

Background: AMPA-type glutamate receptor-mediated synaptic transmission is enhanced in descending pain modulatory circuits under pain conditions.
Results: Epigenetic regulation of BDNF by persistent inflammation triggers AMPA receptor GluA1 phosphorylation.
Conclusion: Synaptic delivery of GluA1 in the brain stem is initiated by BDNF/TrKB activation under pain conditions.
Significance: We investigate how GluA1 is delivered to synapses to understand the molecular mechanisms underlying pain in descending pain modulatory circuits.
Keywords: BDNF, Pain, Epigenetics, GluA1
Abstract
The enhanced AMPA receptor phosphorylation at GluA1 serine 831 sites in the central pain-modulating system plays a pivotal role in descending pain facilitation after inflammation, but the underlying mechanisms remain unclear. We show here that, in the rat brain stem, in the nucleus raphe magnus, which is a critical relay in the descending pain-modulating system of the brain, persistent inflammatory pain induced by complete Freund adjuvant (CFA) can enhance AMPA receptor-mediated excitatory postsynaptic currents and the GluA2-lacking AMPA receptor-mediated rectification index. Western blot analysis showed an increase in GluA1 phosphorylation at Ser-831 but not at Ser-845. This was accompanied by an increase in distribution of the synaptic GluA1 subunit. In parallel, the level of histone H3 acetylation at bdnf gene promoter regions was reduced significantly 3 days after CFA injection, as indicated by ChIP assays. This was correlated with an increase in BDNF mRNA levels and BDNF protein levels. Sequestering endogenous extracellular BDNF with TrkB-IgG in the nucleus raphe magnus decreased AMPA receptor-mediated synaptic transmission and GluA1 phosphorylation at Ser-831 3 days after CFA injection. Under the same conditions, blockade of TrkB receptor functions, phospholipase C, or PKC impaired GluA1 phosphorylation at Ser-831 and decreased excitatory postsynaptic currents mediated by GluA2-lacking AMPA receptors. Taken together, these results suggest that epigenetic up-regulation of BDNF by peripheral inflammation induces GluR1 phosphorylation at Ser-831 sites through activation of the phospholipase C-PKC signaling cascade, leading to the trafficking of GluA1 to pain-modulating neuronal synapses.
Introduction
It is well established that the central glutamatergic system plays a crucial role in developing and maintaining persistent pain, including neuropathic pain and inflammatory pain (1). Targeting the glutamatergic system facilitates the treatment of chronic pain (1–4). In the brain, the majority of fast excitatory synaptic transmissions are mediated by AMPA-type glutamate receptors (5, 6). These are tetrameric assemblies composed of four possible subunits, primarily GluA1 and GluA2 subunits, but there are also some GluA3 and GlA4 subunits (7). Postsynaptic AMPA receptor subunit trafficking, especially GluA1 and GluA2, in the development of synaptic plasticity under pathophysiological conditions may be associated with chronic pain (4, 8–10). For example, in the anterior cingulate cortex and spinal cord, GluA1 was recruited to the plasma membrane under pain conditions to enhance glutamatergic transmission, leading to a behavioral state of sensitized pain (11–13). However, the molecular mechanism underlying dynamic distribution of GluA1 after inflammation in the descending pain modulatory circuitry remains unclear.
BDNF, a member of the neurotrophin family, has diverse trophic effects on structural modifications and the functional plasticity of central synapses in the mammalian brain (14, 15). It has a well documented pronociceptive role in inflammatory and neuropathic pain responses, acting at brain stem-descending pain pathways, including the periaqueductal gray (PAG),2 rostral ventromedial medulla (RVM), and spinal cord (16, 17). High levels of BDNF mRNA and protein have been observed within the PAG and RVM (18, 19). A study by Guo et al. (20) showed that BDNF in the RVM may have originated from BDNF-containing neurons in the PAG and that BDNF activation of TrkB signaling in the RVM induces descending pain facilitation, suggesting that the signaling cascade of BDNF-TrkB receptors in the RVM circuitry plays a critical role in the development of persistent pain after inflammation. Previous studies have shown that persistent inflammation up-regulates BDNF, which decreases the KCC2 function of maintaining the chloride gradient for inhibitory GABA synapses, resulting in impaired GABA inhibition and neuronal hyperexcitability in the nucleus raphe magnus (NRM), which is the major structure of the RVM, contributing to pain sensitization (21). As a pain modulator, BDNF can modulate excitatory glutamatergic and inhibitory GABAergic/glycinergic signals (17, 22). The glutamatergic systems in the PAG and RVM have been shown to be significant analgesic targets. For example, activation of the type 3 metabotropic “glutamate” receptor (mGluR3) in the PAG or RVM has shown analgesic effects in animal models of inflammatory pain (23). However, little is known about how BDNF modulates glutamatergic systems in the supraspinal circuitry underlying persistent inflammatory pain.
Studies have shown that local infusion of BDNF in the tectum can potentiate neurotransmitter secretion at retinotectal synapses and increase the density of postsynaptic AMPA receptors, producing long term excitatory synaptic plasticity (24, 25). A similar effect has been observed in the descending circuitry under pain conditions. Mechanistic investigation in hippocampal and cortical neurons has shown that BDNF up-regulates the total cellular levels of AMPA subunits GluA1-GluA4 (26–28) and promotes GluA1 trafficking to the membrane (26, 29). The binding of BDNF to TrkB receptors activates a series of intracellular signaling pathways that initiate the membrane incorporation of GluA1, including phospholipase Cγ (PLCγ), PI3K/Akt, Scr, CaM-kinase kinase/transient receptor potential canonical, and mammalian target of rapamycin complex 1 (mTOR1) (26, 29–31). The manner in which BDNF regulates AMPA receptor-mediated synaptic transmission at supraspinal levels after inflammation, therefore contributing to persistent pain, is less well understood.
In this study, we demonstrated that activity of the epigenetically up-regulated bdnf gene contributes to the distribution of synaptic GluA1 after inflammation, resulting in enhanced AMPA receptor-mediated synaptic transmission in the rat brain stem NRM. The synaptic delivery of GluA1 is trigged by phosphorylation at Ser-831 sites via PLC-PKC pathways initiated by BDNF/TrkB signaling, which is consistent with previous findings in cultured hippocampal neurons (26).
