

Background: Tight control of cyclin D1 expression is critical for intestinal homeostasis.
Results: Whereas PKCα suppresses cyclin D1 expression, PKCϵ up-regulates cyclin D1 via an ERK and NF-κB/CRE-mediated transcriptional mechanism.
Conclusion: Cyclin D1 levels in intestinal cells reflect a balance between PKCα and PKCϵ signaling.
Significance: The opposing effects of PKCα and PKCϵ on cyclin D1 accumulation reflect their contrasting contributions to intestinal tumorigenesis.
Keywords: Colon Cancer, Cyclin D1, Extracellular Signal-regulated Kinase (ERK), Intestine, NF-kappaB, Protein Kinase C (PKC), Cyclic AMP-response Element (CRE), PKCalpha, PKCepsilon
Abstract
Cellular accumulation of cyclin D1, a key regulator of cell proliferation and tumorigenesis, is subject to tight control. Our previous studies have identified PKCα as a negative regulator of cyclin D1 in the intestinal epithelium. However, treatment of non-transformed IEC-18 ileal crypt cells with PKC agonists has a biphasic effect on cyclin D1 expression. Initial PKCα-mediated down-regulation is followed by recovery and subsequent accumulation of the cyclin to levels markedly higher than those seen in untreated cells. Using protein overexpression strategies, siRNA, and pharmacological inhibitors, we now demonstrate that the recovery and hyperinduction of cyclin D1 reflect the combined effects of (a) loss of negative signals from PKCα due to agonist-induced PKCα down-regulation and (b) positive effects of PKCϵ. PKCϵ-mediated up-regulation of cyclin D1 requires sustained ERK stimulation and transcriptional activation of the proximal cyclin D1 (CCDN1) promoter, without apparent involvement of changes in protein stability or translation. PKCϵ also up-regulates cyclin D1 expression in colon cancer cells, through mechanisms that parallel those in IEC-18 cells. Although induction of cyclin D1 by PKCϵ is dependent on non-canonical NF-κB activation, the NF-κB site in the proximal promoter is not required. Instead, cyclin D1 promoter activity is regulated by a novel interaction between NF-κB and factors that associate with the cyclic AMP-response element adjacent to the NF-κB site. The differential effects of PKCα and PKCϵ on cyclin D1 accumulation are likely to contribute to the opposing tumor-suppressive and tumor-promoting activities of these PKC family members in the intestinal epithelium.
Introduction
Cyclin D1 (CCND1) is a proto-oncogene and critical regulator of cell proliferation, acting as a major mitogen sensor that links cellular signaling networks to the cell cycle machinery (1, 2). The best characterized role of cyclin D1 is as the regulatory subunit of cyclin-dependent kinases 4 and 6. Cyclin D1 both facilitates cyclin-dependent kinase-mediated phosphorylation and inactivation of the tumor-suppressive pocket proteins (retinoblastoma protein pRb, p107, and p130) and sequesters CIP/KIP cyclin-dependent kinase-inhibitory proteins (p21Cip1, p27Kip1, and p57Kip2) away from cyclin·cyclin-dependent kinase 2 complexes (2). In addition, cyclin D1 promotes cell proliferation through interaction with transcription factors such as the estrogen receptor and Sp1 (1, 3). Because of these effects, cell proliferation is extremely sensitive to alterations in the levels of cyclin D1, and even modest changes in its expression can have appreciable effects on cell cycle progression (4–7). Thus, cellular accumulation of cyclin D1 is under tight control, and its expression is regulated at multiple levels, including gene transcription, mRNA transport and stability (8–10), translation (11), and protein degradation (9). Disruption of these regulatory mechanisms is a common feature of multiple cancer types, with overexpression of cyclin D1 being among the most frequent alterations observed in tumors (1). Many oncogenic and tumor-suppressive signals converge on cyclin D1, altering its expression by enhancing transcription through distinct DNA sequences in its promoter as well as through changes in translation and protein degradation (1, 2, 8).
The intestinal epithelium is one of the most rapidly proliferating tissues in the body, and maintenance of mucosal homeostasis is dependent on precise control of cellular proliferation within intestinal crypts. Increased proliferation leads to crypt elongation and, eventually, adenoma formation and tumor progression. Overexpression of cyclin D1 is an early event in intestinal tumorigenesis (12), and robust expression of this cyclin is important for intestinal cell proliferation and maintenance of the transformed phenotype in mouse models and human colon cancer cells (13–15). Although gene amplification drives cyclin D1 overexpression in many cancer types (2), gains and amplifications of CCND1 are rare in colon cancer (16), pointing to disruption of signal transduction pathways upstream of cyclin D1. Thus, the identification of signaling pathways that impact cyclin D1 expression in the normal intestine and in colon cancer cells is critical for understanding of mechanisms underlying intestinal homeostasis and colon cancer development.
In previous studies, we have determined that protein kinase C (PKC) is an important regulator of cyclin D1 expression in the normal intestine and during intestinal tumorigenesis (11, 14, 17–19). PKC comprises a family of at least 10 isozymes that have emerged as key regulators of cell proliferation and tumorigenesis in multiple tissues (20). PKC isozymes have been grouped into subfamilies based on differences in structure and cofactor requirements. Classical PKCs (PKCα, PKCβI, PKCβII, and PKCγ) require diacylglycerol and Ca2+ for activity; novel PKCs (PKCδ, PKCϵ, PKCη, and PKCθ) are activated by diacylglycerol but do not require Ca2+; and atypical PKCs (PKCι/λ and PKCζ) are activated by protein-protein interactions rather than by diacylglycerol (cf. Ref. 21). Physiological activation of classical PKCs and novel PKCs occurs following receptor-mediated activation of phospholipase C, which cleaves phosphatidylinositol 4,5-bisphosphate to generate diacylglycerol and inositol 3-phosphate. This leads to recruitment of these enzymes to the plasma membrane, where they undergo conformational changes that result in activation. A number of pharmacological activators, such as phorbol esters and bryostatins, can also recruit classical PKCs and novel PKCs to the membrane through interaction with the diacylglycerol binding site in their C1 regulatory domain (21). Our previous studies have determined that PKCα signaling rapidly inhibits cyclin D1 expression in intestinal epithelial cells through both translational and transcriptional mechanisms. Translational suppression involves PP2A-dependent activation of the translational repressor 4E-BP14 (11, 18), whereas blockade of transcription involves down-regulation of the transcriptional regulator inhibitor of DNA binding 1 (Id1) and is mediated by promoter elements between −163 and −1745 from the transcriptional start site (14, 19). The physiological relevance of these effects is underscored by the fact that genetic knockout of PKCα in mice leads to increased cyclin D1 expression in intestinal crypts (19). Consistent with this effect, PKCα acts as a tumor suppressor in the intestine (14, 22), and its ability to down-regulate cyclin D1 is an important aspect of its tumor-suppressive activity (14). Other members of the PKC family (e.g. PKCϵ and PKCι) appear to function as oncogenes in the intestine (23, 24), pointing to potential positive regulation of cyclin D1 by PKCs in this tissue.
In the current study, we further analyze the regulation of cyclin D1 in non-transformed intestinal epithelial cells and colon cancer cells and identify PKCϵ as a positive regulator of cyclin D1 accumulation in this system. Our findings demonstrate that the opposing effects of PKCϵ and PKCα on cyclin D1 levels involve distinct mechanisms, with PKCϵ promoting transcriptional up-regulation of the cyclin mediated by an interaction between NF-κB and factors that bind to the cyclic AMP-response element (CRE) in the cyclin D1 gene promoter.
EXPERIMENTAL PROCEDURES
Cell Culture and Drug Treatments
IEC-18 non-transformed rat intestinal epithelial cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum (FBS), 4 mm l-glutamine, and 0.15% insulin. Human colorectal cancer cell lines FET, GEO, and DLD1 (obtained from Dr. M. G. Brattain (University of Nebraska Medical Center) and Dr. Ralph Bernacki (Roswell Park Cancer Institute)) were cultured in RPMI 1640, 10% FBS, and 2 mm l-glutamine. Cells were maintained in a humidified 5% CO2 atmosphere at 37 °C. For PKC agonist treatment, cells were exposed to 100 nm phorbol 12-myristate 13-acetate (PMA) (Sigma), 100 nm bryostatin 1 (Biomol), or 20 μg/ml 1,2-dioctanoyl-sn-glycerol (DiC8) (Sigma). DiC8 was added repeatedly in fresh medium (every hour) to compensate for its rapid metabolism in cells. Where indicated, 5 μm bisindolylmaleimide I (BIM), 1 μm Gö6983, 2 μm Gö6976, 10 μm Rö32-0432 (Calbiochem), 30 μg/ml cycloheximide, 5 μg/ml actinomycin D (Sigma), or 0.1–2.0 μm 3-(1-(3-imidazol-1-ylpropyl)-1H-indol-3-yl)-4-anilino-1H-pyrrole-2,5-dione (Calbiochem) was added 30 min prior to PKC agonists. 100 μm pyrrolidine dithiocarbamate (PDTC) and 100 μm caffeic acid phenethyl ester (CAPE) (Tocris Bioscience) were added 60 min prior to PKC agonist treatment. U0126, used at 10 μm (IEC-18 cells) or 20 μm (DLD1 cells), and 50 μm PD98059 (Alexis) were added 30 or 60 min prior to PMA or at various times during PMA treatment, as indicated. PMA and bryostatin 1 were dissolved in ethanol, and all other drugs were dissolved in DMSO. Control cells were treated with the appropriate vehicle (final concentration of ethanol and DMSO in the medium was ≤0.1 and ≤0.2%, respectively).
Western Blot Analysis
Cells were lysed in 1% SDS, 10 mm Tris-HCl, pH 7.4, and equal amounts of cellular protein were separated by SDS-PAGE, transferred to nitrocellulose membranes, and subjected to immunoblot analysis as we have described (17, 25). Evenness of loading and transfer was routinely confirmed by staining membranes with 0.1% Fast Green (Sigma). Band intensities were quantified by analysis of scanned blots using ImageJ software (National Institutes of Health), and data are presented relative to control (mean ± S.E.).
Antibodies used were as follows: anti-PKCα (Santa Cruz Biotechnology, Inc., sc-8393), 1:1000; anti-PKCα (Epitomics, 1510-1), 1:5000; anti-PKCβII (Santa Cruz Biotechnology, sc-210), 1:1000; anti-PKCδ (Santa Cruz Biotechnology, sc-213), 1:2000; anti-PKCϵ (Santa Cruz Biotechnology, sc-214), 1:2000; anti-phospho-ERK1/2 (Cell Signaling, 9106), 1:2000; anti-total ERK1/2 (Cell Signaling, 9102), 1:2000; anti-cyclin D1 (NeoMarkers, RM-9104-S), 1:500; anti-4E-BP1 (Chemicon, AB3251), 1:2000; anti-β-actin (Sigma-Aldrich, A2066), 1:10,000; anti-eEF2α (Cell Signaling), 1:2000; anti-eIF4E (Cell Signaling), 1:2000; goat anti-rabbit HRP antibody (Millipore, AP132P), 1:2000; goat anti-mouse IgM-HRP antibody (Santa Cruz Biotechnology, sc-2973), 1:5000.
