

Background: Chlamydia trachomatis has a phospholipid composition that resembles its eukaryotic host, but it contains branched-chain fatty acids of chlamydial origin.
Results: The inhibition of the enoyl-acyl carrier protein reductase (FabI) in chlamydial fatty acid synthesis blocks C. trachomatis replication.
Conclusion: Bacterial FASII is required for C. trachomatis proliferation.
Significance: FabI is a therapeutic target against C. trachomatis.
Keywords: Bacterial Metabolism, Chlamydia trachomatis, Enzyme Inhibitor, Fatty Acid Synthase (FAS), Glycerophospholipid, Fatty Acid Synthesis
Abstract
The major phospholipid classes of the obligate intracellular bacterial parasite Chlamydia trachomatis are the same as its eukaryotic host except that they also contain chlamydia-made branched-chain fatty acids in the 2-position. Genomic analysis predicts that C. trachomatis is capable of type II fatty acid synthesis (FASII). AFN-1252 was deployed as a chemical tool to specifically inhibit the enoyl-acyl carrier protein reductase (FabI) of C. trachomatis to determine whether chlamydial FASII is essential for replication within the host. The C. trachomatis FabI (CtFabI) is a homotetramer and exhibited typical FabI kinetics, and its expression complemented an Escherichia coli fabI(Ts) strain. AFN-1252 inhibited CtFabI by binding to the FabI·NADH complex with an IC50 of 0.9 μm at saturating substrate concentration. The x-ray crystal structure of the CtFabI·NADH·AFN-1252 ternary complex revealed the specific interactions between the drug, protein, and cofactor within the substrate binding site. AFN-1252 treatment of C. trachomatis-infected HeLa cells at any point in the infectious cycle caused a decrease in infectious titers that correlated with a decrease in branched-chain fatty acid biosynthesis. AFN-1252 treatment at the time of infection prevented the first cell division of C. trachomatis, although the cell morphology suggested differentiation into a metabolically active reticulate body. These results demonstrate that FASII activity is essential for C. trachomatis proliferation within its eukaryotic host and validate CtFabI as a therapeutic target against C. trachomatis.
Introduction
Chlamydia trachomatis is a Gram-negative, obligate intracellular bacterial parasite that causes a range of human diseases (1). C. trachomatis infections are the most prevalent bacterial sexually transmitted diseases, causing tens of millions of new cases a year, and the leading cause of infectious blindness affecting hundreds of millions of people. C. trachomatis has a dimorphic life cycle (2). The elementary body (EB)2 is the metabolically quiescent, infectious, extracellular form that initiates the infection by attaching to a suitable eukaryotic host cell. Once internalized by the host cell, the EB differentiates into the metabolically active and noninfectious reticulate body (RB) that replicates inside of a vacuole called the chlamydial inclusion. At 16–20 h after the initial chlamydial infection, RBs start differentiating into EBs, and by 48–72 h after the infection, most RBs have converted into EBs. The lysis of the host cell releases the infectious EBs, and the cycle of infection continues.
The major C. trachomatis phospholipid classes are the same as those of the host cell, but they also contain branched-chained fatty acids made by C. trachomatis at the 2-position of some phospholipids (branched chains constitute ∼20% of the total acyl chains in C. trachomatis phospholipids) (3). The branched-chain fatty acids are thought to be produced by chlamydial type II fatty acid synthesis (FASII), but it is unknown whether FASII is required for phospholipid acquisition/synthesis in C. trachomatis. The current model is that C. trachomatis acquires host phospholipids, a portion of which are subsequently deacylated at the 2-position and reacylated with endogenously made anteiso-15:0 (3). The host cPLA2 has been implicated in the deacylation reaction because pharmacological inhibition of the Raf/MEK/ERK/cPLA2 pathway decreases C. trachomatis titers (4). However, recent studies using cPLA2 knockdown human cells found that a decrease in cPLA2 activity has no effect on C. trachomatis growth (5). In mouse cells, cPLA2 knockdown increases chlamydial titers, suggesting that cPLA2 is more likely involved in intracellular immunity (5). One function of FASII is to generate the 3-hydroxy fatty acids for lipopolysaccharide synthesis in Gram-negative bacteria (6). The outer membrane of C. trachomatis is decorated with a similar lipooligosaccharide (LOS) that contains 18 and 20 carbon 3-hydroxy fatty acids (7–9). The inhibition of LOS biosynthesis in C. trachomatis does not block RB proliferation but does prevent the differentiation of RB into EB, resulting in the accumulation of non-infectious RBs within the chlamydial inclusion (10). Thus, one function of FASII would be to provide these 3-hydroxy fatty acids for LOS synthesis in C. trachomatis, but it is unknown whether FASII is needed for phospholipid synthesis.
We used AFN-1252 as a chemical biology tool to specifically inhibit the enoyl-acyl carrier protein (ACP) reductase (FabI) (11–13) and assess the role of FASII in C. trachomatis replication. C. trachomatis is predicted to encode a FASII system that depends on FabI (Fig. 1). AFN-1252 inhibits C. trachomatis FabI (CtFabI) by forming a tight ternary CtFabI·NADH·AFN-1252 complex visualized at 1.8 Å resolution. AFN-1252 treatment arrests the replication of C. trachomatis when added at any time during the infectious cycle, and when administered at the beginning of the infection, it blocks development at a single cell RB-like state. These data show that FASII is required for C. trachomatis replication and validate CtFabI as a suitable target for antichlamydial therapy.
FIGURE 1.
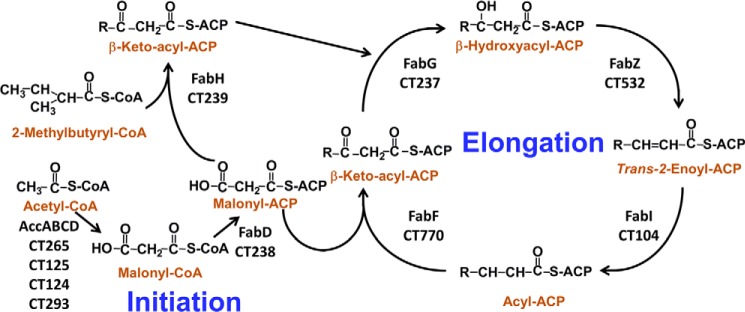
Fatty acid synthesis in C. trachomatis. C. trachomatis encode the genes for FASII. The locus tags of these genes in strain D/UW-3/Cx are annotated below the enzyme names. Acetyl-CoA is converted into malonyl-CoA by the acetyl-CoA carboxylase enzyme complex (AccABCD). Malonyl-CoA is then converted into malonyl-ACP by the enzyme malonyl-CoA-ACP transacylase (FabD). Fatty acid synthesis is initiated by 3-oxoacyl-ACP synthase III (FabH) condensing malonyl-ACP with either acetyl-CoA for straight-chain fatty acid synthesis or 2-methylbutyryl-CoA for anteiso branched-chain fatty acid synthesis. The resulting 3-ketoacyl-ACP is reduced by 3-oxoacyl-ACP reductase (FabG), and the resulting 3-hydroxyacyl-ACP is dehydrated by 3-hydroxyacyl-ACP dehydratase (FabZ) into trans-2-enoyl-ACP. The last step in the cycle is the conversion of trans-2-enoyl-ACP to acyl-ACP by FabI. Known genes for making unsaturated fatty acids are absent in C. trachomatis. Long-chain acyl-ACP is used for phospholipid synthesis, and long-chain 3-hydroxyacyl-ACP is used for LOS synthesis.