MATERIALS AND METHODS
Animals and the Pain Model
Male Wistar rats, 9–14 days old, and adult rats weighing 200–300 g were used (Charles River Laboratories, Wilmington, MA). A rat model of persistent inflammatory pain was induced by a single injection of complete Freund adjuvant (CFA, 20 μl) into a hind paw. Neonatal rats were injected twice with CFA. Then, 20 μl of CFA was injected on the first day, and a second CFA (20-μl) injection was made on the third day to ensure the persistence of inflammatory pain (32, 33). All procedures involving the use of animals conformed to the guidelines of the Institutional Animal Use and Care Committee of the University of Science and Technology of China.
Brian Slice Preparations
Whole-cell voltage clamp recordings of NRM neurons were visualized in slice preparations with general methods as described previously (33, 34). Neonatal rats were used in visualized whole-cell recording experiments because of the limited visibility and quality of cells in NRM slices from older rats. Similarities and differences between the responses of neonatal and adult NRM neurons to pain have been described previously (32). Those differential responses could confound the interpretation of the data. Despite these recognized limitations, cellular recordings from neonatal rats are nonetheless a useful and efficient model to help understand the cellular mechanisms of pain behavior in adult rats (33–37). The rat brain was cut in a vibratome in cold (4 °C) physiological saline to produce brain stem slices (200 μm thick) containing the NRM for whole-cell recording as described previously (36). A single slice was submerged in a shallow recording chamber and perfused with preheated (35 °C) physiological saline containing the following: 126 mm NaCl, 2.5 mm KCl, 1.2 mm NaH2PO4, 1.2 mm MgCl2, 2.4 mm CaCl2, 11 mm glucose, and 25 mm NaHCO3 saturated with 95% O2 and 5% CO2 (pH 7.2–7.4).
Whole-cell Recording and Synaptic Currents
Visualized whole-cell voltage clamp recordings were obtained from identified NRM neurons with a glass pipette (resistance, 3–5 MΩ) filled with a solution containing the following: 126 mm KCl, 10 mm NaCl, 1 mm MgCl2, 11 mm EGTA, 10 mm HEPES, 2 mm ATP, and 0.25 mm GTP, pH adjusted to 7.3 with KOH; osmolarity, 280–290 mosmol. An AxoPatch-700B amplifier and AxoGraph software (Molecular Devices) were used for data acquisition and online and offline data analyses. A seal resistance of 2 GΩ or above and an access resistance of 15 mΩ or below were considered acceptable. Series resistance was optimally compensated, and access resistance was monitored throughout the experiment. Electrical stimuli of constant current (0.25 ms, 0.2–0.4 mA) were used to evoke glutamate excitatory postsynaptic currents (EPSCs) with bipolar stimulating electrodes placed within the nucleus. AMPA EPSCs were recorded in the presence of the N-methyl-d-aspartate-type glutamate receptor antagonist d-(-)-2-amino-5-phosphonopentanoic acid (50 μm) and the GABAA receptor antagonist bicuculline (10 μm). Inhibition by a drug was defined as a reduction to under 90% of control values in the amplitude of synaptic currents. Miniature EPSCs were obtained in the presence of tetrodotoxin (1 μm), and a sliding EPSC template custom, defined using acquisition software, was used to detect and analyze the frequency and amplitude of miniature EPSCs.
Synaptosome Preparations and Western Blot Analysis
The synaptosome preparations and Western blot analysis were performed as described in previous reports (21, 38). For synaptosome preparations, NRM tissues from saline- and CFA-injected rats were homogenized gently in ice-cold 0.32 m sucrose buffer at pH 7.4 and then centrifuged for 10 min at 1000 × g (4 °C). The supernatant was collected and centrifuged for 20 min at 10,000 × g (4 °C). Then, the synaptosomal pellet was resuspended in lysis buffer (0.1% Triton X-100, 150 mm NaCl, 25 mm KCl, and 10 mm Tris-HCl (pH 7.4) with protease inhibitors) at 4 °C for 10 min. The protein concentrations were determined using a Bio-Rad protein assay kit.
For Western blot analysis, total proteins were prepared after tissue lysis and centrifugation for SDS-polyacrylamide gel electrophoresis. The protein was mixed with SDS sample buffer, heated to 95 °C for 10 min, separated under reducing conditions on an 8% or 5% SDS-polyacrylamide gel, and transferred to a nitrocellulose membrane. The membrane was incubated with a polyclonal rabbit antibody for GluA1 (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA), BDNF (1:250, Santa Cruz Biotechnology), β-actin (1:1000, Cell Signaling Technology, Danvers, MA), β-tubulin (1:1000, Cell Signaling Technology), and synapsin 1 (1:5000, Synaptic System, Germany) with agitation overnight at 4 °C and then with horseradish peroxidase-linked secondary antibody (Santa Cruz Biotechnology) for 1 h at room temperature. The bands were detected using enhanced chemiluminescence (GE Healthcare Biosciences). BDNF protein assays were performed with the ChemiKine BDNF sandwich enzyme-linked immunosorbent assay kit (Millipore).
ChIP Assays
ChIP assays were modified from the protocol of the EpiQuik tissue acetyl-histone H3 ChIP kit (Epigentek Group Inc.) as described in previous reports (33, 38). NRM tissues were harvested and immediately cross-linked in 1% formaldehyde for 15–20 min. After washes, the NRM tissue was homogenized through 10–30 strokes in a cell lysis buffer. The homogenate was centrifuged, and the supernatant was removed. The extracted chromatin was sheared by sonication into 200- to 500-bp fragments and diluted 10-fold in ChIP dilution buffer. Then, 10% of the preimmunoprecipitated lysate was used as the input control for normalization. Samples were incubated with an antibody to acetyl histone H3 (Cell Signaling Technology). DNA and histones were dissociated with reverse buffer. Binding buffer was used for DNA precipitation and purification, and elution buffer was used to elute purified DNA from the columns. All buffers were provided in the ChIP kits.
DNA Quantification
Quantitative real-time PCR was performed with a SYBR Green master kit (Applied Biosystems) to measure the amount of cetyl histone H3-associated DNA. Adenine phosphoribosyltransferase (housekeeping mRNA) served as a negative control. Signal difference was calculated as follows: ΔCt = (Nexp − Nave) × Ctave, where Nexp is the normalized Ct value of the target or Cttarget/Ctinput, Nave is the mean N value for control, and Ctave is the mean Ct value for control.