Quantitative Real-time PCR
Total RNA was purified with IllustraSpin (GE Healthcare), Versagene (Gentra), or RNeasy Mini (Qiagen) kits according to the manufacturer's protocol. Analysis was performed using the 1-Step Brilliant II SYBR Green QRT-PCR Master Mix kit (Agilent Technologies) and the 7300 Real-time PCR system (Applied Biosystems). Primers used were as follows: human cyclin D1, GTCTTCCCGCTGGCCATGAACTAC and GGAAGCGTGTGAGGCGGTAGTAGG; rat cyclin D1, GTCTTCCCGCTGGCCATGAACTAC and AAGAAAGTGCGTTGTGCGGTAGCA; 18 S rRNA, CATTGGAGGGCAAGTCTGGTG and CTCCCAAGATCCAACTACGAG.
Adenoviral Transduction
Adenoviruses expressing LacZ, PKCα, or PKCδ were from Drs. T. Kuroki (Kobe University, Japan) and M. G. Kazanietz (University of Pennsylvania), and adenoviruses expressing LacZ or PKCϵ (26) were from Dr. P. J. McDermott (Medical University of South Carolina). Cells were transduced with adenovirus at a multiplicity of infection of 10–20, as we have described (14). Drug treatments and analysis were performed 48 h after transduction.
Small Interfering RNA (siRNA)-mediated Knockdown
siRNAs targeting rat PKCϵ (AAGTGCGCTGGGCTAAAGAAA) and non-targeting siRNA (non-silencing control sequence, 1027281) were purchased from Qiagen. IEC-18 cells were transfected with siRNA at a 100 nm final concentration using Lipofectamine 2000 (Invitrogen), as we have described (27). Cells were treated and harvested 48 h post-transfection.
Metabolic Labeling
Labeling of IEC-18 cells with [35S]methionine/cysteine and analysis of cyclin D1 synthesis by immunoprecipitation, SDS-PAGE, and phosphorimaging were as we have described (11).
Plasmids and Promoter Mutation
The human cyclin D1 promoter-luciferase reporter constructs containing sequences corresponding to the proximal cyclin D1 promoter extending to 1745 bp (−1745CD1Luc), 963 bp (−963CD1Luc), or 163 bp (−163CD1Luc) upstream of the transcription initiation site were from Dr. Richard Pestell (Thomas Jefferson University). The NF-κB activity reporter construct (NF-κB-luciferase) containing three NF-κB binding sites upstream of the minimal CMV promoter and the dominant active IκBα expression and control constructs were obtained from Drs. Andrei Bakin and Andrei Gudkov (Roswell Park Cancer Institute).
Mutant cyclin D1-luciferase constructs were generated using the QuikChange site-directed mutagenesis kit (Agilent). The following binding sites for transcription factors in the promoter were mutated individually: NF-κB, CRE, TCF/LEF, Sp1, Egr1, and E2F. For the promoters containing dual mutation in the NF-κB binding site and the CRE or in both Egr1 sites (Egr1a+b), binding sites were mutated sequentially. The primer used for the Sp1 site mutation inactivates both Sp1 sites in the promoter. Although the Sp1 mutation also introduces a mutation into the Egr1b site, this site is expected to retain the ability to bind Egr1 (28). All mutations were confirmed by sequencing the resultant plasmids. Primers used to introduce the mutations were as follows: NF-κB, CAGTAACGTCACACGGACTACACCCGAGTTTTGTTGAAG and CTTCAACAAAACTCGGGTGTAGTCCGTGTGACGTTACTG; CRE, GCTTAACAACAGTCGCGTCCCCCGGGCTACAGGGGAGTTTTGTTG and CAACAAAACTCCCCTGTAGCCCGGGGGACGCGACTGTTGTTAAGC; SP1, GCCCCCTCCCCCTGCGCATGCCCATGCCCCCCTCCCGCTC and GGGAGCGGGAGGGGGGCATGGGCATGCGCAGGGGGAGGGGGC; E2F, CGCTGCTCCCGGCGTTTCTTGACCGCGCCCCCTCCCCCTGCG and CGCAGGGGGAGGGGGCGCGGTCAAGAAACGCCGGGAGCAGCG; Egr1a, TGGCGCCCGCGTTTTCTCCCCCTGCGCCCGCC and GGCGGGCGCAGGGGGAGAAAACGCGGGCGCCA; Egr1b, CCCCCTGCGCCCGTTTTCGCCCCCCTCCCGC and GCGGGAGGGGGGCGAAAACGGGCGCAGGGGG; TCF/LEF, CCATTCTCTGCCGGGCTTTAGCCTTTGCTTAACAACAGTA and TACTGTTGTTAAGCAAAGGCTAAAGCCCGGCAGAGAATGG.
Promoter Analysis
Transfection of IEC-18 and DLD1 cells using Fugene 6 and measurement of promoter activity using the Dual-Luciferase assay kit (Promega) were as we have described (14, 29). pRL-TK and pRL-CMV plasmids expressing Renilla luciferase (Promega) were routinely included in the transfections to monitor transfection efficiency; however, the thymidine kinase and CMV promoters in these reporters are responsive to PKC agonists. Therefore, the effects of drug treatments were determined from the relative firefly luciferase activity in control and treated cells transfected with the same transfection mixture. When different transfection mixes were used in a single experiment (i.e. those involving promoter mutants or dominant active IκBα), promoter activity was normalized for transfection efficiencies using the respective Renilla luciferase readings for each transcription mixture measured in vehicle-treated cells.
Statistical Analysis
Student's t tests and regression analysis were performed using Microsoft Excel software. Differences with p values of <0.05 were considered statistically significant.
RESULTS
Cyclin D1 Expression Is Subject to Both Negative and Positive Regulation by PKC Isozyme Signaling in Intestinal Epithelial Cells
We have previously demonstrated that treatment of non-transformed IEC-18 rat ileal crypt cells with the PKC agonist PMA has biphasic effects on cyclin D1 expression (17). Fig. 1A further demonstrates that (a) the initial down-regulation of cyclin D1 is maximal at ∼2 h (lane 3), (b) expression of the cyclin recovers to control levels by ∼4 h (Fig. 1A, lane 4), and (c) subsequent accumulation of cyclin D1 to levels markedly higher than those seen in unstimulated cells is evident by 6–8 h (lanes 5 and 6). Up-regulation of cyclin D1 is maintained for at least 24 h in PMA-treated IEC-18 cells (Fig. 1A, lanes 8 and 9). Hyperinduction of cyclin D1 expression is also seen following prolonged treatment with other PKC agonists, including the phorbol ester phorbol 12,13-dibutyrate (data not shown), the macrolide lactone bryostatin 1 (Fig. 1B, lanes 1 and 3), and the synthetic diacylglycerol DiC8 (Fig. 1B, lanes 4 and 5), indicating that the effect is a general response to long term PKC activation and does not reflect a specific response to PMA. In previous studies, we determined that the initial down-regulation of cyclin D1 induced by PKC agonists is mediated by PKCα (11, 14, 17–19); however, the mechanisms underlying the later hyperinduction of the protein remained to be characterized.
FIGURE 1.

PKC signaling has biphasic effects on cyclin D1 expression in IEC-18 cells. A, prolonged PMA treatment leads to hyperinduction of cyclin D1. IEC-18 cells were treated with PMA for the indicated times and analyzed for cyclin D1 and β-actin expression by Western blotting. Lanes in each panel are from the same Western blot; dashed lines show where lanes have been rearranged for clarity. B, prolonged treatment with multiple PKC agonists leads to hyperinduction of cyclin D1. IEC-18 cells were treated with PMA (P), bryostatin 1 (B), DiC8 (D; added in fresh medium every hour to compensate for its rapid metabolism in cells), or the corresponding vehicle (C) for the indicated times, and cyclin D1 and β-actin expression was analyzed by Western blotting. C, PKC dependence of cyclin D1 hyperinduction; as in B except that PMA/control treatment was conducted in the presence of Gö6976, Gö6983, or BIM (or the corresponding vehicle, DMSO). Data are representative of at least three independent experiments.
Prolonged PKC agonist treatment has long been recognized to down-regulate PKC isozymes, and reversal of the growth inhibitory effects of these agents in IEC-18 cells correlates with loss of PKCα (e.g. see Refs. 11, 17, and 18). Therefore, the contribution of loss of PKCα activity to PKC agonist-induced up-regulation of cyclin D1 was tested using the classical PKC inhibitor, Gö6976, which is selective for PKCα in IEC-18 cells (17), as well as the general PKC inhibitors, BIM and Gö6983. In keeping with a restraining effect of PKCα activity on cyclin D1 accumulation (14), all three inhibitors led to increased steady-state levels of cyclin D1 expression in the absence of PKC agonist treatment (Fig. 1C, compare lane 1 with lanes 3, 5, and 7). However, this increase was not as robust as the hyperinduction seen following prolonged (8-h) exposure to PMA (Fig. 1C, compare lanes 3, 5, and 7 with lane 2). General inhibition of PKC activity with BIM or Gö6983 blocked the ability of prolonged PMA treatment to further up-regulate cyclin D1 (Fig. 1C, compare lane 5 with lane 6 and lane 7 with lane 8), confirming that the hyperinduction is PKC-dependent. In contrast, selective blockade of PKCα with Gö6976 failed to prevent PMA-induced up-regulation of cyclin D1 at 8 h (Fig. 1C, compare lanes 3 and 4), indicating that a PKC isozyme other than PKCα must contribute positively to PKC agonist-induced hyperinduction of the molecule in intestinal epithelial cells.
PKCϵ Mediates Positive Effects of PKC Agonists on Cyclin D1 Expression
IEC-18 cells express three PKC isozymes that are responsive to the pharmacological agonists PMA, bryostatin 1, and DiC8: PKCα, PKCδ, and PKCϵ (30). As shown in Fig. 2A, i, PMA down-regulates these isozymes with different kinetics (also see Ref. 31). PKCα is depleted between 3 and 6 h of PMA treatment (Fig 2A, i), correlating with reversal of cyclin D1 loss (see Figs. 1A and 2A (i and ii), and remains barely detectable at later times of treatment (12–24 h), when cyclin D1 hyperinduction is evident (Fig. 2A, ii). PKCδ is down-regulated more rapidly than PKCα (Fig. 2A, i), indicating that it is unlikely to contribute positively to the hyperinduction of cyclin D1 seen at later time points. In contrast, down-regulation of PKCϵ is relatively slow and incomplete (Fig. 2A, i, compare lane 2 with lanes 10 and 12), with the protein remaining readily detectable, albeit at lower levels, even after 24 h of PMA treatment (31). Thus, comparison of the kinetics of PKC isozyme down-regulation and cyclin D1 hyperinduction points to PKCϵ as the positive factor mediating the effect.
FIGURE 2.

PKCϵ mediates cyclin D1 hyperinduction in PKC agonist-treated IEC-18 cells. A, differential agonist-induced down-regulation of PKC isozymes in IEC-18 cells. i, cells were treated with PMA (P) or vehicle control (C) for the indicated times, and whole cell extracts were subjected to Western blot analysis of PKCα, PKCδ, PKCϵ, and cyclin D1 expression. Expression of eEF2α was evaluated as a loading control (L.C.). The individual lanes show analysis of the same whole cell extracts. Note the close correspondence between loss of PKCα expression and recovery of cyclin D1 levels (ii). B, overexpression of PKCϵ increases cyclin D1 levels in IEC-18 cells. Cells were transduced with adenovirus expressing LacZ, PKCα, PKCδ, or PKCϵ, as indicated, and expression of PKCα, PKCδ, PKCϵ, cyclin D1, and β-actin was determined by Western blotting. Data are from the same Western blot, with dashed lines indicating where lanes have been rearranged for clarity. C, effect of PKCϵ knockdown on PKC agonist-induced hyperinduction of cyclin D1. IEC-18 cells were transfected with non-silencing control siRNA (NS) or siRNA directed against rat PKCϵ (PKCϵ). After 48 h, cells were treated with PMA (P) or ethanol vehicle (E) for 6 h prior to analysis of cyclin D1 and PKCϵ expression by Western blotting. Fast Green staining of a representative area of the Western blot membrane is shown as a loading control. D, effects of overexpressing PKCα or PKCϵ on PKC agonist-induced changes in cyclin D1 expression. Cells transduced with adenoviral vectors expressing LacZ, PKCα (i), or PKCϵ (ii) were treated with PMA for the indicated times, and the expression of the indicated proteins was determined by Western blotting. Data are representative of at least three independent experiments.