EXPERIMENTAL PROCEDURES
Molecular Biology
The fabI gene (CT104) of C. trachomatis strain D/UW-3/Cx (NCBI Microbial Genomes Database) was optimized for expression in Escherichia coli through GeneArt gene synthesis (Invitrogen). A NdeI cleavage site was engineered at the 5′-end of the gene with a start codon in the NdeI site, whereas a 6-histidine tag, stop codon, and a EcoRI cleavage site were sequentially engineered at the 3′-end of the gene. The fabI sequence was cloned into the plasmids pET21a (Novagen) and pPJ131, pBluescript plasmid (Stratagene) with a modified multiple cloning site (14), via the NdeI and EcoRI (New England Biolabs) cloning sites. The pET21a-CtfabI expression plasmid was transformed in BL21 Tuner cells (Novagen) for protein expression and purification.
The pPJ131-CtfabI plasmid was transformed into the fabI temperature-sensitive E. coli strain JP1111 to determine complementation. The JP1111 strain is viable at 30 °C but nonviable at 42 °C without fabI gene complementation. The JP1111 cells were transformed with the pPJ131-CtfabI plasmid, the pPJ131 parent plasmid, and pBluescript plasmids expressing fabI from E. coli and Bacillus anthracis and then plated on Luria-Bertani (LB) plates at 30 °C with 100 μg/ml carbenicillin. The transformed cells were restreaked onto LB plates with 100 μg/ml carbenicillin and grown at 30 or 42 °C to determine whether the C. trachomatis fabI complements the E. coli fabI activity.
CtFabI Expression and Purification
BL21 Tuner cells harboring the pET21a-CtfabI plasmid were grown in LB medium with 100 μg/ml carbenicillin at 37 °C and 225 rpm shaking until A600 reached 0.6–0.8. The culture was then induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside and grown at 17 °C and 225 rpm shaking overnight. Cells were pelleted and washed twice with 20 mm Tris, pH 8.0, and finally resuspended in 20 mm Tris, pH 8.0 (40 ml/liter of culture). Cells were lysed via a cell disruptor, and the C-terminal 6-histidine-tagged CtFabI was purified via nickel chelation chromatography. Briefly, the cell lysate was centrifuged at 20,000 × g to remove the cell debris. The resulting supernatant was poured over a nickel-nitrilotriacetic acid resin column (4 ml of resin/liter of cell culture) to bind the protein to the column. The column was washed with 5 column volumes of 20 mm Tris, pH 8.0, and 20 mm imidazole, followed by 5 column volumes of 20 mm Tris, pH 8.0, 500 mm NaCl, and 50 mm imidazole. The protein was eluted from the column with 20 mm Tris pH 8.0 and 250 mm imidazole. The fractions containing protein, as determined by the Bradford reagent, were collected and dialyzed against 20 mm Tris, pH 8.0, 10 mm EDTA, and 150 mm NaCl at 4 °C overnight. A pure protein (>95%) running at ∼32 kDa (theoretical average mass of 32,783.49 with N-terminal methionine) was observed on a NuPAGE 10% BisTris gel. Over 25 mg of purified CtFabI was obtained per liter of culture.
Analytical Ultracentrifugation
Analytical ultracentrifugation experiments and analysis were carried out as described previously by the St. Jude Molecular Interaction Analysis Facility (15, 16). Experiments were performed in 20 mm Tris, pH 7.5, 10 mm EDTA, 150 mm NaCl buffer. Chlamydial FabI has a calculated molecular mass of 32,783 Da.
Enzymology
FabI activity was determined by monitoring the optical density at 340 nm to measure the conversion of NAD(P)H to NAD(P)+. The enzyme reactions were 100 μl in volume and were performed and monitored in Costar UV half-area 96-well plates with a SpectraMax 340 instrument taking 340-nm readings at 10-s intervals at 30 °C. The substrate NADH and NADPH were obtained from Sigma-Aldrich. The substrate crotonyl-ACP from S. aureus was synthesized as described previously (17). For velocity measurements, the CtFabI enzyme was added to 100 μm crotonyl-ACP and a 250 μm concentration of either NADPH or NADH in 20 mm Tris, pH 8.0. Upon finding that CtFabI prefers NADH over NADPH by over 1000-fold, NADH was used for future measurements. For determination of Km of crotonyl-ACP, 100 nm CtFabI was added to 200 μm NADH and 3, 6, 12, 24, 36, or 48 μm crotonyl-ACP. For determination of Km of NADH, 100 nm CtFabI was added to 50 μm crotonyl-ACP and 5, 10, 15, 20, 30, 50, and 75 μm NADH. The reaction was mixed for 10 s by the mix function on the plate reader, and data were acquired at 10-s intervals for 5 min. The initial velocity was calculated from the linear phase of the progress curve, and the initial velocity data were fit using a standard Michaelis-Menten equation to determine the apparent Km. The IC50 of AFN-1252 and triclosan against CtFabI was measured as above at saturating substrate concentrations (50 μm crotonyl-ACP and 200 μm NADH) against different concentrations of AFN-1252 and triclosan (0, 0.156, 0.312, 0.625, 1.25, 2.5, 5, and 10 μm). All kinetic experiments were run in triplicate.
Protein Thermal Shift Analysis
Protein thermal shift analysis was conducted to determine the mode of binding of AFN-1252 to CtFabI. Solutions (30 μl) of CtFabI (10 μm), CtFabI (10 μm) + AFN-1252 (10 μm), CtFabI (10 μm) + NADH (100 μm), CtFabI (10 μm) + NADH (100 μm) + AFN-1252 (10 μm), CtFabI (10 μm) + NAD+ (100 μm), and CtFabI (10 μm) + NAD+ (100 μm) + AFN-1252 (10 μm) in a final buffer of 100 μm Tris, pH 7.0, 0.5% DMSO, and 2.5× Sypro Orange Dye (Sigma-Aldrich) were added to wells of ThermoGrid optically clear PCR plates (Denville Scientific). The plates were centrifuged for 5 min at 1000 × g to remove air bubbles and then subjected to thermal shift analysis in the ABI 7300 real-time PCR system. The temperature was ramped from 25 to 95 °C at 1 °C/min with the fluorescence read six times at each temperature ramp. The resulting data were fit to a Boltzmann sigmoidal equation to determine the melting point of the CtFabI under each particular ligand combination. Each ligand condition was replicated six times, and each replicate was subjected to independent analysis to determine the thermal melting point. The melting points of the six independent runs per ligand condition were averaged to determine the thermal melting point for each ligand condition. Representative thermal shift runs along with the average thermal melting points are presented.
Crystallization and Structure Determination
The CtFabI protein was incubated for 1 h with 0.4 mm NADH and 0.25 mm AFN-1252 at 2 mg/ml and then concentrated to 7 mg/ml for crystallization. Initial trials were performed at 18 ºC against the PEGs Suite and PEGs II Suite (Qiagen), and several conditions produced small crystals that were subsequently optimized. Crystals suitable for x-ray analysis were eventually obtained in 28% (w/v) polyethylene glycol 300, 0.1 m HEPES (pH 7.5), and 0.2 m potassium formate. Crystals were flash-frozen directly from mother liquor in liquid nitrogen and diffracted to 1.8 Å in space group P43 with unit cell dimensions a = 96.2 Å, b = 96.2 Å, c = 263.0 Å. Diffraction data were collected at the SER-CAT beam line ID22 at the Advanced Photon Source and processed using HKL2000 (18). The CtFabI structure was solved by maximum likelihood molecular replacement using the program Phaser (19) and the coordinates of apo-FabI from Brassica napus (Protein Data Bank accession code 1ENO) as the search model. The structure was completed by iterative rounds of refinement using Phenix (20) and manual rebuilding using Coot (21). The refinement was monitored by following the Rfree value calculated from a random subset (5%) of omitted reflections. A summary of the data reduction and structure refinement statistics is provided in Table 1, and the coordinates have been deposited in the Protein Data Bank (accession code 4Q9N). The images of the protein structure were generated with PyMOL (22).
TABLE 1.