Quantitative Real-time PCR
RNA was extracted with a RNAqueous-4 PCR kit and reverse-transcribed with a RETROscript kit (Applied Biosystems). cDNA was quantified using real-time PCR, and specific cDNA regions of the transcripts were amplified with custom-designed primers (Invitrogen). Fold differences of mRNA levels over controls were calculated with ΔCt. The following primers were used: Bdnf, 5′-GAGGGCTCCTGCTTCTCAA-3′ (forward) and 5′-GCCTTCATGCAACCGAAGT-3′ (reverse); Gapdh, 5′-AGGTCGGTGTGAACGGATTTG-3′ (forward) and 5′-TGTAGACCATGTAGTTGAGGTCA-3′ (reverse).
Microinjection and Behavioral Experiments
Adult rats were used for NRM microinjection and behavioral tests as described in previous reports (33, 34). Each rat was implanted with a 26-gauge, double-guide cannula (Plastics One, Roanoke, VA) aimed at the NRM (anteroposterior, −10.0 mm from the bregma; lateral, 0; and dorsoventral, 10.5 mm from the dura). Drugs were microinjected into the NRM in a total volume of 1 μl through a 33-gauge double injector with an infusion pump at a rate of 0.2 μl/min. All NRM microinjection sites were verified histologically after the experiment by injection of 0.5 μl of a blue dye, and controls of off-site injections were performed as described elsewhere (36). The confined effect of an injected drug within the NRM with this microinjection method has been demonstrated in previous studies (21, 33, 34). The pain threshold was measured every 5 min by paw withdrawal test on a freely moving rat using a Hargreaves analgesia instrument (Stoelting, Wood Dale, IL).
Data Analysis and Materials
Analysis of variance (one-way and two-way) and post hoc analysis were used to statistically analyze experimental data between treatment groups with multiple comparisons. Simple comparisons of data between two groups were made with Student's t tests. Behavioral data with multiple measurements were statistically analyzed by two-way analysis of variance for repeated measures with the Bonferroni method for post hoc tests. Data are presented as mean ± S.E., and p < 0.05 was considered to be statistically significant. All statistical analyses were performed with Prism software version 5.04 (GraphPad Software). Drugs were purchased from Sigma-Aldrich or Tocris Bioscience (Ellisville, MO).
RESULTS
Inflammatory Pain Enhances AMPA Receptor-mediated Synaptic Transmission
First, AMPA receptor-mediated EPSCs were examined in NRM neurons from adult rats with persistent inflammatory pain. CFA induced persistent pain sensitization (hyperalgesia), which reached a maximum and plateau 3 days after injection and lasted more than 6 days (Fig. 1A). Whole-cell voltage clamp recordings of input-output curves for AMPA EPSCs were taken in vitro from neonatal rat NRM neurons in brain stem slices at a holding potential of −60 mV, as shown in Fig. 1B. The average amplitude of currents was higher 3 days after CFA injection than in saline injection, suggesting an increase in AMPA receptor-mediated synaptic transmission in the NRM after persistent peripheral inflammation.
FIGURE 1.
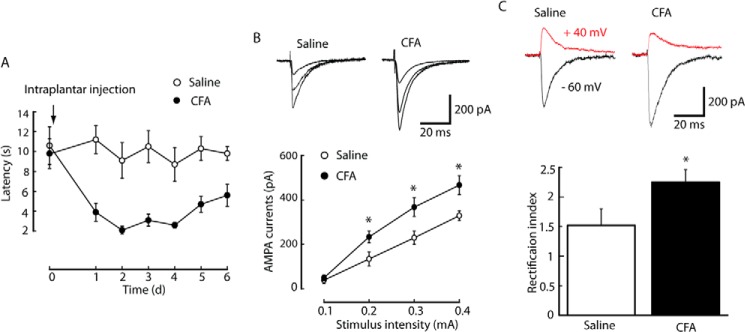
AMPA synaptic transmission in the NRM increases after peripheral inflammation. A, development of persistent pain sensitization induced by CFA and saline controls over time as measured using the paw withdrawal test (n = 5 adult rats/group). d, days. B, representative traces of AMPA EPSCs evoked by increasingly intense stimuli in an NRM neuron (top panel) and an input-output plot of AMPA EPSCs in neurons (bottom panel) in slices from saline-injected (n = 12 neurons) and CFA-injected neonatal rats (n = 18 neurons). C, Representative traces (top panel) and pooled data (bottom panel) of the AMPA EPSC rectification index from the two groups of rats. *, p < 0.05. Data are mean ± S.E.
The inwardly rectifying property of AMPA EPSCs evoked at −60 mV and +40 mV holding potentials was examined to determine whether any alteration of AMPA receptor subunit composition causes the enhanced synaptic transmission observable after inflammation (39). The rectification indices were calculated using the ratio between the currents at −60 mV versus that between the currents at +40 mV. Results showed that the rectification index was significantly higher in NRM neurons from CFA-injected rats (2.25 ± 0.21, n = 26 neurons) than in those from saline-injected control rats (Fig. 1C, 1.52 ± 0.28, n = 18, p < 0.05). The rectification indices may reflect an alteration in the number of GluA1-containing AMPA receptors on the membrane. In this way, this result indicates that the enhanced AMPA receptor-mediated synaptic transmission observable after peripheral inflammation may be caused by changes in synaptic delivery of AMPA receptor subunits (40).
Persistent Inflammation Induces Synaptic Delivery of GluA1
The formation of homomeric AMPA receptors containing GluA1 subunits on the membrane surface is believed to cause an inward rectification of AMPA EPSCs (39). Experiments were conducted to determine the levels of synaptic GluA1 proteins with NRM preparations of synaptosomes, which contain mostly proteins and structures of synaptic membranes with nearly empty cell bodies and greatly reduced intraterminal contents (41). As shown in Fig. 2A, the amount of synaptosomal GluA1 protein was increased to 70% of control in the NRM from CFA-injected rats when compared with that from saline-injected control rats (n = 6 rats/group, p < 0.05). These results suggest that synaptic delivery of GluA1 may contribute to the pain-induced enhancement of AMPA synaptic transmission in the NRM. To test this hypothesis, the phosphorylation level of GluA1 was examined at Ser-831 and Ser-845. These residues were selected because the phosphorylation of both sites has been implicated in synaptic delivery of GluA1 (42, 43). Three days after CFA injection, the total quantity GluA1 protein was similar to that in saline-injected controls, but there was markedly more Ser-831 phosphorylation of GluA1 in the NRM group (Fig. 2B, 145.6 ± 10.8% of control, n = 6, p < 0.05). However, no difference in Ser-845 phosphorylation of GluA1 was observed (Fig. 2C, 112.5 ± 10.6% of control, n = 6, p > 0.05).