The requirement for PKCϵ in cyclin D1 hyperinduction was tested directly by manipulating PKC isozyme expression levels in IEC-18 cells. Transduction of cells with adenovirus expressing PKCα or PKCδ had no effect on cyclin D1 steady-state levels in untreated cells (Fig. 2B, left). In contrast, PKCϵ overexpression led to up-regulation of the cyclin (Fig. 2B, right), indicating that this isozyme is limiting for cyclin D1 expression in IEC-18 cells. The role of PKCϵ was further tested by siRNA-mediated knockdown. Transfection of cells with PKCϵ-targeted siRNA markedly reduced PKCϵ steady-state levels compared with non-targeting siRNA transfection (Fig. 2C, lanes 1 and 3) while having no effect on PKCα or PKCδ (27). In addition, whereas PKCϵ remained readily detectable at 6 h of PMA treatment in non-targeting siRNA-transfected cells (Fig. 2C, lane 2), the enzyme was depleted from PKCϵ siRNA-transfected cells by this time (Fig. 2C, lane 4). Notably, knockdown of PKCϵ completely abrogated the hyperinduction of cyclin D1 promoted by PMA. Indeed, whereas hyperinduced cyclin D1 expression was evident following a 6-h PMA treatment in control cells (Fig. 2C, lanes 1 and 2), levels of the cyclin remained down-regulated at this time point in PKCϵ-silenced cells. Thus, PKCϵ activity is required for both hyperinduction and efficient restoration of cyclin D1 expression following PKCα-mediated down-regulation of the cyclin in PKC agonist-treated cells.
To further examine the contributions of PKCα and PKCϵ to the biphasic effects of PKC agonists on cyclin D1 expression, the consequences of overexpressing these proteins on cyclin D1 down-regulation, recovery, and hyperinduction were tested in IEC-18 cells. Consistent with the inhibitory effects of PKCα, overexpression of this isozyme enhanced PMA-induced down-regulation of cyclin D1 and delayed its initial recovery (Fig. 2D, i, compare lanes 1–4 with lanes 8-11); however, it did not affect the eventual hyperinduction of the cyclin (compare lanes 6 and 7 with lanes 13 and 14). In contrast to PKCα, PKCϵ overexpression reduced the extent and duration of PMA-induced down-regulation of cyclin D1 (Fig. 2D, ii, compare lanes 1–3 with lanes 8–10) while markedly potentiating cyclin D1 hyperinduction by prolonged PKC agonist treatment (Fig. 2D, ii, compare lanes 5–7 with lanes 12–14).
Collectively, these data indicate that regulation of cyclin D1 expression by PKC agonists is dictated by opposing activities of two members of the PKC family, PKCα and PKCϵ. The initial down-regulation results from suppressive effects of PKCα; as these negative effects wane due to agonist-induced down-regulation of PKCα signaling, positive effects of PKCϵ can predominate, leading to recovery and eventual hyperinduction of cyclin D1 expression.
Sustained ERK Activity Is Required for PKCϵ-mediated Up-regulation of Cyclin D1
The MEK-ERK signal transduction pathway is an important downstream mediator of PKC function and can act as a positive or negative regulator of cyclin D1 expression (27, 32, 33). We have determined that sustained ERK activation during the first hour of PKC agonist treatment is required and sufficient for the growth-inhibitory functions of PKCα in IEC-18 cells (27). However, the activation of ERK induced by PKC agonists extends well beyond 1 h (27), pointing to the possibility that later ERK activation contributes to the PKCϵ-dependent up-regulation of cyclin D1 expression. To test this possibility, the duration of PKC agonist-induced ERK activity was manipulated by the addition of the MEK inhibitor, U0126, at various times after the initiation of PMA or vehicle treatment (Fig. 3A). In control cells, treatment with U0126 for 2–3 h had minimal effects on basal expression of cyclin D1 (Fig. 3B, lanes 1, 7, and 9); however, with longer ERK inhibition, expression of the cyclin dropped off and became barely detectable following 6–8 h of U0126 treatment (lanes 3 and 5) (data not shown). Based on these findings, a 6-h PMA treatment was selected for these experiments; 6 h was sufficiently long to observe hyperinduction of cyclin D1 (Figs. 1, 2A, and 3B, lanes 1 and 2) while allowing for its detection at the longest times of MEK inhibitor treatment (5 h, lane 3). The addition of U0126 after 4 h of PMA treatment, a time when levels of cyclin D1 have recovered to control levels (Fig. 1A), blocked the hyperinduction of cyclin D1 seen at 6 h of PMA treatment (Fig. 3B, compare lane 1 with lane 2 and lane 9 with lane 10). Interestingly, when U0126 was added at times when cyclin D1 was down-regulated (1–3 h), cyclin D1 failed to recover by 6 h, with levels remaining lower in cells treated with PMA and U0126 than in cells treated with U0126 alone (Fig. 3B, compare lane 3 with lane 4, lane 5 with lane 6, and lane 7 with lane 8). Similar results were observed with the structurally unrelated MEK inhibitor PD98059 (data not shown), indicating that the effects were specific for inhibition of ERK signaling. Thus, ongoing ERK activity is required both for efficient recovery of cyclin D1 expression and for its eventual hyperinduction in PKC agonist-treated IEC-18 cells. Notably, these findings mirror the effects of PKCϵ knockdown, which also blocked both recovery and hyperinduction of cyclin D1 expression following a 6-h PMA treatment (Fig. 2C). Taken together, these data indicate that ERK signaling mediates opposing effects of PKC isozymes on cyclin D1 expression in PKC agonist-treated cells. Early (≤1 h) PKCα-mediated ERK activation negatively regulates cyclin D1 expression, whereas later ERK activity is required for the positive effects of PKCϵ on expression of this cyclin.
FIGURE 3.

Sustained ERK signaling is required for hyperinduction of cyclin D1 in PMA-treated IEC-18 cells. A, experimental scheme for PMA and U0126 treatment. Cells were treated with PMA or vehicle (EtOH) for a total of 6 h, with the addition of 10 μm U0126 to inhibit ERK activity 1, 2, 3, or 4 h into the PMA treatment. The duration of ERK activity after PMA addition is indicated on the right. B, effect of U0126 on cyclin D1 recovery and hyperinduction. IEC-18 cells were treated with PMA (P) or vehicle (C), and U0126 (10 μm) or vehicle (DMSO) was added at the times indicated in A. After 6 h of PMA treatment, cells were harvested, and protein extracts were subjected to Western blot analysis for cyclin D1, phospho-ERK, and total ERK. Data are representative of at least three independent experiments.
PKCϵ Up-regulates Cyclin D1 mRNA
Cyclin D1 expression is controlled at transcriptional, translational, and post-translational levels. Analysis of the rate of disappearance of the protein following cycloheximide treatment excluded a role for alterations in protein stability in the hyperinduction of cyclin D1 observed in long-term PMA-treated cells (Fig. 4A). These studies confirmed that cyclin D1 is a relatively unstable protein in IEC-18 cells (11), with a half-life of about 15 min (Fig. 4A). However, although levels of cyclin D1 are elevated in cells exposed to PMA for 6 h (compare lanes 1 and 5), long term PMA treatment did not significantly change the rate of cyclin D1 protein disappearance following the addition of cycloheximide (p = 0.41).
FIGURE 4.

Effects of long term PMA treatment on cyclin D1 protein stability and translation in IEC-18 cells. A, PMA does not affect cyclin D1 protein stability. Cells were treated with PMA (P) or vehicle (C) for 6 h, followed by the addition of 30 μg/ml cycloheximide (CHX) for the indicated times and analysis of cyclin D1 expression by Western blotting. Fast Green staining of the Western blot membrane is shown as a loading control. Graph, relative band intensities for cyclin D1 in PMA- and vehicle (EtOH)-treated cells expressed relative to vehicle-treated control cells (average ± S.E. (error bars) of three independent experiments). B, PMA treatment increases cyclin D1 protein synthesis. Cells were treated with PMA (■) or vehicle (control, ♦) in methionine-free medium. [35S]Methionine/cysteine was added 5.5 h later, and cells were harvested at the indicated times of PMA treatment. Cyclin D1 was immunoprecipitated, and 35S incorporation was quantified by SDS-PAGE and phosphorimaging. C, effects of PMA treatment on 4E-BP1 phosphorylation. Cells were treated with PMA for the indicated times, and 4E-BP1 phospho-forms were detected by Western blotting (eIF4E expression is shown as a loading control; L.C.). Data are from the same Western blot, with dashed lines indicating where lanes have been rearranged for clarity. Note the marked accumulation of the active α and β forms of 4E-BP1, which persist for >6 h. Numbers at the bottom show relative band intensities for the α and β forms of 4E-BP1 (averages ± S.E. of three independent experiments). Data are representative of three independent experiments.
Having excluded protein stabilization as the mechanism underlying PMA-induced hyperinduction of cyclin D1, the role of alterations in protein synthesis was explored. Metabolic labeling experiments demonstrated increased incorporation of [35S]methionine into cyclin D1 in IEC-18 cells treated with PMA for >6 h (Fig. 4B). Our previous studies identified inhibition of cap-dependent translation as a major mechanism underlying PKCα-mediated down-regulation of cyclin D1 in intestinal epithelial cells (11, 14). This effect is the result of PKCα-induced accumulation of the active, translationally repressive, dephosphorylated α and β forms of 4E-BP1, as can be clearly seen in IEC-18 cells at 1–4 h of PMA treatment (Fig. 4C, arrows). However, although levels of these faster migrating repressive forms of 4E-BP1 begin to fall by 6 h, they remain above control levels up to 10 h of PMA treatment (Fig. 4C, compare lane 1 with lanes 5 and 6). Therefore, changes in the efficiency of cap-dependent translation are unlikely to account for the hyperinduction of cyclin D1 expression seen at these times.
To determine whether enhanced translation of cyclin D1 reflects alterations in levels of its mRNA, quantitative RT-PCR analysis was performed on cells treated with PMA for various times (Fig. 5A, i). As noted in our previous studies (11), PMA treatment led to an initial down-regulation of cyclin D1 mRNA (Fig. 5A, i, black bars, 2 h). However, as with the protein, expression of cyclin D1 mRNA subsequently recovered and rose to levels higher than those seen in control cells by 6 h of PMA treatment (Fig. 5A, i, black bars, 6 and 8 h). This effect was maintained for at least 24 h, as confirmed by Northern blot analysis (Fig. 5A, ii). The PKC dependence of these effects was confirmed by the ability of Rö32-0432 to block both the down-regulation and the subsequent hyperinduction of cyclin D1 mRNA in PMA-treated cells (Fig. 5A, i, gray bars). Notably, as seen at the protein level, overexpression of PKCϵ enhanced the accumulation of cyclin D1 mRNA, whereas overexpression of PKCα or PKCδ had no effect (Fig. 5B). Thus, PKCϵ positively regulates cyclin D1 expression in IEC-18 cells through up-regulation of its mRNA.