X-ray crystallographic data and refinement statistics for CtFabI·NADH·AFN1252 complex (Protein Data Bank entry 4Q9N)
| Crystal | CtFabI·NADH· AFN-1252 |
|---|---|
| Data collection | |
| Space group | P43 |
| Wavelength (Å) | 1.0 |
| Unit cell dimensions | |
| a (Å) | 96.15 |
| b (Å) | 96.15 |
| c (Å) | 263.08 |
| α, β, γ (degrees) | 90, 90, 90 |
| Molecules/ASUa | 8 |
| Resolution (Å) b | 1.8 (1.85–1.80)d |
| Completeness (%)b | 98.8 (95.1) |
| Redundancyb | 6.3 (3.4) |
| No. of total reflections | 335,128 |
| No. of unique reflections | 221,426 |
| I/σb | 15.5 (1.57) |
| Rsymb,c | 12.2 (61.6) |
| Refinement statistics | |
| Resolution (Å) | 1.8 |
| No. of reflections | 218,306 |
| Rwork/Rfree (%)d,e | 0.1863/0.2292 |
| No. of atoms | 19,232 |
| Protein | 17,700 |
| Ligand/ion | 576 |
| Water | 956 |
| B-Factors (Å2) | |
| Protein | 25.4 |
| Ligand/ion | 22.7 |
| Water | 28.2 |
| Root mean square deviations | |
| Bond length (Å) | 0.006 |
| Bond angle (degrees) | 1.057 |
| Ramachandran analysis | |
| Most favored (%) | 97.5 |
| Allowed (%) | 3.3 |
| Disallowed (%) | 0.2 |
a Asymmetric unit.
b Values in parentheses are for the highest resolution shell.
c Rsym = Σ|I − 〈I〉|/Σ〈I〉, where I is the observed intensity, and 〈I〉 is the average intensity of multiple observations of symmetry-related reflections.
d r = Σhkl‖Fo| − |Fc‖/Σhkl|Fo|.
e Rfree is calculated from 5% of the reflections excluded from refinement.
C. trachomatis Strains and Propagation
C. trachomatis serovar L2 (strain 434/Bu) was used for most experiments. C. trachomatis was propagated by standard infection through centrifugation protocols in HeLa cells grown in DMEM containing 10% fetal bovine serum (23). Cell cultures were grown in Costar 6-well cell culture plates (Corning) with 3 ml of medium. Depending on the specific experimental conditions, AFN-1252 was added either immediately postinfection or some number of hours postinfection. Infected cells were harvested at 48 h postinfection, and the number of infectious units was determined through reinfection. Serovar D was propagated similarly but harvested 72 h after infection.
Genome Copy Number Quantitation
HeLa 229 cells were infected with C. trachomatis 434/Bu (NCBI Reference Sequence: NC_010287.1) at a multiplicity of infection of 1 in 6-well plates. DNA was collected at 0, 7, 14, 27, and 53 h postinfection. For some samples, AFN-1252 was added at the time of infection at a concentration of 10 μm. Nucleic acid was collected using MasterPure (Epicenter), and samples were processed according to the manufacturer's instructions. After RNA was removed by digestion with RNase A, the DNA was repurified by isopropyl alcohol precipitation. DNA quantification was carried out using a LightCycler 480 system (Roche Applied Science). A standard curve was generated using purified chromosomal template DNA at concentrations ranging from 10 to 0.001 ng/well, and samples (2 ng/well) were then analyzed in triplicate according to the manufacturer's instructions. The gene for 16 S RNA was selected for quantification, and primer design and Roche Universal probe library selection were carried out using Web-based ProbeFinder software version 2.50 (Roche Applied Science). Forward and reverse Primer sequences were 5′-GAGATGGAGCAAATCCTCAAA and 5′-ACTTCATGTAGTCGAGTTGCAGA, respectively. Data analysis and standard curve construction were carried out using LightCycler 480 software. Measurements were made in triplicate.
Localization Analyses
Mouse monoclonal antibodies directed against chlamydial Hsp60 were provided by Dr. R. P. Morrison, and antibodies against the chlamydial major outer membrane protein (MOMP) were obtained from Meridian Life Sciences. AlexaFluor-conjugated secondary antibodies and Hoechst were obtained from Invitrogen. HeLa cells infected with C. trachomatis serovar D or C. trachomatis serovar L2 in the absence or presence of AFN-1252 were fixed at 48 h postinfection by incubation in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min. Following rinsing in PBS, the cells were permeabilized by incubation in methanol for 10 s and rinsed again in PBS. The cells were then incubated with monoclonal antibodies directed against chlamydial Hsp60 followed by secondary antibodies conjugated to AlexaFluor 488 prior to analysis on a Zeiss Axioplan 2 epifluorescent microscope. In some cases, cells were double-stained with antibodies directed against MOMP and Hsp60.
C. trachomatis Metabolic Labeling
Labeling experiments were performed on C. trachomatis cultures 20 h postinfection. Experiments were conducted in Costar 6-well cell culture plates with 3 ml of medium in each well. Radiolabeled chemicals were from American Radiolabeled Chemicals. For [U-14C]isoleucine labeling experiments, the cell culture was washed with 1 ml of PBS and then incubated in 3 ml of fresh DMEM with 10% FBS. For [U-14C]glucose labeling experiments, the cell culture was washed with 1 ml of PBS and then incubated in 3 ml of fresh DMEM with 10% FBS containing only 0.45 g/liter glucose to increase the specific activity of the labeled glucose added later. AFN-1252 was added to the culture for 30 min before the radioactive labels (10 μCi of either [U-14C]isoleucine or [U-14C]glucose) were added. DMSO (equal volume to AFN-1252 additions) was added to the infected culture for 30 min for the no drug controls. Labeling was allowed for 4 h at 37 °C and 5% CO2 before cells were harvested and extracted for lipids via the Bligh and Dyer protocol (24). Radioactive incorporation was measured by counting the lipid extracts on an LS6500 multipurpose scintillation counter. Measurements were made in triplicates, and the averages with S.E. were reported. For the HeLa cell controls, a mock infection (same infection protocol with no C. trachomatis) was performed on the cells, and the same labeling procedure was performed. Cell viability was measured with the NucleoCounter (New Brunswick Scientific) following the manufacturer's protocol.
RESULTS
C. trachomatis FASII
A bioinformatics analysis of the C. trachomatis genome suggests that it encodes a complete FASII (Fig. 1). Branched-chain fatty acids arise from the utilization of 2-methylbutyryl-CoA by FabH to prime FASII (25). 2-Methylbutyryl-CoA is generated from isoleucine by the branched-chain α-ketoacid dehydrogenase complex. The structure and mechanism of the branched-chain α-ketoacid dehydrogenase complex is similar to the pyruvate dehydrogenase complex. The genes encoding both the branched-chain α-ketoacid dehydrogenase complex (CT340, CT400, and CT557) and the pyruvate dehydrogenase complex (CT245 to -247) were identified in the C. trachomatis genome. This bioinformatics analysis predicts that C. trachomatis has the gene set for the synthesis of branched-chain and hydroxy fatty acids that cannot be obtained from the host. Branched-chain fatty acids are incorporated into chlamydial phospholipids (26), and the straight-chain 3-hydroxy fatty acids are found in the LOS of this organism (27–29). The gene(s) for unsaturated fatty acid synthesis are absent.
CtFabI Is an Enoyl-ACP Reductase
The enoyl-ACP reductase plays a determinant role in establishing the rate of FASII (30, 31). The predicted FabI sequences from different chlamydial strains have greater than 99% amino acid identity and are homologous to the SaFabI (31% identity, 49% similarity). The function of CtFabI in FASII was assessed by determining whether CtFabI expression complemented the growth phenotype of an E. coli fabI(Ts) mutant. The E. coli codon-optimized fabI gene from C. trachomatis D/UW-3/Cx was cloned into the pBluescript expression vector and transformed into E. coli strain JP1111 (fabI(Ts)) (32). At the permissive temperature (30 °C), strain JP1111 and all its transformants exhibited robust growth (Fig. 2A). At the non-permissive temperature (42 °C), strain JP1111 was unable to grow without a complementing fabI gene being present in the plasmid (Fig. 2B). The E. coli, B. anthracis, and C. trachomatis fabI genes all complemented growth at the non-permissive temperature, confirming that the C. trachomatis fabI gene product functioned as an enoyl-ACP reductase in the heterologous E. coli FASII.