FIGURE 2.
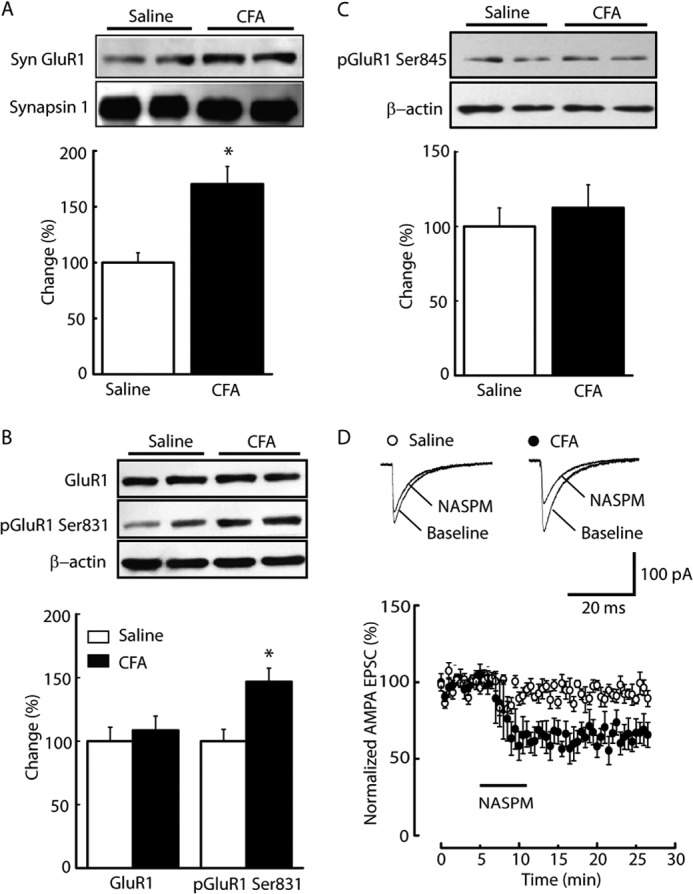
Persistent inflammatory pain increases the level of synaptic GluA1 subunits. A, representative Western blot lanes of synaptosomal (Syn) GluA1 proteins (top panel) in NRM tissues harvested from saline- and CFA-treated groups 3 days after injection (n = 6 rats/group) and pooled data (bottom panel) normalized to synapsin 1. B and C, Western blot analyses of total and phosphorylated GluA1 at Ser-831 (B, pGluA1 Ser-831) and Ser-845 (C, pGluA1 Ser-845) 3 days after CFA or saline injection (n = 6 rats/group). D, AMPA EPSCs (top panel) and summarized results (bottom panel) before and during application of the GluA2-lacking AMPA receptor antagonist 1-naphthyl acetyl spermine (NASPM, 100 μm) in the indicated animal groups (n = 6–10 neurons/group). Data are expressed as mean ± S.E. *, p < 0.05.
To further demonstrate the changes in surface GluA1-containing AMPA receptors after peripheral inflammation, pharmacologic manipulations were performed by inhibiting GluA2-lacking AMPA receptors. Blocking GluA2-lacking AMPA receptors with a bath application of 1-naphthyl acetyl spermine (100 μm) significantly decreased the EPSC amplitude in NRM neurons from CFA-injected rats but not in those from saline-injected rats (Fig. 2D).
BDNF Contributes to Inflammation-induced Enhancement of AMPA EPSCs
One possible factor that causes the enhancement of AMPA receptor-mediated synaptic transmission under pain conditions is BDNF. Exogenous BDNF has been shown to strongly affect AMPA receptor-mediated synaptic transmission (26, 44). The results of our Western blot analysis and ELISA showed there to be statistically significant higher levels of BDNF proteins in the NRM 3 days after CFA injection than in saline-injected control rats (Fig. 3A, 176.9 ± 16.4% of control, n = 6, p < 0.05; ELISA: control, 18.96 ± 5.13 pg/mg; CFA, 35.84 ± 7.16 pg/mg, n = 6, p < 0.05). A similar difference was observed in the BDNF mRNA level (Fig. 3B, 1.92- ± 0.56-fold that of control, n = 5, p < 0.05). Using ChIP assays, the level of histone H3 acetylation at the Bdnf P3 and P4 promoters, two of four Bdnf promoters (P1-P4) were also increased 3 days after CFA injection (Fig. 3C). In this way, persistent peripheral inflammation epigenetically enhances Bdnf transcriptional activity.
FIGURE 3.

Persistent inflammation epigenetically activates the Bdnf gene, and exogenous BDNF increases GluA1-containing AMPA EPCSs in NRM neurons. A and B, levels of BDNF proteins (A) and mRNAs (B) analyzed by Western blotting, ELISA, and RT-PCR in NRM tissues from saline- and CFA-injected rats (n = 5–6 rats/group). GAPDH was used as a control in RT-PCR, and no difference was observed in NRM tissues from saline- and CFA-injected rats. C, summarized data of AcH3 levels on different Bdnf promoter (P1-P4) regions using ChIP assay in NRM tissues from the two groups of rats (n = 6 rats/group). D—F, mEPSCs (D), input-output (E), and rectification index (F) of AMPA EPSCs in NRM neurons from normal rat slices incubated with vehicle or 50 ng/ml BDNF (n = 16–18 neurons/group). The top panels show the representative AMPA current traces, and the bottom panels show the summarized data. Data are mean ± S.E. *, p < 0.05; **, p < 0.01.