FIGURE 5.

PKCϵ induces cyclin D1 mRNA in IEC-18 cells through activation of the proximal cyclin D1 promoter. A, effects of PMA on cyclin D1 mRNA expression. i, cells were treated with PMA in the presence or absence of the PKC inhibitor Rö32-0432 (5 μm) for the indicated times, and cyclin D1 mRNA levels were analyzed by quantitative real-time PCR. Data were normalized to 18 S rRNA and expressed as -fold change relative to that in cells treated with vehicle (ethanol). Data are averages ± S.E. (error bars) of three independent experiments. ii, cells were treated with PMA or vehicle for the indicated times and analyzed for cyclin D1 mRNA and protein levels by Northern and Western blotting, respectively. B, overexpression of PKCϵ induces cyclin D1 mRNA expression. Cells were transduced with adenoviral vectors (multiplicity of infection 20) expressing either LacZ, PKCα, PKCδ, or PKCϵ, and expression of cyclin D1 mRNA was assessed and normalized to 18 S rRNA as in A. Data are expressed relative to levels in LacZ-transduced cells and are averages ± S.E. of more than three independent experiments. *, statistically different from LacZ-transduced cells (p < 0.05). C, hyperinduction of cyclin D1 requires new transcription. Cells were pretreated with 5 μg/ml actinomycin D (Act D) or vehicle (DMSO) for 30 min prior to the addition of PMA or vehicle (EtOH) for the indicated times. Cyclin D1 and β-actin protein levels were analyzed by Western blotting. Data are representative of more than three independent experiments. D, PMA activates the proximal (−163 bp) cyclin D1 promoter. Cells were transfected with promoter-luciferase constructs representing the proximal 1745 (−1745CD1Luc), 963 (−936CD1Luc), or 163 bp (−163CD1Luc) of the cyclin D1 promoter and treated with PMA or vehicle for 22 h. Promoter activity was assessed from luciferase expression and is expressed relative to that in control (vehicle-treated) cells for each construct. Data are averages ± S.E. of three independent experiments. *, statistically different from vehicle (EtOH)-treated control cells (p < 0.05).
PKCϵ-mediated Up-regulation of Cyclin D1 Involves Transcriptional Activation through Proximal Promoter Sequences
To determine the mechanism(s) underlying PKC agonist-induced up-regulation of cyclin D1 mRNA, the involvement of transcriptional alterations was tested using the RNA polymerase inhibitor, actinomycin D. As expected, actinomycin D led to a reduction in the overall levels of cyclin D1 expression in IEC-18 cells (Fig. 5C, compare lanes 1–3 with lanes 4–6). Actinomycin D also abrogated PMA-induced up-regulation of cyclin D1 at 6 and 8 h (Fig. 5C, compare lanes 8 and 9 with lanes 11 and 12). Thus, the PKCϵ-dependent up-regulation of cyclin D1 expression seen following sustained PKC agonist treatment is dependent on new transcription.
The main regulatory elements in the cyclin D1 promoter are located within a region 1745 bp upstream of the transcriptional start site (8). To test whether the transcriptional dependence reflected direct activation of the cyclin D1 promoter, cells were transfected with reporter constructs in which luciferase expression is driven by 1745 bp of the cyclin D1 promoter (−1745CD1Luc) or by a promoter truncated at −963 bp (−963CD1Luc) or −163 bp (−163CD1Luc). Treatment of transfected cells with PMA for 22 h led to a ∼2-fold increase in the activity of all three of these promoter constructs (Fig. 5D). These data show that long term PMA treatment induces cyclin D1 transcription through promoter sequences within 163 base pairs upstream of the transcriptional start site.
PKCϵ Up-regulates Cyclin D1 in DLD1 Colon Cancer Cells
To determine whether PKCϵ signaling also up-regulates cyclin D1 in colon cancer cells, the effects of long term PMA treatment were tested in FET, GEO, and DLD1 cells. In contrast to IEC-18 cells, colon cancer cells lack functional PKCα signaling (14) and do not repress cyclin D1 levels in response to PKC agonists (Fig. 6A, compare lanes 1 and 2 with lanes 3 and 4; see also Ref. 14). However, longer PMA treatments led to a sustained increase in cyclin D1 levels in these cells (Fig. 6A, compare lanes 1 and 2 with lanes 4–7). Notably, consistent with the absence of repressive PKCα signaling in colon cancer cells, this induction occurred more rapidly than in IEC-18 cells, with increased accumulation observed as early as 4 h after the PMA addition (compare lanes 1, 2, and 4 in Figs. 1A and 6A).
FIGURE 6.

PKC agonist treatment induces cyclin D1 expression in human colon cancer cells. A, PMA induces cyclin D1 expression in multiple colon cancer cell lines. FET, GEO, and DLD1 cells were treated with PMA for the indicated times, and the expression of cyclin D1 was analyzed by Western blotting. Even loading on blots was confirmed by immunoblotting for β-actin and/or fast green staining. B, the synthetic diacylglycerol DiC8 induces cyclin D1 expression in colon cancer cells. DLD1 cells were treated with DiC8 (D) or vehicle (ethanol; C) for 4 or 6 h, and expression of cyclin D1 and β-actin was determined by Western blotting. DiC8 was added repeatedly in fresh medium (every hour) to compensate for its rapid metabolism in cells. C, induction of cyclin D1 expression by PMA in DLD1 cells is PKC-dependent. DLD1 cells were treated with PMA (P) or vehicle (C) for 5 h in the presence of the general PKC inhibitor BIM or vehicle (DMSO), as indicated, and analyzed as in B. D, PMA induction of cyclin D1 in colon cancer cells is dependent on ERK activity; as in C except that 10 or 20 μm U0126 was used in place of BIM. E, DLD1 cells express PKCϵ and PKCβII as PKC agonist-responsive isozymes. IEC-18 and DLD1 cells were analyzed for the expression of the indicated PKC isozymes by Western blotting. All panels represent analysis of the same protein extracts from each cell line. F, PKCβII activity is not required for PMA induction of cyclin D1 expression in DLD1 cells. Cells were treated with PMA (P) or vehicle (C) in the presence of the indicated concentrations of the PKCβ inhibitor 3-(1-(3-imidazol-1-ylpropyl)-1H-indol-3-yl)-4-anilino-1H-pyrrole-2,5-dione and analyzed for cyclin D1, PKCβII, and β-actin expression by immunoblotting. Data are representative of at least three independent experiments.
Having established that PKC agonists up-regulate cyclin D1 in colon cancer cells, the molecular mechanisms underlying this effect were further characterized using DLD1 cells. Prolonged DiC8 treatment also led to hyperinduction of cyclin D1 in these cells (Fig. 6B), and the PKC dependence of the effect was further confirmed by the ability of the general PKC inhibitor, BIM, to block the PMA-induced increase in cyclin D1 expression (Fig. 6C). Thus, as seen in IEC-18 cells, cyclin D1 hyperinduction is a result of PKC activation and not a specific response to phorbol esters. Also paralleling the induction in IEC-18 cells, the ability of PKC agonists to hyperinduce cyclin D1 in DLD1 cells was dependent on ERK activity because it could be blocked with U0126 or PD98059 (Fig. 6D) (data not shown). DLD1 cells lack expression of PKCα and PKCδ but express PKCβII in addition to PKCϵ as the only two PMA-responsive isozymes (Fig. 6E) (14). Therefore, the contribution of PKCβII to the effects of PKC agonists was tested using the PKCβ-selective inhibitor 3-(1-(3-imidazol-1-ylpropyl)-1H-indol-3-yl)-4-anilino-1H-pyrrole-2,5-dione (34). Even at concentrations as high as 1 μm, this inhibitor failed to affect either the basal levels of cyclin D1 or the ability of PMA to induce its expression (Fig. 6F), arguing that PKCβII activity is not required for PMA-induced up-regulation of cyclin D1 in these cells. Because PMA treatment failed to down-regulate PKCβII in DLD1 cells, the efficacy of the inhibitor could not be tested by its ability to block activation-dependent down-regulation of the enzyme (cf. Ref. 25). However, even at concentrations as low as 100 nm, the PKCβ inhibitor potently blocked down-regulation of PKCβII in SK-UT-1B cells (data not shown), confirming the efficacy of this compound as an inhibitor of this PKC isozyme. Because PKCβII and PKCϵ are the only PMA-responsive isozymes in DLD1 cells, these findings indicate that PKCϵ can positively regulate cyclin D1 expression in both colon cancer cells and non-transformed IEC-18 cells.
Consistent with findings in IEC-18 cells, PKC agonist-induced up-regulation of cyclin D1 in DLD1 cells is dependent on new transcription, as confirmed by the failure of PMA to increase cyclin D1 levels in the presence of actinomycin D (Fig. 7A). Transcriptional activation in DLD1 cells occurs through the same proximal promoter region responsible for PKCϵ-mediated up-regulation of cyclin D1 in IEC-18 cells, as demonstrated by the ability of PMA to enhance the activity of the −963CD1Luc and −163CD1Luc constructs in transient transfection assays (Fig. 7B). Collectively, these data indicate not only that PKCϵ signaling is able to induce cyclin D1 expression in colon cancer cells but that it utilizes the same mechanisms for this up-regulation in both non-transformed and transformed intestinal epithelial cells.
FIGURE 7.

PMA induces cyclin D1 transcription in colon cancer cells. A, induction of cyclin D1 by PMA in DLD1 cells requires new transcription. DLD1 cells were pretreated with 5 μg/ml actinomycin D (Act D) or vehicle (DMSO) for 30 min prior to the addition of PMA (P) or vehicle (C) for 4 h, and levels of cyclin D1 and β-actin were determined by Western blot analysis. Data are representative of more than three independent experiments. B, PMA activates the proximal cyclin D1 promoter in colorectal cancer cells. DLD1 cells were transfected with the −963CD1Luc or −163CD1Luc cyclin D1 promoter-luciferase constructs and treated with PMA for 5 h. The activity of cyclin D1 promoter fragments was measured by the Dual-Luciferase assay and is normalized to the activity in control cells treated with vehicle for 5 h. Data are averages ± S.E. (error bars) of nine independent experiments for the −163 bp CD1-Luc construct and of two independent experiments for the −963 bp CD1-Luc construct. *, statistically different from vehicle (EtOH)-treated control cells (p < 0.05).
PKC Agonists Activate NF-κB in Intestinal Epithelial Cells
The NF-κB site at −38 bp in the PKCϵ-responsive cyclin D1 proximal promoter region (Fig. 8A) is a strong regulator of cyclin D1 transcription in multiple contexts (35). Because PKC signaling can elicit sustained NF-κB activation in various systems, including colon cancer cells (e.g. see Refs. 35–39), the ability of PMA to stimulate NF-κB in IEC-18 cells was tested using an NF-κB reporter construct. PMA treatment increased NF-κB activity about 2–2.5-fold in IEC-18 cells (Fig. 8, B and C (i)). Increased NF-κB activity was first seen at 2–4 h, a time that slightly precedes the up-regulation of cyclin D1 mRNA levels, and the activity remained elevated for at least 24 h (Fig. 8B). This level of induction is close to that seen in DLD1 cells, which also occurred by 3 h of PMA treatment (Fig. 8C, ii) (data not shown).