FIGURE 2.
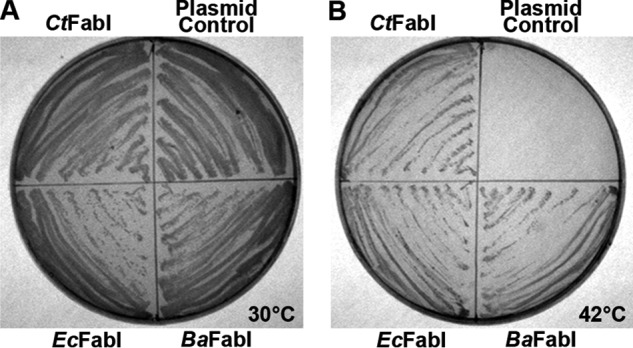
The CT104 gene functions as a FabI. A, growth of the temperature-sensitive E. coli strain JP1111 (fabI(Ts)) transformed with plasmids expressing either the C. trachomatis, E. coli (EcFabI), or B. anthracis FabI (BaFabI) or the empty vector control under the permissive growth condition (30 °C). B. anthracis was used as a control to illustrate that a FabI from an organism that makes branched-chain fatty acids can complement the E. coli enzyme. B, growth of the same strain series at the non-permissive temperature (42 °C). Complementation of the growth phenotype at 42 °C indicates the expression of a functional enoyl-ACP reductase.
CtFabI was expressed, and the protein was purified by affinity chromatography (Fig. 3A). CtFabI sedimented as a 128-kDa complex in analytical centrifugation experiments (Fig. 3B), which was consistent with CtFabI as a homotetramer (131 kDa expected), like other characterized FabI proteins (33). The enzymatic reaction of CtFabI was assessed by measuring NADH/NADPH conversion to NAD+/NADP+ at 340 nm. The CtFabI catalyzed the turnover of NADH (Km = 22.93 ± 3.16 μm) (Fig. 3C) into NAD+ in the presence of crotonyl-ACP (Km = 18.18 ± 2.67 μm) (Fig. 3D). NADPH was >1000-fold less efficient than NADH in supporting the reaction, consistent with CtFabI preferring NADH like most bacterial FabIs, with the sole exception of SaFabI (30, 34). The kcat of CtFabI with NADH and crotonyl-ACP was 15.6 ± 0.05 min−1, which is similar in velocity to other characterized FabI enzymes (35). These biochemical experiments confirmed that CtFabI was an enoyl-ACP reductase.
FIGURE 3.

Purification and biochemistry of CtFabI. A, CtFabI was purified by affinity and gel filtration chromatography. Gel electrophoresis shows the purity of CtFabI. Lane 1, standards; lane 2, CtFabI (32.7 kDa). B, the sedimentation velocity profiles (fringe displacement) were fitted to a continuous sedimentation coefficient distribution model c(s). CtFabI sedimented as a 128-kDa tetramer (131 kDa theoretical mass). C, initial velocities were determined in triplicate as a function of NADH concentration. D, initial velocities were determined in triplicate as a function of crotonyl-ACP concentration. The data points were fit to the Michaelis-Menten nonlinear regression fit in GraphPad Prism version 5. The fitted line is shown on the graph. The fitted Km values with the S.E. values (error bars) are reported.
AFN-1252 Inhibition of CtFabI
The chemical biology approach was validated by determining the affinity and the mode of inhibition of AFN-1252 for CtFabI. AFN-1252 exhibited an IC50 of 0.95 ± 0.21 μm against CtFabI at saturating substrate concentrations (Fig. 4A). The IC50 against the prototypical FabI inhibitor, triclosan, was also determined (0.32 ± 0.08 μm). AFN-1252 inhibited SaFabI by forming a FabI·NADPH·AFN-1252 complex (12, 17). The formation of this ternary complex on CtFabI was demonstrated by examining the effects of AFN-1252 on the CtFabI melting temperature using a protein thermal denaturation assay (Fig. 4B). CtFabI had a melting temperature of 48 °C in the absence of any ligand. The addition of AFN-1252 did not shift the melting temperature, indicating the absence of a CtFabI·AFN-1252 complex. The addition of NADH stabilized CtFabI, as indicated by a shift in the melting temperature to 50.2 °C. The addition of NADH and AFN-1252 increased stability substantially by shifting the melting temperature to 65.8 °C. The addition of NAD+ or NAD+ and AFN-1252 to CtFabI did not significantly increase the thermal stability, indicating the absence of a CtFabI·NAD+·AFN-1252 complex. These data indicated that AFN-1252 inhibited CtFabI by binding to the CtFabI·NADH complex. Thus, AFN-1252 is a sharp tool to study the effects of FASII inhibition on C. trachomatis replication in light of its selectivity, its binding affinity, and the absence of off-target effects on human cells (11, 36).
FIGURE 4.
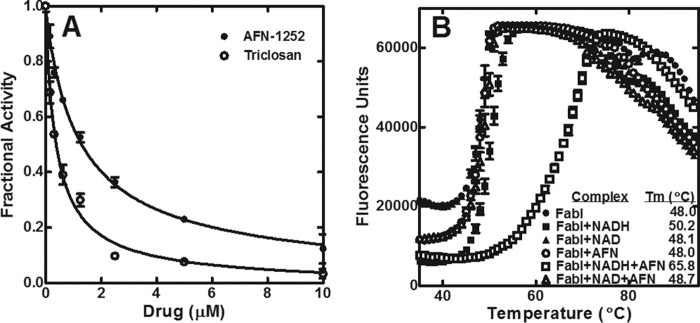
AFN-1252 inhibition of CtFabI. A, initial velocities were determined in triplicate as a function of AFN-1252 (●) or triclosan (○) concentration. The data points were fit to the IC50 equation of the form, vi/v0 = 1/(1 + [I]/IC50)), using custom equation fit in GraphPad Prism 5. The fitted line is shown on the graph. The fitted IC50 values and S.E. values (error bars) are reported. B, protein thermal denaturation analysis was used to determine the melting point of FabI alone (●), FabI + NADH (■), FabI + NAD+ (▴), FabI + AFN-1252 (○), FabI + NADH + AFN-1252 (□), and FabI + NAD+ + AFN-1252 (▵).
The FabI·NADH·AFN-1252 Ternary Complex
A 1.8 Å crystal structure of the ternary complex was solved to validate the biochemical characterization (Table 1). There were eight molecules of the ternary complex in the crystal asymmetric unit that represented two copies of the biological tetramer. The protomers were all structurally similar with a root mean square deviation between all main chain atoms of 0.17 Å. The electron densities of the co-factor NADH and the small molecule inhibitor in each of the eight active sites were well resolved. The comparison of the overall fold of CtFabI with that of SaFabI shows that both proteins have a seven-stranded parallel β-sheet flanked by 11 α-helices and several loops that are typical for FabIs (Fig. 5) (33). The active site contains the signature catalytic tyrosine and lysine residues (Tyr-188 and Lys-195) directly adjacent to the bound cofactor (33). CtFabI has two additional loops comprising residues 56–69 and 84–104, but they are located at the surface and do not impact the conserved FabI fold or the active site (Fig. 5). AFN-1252 consists of oxotetrahydronaphthyridine and 3-methylbenzofuran groups linked by a cis-amide. The carbonyl oxygen atom of the cis-amide makes hydrogen bond interactions with the 2′-hydroxyl of the nicotinamide ribose and the hydroxyl of Tyr-188, whereas the pyridyl nitrogen and the N-acyl hydrogen of the oxotetrahydronaphthyridine moiety make hydrogen bonds with the peptide backbone carbonyl and amide of Ser-130, respectively (Fig. 6A). The 3-methylbenzofuran group forms an edge-to-face π-interaction with the side chain of Phe-235. FabI is characterized by a flexible substrate-binding loop, called the “flipping loop,” that “closes” to orient the substrate with respect to the cofactor nicotinamide ring for reduction (37–39). This loop (residues 228–240) adopts the “closed” conformation that has been observed with other FabI·inhibitor complexes (12, 40, 41), and the cis-amide stacks onto the nicotinamide ring to apparently mimic the targeted double bond of the bound substrate (33).