Given the critical role of supraspinal BDNF in the descending facilitation of inflammatory pain, the effect of BDNF on AMPA EPSCs was determined in these NRM neurons (20). Both the frequency and amplitude of AMPA miniature EPSCs (mEPSCs) increased after incubation of brain slices from vehicle-injected rats in either 50 ng/ml BDNF or vehicle control for at least 1 h (Fig. 3D; frequency, 178.9 ± 21.2% of control, p < 0.01; amplitude, 146.7 ± 5.8% of control, p < 0.05; n = 18–25 neurons/group). Similar increases were observed in both the averaged amplitude of currents of input-output curves for AMPA EPSCs (Fig. 3E) and the rectification index (Fig. 3F; vehicle, 1.59 ± 0.33, n = 16 neurons; BDNF, 2.52 ± 0.38, n = 18 neurons, p < 0.05).
Next, the effect of BDNF was tested on AMPA EPSCs under pain conditions. Unlike the BDNF effect on vehicle-injected rats, the average amplitude of currents of input-output curves and the rectification index of AMPA EPSCs were not affected by BDNF (50 ng/ml) in NRM neurons from CFA-injected rats (Fig. 4A). However, the TrkB-IgG fusion protein (TrkB-IgG, 1 μg/ml), which acts as a false TrkB receptor to sequester endogenous BDNF, inhibited AMPA EPSCs (Fig. 4A) and the rectification index (Fig. 4B; vehicle, 2.41 ± 0.25, n = 18; BDNF, 2.68 ± 0.31, n = 18; TrkB-IgG, 1.76 ± 0.28, n = 18, p < 0.05 relative to the vehicle group). These results suggest that inflammatory pain-induced up-regulation of BDNF contributes to the enhancement of GluA1-containg AMPA receptor-mediated synaptic transmission in the NRM by activation of TrkB receptors.
FIGURE 4.

Sequestering endogenous extracellular BDNF reduces synaptic delivery of GluA1 under pain conditions. A, representative traces of AMPA EPSCs evoked by increasingly intense stimuli in an NRM neuron (top panel) and an input-output plot of AMPA EPSCs in neurons (bottom panel) from CFA-injected rat slices incubated with vehicle, BDNF (50 ng/ml), or TrkB-IgG fusion proteins (TrkB-IgG, 1 μg/ml). B, the rectification index of AMPA EPSCs in NRM neurons from the three treatment groups in A. Data are mean ± S.E. *, p < 0.05 (n = 18 neurons/group).
Signaling Pathways of Inflammation-induced Synaptic Delivery of AMPA Receptors
A series of intracellular signaling pathways, including PLCγ, PI3K/Akt, Scr, CaM-kinase kinase/transient receptor potential canonical, and mTOR1 have been shown to initiate the membrane incorporation of GluA1 (26, 29–31). Next, signaling pathways that were predominantly involved in GluA1 phosphorylation were identified using TrkB receptor activation under pain conditions by treating slices with a specific inhibitor for a long period (2–4 h). In slices from CFA-injected rats treated with the PLC inhibitor U73122 (2 μm), the maximum number of AMPA EPSCs was lower (vehicle, 642.9 ± 43.1, n = 8; U73122, 468.2 ± 32.4, n = 12, p < 0.05) (Fig. 5, A and B). The same was true of treatment of the slices with the PKC inhibitor GF109203X (3 μm, 410.6 ± 49.8, n = 11, p < 0.05) or the diacylglycerol (DAG) antagonist 1-O-hexadecyl-2-O-acetyl-glycerol (100 μm, 434.7 ± 50.3, n = 12, p < 0.05). A similar increase was observed in the rectification index of AMPA EPSC (vehicle, 2.36 ± 2.3, n = 8; U73122, 1.46 ± 0.51, n = 12, p < 0.05; GF109203X, 1.55 ± 0.24, n = 11, p < 0.05; 1-O-hexadecyl-2-O-acetyl-glycerol, 1.62 ± 0.35, n = 12, p < 0.05) (Fig. 5, C and D). This suggests that PLCγ-PKC pathways may be involved in the enhancement of AMPA receptor-mediated synaptic transmission after inflammation. To test this hypothesis, changes in GluA1 phosphorylation after NRM infusion of the inhibitor from CFA-injected rats were assessed. As shown in Fig. 6, the level of GluA1 phosphorylation at Ser-831 was significantly lower than in the vehicle group 6 h after microinjection of TrkB-IgG (100 ng, 70.9 ± 12.7%, n = 6, p < 0.05), GF109203 (0.5 ng, 63.7 ± 13.9%, n = 6, p < 0.05) or GF109203 (100 ng, 47.6 ± 15.8%, n = 6, p < 0.05) but the total level of GluA1 was not.
FIGURE 5.

PLC-PKC pathways are required for BDNF-induced synaptic delivery of GluA1 after inflammation. A and B, representative input-output (A) and rectification index (B) of AMPA EPSCs (left panels) and group data (right panels) in NRM neurons from CFA-injected rat slices incubated with vehicle, the PLC inhibitor U73122 (2 μm), the PKC inhibitor GF109203X (3 μm), or the DAG antagonist 1-O-hexadecyl-2-O-acetyl-glycerol (HAG, 100 μm) (n = 11–12 neurons/group). Data are mean ± S.E. *, p < 0.05.
FIGURE 6.
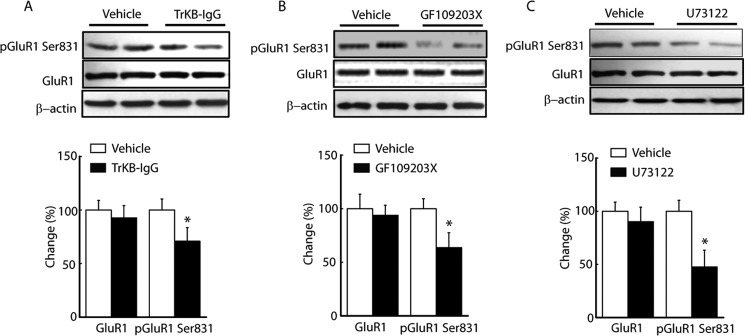
BDNF-induced GluA1 phosphorylation after inflammation requires PLC-PKC pathway activation. A–C, Western blot (top panels) and summarized data (bottom panel) of NRM GluA1 and pGluR1 Ser-831 in CFA-injected rats treated by NRM infusion of TrkB-IgG (A, 100 ng), the PLC inhibitor U73122 (B, 0.5 ng), or the PKC inhibitor GF109203X (C, 100 ng) (n = 6 rats/group). Data are mean ± S.E. *, p < 0.05.