FIGURE 8.

Prolonged treatment with PMA stimulates canonical and non-canonical NF-κB activity. A, transcription factor binding sites in the −163 bp region of the cyclin D1 promoter. The position of the indicated transcription factor binding sites in the cyclin D1 promoter is given as the number of bp from the transcriptional start site. Note that there are two binding sites for SP1 and Egr1. This figure is modified from Ref. 70. B, time course for PMA induction of NF-κB activity in IEC-18 cells. Cells transfected with an NF-κB reporter construct containing three NF-κB sites driving expression of firefly luciferase were treated with PMA for various times as indicated. NF-κB activity at each time point was measured by the Dual-Luciferase activity assay and normalized to that in cells treated with vehicle (EtOH) for the equivalent time. *, statistically different from vehicle (EtOH)-treated control cells (p < 0.05). C, effect of dominant active IκBα on PMA-induced NF-κB activity. IEC-18 (i) or DLD1 (ii) cells were transfected with the NF-κB reporter and treated with PMA or vehicle for 8 h. Where indicated, cells were also transfected with a vector expressing dominant active IκB (D.A. IκB) or empty vector (Con). pRL-TK was included in all transfections to control for transfection efficiency; promoter activities in the different transfection conditions were normalized using pRL-TK activity in vehicle-treated cells and are expressed relative to control NF-κB reporter/empty vector-transfected cells. *, statistically different from vehicle (EtOH)-treated control cells (p < 0.05). Data are representative of three independent experiments. Error bars, S.E.
PMA has been shown to promote NF-κB activation in colon cancer cells through the canonical pathway, which requires the ubiquitination and degradation of IκB, and through mechanisms that are independent of IκB degradation (38–40). Therefore, the effects of expressing a mutant IκBα protein that is refractory to ubiquitination and degradation (DA IκB) were tested. When DA IκB was expressed in the NF-κB activity assay, basal NF-κB activity was markedly reduced in both IEC-18 and DLD1 cells, confirming the activity of DA IκB under the experimental conditions used (Fig. 8C). However, although overall NF-κB activity was decreased, PMA treatment was able to promote induction of NF-κB >3-fold in the presence of DA IκB, indicating that PKC agonists activate NF-κB through pathways that are both sensitive and resistant to IκB.
Inhibition of NF-κB Activity Blocks PKC Agonist-induced Up-regulation of Cyclin D1
Because PKC agonists increased NF-κB activity in IEC-18 and DLD1 cells through mechanisms that are not sensitive to IκB, the requirement for NF-κB in PKCϵ-mediated induction of cyclin D1 was evaluated using PDTC. This compound blocks NF-κB activation by inhibiting the SCF/β-TrCP ubiquitin ligase (41) and IKK activity (42) and can thus suppress both the canonical and non-canonical NF-κB activation pathways (42, 43). Based on analysis of the concentration of this compound required to inhibit PMA-induced NF-κB activity (data not shown) and on published data (44–46), 100 μm PDTC was used in these studies. At this concentration, PDTC blocked PMA-induced up-regulation of cyclin D1 protein in both IEC-18 and DLD1 cells (Fig. 9, A (i) and B (i)). In addition to preventing the hyperinduction of cyclin D1 in IEC-18 cells, treatment with PDTC unexpectedly led to an increase in steady-state levels of cyclin D1 (Fig. 9A, i). This effect, which may relate to the antioxidant properties of PDTC (47–49), is posttranscriptional because the inhibitor decreased cyclin D1 mRNA expression in IEC-18 (Fig. 9C, i) and is thus unrelated to the transcriptional effects of PKCϵ. The involvement of NF-κB was also investigated using CAPE, an NF-κB inhibitor that differs from PDTC in structure and mechanism of action (50) and that also inhibits both the canonical and non-canonical pathways (e.g. see Ref. 51). Although CAPE was mildly toxic to both IEC-18 and DLD1 cells under the conditions used (see the legend to Fig. 9) and resulted in down-regulation of basal cyclin D1 expression in both cell types (Fig. 9, A–D), this inhibitor (10–100 μm) also prevented cyclin D1 up-regulation by prolonged PMA treatment (Fig. 9, A (ii) and B (ii)). PDTC and CAPE also blocked PMA-induced up-regulation of cyclin D1 mRNA (Fig. 9C) and activation of the −163CD1 promoter (Fig. 9D). In contrast to the effects of PDTC and CAPE, activation of cyclin D1 transcription by PMA in intestinal epithelial cells was not affected by expression of dominant active IκB (Fig. 9E), indicating that the effect is not dependent on the canonical pathway of NF-κB activation.
FIGURE 9.

NF-κB inhibitors block PKCϵ-induced cyclin D1 up-regulation. A, NF-κB inhibitors block up-regulation of cyclin D1 by PMA in IEC-18 cells. Cells were treated with PMA (P) or vehicle (C) for 8 h in the presence of 100 μm PDTC (i), CAPE (ii), or vehicle (DMSO), and levels of cyclin D1 and β-actin were determined by Western blot analysis. Data are representative of at least three independent experiments. In each panel, data are from the same Western blot, with dashed lines indicating where lanes have been rearranged for clarity. PDTC and CAPE showed some mild toxicity under the conditions used, with protein recovery relative to control as follows: 100 μm PDTC, 93 ± 6%; 10 μm CAPE, 91 ± 3%; 100 μm CAPE, 80 ± 7%. These values were not affected by PMA treatment. B, NF-κB inhibitors block up-regulation of cyclin D1 by PMA in DLD1 colon cancer cells; as in A except that DLD1 cells were used, and PMA treatment was for 4 h. CAPE showed some mild toxicity under the conditions used, with protein recoveries for samples treated with PDTC and CAPE relative to control as follows: 100 μm PDTC, 98 ± 3%; 10 μm CAPE, 91 ± 3%; 100 μm CAPE, 77 ± 7%. These values were not affected by PMA treatment. C, NF-κB inhibitors block PKC-induced up-regulation of cyclin D1 mRNA. IEC-18 (i) or DLD1 (ii) cells were treated with PMA or vehicle (EtOH) in the presence of 100 μm PDTC, 100 μm CAPE, or vehicle (DMSO). After 8 h (i) or 4 h (ii) of PMA treatment, cells were harvested and analyzed by quantitative RT-PCR. Cyclin D1 mRNA expression was normalized to 18 S rRNA levels and is shown relative to that in control (vehicle only-treated) cells. Data are averages ± S.E. (error bars) of at least four independent experiments except for data for CAPE-treated DLD1 cells, which are from two experiments. *, statistically different from vehicle (EtOH)-treated control cells (p < 0.05). #, statistically different from corresponding treatment of cells not exposed to PDTC or CAPE (p < 0.05). D, NF-κB inhibitors block PMA-induced increase in cyclin D1 promoter activity. DLD1 cells were transfected with the −963CD1Luc reporter and treated with PMA or vehicle (EtOH) for 4 h in the presence of 100 μm PDTC, 100 μm CAPE, or vehicle. Promoter activity was measured by the Dual-Luciferase assay and is normalized to the activity in control cells treated with vehicle only. Data are averages ± S.E. of four independent experiments. *, statistically different from vehicle (EtOH)-treated cells (p < 0.05). #, statistically different from corresponding treatment of cells not exposed to PDTC or CAPE (p < 0.05). E, up-regulation of cyclin D1 transcription by PMA does not require canonical activation of NF-κB. IEC-18 (i) or DLD1 (ii) cells, transfected with the NF-κB reporter along with pRL-TK and dominant active IκBα-expressing vector (DA IκB) or empty vector (Con), were treated with PMA or vehicle (EtOH) for 8 h (i) or 5 h (ii). Firefly and Renilla luciferase activities were determined using the Dual-Luciferase assay. Differences in transfection efficiency were controlled for by normalizing against pRL-TK activity in vehicle-treated cells. These data are expressed relative to empty vector-transfected, vehicle-treated cells and are the averages ± S.E. of at least three independent experiments. *, statistically different from corresponding vehicle (EtOH)-treated control cells (p < 0.05).
PKCϵ-induced Up-regulation of Cyclin D1 Transcription Involves Cooperation between NF-κB- and CRE-interacting Factors
To identify cyclin D1 promoter elements that mediate the transcriptional effects of PKCϵ-NF-κB signaling, transcription factor binding sites in the proximal 163-bp promoter region (Fig. 8A) were individually mutated, and the resulting constructs were tested in DLD1 cells. Mutation of the majority of these sites had little effect on either the basal level of cyclin D1 promoter activity or the extent of its induction following PMA treatment (Fig. 10A). Interestingly, despite the evidence that NF-κB activity is required for PKCϵ-mediated up-regulation of cyclin D1 (Fig. 9), this was true even for the NF-κB site, indicating that an additional promoter element(s) is involved.
FIGURE 10.

PKCϵ-mediated induction of cyclin D1 transcription involves cooperation between NF-κB and CRE-interacting proteins. A, effect of transcription factor binding site mutation on induction of cyclin D1 promoter activity by PKCϵ. The binding sites for each transcription factor in the −163 bp cyclin D1 promoter construct were mutated individually. The two binding sites for Sp1 were mutated simultaneously due to the proximity of their location, and the two sites for Egr1, designated Egr1a (−118) and Egr1b (−137) were mutated individually and together. DLD1 cells were transfected with cyclin D1 promoter fragments containing the indicated mutations along with pRL-TK and treated with PMA or vehicle (EtOH) for 5 h. The activity of each cyclin D1 promoter mutant, measured by the Dual-Luciferase assay, is normalized for differences in transfection efficiency using the activity of pRL-TK in the corresponding vehicle-treated cells. Data, expressed relative to the activity of the wild-type (w.t.) promoter, are averages ± S.E. (error bars) of at least three independent experiments. *, statistically different (p < 0.05) from wild-type promoter construct. B, effect of mutating the NF-κB site and CRE on induction of cyclin D1 promoter activity by PMA in IEC-18 cells; as in A, except that IEC-18 cells were used, and PMA/vehicle treatment was for 24 h. *, statistically different (p < 0.05) from wild-type promoter construct. #, statistically different (p < 0.05) from CRE mutant promoter construct.
Mutation of three sites, the Egr1 site at −137 bp (Egr1b), the TCF/LEF site, and the CRE, did affect activity of the promoter. Mutation of the Egr1b site or the TCF/LEF site decreased the basal level of cyclin D1 promoter activity in DLD1 cells (Fig. 10A) but did not significantly affect the relative induction of promoter activity by PMA (wild-type, 1.9 ± 0.1-fold; Egr1b mutant, 1.6 ± 0.2-fold; TCF/LEF mutant, 2.0 ± 0.1-fold). In contrast, mutation of the CRE not only led to a reduction in basal activity (Fig. 10A), but also significantly reduced the ability of PMA treatment (4 h) to enhance activity of the promoter (-fold induction = 1.4 ± 0.1 for the CRE mutant versus 1.9 ± 0.1 for the wild type, p = 0.004, n = 9). Thus, although the TCF/LEF, Egr1b, and CRE sites support basal cyclin D1 promoter activity in DLD1 cells, factors that interact with the CRE are also involved in mediating the effects of PMA.