FIGURE 5.

Comparison of the primary and secondary structures of CtFabI and SaFabI. The primary and overall secondary structures of CtFabI and SaFabI are conserved. The two substrate binding loop regions are surrounded by boxes with colors corresponding to the structural elements depicted in Fig. 6. Notably, the helical loop η3 in SaFabI that contains the critical Met-99 residue is replaced in CtFabI with a less structured loop region containing a two-residue deletion. Red, identical residues. Structure-based sequence alignments were performed using Salign (47) and analyzed using ESPript3 (48).
FIGURE 6.
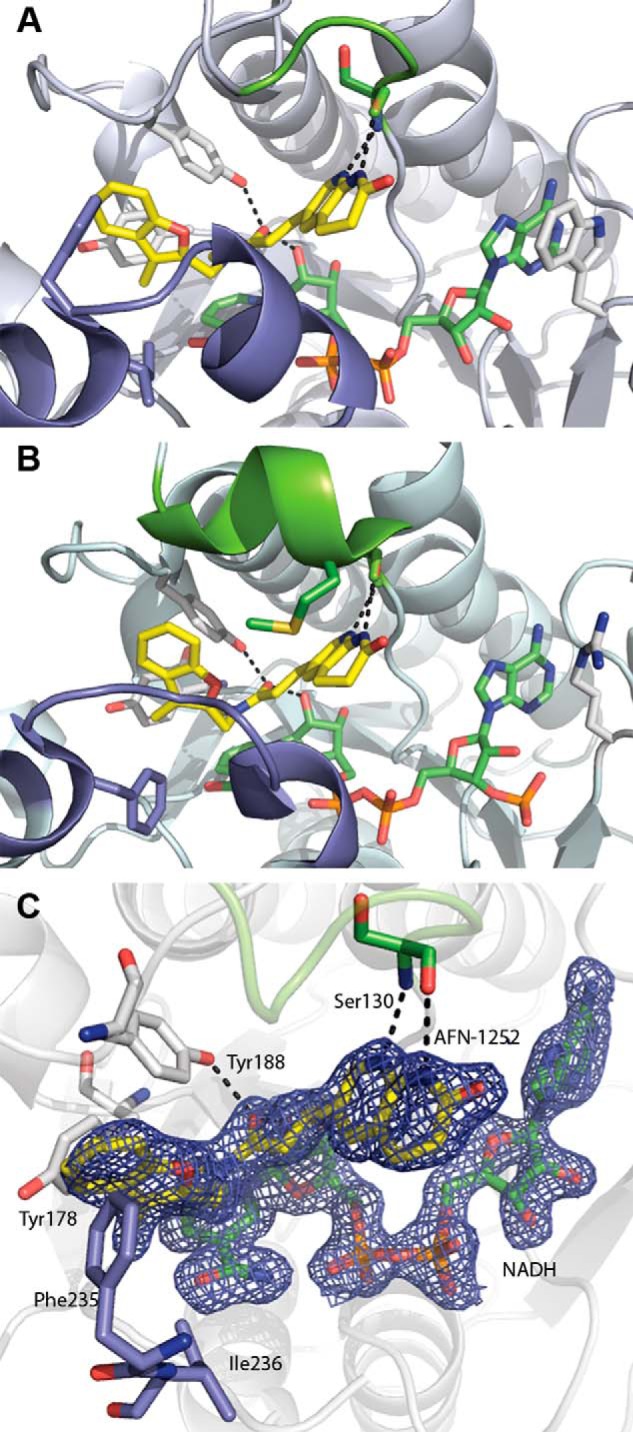
FabI·NADH·AFN ternary complex structure. A, CtFabI·NADH·AFN-152 ternary complex (Protein Data Bank accession code 4Q9N). B, SaFabI·NADPH·AFN-1252 ternary complex (Protein Data Bank accession code 4FS3). The color schemes are the same in A and B. AFN-1252 is shown in yellow, and NADPH/NADH is shown in green. The flexible, substrate-binding flipping loop is colored slate, and the poorly conserved lid loop is colored chartreuse. Black dashes show the conserved hydrogen bonding network for AFN-1252. In SaFabI, the substrate lid adopts a more ordered, helical structure and orients Met-99 into the active site toward the inhibitor. In contrast, in CtFabI, the active site is more open, due to the less structured conformation of the lid. C, active site of the CtFabI·NADH·AFN-1252 ternary complex. AFN-1252 binds within a hydrophobic pocket composed of Tyr-178, Phe-235, and Ile-236, with Phe-235 positioned for π-interactions with the 3-methylbenzofuran of AFN-1252. The 2′-hydroxyl of NADH and the hydroxyl of Tyr-188 make hydrogen bonds with the carbonyl of the linking cis-amide, and the Ser-130 peptide backbone carbonyl and amide make hydrogen bonds with the pyridyl nitrogen and the N-acyl hydrogen of the oxotetrahydronaphthyridine of AFN-1252. σA-weighted 2Fo − Fc electron density is shown for AFN-1252 and NADH contoured at 1.5 σ.
The CtFabI and SaFabI (Protein Data Bank accession code 4FS3) structures are very similar, with a root mean square deviation on main chain atoms of 1.4 Å, and the modes of binding of the cofactor and inhibitor are practically identical (Fig. 6). One difference is that Phe-204 on the SaFabI substrate-binding loop (residues 195–207) is replaced by Ile-236 in CtFabI; however, the adjacent Phe-235 of CtFabI remains positioned to make the edge-to-face π-interaction with the 3-methylbenzofuran group of the inhibitor. FabI proteins have a relatively unconserved and conformationally flexible substrate-binding α-helical loop that creates a “lid” over the active site following the binding of the cofactor and substrate (17, 40). In SaFabI, Met-99 within this segment (residues 97–102) extends into the active site pocket toward the oxotetrahydronaphthyridine group of AFN-1252, and this key hydrophobic interaction is critical for the high-affinity binding of AFN-1252 to SaFabI (Fig. 6B) (17). Comparison of the CtFabI and SaFabI structures revealed not only that CtFabI contains a two-residue deletion, including this methionine, but that the loop (residues 130–133) was apparently more flexible and lacked the ordered α-helical segment found in SaFabI (Fig. 6). Most significantly, the loop is flipped away from the inhibitor-binding site and, as a result, no residues of CtFabI are positioned to replace the lost hydrophobic interaction. In addition, the movement of this loop reduces the binding surface of AFN-1252 such that the oxotetranapthyridine moiety occupies a solvent-exposed cavity (Fig. 6A). In comparison, the lid adopts a more tightly closed conformation around the inhibitor in the SaFabI structure (Fig. 6B). These differences provide a structural explanation for why AFN-1252 exhibits lower affinity against CtFabI compared with SaFabI. However, the key interactions between AFN-1252 and the peptide backbone and cofactor are maintained in the CtFabI·NADH·AFN-1252 complex, offering opportunities for the structure-based design of modified AFN-1252 derivatives that can exploit the larger substrate pocket in CtFabI.