DISCUSSION
The NRM is a supraspinal site on the brain stem-descending pathways and is crucial to pain modulation. It plays an important role in the development and maintenance of the behavioral state of sensitized pain and opioid analgesia under chronic pain conditions (45, 46). It functions by receiving integrated inputs from higher brain sites through connections with the PAG and by projecting directly to pain-transmitting neurons in the dorsal horn of the spinal cord (47). In this way, neurons in RVM, including the NRM, can either inhibit or facilitate spinal pain transmission in different behavioral states. Regarding excitatory glutamatergic systems, AMPA receptor-mediated synaptic transmission is enhanced in response to inflammatory hyperalgesia in the RVM, contributing to enhanced descending pain modulation (48–50). Hypofunction of AMPA synaptic transmission has also been observed in the PAG (51, 52). Our results support this hypothesis and demonstrate that inflammatory pain-induced up-regulation of BDNF contributes to the enhancement of excitatory synaptic transmission in the NRM.
Synaptic delivery of GluA1 into the postsynaptic membrane facilitates AMPA synaptic transmission by increasing single-channel conductance. It also plays a role in synaptic plasticity (53, 54). In this study, a significant increase in AMPA inward current rectification and GluA1 protein levels was observed in the synaptosome preparations 3 days after inflammation (Figs. 1C and Fig. 2A). This indicated that recruitment of GluA1-containing calcium-permeable AMPA receptors on the surface of the postsynaptic membrane may cause enhancement of AMPA synaptic strength in the NRM under pain conditions (39, 40). Pharmacological results showed that the GluA2-lacking AMPA receptor inhibitor 1-naphthyl acetyl spermine produced more inhibition of AMPA EPSCs in CFA-injected rats than that in saline-injected rats (Fig. 2B). This supports the conclusion that inflammatory pain can induce synaptic delivery of GluA1.
The delivery of GluA1 subunits to synapse is promoted by phosphorylation in the C-terminal region (42, 43). The present study shows that inflammation can increase the phosphorylation of GluA1 Ser-831, but not GluA1 Ser-845, which facilitates the trafficking of GluA1 in NRM pain modulatory circuits under pain conditions. This is consistent with the results of previous works showing that both 30 min and 7 days of inflammation increase GluA1 phosphorylation at Ser-831 (55). The mechanism underlying the critical role of GluA1 phosphorylation associated with the functional AMPA receptors in NRM neurons has not yet been addressed under inflammatory pain conditions.
BDNF can up-regulate the total levels of GluA1-GluA4 and promote the trafficking of GluA1 into the synapse (26, 28, 31, 56). In the spinal cord, BDNF released from microglia acts as a signaling link between microglia and dorsal horn neurons for neuronal hyperexcitability under neuropathic pain conditions (57, 58). In the brain stem-descending pain-modulating pathway, the BDNF in the RVM is likely originated from BDNF-containing neurons in the PAG. BDNF activation of TrkB signaling in the RVM underlies the development of persistent pain (20). From a molecular standpoint, persistent inflammation was found to epigenetically up-regulate BDNF protein expression (Fig. 3). A previous study showed that the characteristics of chronic pain are strongly suggestive of epigenetic modulations in the NRM (33). This indicates a positive correlation with the enhanced AMPA synaptic transmission. Under normal conditions, exogenous BDNF can cause increases in the number of AMPA EPSCs and rectification in NRM neurons. There was no detectable effect under persistent inflammation conditions. Interestingly, under pain conditions, chelation of endogenous extracellular BDNF decreased AMPA EPSCs and rectification in vitro and GluA1 phosphorylation in vivo (Figs. 4–6), suggesting a positive correlation between BDNF and GluA1 delivery after inflammation. Previous studies confirmed that synaptic delivery of GluA1 induced by BDNF could be blocked by inactivation of BDNF/TrkB signaling in cultured hippocampal neurons (26). These results also suggest that persistent inflammation up-regulates the activity of endogenous BDNF to some extent, thereby preventing exogenous addition of BDNF under pain conditions.
Many intracellular signaling cascades related to GluA1 phosphorylation are initiated after activation of BDNF/TrkB signaling. These include PLCγ, PI3K/Akt, Scr, CaM-kinase kinase/transient receptor potential canonical, calmodulin-dependent protein kinase II (CaMKII), and mTOR (26, 29–31, 59). In the RVM, under pain conditions, inositol 1,4,5-trisphosphate and PKC are involved in BDNF activation of TrkB (20). This event is probably initiated by inositol 1,4,5-trisphosphate (IP3) and DAG formation through PLCγ activation, the release of intracellular calcium, and PKC activation (16). In this study, electrophysiological studies show that inflammation-induced increases in AMPA currents and rectification were both inhibited by blockage of PLC, PKC, and DAG (Fig. 5). Microinjection of these inhibitors into the NRM in vivo decreased GluA1 phosphorylation at Ser-831 under pain conditions (Fig. 6). These results suggest that synaptic delivery of GluA1 induced by BDNF is mediated by PLC-PKC signaling in descending pain modulatory circuitry after inflammation. In summary, these results demonstrate that persistent inflammation can epigenetically up-regulate BDNF expression, therefore initiating the delivery of GluA1 subunits to the synapse in the NRM via PLC-PKC signaling after TrkB activation.
This work was supported by 973 Program Grant 2014CB548100 and by National Natural Science Foundation of China Grants 31100802 and 91332109 (to Z. Z.).
- PAG
- periaqueductal gray
- RVM
- rostral ventromedial medulla
- NRM
- nucleus raphe mangus
- PLC
- phospholipase C
- mTOR
- mammalian target of rapamycin
- CFA
- complete Freund adjuvant
- EPSC
- excitatory postsynaptic current.