The CRE in the cyclin D1 promoter is adjacent to the NF-κB site (Fig. 8A); therefore, the effect of simultaneous mutation of these sites was investigated (Fig. 10A). Mutating both sites had only a slight effect on basal activity of the cyclin D1 promoter in DLD1 cells compared with mutation of the CRE alone; however, double mutation of these sites abrogated the increased activity of the cyclin D1 promoter seen with prolonged PMA treatment (-fold induction = 1.1 ± 0.2, n = 6). The added effect of mutating both sites confirms a role for NF-κB at the promoter and indicates that cooperation between the CRE and NF-κB is integral to the ability of PKCϵ to enhance cyclin D1 expression in DLD1 colon cancer cells.
Analysis of the CRE and NF-κB mutant promoters in IEC-18 cells also revealed cooperation between these sites, although differences in the roles of each site were noted in these cell lines. Mutation of the NF-κB site increased basal promoter activity in IEC-18 cells (Fig 10B), pointing to a repressive role of this site in regulating basal expression of cyclin D1 in non-transformed intestinal epithelial cells. However, as seen in DLD1 cells, the NF-κB site was dispensable for cyclin D1 hyperinduction because mutation of this site did not affect PMA-induced up-regulation of promoter activity. In contrast to the effects in DLD1 cells, mutation of the CRE alone did not affect basal levels of cyclin D1 promoter activity in IEC-18 cells, although CRE inactivation did reduce PMA-induced stimulation of the promoter (Fig. 10B). Notably, mutation of the CRE converted the NF-κB site from a repressive to an activating promoter element; whereas mutation of the NF-κB site increased basal activity of the wild-type promoter, the activity of the CRE/NF-κB double mutant was less than that of the promoter with only the CRE mutated. Thus, the repressive activity of the NF-κB site in IEC-18 cells requires interaction between the NF-κB site and the CRE. Collectively, these data indicate that, although there are subtle differences between the regulation of the cyclin D1 promoter in DLD1 and IEC-18 cells, cooperation between factors that bind to the CRE and NF-κB site mediates the up-regulation of cyclin D1 transcription induced by prolonged PKC agonist treatment in both non-transformed intestinal epithelial cells and colon cancer cells.
DISCUSSION
The findings presented herein cast new light on the regulation of cyclin D1 by members of the PKC family. PKCα and PKCϵ have opposing effects on intestinal tumorigenesis, with PKCα acting as a tumor suppressor (14, 22) and PKCϵ exhibiting tumor-promoting activity (24, 52). Our previous studies have determined that PKCα potently down-regulates cyclin D1 in intestinal epithelial cells and that loss of cyclin D1 represents an important component of the growth and tumor-suppressive effects of this isozyme (11, 14, 17, 19). The present study indicates that these negative effects of PKCα are opposed by PKCϵ, which up-regulates cyclin D1 transcription in both non-transformed IEC-18 cells and colon cancer cells. The localization of PKCϵ activity in intestinal tissues supports this function. In contrast to other PKC isozymes, which are predominantly activated in postmitotic cells of the intestinal villus and colon surface mucosa, activated (membrane-associated) PKCϵ is also detected in proliferating crypt cells (30). Although activation of PKCϵ in villus/surface mucosa cells presumably reflects its role in differentiated functions, such as mucin expression and chloride transport (53, 54), the PKCϵ activity in crypt cells coincides with expression of cyclin D1 in this tissue (19). Furthermore, whereas several PKCs, such as PKCα and PKCδ, tend to be lost during intestinal tumorigenesis, PKCϵ is retained in colon cancer cells (14). Collectively, these findings point to PKCϵ as a physiologically relevant regulator of cyclin D1 both in intestinal homeostasis and during tumor development.
Analysis of downstream signaling pathways determined that efficient recovery of cyclin D1 expression from PKCα-induced down-regulation as well as its eventual hyperinduction require sustained ERK activity (Fig. 3). ERK activation is a common downstream effect of PKCϵ (24, 55) that has been linked to its oncogenic effects in intestinal epithelial cells (52) and the prostate (56). Interestingly, PKCα-mediated down-regulation of cyclin D1 is also ERK-dependent (18, 27). However, the temporal requirement for ERK activation in the effects of these two isozymes differs; whereas PKCϵ-dependent hyperinduction of cyclin D1 requires ERK activity for >4 h following PMA addition (Fig. 3), 1 h of ERK activity is sufficient to elicit the full growth-inhibitory effects of PKCα (27). Divergent effects of ERK signaling can result from differential timing and localization of activated ERK (33). In this regard, it is noteworthy that PMA treatment of IEC-18 cells promotes nuclear and cytoplasmic accumulation of phospho-ERK (27). Nuclear accumulation of active ERK, which is linked to the growth-inhibitory effects of PKCα, is short-lived. In contrast, cytoplasmic ERK activity is more prolonged, consistent with the duration of ERK activation required for PKCϵ-dependent induction of cyclin D1 expression. Because divergent effects of PKCα and PKCϵ on the localization and timing of ERK activation have been noted in other cell types (e.g. see Refs. 57 and 58), differential regulation of ERK signaling is likely to be a common mechanism by which individual PKC isozymes elicit distinct responses.
Consistent with these differences in downstream signaling, PKCα and PKCϵ are not directly antagonistic but appear to modulate cyclin D1 expression via distinct mechanisms (see Fig. 11). The inhibitory activity of PKCα involves both translational and transcriptional repression (11, 14), whereas the stimulatory effects of PKCϵ appear to be predominantly transcriptional (Figs. 4, 5, and 7). PKCα signaling blocks cyclin D1 translation through rapid (within 30 min) PP2A-dependent dephosphorylation of 4E-BP1 (11, 18); however, PKC agonists do not elicit changes in 4E-BP1 phosphorylation or PP2A activity when PKCα activity is selectively inhibited, excluding a role for PKCϵ in modulation of this pathway. At the transcriptional level, PKCα exerts its inhibitory effects through cyclin D1 promoter sequences between 1745 and 163 bp upstream of the transcriptional start site (14), whereas the effects of PKCϵ are mediated by sequences in the proximal 163 bp of the promoter (Figs. 5 and 7).
FIGURE 11.
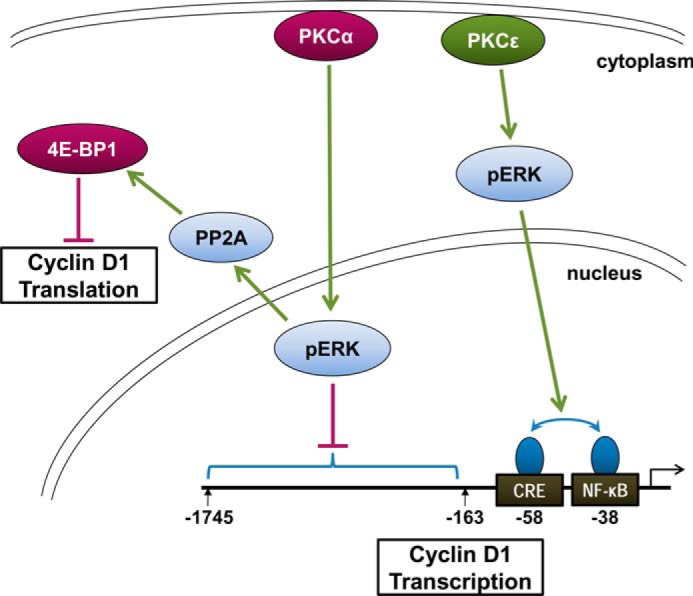
Model of the differential effects of PKCα and PKCϵ signaling on cyclin D1 expression in intestinal epithelial cells. We have previously demonstrated that PKCα signaling mediates translational and transcriptional repression of cyclin D1 in intestinal epithelial cells via ERK-dependent mechanisms (14, 18, 27). Translational repression involves PP2A-mediated activation of 4E-BP1, and transcriptional inhibition is mediated by an element(s) between −163 and −1745 bp of the cyclin D1 promoter. The current study further demonstrates that PKCϵ promotes ERK-dependent transcriptional activation of the cyclin D1 promoter, via positive cooperation between NF-κB- and CRE-interacting protein(s) near the transcriptional start site. The effects of PKCα are dominant over those of PKCϵ in these cells. Ongoing studies are investigating the mechanisms underlying the effects of PKCα on PP2A and the B subunit involved, and a role for α4 protein has been excluded.
The opposing signals from PKCα and PKCϵ result in biphasic effects of PKC agonists on cyclin D1 expression in IEC-18 cells. The initial agonist-induced down-regulation indicates that the repressive effects of PKCα are dominant over those of PKCϵ at early time points. However, PKCα-mediated repression of cyclin D1 is transient, and reversal of the effect correlates with agonist-induced down-regulation of the isozyme (Fig. 2A) (17, 31). Although the recovery and increased expression of cyclin D1 can be explained, in part, by loss of repressive effects of PKCα, positive effects of PKCϵ play an important role both in the timing of the recovery and in the extent of cyclin D1 up-regulation. Hyperinduction of cyclin D1 occurs at times when repressive effects of PKCα are still evident, albeit more weakly, in IEC-18 cells. For example, cyclin D1 is hyperinduced at 6 h of PMA treatment despite the continued presence of elevated levels of the active α and β forms of 4E-BP1 (Fig. 4C). The involvement of PKCϵ in overcoming these persistent repressive effects is reflected in the fact that cyclin D1 remains below basal levels at 6 h of PMA treatment in cells in which PKCϵ signaling is abrogated by siRNA-mediated knockdown or by inhibition of downstream ERK activation (Figs. 2C and 3). A requirement for PKCϵ in cyclin D1 hyperinduction is also seen in (a) the ability of general PKC inhibitors to abrogate PMA-induced cyclin D1 up-regulation and (b) the fact that the effect is still seen when PKCα activity is selectively inhibited with Gö6976 and in DLD1 colon cancer cells that lack PKCα activity (14). Furthermore, whereas overexpression of PKCα enhanced and prolonged cyclin D1 down-regulation in agonist-treated cells, PKCϵ overexpression attenuated PKCα-induced cyclin D1 suppression while increasing the extent of its eventual hyperinduction. Thus, the balance of the relative strengths of signaling from these isozymes dictates the levels of cyclin D1 in intestinal cells. The consequences of disrupting this balance are evident in colon cancer cells, where the absence of functional PKCα signaling not only prevents the initial down-regulation of cyclin D1 in response to PKC-agonist treatment but is also associated with earlier cyclin D1 induction (Figs. 1A, 2A, and 6A). Restoration of PKCα in colon cancer cells, on the other hand, decreases basal expression of cyclin D1, re-establishes the ability of PKC agonists to down-regulate the cyclin (14), and delays its eventual PKCϵ-mediated hyperinduction.
Our data in IEC-18 and DLD1 cells demonstrate that NF-κB activity is required for PKCϵ-mediated up-regulation of cyclin D1 in intestinal epithelial cells. Studies from a number of systems have established the ability of PKC signaling, and PKCϵ in particular, to activate NF-κB through the canonical IκB-regulated pathway (e.g. see Refs. 59–61). However, PKC agonist-induced up-regulation of cyclin D1 promoter activity in intestinal epithelial cells was insensitive to expression of DA IκB, excluding involvement of this pathway in the effects seen here. This finding is consistent with the report that activation of NF-κB by PMA can be independent of IκB degradation in DLD1 cells (39). In contrast to the canonical pathway, the non-canonical pathway generally gives rise to prolonged NF-κB activation (62), further supporting a role for the latter in PKCϵ-mediated hyperinduction of cyclin D1 transcription. Involvement of non-canonical activation of NF-κB is also consistent with the finding that p52·RelB (63, 64) and p52/p52·Bcl3 complexes (51, 65) can activate the cyclin D1 promoter. Current efforts are determining the involvement of these mechanisms in the observed effects of PKCϵ on cyclin D1 transcription.