AFN-1252 Inhibits Fatty Acid Synthesis
The effect of AFN-1252 on branched-chain fatty acid synthesis was determined to verify that the drug blocked bacterial FASII. The branched-chain fatty acids are synthesized by initiating FASII with a short-chain branched-chain acyl-CoA, followed by five rounds of elongation using malonyl-ACP derived from acetyl-CoA (Fig. 1) (25). Straight-chain fatty acids are built from acetyl-CoA molecules entirely (25, 42). Isoleucine is the precursor to 2-methylbutyryl-CoA via the bacterially encoded branched-chain α-ketoacid dehydrogenase, whereas glucose is a precursor to acetyl-CoA in both mammalian and bacterial metabolic schemes. Incorporation of the radioactivity from [U-14C]glucose into the lipid fraction indicated total fatty acid synthesis activity in both the host and the parasite. Because the eukaryotic host cannot make branched-chain fatty acids, incorporation of radioactive [U-14C]isoleucine into the lipid fraction primarily reflected the FASII activity of C. trachomatis. One caveat was that isoleucine is broken down into acetyl-CoA by the mammalian host (43), which can then be incorporated into fatty acids derived from both host and parasite. Therefore, we also measured the amount of [U-14C]isoleucine incorporated into uninfected HeLa cells to serve as an estimate for the extent of this metabolic conversion.
Cell viability and [U-14C]glucose and [U-14C]isoleucine incorporation into the uninfected HeLa cells when treated with 50 μm of AFN-1252 were determined to ensure that AFN-1252 does not exhibit off-target effects on the host cell. Treatment of the host cell with 50 μm AFN-1252 did not affect radioactive incorporation from [U-14C]glucose (Fig. 7A) or cell viability (>95%). Infected HeLa cells were pulse-labeled for 4 h with [U-14C]glucose 20 h following the initial infection to measure the effect of AFN-1252 on the total fatty acid biosynthetic activity in the infection model. The infected cell population incorporated 7.5 times more radioactivity compared with uninfected HeLa cells (Fig. 7A). AFN-1252 treatment reduced [U-14C]glucose incorporation by over 70% compared with the untreated infected cells. Similar labeling experiments with [U-14C]isoleucine revealed that infected HeLa cells incorporated over 6 times more radioactivity into the lipid fraction than did uninfected HeLa cells, consistent with an increased synthesis of branched-chain fatty acids in the infected cells (Fig. 7B). AFN-1252 treatment of infected HeLa cells caused a dose-dependent decrease in [U-14C]isoleucine incorporation (Fig. 7C). In cells treated with 50 μm AFN-1252, [U-14C]isoleucine incorporation was the same as in uninfected cells (Fig. 7, B and C), demonstrating that AFN-1252 blocked the increase in [U-14C]isoleucine incorporation resulting from the infection. The incorporation of [U-14C]isoleucine into uninfected cells and the residual incorporation in AFN-1252-treated cells were attributed to the conversion of isoleucine to acetyl-CoA (43), which was used for host fatty acid synthesis. These results demonstrated that AFN-1252 inhibited C. trachomatis fatty acid synthesis in the infection model.
FIGURE 7.
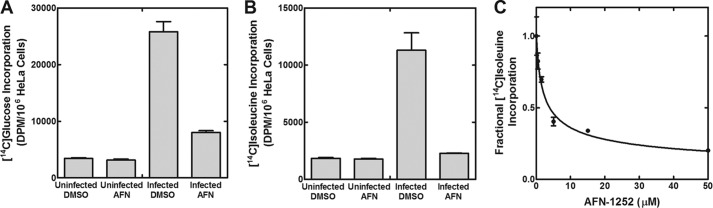
AFN-1252 inhibits C. trachomatis FASII. A, [U-14C]glucose incorporation into the lipids of uninfected HeLa cells, uninfected HeLa cells treated with 50 μm AFN-1252, infected HeLa cells, and infected HeLa cells treated with AFN-1252. B, [U-14C]isoleucine incorporation into the lipids of uninfected HeLa cells, uninfected HeLa cells treated with AFN-1252, infected HeLa cells, and infected HeLa cells treated with AFN-1252. C, [U-14C]isoleucine incorporation into the lipids of infected HeLa cells as a function of increasing concentrations of AFN-1252. HeLa cells were infected at a multiplicity of infection of 5 at time 0. All labeling experiments were conducted by adding AFN-1252 to the cell cultures for 30 min at 20 h postinfection, followed by radioactive pulse labeling for 4 h with the indicated radiochemical. Error bars, S.E.
AFN-1252 Inhibits C. trachomatis Replication
The role of endogenous fatty acid synthesis in regulating C. trachomatis growth was determined by treating the Chlamydia-HeLa cell infection model with AFN-1252. HeLa cells infected with C. trachomatis serovar L2 were incubated with increasing concentrations of AFN-1252 for 48 h. The number of inclusions in an infected monolayer (Fig. 8A) and the number of infectious units (EBs) formed (Fig. 8B) were both decreased in a dose-dependent manner by AFN-1252. At >6 μm AFN-1252, no infectious particles were generated. AFN-1252 effectively decreased the infectious titers when added as late as 32 h postinfection, consistent with a critical role for FASII throughout the entire developmental cycle (Fig. 8C). The number of chlamydial genomes per cell decreased over time when infected cells were treated with 10 μm AFN-1252, suggesting that AFN-1252 arrested the replication of C. trachomatis, and the host cell cleared the organisms that had been internalized (Fig. 8D).
FIGURE 8.

AFN-1252 inhibits C. trachomatis replication. A, number of chlamydial inclusions observed per ×40 field at 42 h postinfection was determined as a function of increasing AFN-1252 concentrations added immediately after the infection of HeLa cells with C. trachomatis serovar L2. B, number of infectious units of C. trachomatis serovar L2 generated in the presence of increasing AFN-1252 concentrations added immediately after the initial infection. C, number of infectious units of C. trachomatis serovar L2 produced when 6 μm AFN-1252 was added at 0, 8, 16, 24, and 32 h after the initiating infection. The number of infectious particles was counted at the end of 48 h in all cases. Six measurements were made in each of two experiments. Significance was determined using Student's t test: *, p < 0.05; **, p < 0.01; ***, p < 0.001. D, number of C. trachomatis serovar L2 genomes during the course of an infection cycle in control and cells treated with 10 μm AFN-1252. Error bars, S.E.
The stage in the developmental cycle where AFN-1252 arrested chlamydial growth was determined by double-staining individual cells with a mouse monoclonal antibody directed against chlamydial Hsp60 (cytoplasmic marker, green) and Hoechst (blue) to mark host cell nuclei. AFN-1252 treatment caused a dose-dependent decrease in the size of the chlamydial inclusion in HeLa cells infected with C. trachomatis serovar L2 (Fig. 9A). At concentrations exceeding 4 μm AFN-1252, only a single bacterium was detected in each infected cell (Fig. 9A). The effect of AFN-1252 on C. trachomatis serovar D was also examined to verify that the ability of AFN-1252 to arrest C. trachomatis replication was not strain-dependent (Fig. 9B). In the D serovar, at AFN-1252 concentrations exceeding 4 μm, only a single bacterium was detected in each infected cell. These data also showed that AFN-1252 prevented the first cell division of C. trachomatis following infection. Double staining with Hsp60 (red) and a polyclonal antibody against the chlamydial MOMP (green) was performed to determine whether EBs differentiated into RBs in AFN-1252-treated cells. EBs were characterized by a small area of Hsp60 staining completely enclosed by MOMP indicated by a yellow cytoplasmic dot surrounded by green (Fig. 9C). Treatment of C. trachomatis-infected cells with rifampicin (RNA synthesis inhibitor) or chloramphenicol (protein synthesis inhibitor) arrested cells in an EB-like state with no change in size or pattern of MOMP or Hsp60 staining compared with EBs (Fig. 9C), consistent with RNA and protein synthesis being required to support differentiation into an RB. In the normal developmental cycle of C. trachomatis, the endocytosed EB differentiates into a slightly larger RB characterized by an expanded, intense cytoplasmic Hsp60 (red) staining (Fig. 9C). In cells treated with AFN-1252, the internalized bacterium had the characteristics of an RB indicated by an increase in size and the intensity of Hsp60 staining (Fig. 9C). The staining of MOMP in AFN-1252-arrested cells was also polarized to one side of the cell. Whether the staining pattern in the presence of AFN-1252 reflected the accumulation of a normal intermediate in the developmental cycle or was due to an effect of the drug is not clear. These data indicated that AFN-1252 arrested C. trachomatis after the differentiation into RBs was initiated but before the first bacterial cell division.