REFERENCES
- 1. Osikowicz M., Mika J., Przewlocka B. (2013) Complement-derived anaphylatoxin C5a protects against glutamate-mediated neurotoxicity. Exp. Physiol. 98, 372–38423002244 [Google Scholar]
- 2. Block F., Habermeyer B. (2003) Glutamate antagonists for treatment of neuropathic pain. Schmerz 17, 261–267 [DOI] [PubMed] [Google Scholar]
- 3. Bleakman D., Alt A., Nisenbaum E. S. (2006) Glutamate receptors and pain. Semin. Cell Dev. Biol. 17, 592–604 [DOI] [PubMed] [Google Scholar]
- 4. Descalzi G., Kim S., Zhuo M. (2009) Presynaptic and postsynaptic cortical mechanisms of chronic pain. Mol. Neurobiol. 40, 253–259 [DOI] [PubMed] [Google Scholar]
- 5. Sah P., Nicoll R. A. (1991) Mechanisms underlying potentiation of synaptic transmission in rat anterior cingulate cortex in vitro. J. Physiol. 433, 615–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wei F., Li P., Zhuo M. (1999) Loss of synaptic depression in mammalian anterior cingulate cortex after amputation. J. Neurosci. 19, 9346–9354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wenthold R. J., Petralia R. S., Blahos J., 2nd, Niedzielski A. S. (1996) Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J. Neurosci. 16, 1982–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu B., Descalzi G., Ye H. R., Zhuo M., Wang Y. W. (2012) Translational investigation and treatment of neuropathic pain. Mol. Pain 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tao Y. X. (2012) AMPA receptor trafficking in inflammation-induced dorsal horn central sensitization. Neurosci. Bull. 28, 111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garry E. M., Fleetwood-Walker S. M. (2004) A new view on how AMPA receptors and their interacting proteins mediate neuropathic pain. Pain 109, 210–213 [DOI] [PubMed] [Google Scholar]
- 11. Bie B., Brown D. L., Naguib M. (2011) Increased synaptic GluR1 subunits in the anterior cingulate cortex of rats with peripheral inflammation. Eur. J. Pharmacol. 653, 26–31 [DOI] [PubMed] [Google Scholar]
- 12. Xu H., Wu L. J., Wang H., Zhang X., Vadakkan K. I., Kim S. S., Steenland H. W., Zhuo M. (2008) Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J. Neurosci. 28, 7445–7453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Galan A., Laird J. M., Cervero F. (2004) In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain 112, 315–323 [DOI] [PubMed] [Google Scholar]
- 14. Park H., Poo M. M. (2013) Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23 [DOI] [PubMed] [Google Scholar]
- 15. Chao M. V., Bothwell M. (2002) Neurotrophins: to cleave or not to cleave. Neuron 33, 9–12 [DOI] [PubMed] [Google Scholar]
- 16. Pezet S., McMahon S. B. (2006) Neurotrophins: mediators and modulators of pain. Annu. Rev. Neurosci. 29, 507–538 [DOI] [PubMed] [Google Scholar]
- 17. Merighi A., Salio C., Ghirri A., Lossi L., Ferrini F., Betelli C., Bardoni R. (2008) BDNF as a pain modulator. Prog. Neurobiol. 85, 297–317 [DOI] [PubMed] [Google Scholar]
- 18. Conner J. M., Lauterborn J. C., Yan Q., Gall C. M., Varon S. (1997) Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J. Neurosci. 17, 2295–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. King V. R., Michael G. J., Joshi R. K., Priestley J. V. (1999) trkA, trkB, and trkC messenger RNA expression by bulbospinal cells of the rat. Neuroscience 92, 935–944 [DOI] [PubMed] [Google Scholar]
- 20. Guo W., Robbins M. T., Wei F., Zou S., Dubner R., Ren K. (2006) Supraspinal brain-derived neurotrophic factor signaling: a novel mechanism for descending pain facilitation. J. Neurosci. 26, 126–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Z., Wang X., Wang W., Lu Y. G., Pan Z. Z. (2013) Brain-derived neurotrophic factor-mediated downregulation of brainstem K+-Cl- cotransporter and cell-type-specific GABA impairment for activation of descending pain facilitation. Mol. Pharmacol. 84, 511–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trang T., Beggs S., Salter M. W. (2011) Brain-derived neurotrophic factor from microglia: a molecular substrate for neuropathic pain. Neuron Glia Biol. 7, 99–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamada T., Zuo D., Yamamoto T., Olszewski R. T., Bzdega T., Moffett J. R., Neale J. H. (2012) NAAG peptidase inhibition in the periaqueductal gray and rostral ventromedial medulla reduces flinching in the formalin model of inflammation. Mol. Pain 8, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gärtner A., Polnau D. G., Staiger V., Sciarretta C., Minichiello L., Thoenen H., Bonhoeffer T., Korte M. (2006) Hippocampal long-term potentiation is supported by presynaptic and postsynaptic tyrosine receptor kinase B-mediated phospholipase Cγ signaling. J. Neurosci. 26, 3496–3504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Du J. L., Wei H. P., Wang Z. R., Wong S. T., Poo M. M. (2009) Long-range retrograde spread of LTP and LTD from optic tectum to retina. Proc. Natl. Acad. Sci. U.S.A. 106, 18890–18896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Caldeira M. V., Melo C. V., Pereira D. B., Carvalho R., Correia S. S., Backos D. S., Carvalho A. L., Esteban J. A., Duarte C. B. (2007) Brain-derived neurotrophic factor regulates the expression and synaptic delivery of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J. Biol. Chem. 282, 12619–12628 [DOI] [PubMed] [Google Scholar]
- 27. Slipczuk L., Bekinschtein P., Katche C., Cammarota M., Izquierdo I., Medina J. H. (2009) BDNF activates mTOR to regulate GluR1 expression required for memory formation. PloS ONE 4, e6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schratt G. M., Nigh E. A., Chen W. G., Hu L., Greenberg M. E. (2004) BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J. Neurosci. 24, 7366–7377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakata H., Nakamura S. (2007) Brain-derived neurotrophic factor regulates AMPA receptor trafficking to post-synaptic densities via IP3R and TRPC calcium signaling. FEBS Lett. 581, 2047–2054 [DOI] [PubMed] [Google Scholar]
- 30. Huang E. J., Reichardt L. F. (2003) Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642 [DOI] [PubMed] [Google Scholar]
