google Scholar]
- 2. Jones S. A., Scheller J., Rose-John S. (2011) Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Invest. 121, 3375–3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waetzig G. H., Rose-John S. (2012) Hitting a complex target: an update on interleukin-6 trans-signalling. Expert Opin. Ther. Targets 16, 225–236 [DOI] [PubMed] [Google Scholar]
- 4. Narazaki M., Yasukawa K., Saito T., Ohsugi Y., Fukui H., Koishihara Y., Yancopoulos G. D., Taga T., Kishimoto T. (1993) Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood 82, 1120–1126 [PubMed] [Google Scholar]
- 5. Tanaka M., Kishimura M., Ozaki S., Osakada F., Hashimoto H., Okubo M., Murakami M., Nakao K. (2000) Cloning of novel soluble gp130 and detection of its neutralizing autoantibodies in rheumatoid arthritis. J. Clin. Invest. 106, 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang J. G., Zhang Y., Owczarek C. M., Ward L. D., Moritz R. L., Simpson R. J., Yasukawa K., Nicola N. A. (1998) Identification and characterization of two distinct truncated forms of gp130 and a soluble form of leukemia inhibitory factor receptor α-chain in normal human urine and plasma. J. Biol. Chem. 273, 10798–10805 [DOI] [PubMed] [Google Scholar]
- 7. Sharkey A. M., Dellow K., Blayney M., Macnamee M., Charnock-Jones S., Smith S. K. (1995) Stage-specific expression of cytokine and receptor messenger ribonucleic acids in human preimplantation embryos. Biol. Reprod. 53, 974–981 [DOI] [PubMed] [Google Scholar]
- 8. Diamant M., Rieneck K., Mechti N., Zhang X. G., Svenson M., Bendtzen K., Klein B. (1997) Cloning and expression of an alternatively spliced mRNA encoding a soluble form of the human interleukin-6 signal transducer gp130. FEBS Lett. 412, 379–384 [DOI] [PubMed] [Google Scholar]
- 9. Tian B., Pan Z., Lee J. Y. (2007) Widespread mRNA polyadenylation events in introns indicate dynamic interplay between polyadenylation and splicing. Genome Res. 17, 156–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vorlová S., Rocco G., Lefave C. V., Jodelka F. M., Hess K., Hastings M. L., Henke E., Cartegni L. (2011) Induction of antagonistic soluble decoy receptor tyrosine kinases by intronic polyA activation. Mol. Cell 43, 927–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sommer J., Effenberger T., Volpi E., Waetzig G. H., Bernhardt M., Suthaus J., Garbers C., Rose-John S., Floss D. M., Scheller J. (2012) Constitutively active mutant gp130 receptor protein from inflammatory hepatocellular adenoma is inhibited by an anti-gp130 antibody that specifically neutralizes interleukin 11 signaling. J. Biol. Chem. 287, 13743–13751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fischer M., Goldschmitt J., Peschel C., Brakenhoff J. P., Kallen K. J., Wollmer A., Grötzinger J., Rose-John S. (1997) I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat. Biotechnol. 15, 142–145 [DOI] [PubMed] [Google Scholar]
- 13. Schroers A., Hecht O., Kallen K. J., Pachta M., Rose-John S., Grötzinger J. (2005) Dynamics of the gp130 cytokine complex: a model for assembly on the cellular membrane. Protein Sci. 14, 783–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Floss D. M., Mrotzek S., Klöcker T., Schröder J., Grötzinger J., Rose-John S., Scheller J. (2013) Identification of canonical tyrosine-dependent and non-canonical tyrosine-independent STAT3 activation sites in the intracellular domain of the interleukin 23 receptor. J. Biol. Chem. 288, 19386–19400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tenhumberg S., Waetzig G. H., Chalaris A., Rabe B., Seegert D., Scheller J., Rose-John S., Grötzinger J. (2008) Structure-guided optimization of the interleukin-6 trans-signaling antagonist sgp130. J. Biol. Chem. 283, 27200–27207 [DOI] [PubMed] [Google Scholar]