FIGURE 9.

AFN-1252 arrests C. trachomatis prior to the first bacterial cell division. A, immunofluorescence staining (antichlamydial Hsp60 antibody in green and Hoechst in blue) of HeLa cells infected with C. trachomatis serovar L2 and treated with increasing concentrations of AFN-1252. Size bar, 10 μm. B, immunofluorescence staining of HeLa cells infected with C. trachomatis serovar D and treated with increasing concentrations of AFN-1252. Size bar, 10 μm. C, morphology of AFN-1252-treated C. trachomatis serovar L2 infection model compared with a typical EB, a typical RB found 6 h after infection, and the appearance of the infection model treated with rifampicin or chloramphenicol at the time of infection. Drug-treated cells were fixed at 24 h postinfection and stained with antibodies directed against MOMP (green) and chlamydial Hsp60 (red). The EBs were stained at 1 h postinfection, whereas the RB was stained at 14 h postinfection. Yellow, overlapping staining. Size bars, 0.5 μm.
DISCUSSION
Our results show that FASII is essential for the replication of C. trachomatis within its eukaryotic host. FASII is essential for the synthesis of two major membrane constituents in free-living Gram-negative bacteria. First, FASII supplies the fatty acids required for the synthesis of membrane phospholipids, and second, FASII generates the 3-hydroxy fatty acids required for LOS synthesis. Previous work showed that the selective inhibition of LOS synthesis at the LpxC step permitted the proliferation of C. trachomatis within its cytoplasmic inclusion but blocked the terminal differentiation of RBs into EBs (10). If FASII is only required for generating 3-hydroxy fatty acids for LOS synthesis and the bacterium obtains its phospholipids from the host, then AFN-1252 inhibition would have the same effect on the infection cycle as a LOS inhibitor. However, blocking FASII arrests cell replication whenever it is added over the entire developmental cycle, which is consistent with phospholipid synthesis using products derived from FASII as essential for C. trachomatis replication. Inhibitors of RNA synthesis (rifampicin) and protein synthesis (chloramphenicol) blocked C. trachomatis development at the EB stage, showing that these processes are necessary for EB to RB conversion. C. trachomatis had an enlarged, RB-like morphology in AFN-1252-treated cells, indicating that FASII inhibition arrested development after the differentiation from EB to RB is initiated but before the first cell division. The phospholipid classes of C. trachomatis reflect the host cell composition (26). Previous work suggests that C. trachomatis obtains its phospholipids from the host and modifies a subset of these lipids by introducing a branched-chain fatty acid into the 2-position (3, 26). Additional work is needed to determine whether C. trachomatis synthesizes an important class of phospholipids de novo, using the products of FASII, or whether the acylation/deacylation pathway is critical for the modification of host phospholipids with branched-chain fatty acids. Nonetheless, it is clear from this study that FASII products are critical for cell division in this parasitic bacterium.
This work identifies FASII, and FabI in particular, as a therapeutic target in C. trachomatis. As an intracellular pathogen that acquires vital nutrients from the host, C. trachomatis lacks many biochemical pathways that are traditional antibacterial drug targets (44, 45). Although previous work suggested that fatty acids and phospholipids of chlamydial membranes are obtained from the host (45), our study shows that FASII is nonetheless essential for chlamydial replication. AFN-1252 is effective in cell culture against C. trachomatis and has pharmacological properties that make it an effective agent against S. aureus infections (36, 46). However, its potency may have to be improved in order for it to be effectively deployed to treat C. trachomatis. Our CtFabI·NADH·AFN-1252 crystal structure shows that AFN-1252 inhibits CtFabI by making similar contacts to the backbone amides and nucleotide cofactor as SaFabI (12). Our structure characterization also clearly points to the absence of the Met-99 side chain interaction with the oxotetrahydronaphthyridine ring of AFN-1252 as the primary reason for the lower affinity of AFN-1252 binding to CtFabI compared with SaFabI. This interaction with Met-99 in SaFabI is established to be critical for high-affinity AFN-1252 binding in SaFabI based on the finding that a missense mutation giving rise to a M99T mutation in FabI accounts for acquired resistance to AFN-1252 in S. aureus (17). Structure modifications to AFN-1252 that compensate for this interaction should allow for the design of a potent and selective CtFabI inhibitor.
Acknowledgments
We thank Nirun Naher for expert technical assistance and Amanda Nourse (St. Jude Molecular Interaction Analysis Facility) for the analytical ultracentrifugation experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants GM034496 (to C. O. R.), AI070693 (to R. J. B), and Cancer Center Support Grant CA21765. This work was also supported by the American Lebanese Syrian Associated Charities.
The atomic coordinates and structure factors (code 4Q9N) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- EB
- elementary body
- ACP
- acyl carrier protein
- FabI
- enoyl-ACP reductase
- CtFabI
- enoyl-ACP reductase of C. trachomatis
- SaFabI
- enoyl-ACP reductase of S. aureus
- FASII
- bacterial type II fatty acid synthesis
- LOS
- lipooligosacchride
- RB
- reticulate body
- MOMP
- chlamydial major outer membrane protein
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Belland R., Ojcius D. M., Byrne G. I. (2004) Focus: Chlamydia. Nat. Rev. Microbiol. 2, 530–531 [DOI] [PubMed] [Google Scholar]
- 2. Becker Y. (1978) The Chlamydia: molecular biology of procaryotic obligate parasites of eucaryocytes. Microbiol. Rev. 42, 274–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wylie J. L., Hatch G. M., McClarty G. (1997) Host cell phospholipids are trafficked to and then modified by Chlamydia trachomatis. J. Bacteriol. 179, 7233–7242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Su H., McClarty G., Dong F., Hatch G. M., Pan Z. K., Zhong G. (2004) Activation of Raf/MEK/ERK/cPLA2 signaling pathway is essential for chlamydial acquisition of host glycerophospholipids. J. Biol. Chem. 279, 9409–9416 [DOI] [PubMed] [Google Scholar]
- 5. Vignola M. J., Kashatus D. F., Taylor G. A., Counter C. M., Valdivia R. H. (2010) cPLA2 regulates the expression of type I interferons and intracellular immunity to Chlamydia trachomatis. J. Biol. Chem. 285, 21625–21635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raetz C. R., Reynolds C. M., Trent M. S., Bishop R. E. (2007) Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 76, 295–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rund S., Lindner B., Brade H., Holst O. (1999) Structural analysis of the lipopolysaccharide from Chlamydia trachomatis serotype L2. J. Biol. Chem. 274, 16819–16824 [DOI] [PubMed] [Google Scholar]
- 8. Heine H., Müller-Loennies S., Brade L., Lindner B., Brade H. (2003) Endotoxic activity and chemical structure of lipopolysaccharides from Chlamydia trachomatis serotypes E and L2 and Chlamydophila psittaci 6BC. Eur. J. Biochem. 270, 440–450 [DOI] [PubMed] [Google Scholar]
- 9. Heine H., Gronow S., Zamyatina A., Kosma P., Brade H. (2007) Investigation on the agonistic and antagonistic biological activities of synthetic Chlamydia lipid A and its use in in vitro enzymatic assays. J. Endotoxin Res. 13, 126–132 [DOI] [PubMed] [Google Scholar]