Although PKCϵ-mediated transcriptional up-regulation of cyclin D1 is dependent on NF-κB activity, the NF-κB site in the proximal promoter is not required for the effect. Rather, induction of cyclin D1 transcription involves interaction between NF-κB activity and the neighboring CRE. Although the precise mechanisms underlying this cooperation remain to be established, the finding that either the CRE or NF-κB site can support the induction of cyclin D1 transcription (Fig. 10) points to a role for physical interaction between NF-κB proteins and factors that bind to the CRE. This CRE binds multiple proteins that have been shown to physically interact with NF-κB family members, including JunD, ATF, c-Jun, and c-Fos (e.g. see Refs. 66–68). As with the cooperation seen here, interaction of NF-κB with c-Jun or c-Fos can support synergistic activation of transcription through promoter sites for either factor (67). Furthermore, in immortalized human hepatocyte cells, c-Jun can activate cyclin D1 promoter activity through the NF-κB site in a DA IκB-insensitive manner, pointing to c-Jun as a potential mediator of the observed cooperation in intestinal cells.
In addition to further characterizing regulation of cyclin D1 by PKC isozymes, the current study has revealed aspects of its transcriptional regulation that may be of direct relevance to the up-regulation of cyclin D1 in colon cancer cells. Mutational analysis determined that the NF-κB site in the cyclin D1 promoter is a repressive element in IEC-18 cells (Fig. 10B). Notably, this NF-κB site is also repressive in mammary epithelial and smooth muscle cells (37, 64), indicating that negative regulation of cyclin D1 transcription through the NF-κB site might be common in non-transformed cells. However, this site is not repressive in colon cancer cells (Fig. 10A) (e.g. see Ref. 69); thus, loss of the repressive effects of the NF-κB site would appear to contribute to the up-regulation of cyclin D1 expression seen during intestinal tumorigenesis. Conversely, the CRE gains positive effects in colon cancer cells (Fig. 10, A and B), indicating that changes in factors that bind this site also play a role in increasing cyclin D1 expression in these tumors. These two phenomena appear to be related because the negative effects of the NF-κB site in IEC-18 cells require interaction with an intact CRE (Fig. 10B). Thus, the ability of PKCϵ to promote positive cooperation between NF-κB and CRE-interacting protein(s) at the cyclin D1 promoter positions this isozyme to be an important player in intestinal transformation and is likely to account, in part, for its oncogenic activity.
In conclusion, the current study complements our previous findings that cyclin D1 is an important target for the tumor-suppressive effects of PKCα. Given the importance of cyclin D1 in intestinal homeostasis and tumorigenesis, the antagonistic effects of PKCα and PKCϵ on expression of this cyclin would, at least partially, explain their contrasting effects on intestinal tumor development. These findings highlight a role for opposing actions of PKC family members, which converge on cyclin D1 expression, as important regulators of tissue homeostasis and tumorigenesis in the intestine (Fig. 11).
Acknowledgments
We thank Kathryn J. Curry, Misty Pocwierz, and Guishen He for expert technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants DK60632, DK54909, CA16056, and CA036727 and National Institutes of Health Postdoctoral Fellowship CA113048 (to M. A. P.). This work was also supported by National Science Foundation Grant DBI-0353490.
- 4E-BP1
- 4E-binding protein 1
- BIM
- bisindolylmaleimide I
- DA IκB
- dominant active IκBα
- DiC8
- 1,2-dioctanoyl-sn-glycerol
- CAPE
- caffeic acid phenethyl ester
- CRE
- cyclic AMP-response element
- PDTC
- pyrrolidine dithiocarbamate
- PMA
- phorbol 12-myristate 13-acetate.
REFERENCES
- 1. Fu M., Wang C., Li Z., Sakamaki T., Pestell R. G. (2004) Minireview: cyclin D1: normal and abnormal functions. Endocrinology 145, 5439–5447 [DOI] [PubMed] [Google Scholar]
- 2. Kim J. K., Diehl J. A. (2009) Nuclear cyclin D1: an oncogenic driver in human cancer. J. Cell Physiol. 220, 292–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Musgrove E. A., Caldon C. E., Barraclough J., Stone A., Sutherland R. L. (2011) Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 11, 558–572 [DOI] [PubMed] [Google Scholar]
- 4. Sherr C. J., Roberts J. M. (1995) Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 9, 1149–1163 [DOI] [PubMed] [Google Scholar]
- 5. Musgrove E. A., Lee C. S., Buckley M. F., Sutherland R. L. (1994) Cyclin D1 induction in breast cancer cells shortens G1 and is sufficient for cells arrested in G1 to complete the cell cycle. Proc. Natl. Acad. Sci. U.S.A. 91, 8022–8026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ng R., Song G., Roll G. R., Frandsen N. M., Willenbring H. (2012) A microRNA-21 surge facilitates rapid cyclin D1 translation and cell cycle progression in mouse liver regeneration. J. Clin. Invest. 122, 1097–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kilker R. L., Planas-Silva M. D. (2006) Cyclin D1 is necessary for tamoxifen-induced cell cycle progression in human breast cancer cells. Cancer Res. 66, 11478–11484 [DOI] [PubMed] [Google Scholar]
- 8. Klein E. A., Assoian R. K. (2008) Transcriptional regulation of the cyclin D1 gene at a glance. J. Cell Sci. 121, 3853–3857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guo Y., Harwalkar J., Stacey D. W., Hitomi M. (2005) Destabilization of cyclin D1 message plays a critical role in cell cycle exit upon mitogen withdrawal. Oncogene 24, 1032–1042 [DOI] [PubMed] [Google Scholar]
- 10. Rousseau D., Kaspar R., Rosenwald I., Gehrke L., Sonenberg N. (1996) Translation initiation of ornithine decarboxylase and nucleocytoplasmic transport of cyclin D1 mRNA are increased in cells overexpressing eukaryotic initiation factor 4E. Proc. Natl. Acad. Sci. U.S.A. 93, 1065–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hizli A. A., Black A. R., Pysz M. A., Black J. D. (2006) Protein kinase C α signaling inhibits cyclin D1 translation in intestinal epithelial cells. J. Biol. Chem. 281, 14596–14603 [DOI] [PubMed] [Google Scholar]
- 12. Arber N., Hibshoosh H., Moss S. F., Sutter T., Zhang Y., Begg M., Wang S., Weinstein I. B., Holt P. R. (1996) Increased expression of cyclin D1 is an early event in multistage colorectal carcinogenesis. Gastroenterology 110, 669–674 [DOI] [PubMed] [Google Scholar]
- 13. Hulit J., Wang C., Li Z., Albanese C., Rao M., Di Vizio D., Shah S., Byers S. W., Mahmood R., Augenlicht L. H., Russell R., Pestell R. G. (2004) Cyclin D1 genetic heterozygosity regulates colonic epithelial cell differentiation and tumor number in ApcMin mice. Mol. Cell Biol. 24, 7598–7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pysz M. A., Leontieva O. V., Bateman N. W., Uronis J. M., Curry K. J., Threadgill D. W., Janssen K. P., Robine S., Velcich A., Augenlicht L. H., Black A. R., Black J. D. (2009) PKCα tumor suppression in the intestine is associated with transcriptional and translational inhibition of cyclin D1. Exp. Cell Res. 315, 1415–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilding J., Straub J., Bee J., Churchman M., Bodmer W., Dickson C., Tomlinson I., Ilyas M. (2002) Cyclin D1 is not an essential target of β-catenin signaling during intestinal tumorigenesis, but it may act as a modifier of disease severity in multiple intestinal neoplasia (Min) mice. Cancer Res. 62, 4562–4565 [PubMed] [Google Scholar]
- 16. Toncheva D., Petrova D., Tzenova V., Dimova I., Yankova R., Yordanov V., Damjanov D., Todorov T., Zaharieva B. (2004) Tissue microarray analysis of cyclin D1 gene amplification and gain in colorectal carcinomas. Tumor Biol. 25, 157–160 [DOI] [PubMed] [Google Scholar]
- 17. Frey M. R., Clark J. A., Leontieva O., Uronis J. M., Black A. R., Black J. D. (2000) Protein kinase C signaling mediates a program of cell cycle withdrawal in the intestinal epithelium. J. Cell Biol. 151, 763–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guan L., Song K., Pysz M. A., Curry K. J., Hizli A. A., Danielpour D., Black A. R., Black J. D. (2007) Protein kinase C-mediated down-regulation of cyclin D1 involves activation of the translational repressor 4E-BP1 via a phosphoinositide 3-kinase/Akt-independent, protein phosphatase 2A-dependent mechanism in intestinal epithelial cells. J. Biol. Chem. 282, 14213–14225 [DOI] [PubMed] [Google Scholar]
- 19. Hao F., Pysz M. A., Curry K. J., Haas K. N., Seedhouse S. J., Black A. R., Black J. D. (2011) Protein kinase Cα signaling regulates inhibitor of DNA binding 1 in the intestinal epithelium. J. Biol. Chem. 286, 18104–18117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kazanietz M. G. (ed) (2010) Protein Kinase C in Cancer Signaling and Therapy, Humana Press, New York [Google Scholar]
- 21. Black A. R., Black J. D. (2012) Protein kinase C signaling and cell cycle regulation. Front. Immunol. 3, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oster H., Leitges M. (2006) Protein kinase C α but not PKCζ suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res. 66, 6955–6963 [DOI] [PubMed] [Google Scholar]
- 23. Murray N. R., Weems J., Braun U., Leitges M., Fields A. P. (2009) Protein kinase C βII and PKCι/λ: collaborating partners in colon cancer promotion and progression. Cancer Res. 69, 656–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gorin M. A., Pan Q. (2009) Protein kinase Cϵ: an oncogene and emerging tumor biomarker. Mol. Cancer 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lum M. A., Balaburski G. M., Murphy M. E., Black A. R., Black J. D. (2013) Heat shock proteins regulate activation-induced proteasomal degradation of the mature phosphorylated form of protein kinase C. J. Biol. Chem. 288, 27112–27127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rao V. U., Shiraishi H., McDermott P. J. (2004) PKC-ϵ regulation of extracellular signal-regulated kinase: a potential role in phenylephrine-induced cardiocyte growth. Am. J. Physiol. Heart Circ. Physiol. 286, H2195–H2203 [DOI] [PubMed] [Google Scholar]
- 27. Clark J. A., Black A. R., Leontieva O. V., Frey M. R., Pysz M. A., Kunneva L., Woloszynska-Read A., Roy D., Black J. D. (2004) Involvement of the ERK signaling cascade in protein kinase C-mediated cell cycle arrest in intestinal epithelial cells. J. Biol. Chem. 279, 9233–9247 [DOI] [PubMed] [Google Scholar]