- 31. Fortin D. A., Srivastava T., Dwarakanath D., Pierre P., Nygaard S., Derkach V. A., Soderling T. R. (2012) Brain-derived neurotrophic factor activation of CaM-kinase kinase via transient receptor potential canonical channels induces the translation and synaptic incorporation of GluA1-containing calcium-permeable AMPA receptors. J. Neurosci. 32, 8127–8137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Z., Pan Z. Z. (2010) Synaptic mechanism for functional synergism between δ- and μ-opioid receptors. J. Neurosci. 30, 4735–4745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Z., Cai Y. Q., Zou F., Bie B., Pan Z. Z. (2011) Epigenetic suppression of GAD65 expression mediates persistent pain. Nat. Med. 17, 1448–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang L., Hammond D. L. (2010) Cellular basis for opioid potentiation in the rostral ventromedial medulla of rats with persistent inflammatory nociception. Pain 149, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma J., Pan Z. Z. (2006) Contribution of brainstem GABAA synaptic transmission to morphine analgesic tolerance. Pain 122, 163–173 [DOI] [PubMed] [Google Scholar]
- 36. Bie B., Peng Y., Zhang Y., Pan Z. Z. (2005) cAMP-mediated mechanisms for pain sensitization during opioid withdrawal. J. Neurosci. 25, 3824–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pan Z. Z., Tershner S. A., Fields H. L. (1997) Cellular mechanism for anti-analgesic action of agonists of the κ-opioid receptor. Nature 389, 382–385 [DOI] [PubMed] [Google Scholar]
- 38. Bie B., Wang Y., Cai Y. Q., Zhang Z., Hou Y. Y., Pan Z. Z. (2012) Upregulation of nerve growth factor in central amygdala increases sensitivity to opioid reward. Neuropsychopharmacology 37, 2780–2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hayashi Y., Shi S. H., Esteban J. A., Piccini A., Poncer J. C., Malinow R. (2000) Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287, 2262–2267 [DOI] [PubMed] [Google Scholar]
- 40. Shi S., Hayashi Y., Esteban J. A., Malinow R. (2001) Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105, 331–343 [DOI] [PubMed] [Google Scholar]
- 41. Dunkley P. R., Jarvie P. E., Robinson P. J. (2008) A rapid Percoll gradient procedure for preparation of synaptosomes. Nat. Protoc. 3, 1718–1728 [DOI] [PubMed] [Google Scholar]
- 42. Lee H. K., Barbarosie M., Kameyama K., Bear M. F., Huganir R. L. (2000) Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405, 955–959 [DOI] [PubMed] [Google Scholar]
- 43. Lee H. K., Takamiya K., Han J. S., Man H., Kim C. H., Rumbaugh G., Yu S., Ding L., He C., Petralia R. S., Wenthold R. J., Gallagher M., Huganir R. L. (2003) Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112, 631–643 [DOI] [PubMed] [Google Scholar]
- 44. Boulanger L., Poo M. M. (1999) Gating of BDNF-induced synaptic potentiation by cAMP. Science 284, 1982–1984 [DOI] [PubMed] [Google Scholar]
- 45. Fields H. (2004) State-dependent opioid control of pain. Nat. Rev. Neurosci. 5, 565–575 [DOI] [PubMed] [Google Scholar]
- 46. Porreca F., Ossipov M. H., Gebhart G. F. (2002) Chronic pain and medullary descending facilitation. Trends Neurosci. 25, 319–325 [DOI] [PubMed] [Google Scholar]
- 47. Basbaum A. I., Bautista D. M., Scherrer G., Julius D. (2009) Cellular and molecular mechanisms of pain. Cell 139, 267–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guan Y., Terayama R., Dubner R., Ren K. (2002) Plasticity in excitatory amino acid receptor-mediated descending pain modulation after inflammation. J. Pharmacol. Exp. Ther. 300, 513–520 [DOI] [PubMed] [Google Scholar]
- 49. Guan Y., Guo W., Zou S. P., Dubner R., Ren K. (2003) Changes in AMPA receptor phosphorylation in the rostral ventromedial medulla after inflammatory hyperalgesia in rats. Pain 104, 401–413 [DOI] [PubMed] [Google Scholar]
- 50. Radhakrishnan R., Sluka K. A. (2009) Increased glutamate and decreased glycine release in the rostral ventromedial medulla during induction of a pre-clinical model of chronic widespread muscle pain. Neurosci. Lett. 457, 141–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ho Y. C., Cheng J. K., Chiou L. C. (2013) Hypofunction of glutamatergic neurotransmission in the periaqueductal gray contributes to nerve-injury-induced neuropathic pain. J. Neurosci. 33, 7825–7836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Starowicz K., Maione S., Cristino L., Palazzo E., Marabese I., Rossi F., de Novellis V., Di Marzo V. (2007) Tonic endovanilloid facilitation of glutamate release in brainstem descending antinociceptive pathways. J. Neurosci. 27, 13739–13749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barria A., Muller D., Derkach V., Griffith L. C., Soderling T. R. (1997) Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 276, 2042–2045 [DOI] [PubMed] [Google Scholar]
- 54. Mammen A. L., Kameyama K., Roche K. W., Huganir R. L. (1997) Phosphorylation of the α-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J. Biol. Chem. 272, 32528–32533 [DOI] [PubMed] [Google Scholar]
- 55. Guan Y., Guo W., Robbins M. T., Dubner R., Ren K. (2004) Inflammation-induced upregulation of AMPA receptor subunit expression in brain stem pain modulatory circuitry. Neurosci. Lett. 366, 201–205 [DOI] [PubMed] [Google Scholar]
- 56. Narisawa-Saito M., Carnahan J., Araki K., Yamaguchi T., Nawa H. (1999) Brain-derived neurotrophic factor regulates the expression of AMPA receptor proteins in neocortical neurons. Neuroscience 88, 1009–1014 [DOI] [PubMed] [Google Scholar]
- 57. Biggs J. E., Lu V. B., Stebbing M. J., Balasubramanyan S., Smith P. A. (2010) Is BDNF sufficient for information transfer between microglia and dorsal horn neurons during the onset of central sensitization?. Mol. Pain 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ferrini F., De Koninck Y. (2013) Microglia control neuronal network excitability via BDNF signalling. Neural Plast. 2013, 429815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gähwiler B. H., Capogna M., Debanne D., McKinney R. A., Thompson S. M. (1997) Organotypic slice cultures: a technique has come of age. Trends Neurosci. 20, 471–477 [DOI] [PubMed] [Google Scholar]