- 16. Lateef S. S., Gupta S., Jayathilaka L. P., Krishnanchettiar S., Huang J. S., Lee B. S. (2007) An improved protocol for coupling synthetic peptides to carrier proteins for antibody production using DMF to solubilize peptides. J. Biomol. Tech. 18, 173–176 [PMC free article] [PubMed] [Google Scholar]
- 17. Clement H. W., Vazquez J. F., Sommer O., Heiser P., Morawietz H., Hopt U., Schulz E., von Dobschütz E. (2010) Lipopolysaccharide-induced radical formation in the striatum is abolished in Nox2 gp91phox-deficient mice. J. Neural. Transm. 117, 13–22 [DOI] [PubMed] [Google Scholar]
- 18. Garbers C., Monhasery N., Aparicio-Siegmund S., Lokau J., Baran P., Nowell M. A., Jones S. A., Rose-John S., Scheller J. (2014) The interleukin-6 receptor Asp358Ala single nucleotide polymorphism rs2228145 confers increased proteolytic conversion rates by ADAM proteases. Biochim. Biophys. Acta 1842, 1485–1494 [DOI] [PubMed] [Google Scholar]
- 19. Garbers C., Thaiss W., Jones G. W., Waetzig G. H., Lorenzen I., Guilhot F., Lissilaa R., Ferlin W. G., Grötzinger J., Jones S. A., Rose-John S., Scheller J. (2011) Inhibition of classic signaling is a novel function of soluble glycoprotein 130 (sgp130), which is controlled by the ratio of interleukin 6 and soluble interleukin 6 receptor. J. Biol. Chem. 286, 42959–42970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Usón J., Balsa A., Pascual-Salcedo D., Cabezas J. A., Gonzalez-Tarrio J. M., Martín-Mola E., Fontan G. (1997) Soluble interleukin 6 (IL-6) receptor and IL-6 levels in serum and synovial fluid of patients with different arthropathies. J. Rheumatol. 24, 2069–2075 [PubMed] [Google Scholar]
- 21. Keul R., Heinrich P. C., Müller-newen G., Muller K., Woo P. (1998) A possible role for soluble IL-6 receptor in the pathogenesis of systemic onset juvenile chronic arthritis. Cytokine 10, 729–734 [DOI] [PubMed] [Google Scholar]
- 22. Robak T., Gladalska A., Stepień H., Robak E. (1998) Serum levels of interleukin-6 type cytokines and soluble interleukin-6 receptor in patients with rheumatoid arthritis. Mediators Inflamm. 7, 347–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jones S. A., Richards P. J., Scheller J., Rose-John S. (2005) IL-6 transsignaling: the in vivo consequences. J. Interferon Cytokine Res. 25, 241–253 [DOI] [PubMed] [Google Scholar]
- 24. Atreya R., Mudter J., Finotto S., Müllberg J., Jostock T., Wirtz S., Schütz M., Bartsch B., Holtmann M., Becker C., Strand D., Czaja J., Schlaak J. F., Lehr H. A., Autschbach F., Schürmann G., Nishimoto N., Yoshizaki K., Ito H., Kishimoto T., Galle P. R., Rose-John S., Neurath M. F. (2000) Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat. Med. 6, 583–588 [DOI] [PubMed] [Google Scholar]
- 25. Tenhumberg S., Schuster B., Zhu L., Kovaleva M., Scheller J., Kallen K. J., Rose-John S. (2006) gp130 dimerization in the absence of ligand: preformed cytokine receptor complexes. Biochem. Biophys. Res. Commun. 346, 649–657 [DOI] [PubMed] [Google Scholar]
- 26. Lemmon M. A., Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shawver L. K., Slamon D., Ullrich A. (2002) Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell 1, 117–123 [DOI] [PubMed] [Google Scholar]