- 28. Swirnoff A. H., Milbrandt J. (1995) DNA-binding specificity of NGFI-A and related zinc finger transcription factors. Mol. Cell Biol. 15, 2275–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bateman N. W., Tan D., Pestell R. G., Black J. D., Black A. R. (2004) Intestinal tumor progression is associated with altered function of KLF5. J. Biol. Chem. 279, 12093–12101 [DOI] [PubMed] [Google Scholar]
- 30. Saxon M. L., Zhao X., Black J. D. (1994) Activation of protein kinase C isozymes is associated with post-mitotic events in intestinal epithelial cells in situ. J. Cell Biol. 126, 747–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frey M. R., Saxon M. L., Zhao X., Rollins A., Evans S. S., Black J. D. (1997) Protein kinase C isozyme-mediated cell cycle arrest involves induction of p21waf1/cip1 and p27kip1 and hypophosphorylation of the retinoblastoma protein in intestinal epithelial cells. J. Biol. Chem. 272, 9424–9435 [DOI] [PubMed] [Google Scholar]
- 32. Chambard J.-C., Lefloch R., Pouysségur J., Lenormand P. (2007) ERK implication in cell cycle regulation. Biochim. Biophys. Acta 1773, 1299–1310 [DOI] [PubMed] [Google Scholar]
- 33. Roovers K., Assoian R. K. (2000) Integrating the MAP kinase signal into the G1 phase cell cycle machinery. BioEssays 22, 818–826 [DOI] [PubMed] [Google Scholar]
- 34. Tanaka M., Sagawa S., Hoshi J., Shimoma F., Matsuda I., Sakoda K., Sasase T., Shindo M., Inaba T. (2004) Synthesis of anilino-monoindolylmaleimides as potent and selective PKCβ inhibitors. Bioorg. Med. Chem. Lett. 14, 5171–5174 [DOI] [PubMed] [Google Scholar]
- 35. Witzel I. I., Koh L. F., Perkins N. D. (2010) Regulation of cyclin D1 gene expression. Biochem. Soc. Trans. 38, 217–222 [DOI] [PubMed] [Google Scholar]
- 36. Holden N. S., Squires P. E., Kaur M., Bland R., Jones C. E., Newton R. (2008) Phorbol ester-stimulated NF-κB-dependent transcription: roles for isoforms of novel protein kinase C. Cell. Signal. 20, 1338–1348 [DOI] [PubMed] [Google Scholar]
- 37. Page K., Li J., Corbit K. C., Rumilla K. M., Soh J.-W., Weinstein I. B., Albanese C., Pestell R. G., Rosner M. R., Hershenson M. B. (2002) Regulation of Airway Smooth Muscle Cyclin D1 Transcription by Protein Kinase C-δ. Am. J. Respir. Cell Mol. Biol. 27, 204–213 [DOI] [PubMed] [Google Scholar]
- 38. Kang D. W., Park M. H., Lee Y. J., Kim H. S., Kwon T. K., Park W.-S., Min D. S. (2008) Phorbol ester up-regulates phospholipase D1 but not phospholipase D2 expression through a PKC/Ras/ERK/NFκB-dependent pathway and enhances matrix metalloproteinase-9 secretion in colon cancer cells. J. Biol. Chem. 283, 4094–4104 [DOI] [PubMed] [Google Scholar]
- 39. Wilson L., Szabó C., Salzman A. L. (1999) Protein kinase C-dependent activation of NF-κB in enterocytes is independent of IκB degradation. Gastroenterology 117, 106–114 [DOI] [PubMed] [Google Scholar]
- 40. Wang X., Wang Q., Hu W., Evers B. M. (2004) Regulation of phorbol ester-mediated TRAF1 induction in human colon cancer cells through a PKC/RAF/ERK/NF-κB-dependent pathway. Oncogene 23, 1885–1895 [DOI] [PubMed] [Google Scholar]
- 41. Hayakawa M., Miyashita H., Sakamoto I., Kitagawa M., Tanaka H., Yasuda H., Karin M., Kikugawa K. (2003) Evidence that reactive oxygen species do not mediate NF-κB activation. EMBO J. 22, 3356–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morais C., Pat B., Gobe G., Johnson D. W., Healy H. (2006) Pyrrolidine dithiocarbamate exerts anti-proliferative and pro-apoptotic effects in renal cell carcinoma cell lines. Nephrol. Dial. Transplant. 21, 3377–3388 [DOI] [PubMed] [Google Scholar]
- 43. Wang B., Parobchak N., Rosen T. (2012) RelB/NF-κB2 regulates corticotropin-releasing hormone in the human placenta. Mol. Endocrinol. 26, 1356–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hellmuth M., Wetzler C., Nold M., Chang J.-H., Frank S., Pfeilschifter J., Mühl H. (2002) Expression of interleukin-8, heme oxygenase-1 and vascular endothelial growth factor in DLD-1 colon carcinoma cells exposed to pyrrolidine dithiocarbamate. Carcinogenesis 23, 1273–1279 [DOI] [PubMed] [Google Scholar]
- 45. Cavicchi M., Whittle B. J. (1999) Potentiation of cytokine induced iNOS expression in the human intestinal epithelial cell line, DLD-1, by cyclic AMP. Gut 45, 367–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. De Plaen I. G., Han X. B., Liu X., Hsueh W., Ghosh S., May M. J. (2006) Lipopolysaccharide induces CXCL2/macrophage inflammatory protein-2 gene expression in enterocytes via NF-κB activation: independence from endogenous TNF-α and platelet-activating factor. Immunology 118, 153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fasanaro P., Magenta A., Zaccagnini G., Cicchillitti L., Fucile S., Eusebi F., Biglioli P., Capogrossi M. C., Martelli F. (2006) Cyclin D1 degradation enhances endothelial cell survival upon oxidative stress. FASEB J. 20, 1242–1244 [DOI] [PubMed] [Google Scholar]
- 48. Pervin S., Singh R., Chaudhuri G. (2001) Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): potential role of cyclin D1. Proc. Natl. Acad. Sci. U.S.A. 98, 3583–3588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lim J.-H., Lee Y.-M., Chun Y.-S., Park J.-W. (2008) Reactive oxygen species-mediated cyclin D1 degradation mediates tumor growth retardation in hypoxia, independently of p21cip1 and hypoxia-inducible factor. Cancer Sci. 99, 1798–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Natarajan K., Singh S., Burke T. R., Jr., Grunberger D., Aggarwal B. B. (1996) Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-κB. Proc. Natl. Acad. Sci. U.S.A. 93, 9090–9095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Westerheide S. D., Mayo M. W., Anest V., Hanson J. L., Baldwin A. S. (2001) The putative oncoprotein Bcl-3 induces cyclin D1 to stimulate G1 transition. Mol. Cell Biol. 21, 8428–8436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Perletti G. P., Concari P., Brusaferri S., Marras E., Piccinini F., Tashjian A. H., Jr. (1998) Protein kinase Cϵ is oncogenic in colon epithelial cells by interaction with the ras signal transduction pathway. Oncogene 16, 3345–3348 [DOI] [PubMed] [Google Scholar]
- 53. Tang J., Bouyer P., Mykoniatis A., Buschmann M., Matlin K. S., Matthews J. B. (2010) Activated PKCδ and PKCϵ inhibit epithelial chloride secretion response to cAMP via inducing internalization of the Na+-K+-2Cl- cotransporter NKCC1. J. Biol. Chem. 285, 34072–34085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hong D.-H., Petrovics G., Anderson W. B., Forstner J., Forstner G. (1999) Induction of mucin gene expression in human colonic cell lines by PMA is dependent on PKC-ϵ. Am. J. Physiol. 277, G1041–G1047 [DOI] [PubMed] [Google Scholar]
- 55. Yan S., Wenner C. E. (2001) Modulation of cyclin D1 and its signaling components by the phorbol ester TPA and the tyrosine phosphatase inhibitor vanadate. J. Cell Physiol. 186, 338–349 [DOI] [PubMed] [Google Scholar]
- 56. Benavides F., Blando J., Perez C. J., Garg R., Conti C. J., DiGiovanni J., Kazanietz M. G. (2011) Transgenic overexpression of PKCϵ in the mouse prostate induces preneoplastic lesions. Cell Cycle 10, 268–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cheng J.-J., Wung B.-S., Chao Y.-J., Wang D. L. (2001) Sequential activation of protein kinase C (PKC)-α and PKC-ϵ contributes to sustained Raf/ERK1/2 activation in endothelial cells under mechanical strain. J. Biol. Chem. 276, 31368–31375 [DOI] [PubMed] [Google Scholar]
- 58. Besson A., Davy A., Robbins S. M., Yong V. W. (2001) Differential activation of ERKs to focal adhesions by PKCϵ is required for PMA-induced adhesion and migration of human glioma cells. Oncogene 20, 7398–7407 [DOI] [PubMed] [Google Scholar]
- 59. Garg R., Blando J., Perez C. J., Wang H., Benavides F. J., Kazanietz M. G. (2012) Activation of nuclear factor κB (NF-κB) in prostate cancer is mediated by protein kinase Cϵ (PKCϵ). J. Biol. Chem. 287, 37570–37582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tojima Y., Fujimoto A., Delhase M., Chen Y., Hatakeyama S., Nakayama K., Kaneko Y., Nimura Y., Motoyama N., Ikeda K., Karin M., Nakanishi M. (2000) NAK is an IκB kinase-activating kinase. Nature 404, 778–782 [DOI] [PubMed] [Google Scholar]
- 61. Faisal A., Saurin A., Gregory B., Foxwell B., Parker P. J. (2008) The scaffold MyD88 acts to couple protein kinase Cϵ to Toll-like receptors. J. Biol. Chem. 283, 18591–18600 [DOI] [PubMed] [Google Scholar]
- 62. Shih V. F., Tsui R., Caldwell A., Hoffmann A. (2011) A single NFκB system for both canonical and non-canonical signaling. Cell Res. 21, 86–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fusco A. J., Huang D. B., Miller D., Wang V. Y., Vu D., Ghosh G. (2009) NF-κB p52:RelB heterodimer recognizes two classes of κB sites with two distinct modes. EMBO Rep. 10, 152–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang J., Warren M. A., Shoemaker S. F., Ip M. M. (2007) NFκB1/p50 is not required for tumor necrosis factor-stimulated growth of primary mammary epithelial cells: implications for NFκB2/p52 and RelB. Endocrinology 148, 268–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang F., Shi Y., Yadav S., Wang H. (2010) p52-Bcl3 complex promotes cyclin D1 expression in BEAS-2B cells in response to low concentration arsenite. Toxicology 273, 12–18 [DOI] [PubMed] [Google Scholar]
- 66. Toualbi-Abed K., Daniel F., Güller M. C., Legrand A., Mauriz J.-L., Mauviel A., Bernuau D. (2008) Jun D cooperates with p65 to activate the proximal κB site of the cyclin D1 promoter: role of PI3K/PDK-1. Carcinogenesis 29, 536–543 [DOI] [PubMed] [Google Scholar]
- 67. Stein B., Baldwin A. S., Jr., Ballard D. W., Greene W. C., Angel P., Herrlich P. (1993) Cross-coupling of the NF-κB p65 and Fos/Jun transcription factors produces potentiated biological function. EMBO J. 12, 3879–3891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li X., LuValle P. (2011) Activating transcription factor 2 targets c-Fos, but not c-Jun, in growth plate chondrocytes. J. Cell Biochem. 112, 211–216 [DOI] [PubMed] [Google Scholar]
- 69. Guo X., Li W., Wang Q., Yang H.-S. (2011) AKT activation by Pdcd4 knockdown up-regulates cyclin D1 expression and promotes cell proliferation. Genes Cancer 2, 818–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Guo Z.-Y., Hao X.-H., Tan F.-F., Pei X., Shang L.-M., Jiang X.-L., Yang F. (2011) The elements of human cyclin D1 promoter and regulation involved. Clin. Epigenetics 2, 63–76 [DOI] [PMC free article] [PubMed] [Google Scholar]