- 10. Nguyen B. D., Cunningham D., Liang X., Chen X., Toone E. J., Raetz C. R., Zhou P., Valdivia R. H. (2011) Lipooligosaccharide is required for the generation of infectious elementary bodies in Chlamydia trachomatis. Proc. Natl. Acad. Sci. U.S.A. 108, 10284–10289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaplan N., Garner C., Hafkin B. (2013) AFN-1252 in vitro absorption studies and pharmacokinetics following microdosing in healthy subjects. Eur. J. Pharm. Sci. 50, 440–446 [DOI] [PubMed] [Google Scholar]
- 12. Kaplan N., Albert M., Awrey D., Bardouniotis E., Berman J., Clarke T., Dorsey M., Hafkin B., Ramnauth J., Romanov V., Schmid M. B., Thalakada R., Yethon J., Pauls H. W. (2012) Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob. Agents Chemother. 56, 5865–5874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Girard G., Barends S., Rigali S., van Rij E. T., Lugtenberg B. J. J., Bloemberg G. V. (2006) Pip, a novel activator of phenazine biosynthesis in Pseudomonas chlororaphis PCL1391. J. Bacteriol. 188, 8283–8293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paoletti L., Lu Y.-J., Schujman G. E., de Mendoza D., Rock C. O. (2007) Coupling of fatty acid and phospholipid synthesis in Bacillus subtilis. J. Bacteriol. 189, 5816–5824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pemble C. W., 4th, Mehta P. K., Mehra S., Li Z., Nourse A., Lee R. E., White S. W. (2010) Crystal structure of the 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase·dihydropteroate synthase bifunctional enzyme from Francisella tularensis. PLoS One 5, e14165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moldoveanu T., Grace C. R., Llambi F., Nourse A., Fitzgerald P., Gehring K., Kriwacki R. W., Green D. R. (2013) BID-induced structural changes in BAK promote apoptosis. Nat. Struct. Mol. Biol. 20, 589–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yao J., Maxwell J. B., Rock C. O. (2013) Resistance to AFN-1252 arises from missense mutations in Staphylococcus aureus enoyl-acyl carrier protein reductase (FabI). J. Biol. Chem. 288, 36261–36271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 19. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Echols N., Headd J. J., Hung L. W., Jain S., Kapral G. J., Grosse Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R. D., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2011) The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 22. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.1, Schroedinger, LLC, New York [Google Scholar]
- 23. Scidmore M. A. (2005) Cultivation and laboratory maintenance of Chlamydia trachomatis. Curr. Protoc. Microbiol. Chapter 11, Unit 11A.1 [DOI] [PubMed] [Google Scholar]
- 24. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 25. Kaneda T. (1991) Iso- and anteiso-fatty acids in bacteria: biosynthesis, function, and taxonomic significance. Microbiol. Rev. 55, 288–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hatch G. M., McClarty G. (1998) Phospholipid composition of purified Chlamydia trachomatis mimics that of the eucaryotic host cell. Infect. Immun. 66, 3727–3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sweet C. R., Lin S., Cotter R. J., Raetz C. R. (2001) A Chlamydia trachomatis UDP-N-acetylglucosamine acyltransferase selective for myristoyl-acyl carrier protein. Expression in Escherichia coli and formation of hybrid lipid A species. J. Biol. Chem. 276, 19565–19574 [DOI] [PubMed] [Google Scholar]
- 28. Kosma P. (1999) Chlamydial lipopolysaccharide. Biochim. Biophys. Acta 1455, 387–402 [DOI] [PubMed] [Google Scholar]
- 29. Qureshi N., Kaltashov I., Walker K., Doroshenko V., Cotter R. J., Takayama K., Sievert T. R., Rice P. A., Lin J. S., Golenbock D. T. (1997) Structure of the monophosphoryl lipid A moiety obtained from the lipopolysaccharide of Chlamydia trachomatis. J. Biol. Chem. 272, 10594–10600 [DOI] [PubMed] [Google Scholar]
- 30. Heath R. J., Rock C. O. (1995) Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J. Biol. Chem. 270, 26538–26542 [DOI] [PubMed] [Google Scholar]
- 31. Heath R. J., Rock C. O. (1996) Regulation of fatty acid elongation and initiation by acyl-acyl carrier protein in Escherichia coli. J. Biol. Chem. 271, 1833–1836 [DOI] [PubMed] [Google Scholar]
- 32. Bergler H., Högenauer G., Turnowsky F. (1992) Sequences of the envM gene and two mutated alleles in Escherichia coli. J. Gen. Microbiol. 138, 2093–2100 [DOI] [PubMed] [Google Scholar]
- 33. White S. W., Zheng J., Zhang Y.-M., Rock C. O. (2005) The structural biology of type II fatty acid biosynthesis. Annu. Rev. Biochem. 74, 791–831 [DOI] [PubMed] [Google Scholar]
- 34. Schiebel J., Chang A., Lu H., Baxter M. V., Tonge P. J., Kisker C. (2012) Staphylococcus aureus FabI: inhibition, substrate recognition, and potential implications for in vivo essentiality. Structure 20, 802–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu H., Sullivan T. J., Sekiguchi J., Kirikae T., Ojima I., Stratton C. F., Mao W., Rock F. L., Alley M. R., Johnson F., Walker S. G., Tonge P. J. (2008) Mechanism and inhibition of saFabI, the enoyl reductase from Staphylococcus aureus. Biochemistry 47, 4228–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Banevicius M. A., Kaplan N., Hafkin B., Nicolau D. P. (2013) Pharmacokinetics, pharmacodynamics, and efficacy of novel FabI inhibitor AFN-1252 against MSSA and MRSA in the murine thigh infection model. J. Chemother. 25, 26–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rozwarski D. A., Vilchèze C., Sugantino M., Bittman R., Sacchettini J. C. (1999) Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J. Biol. Chem. 274, 15582–15589 [DOI] [PubMed] [Google Scholar]
- 38. Rafi S., Novichenok P., Kolappan S., Zhang X., Stratton C. F., Rawat R., Kisker C., Simmerling C., Tonge P. J. (2006) Structure of acyl carrier protein bound to FabI, the FASII enoyl reductase from Escherichia coli. J. Biol. Chem. 281, 39285–39293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Suh M. C., Schultz D. J., Ohlrogge J. B. (1999) Isoforms of acyl carrier protein involved in seed-specific fatty acid synthesis. Plant J. 17, 679–688 [DOI] [PubMed] [Google Scholar]
- 40. Priyadarshi A., Kim E. E., Hwang K. Y. (2010) Structural insights into Staphylococcus aureus enoyl-ACP reductase (FabI), in complex with NADP and triclosan. Proteins 78, 480–486 [DOI] [PubMed] [Google Scholar]
- 41. Qiu X., Janson C. A., Court RI, Smyth M. G., Payne D. J., Abdel-Meguid S. S. (1999) Molecular basis for triclosan activity involves a flipping loop in the active site. Protein Sci. 8, 2529–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rock C. O., Jackowski S. (2002) Forty years of fatty acid biosynthesis. Biochem. Biophys. Res. Commun. 292, 1155–1166 [DOI] [PubMed] [Google Scholar]
- 43. Hutson S. M., Sweatt A. J., Lanoue K. F. (2005) Branched-chain amino acid metabolism: implications for establishing safe intakes. J. Nutr. 135, 1557S–1564S [DOI] [PubMed] [Google Scholar]
- 44. Stephens R. S., Kalman S., Lammel C., Fan J., Marathe R., Aravind L., Mitchell W., Olinger L., Tatusov R. L., Zhao Q., Koonin E. V., Davis R. W. (1998) Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282, 754–759 [DOI] [PubMed] [Google Scholar]
- 45. Saka H. A., Valdivia R. H. (2010) Acquisition of nutrients by Chlamydiae: unique challenges of living in an intracellular compartment. Curr. Opin. Microbiol. 13, 4–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Parsons J. B., Kukula M., Jackson P., Pulse M., Simecka J. W., Valtierra D., Weiss W. J., Kaplan N., Rock C. O. (2013) Perturbation of Staphylococcus aureus gene expression by the enoyl-acyl carrier protein reductase inhibitor AFN-1252. Antimicrob. Agents Chemother. 57, 2182–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Braberg H., Webb B. M., Tjioe E., Pieper U., Sali A., Madhusudhan M. S. (2012) SALIGN: a web server for alignment of multiple protein sequences and structures. Bioinformatics 28, 2072–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gouet P., Courcelle E., Stuart D. I., Métoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]