

Background: Sphingolipids are important in multiple biological processes, but there is a lack of understanding of the pathways that mediate these effects.
Results: Acid sphingomyelinase is involved in p38 activation, interleukin-6 production, and cancer cell invasion.
Conclusion: Acid sphingomyelinase plays a role in inflammatory, proinvasive signaling in cancer cells.
Significance: This work expands the understanding of acid sphingomyelinase signaling and downstream biology.
Keywords: Cancer Biology, Cell Signaling, Interleukin 6 (IL-6), p38 MAPK, Sphingolipid, Acid Sphingomyelinase
Abstract
Acid sphingomyelinase (ASM) is one of the key enzymes involved in regulating the metabolism of the bioactive sphingolipid ceramide in the sphingolipid salvage pathway, yet defining signaling pathways by which ASM exerts its effects has proven difficult. Previous literature has implicated sphingolipids in the regulation of cytokines such as interleukin-6 (IL-6), but the specific sphingolipid pathways and mechanisms involved in inflammatory signaling need to be further elucidated. In this work, we sought to define the role of ASM in IL-6 production because our previous work showed that a parallel pathway of ceramide metabolism, acid β-glucosidase 1, negatively regulates IL-6. First, silencing ASM with siRNA abrogated IL-6 production in response to the tumor promoter, 4β-phorbol 12-myristate 13-acetate (PMA), in MCF-7 cells, in distinction to acid β-glucosidase 1 and acid ceramidase, suggesting specialization of the pathways. Moreover, treating cells with siRNA to ASM or with the indirect pharmacologic inhibitor desipramine resulted in significant inhibition of TNFα- and PMA-induced IL-6 production in MDA-MB-231 and HeLa cells. Knockdown of ASM was found to significantly inhibit PMA-dependent IL-6 induction at the mRNA level, probably ruling out mechanisms of translation or secretion of IL-6. Further, ASM knockdown or desipramine blunted p38 MAPK activation in response to TNFα, revealing a key role for ASM in activating p38, a signaling pathway known to regulate IL-6 induction. Last, knockdown of ASM dramatically blunted invasion of HeLa and MDA-MB-231 cells through Matrigel. Taken together, these results demonstrate that ASM plays a critical role in p38 signaling and IL-6 synthesis with implications for tumor pathobiology.
Introduction
In addition to its physiological roles in inflammation and immunology, interleukin-6 (IL-6) has been widely implicated in cancer biology (1). IL-6 is known to stimulate proliferation, invasion, and metastasis of tumors (2–4). Moreover, there is a strong correlation between invasiveness of cancer cell lines and the level of IL-6 production (5), suggesting that although IL-6 is capable of being produced by many cell types, tumor-derived IL-6 may be a critical determinant in a tumor's intrinsic invasive capability.
Bioactive sphingolipids have been implicated as regulators of IL-6 formation, but the specific sphingolipid pathways involved are not clearly defined. Sphingolipid metabolism is broadly divided into either de novo or hydrolytic/salvage pathways (6, 7). In the salvage pathway, sphingomyelin (SM)3 and glucosylceramide are hydrolyzed into ceramide by acid sphingomyelinase (ASM) and acid β-glucosidase 1 (GBA1), respectively. Ceramide can then be cleaved to form sphingosine by acid ceramidase (ACD). Thus, the salvage pathway is poised to make rapid changes in downstream metabolites, including ceramide and sphingosine due to the relative abundance of the complex sphingolipids, such as SM and glucosylceramide, and also the energetically favorable process of hydrolysis. Consistent with this, activation of PKC∂ stimulates the hydrolysis of complex sphingolipids, leading to the production of ceramide from either GBA1 or ASM, leading to flux through the sphingolipid salvage pathway (8–10).
Insofar as evidence for involvement of sphingolipids in IL-6 production, early work by Laulederkind et al. (11) demonstrated that exogenous treatment of dermal fibroblasts with bacterial sphingomyelinase was sufficient to induce IL-6 similarly to IL-1β treatment, suggesting that a pool of ceramide at the plasma membrane could be involved in triggering signaling to IL-6. Conversely, previous work from our laboratory has demonstrated that IL-6 production and p38 activation are negatively regulated by GBA1-derived ceramide in MCF-7 cells (12). Literature related to ASM has shown that ASM is not required for p38 signaling in ASM−/− murine macrophages (13), whereas other work has indicated a role for ASM in cytokine production, including IL-6, with the use of an SM-based ASM inhibitor (14). While this work was in progress, Kumagai et al. (15) showed that ASM is involved in IL-6 production in bladder cancer cells; however, a signaling pathway leading to IL-6 was not identified, underscoring the need to identify signaling pathways that ASM regulates to affect IL-6 secretion.
This work provides evidence for the involvement of ASM in the production of IL-6 and the phosphorylation of p38, in distinction to GBA1, revealing functional specificity within the sphingolipid salvage pathway. Furthermore, studies were performed that implicate ASM in IL-6 mRNA regulation by multiple mechanisms, including transcription and message stabilization, and that reveal distinct RNA dynamics among MCF-7, MDA-MB-231, and HeLa carcinoma cell lines. This study also provides novel evidence that ASM is required for invasion of aggressive carcinoma cells. The implications of these findings for sphingolipid signaling and cancer biology are further discussed.
EXPERIMENTAL PROCEDURES
Materials
Active phospho-p38 antibody and p38∂ antibodies were from Promega (Madison, WI) and R&D Systems (Minneapolis, MN), respectively. PMA was from Calbiochem. TNFα was from PeproTech. HRP-linked secondary antibodies were from Santa Cruz Biotechnology, Inc. Actinomycin D and myriocin were purchased from Sigma. Invasion wells were from BD Biosciences. Fumonisin B1 was from Enzo Life Sciences (Farmingdale, NY).
Cell Culture
MCF-7, MDA-MB-231, HeLa, control fibroblasts, and Niemann-Pick disease (NPD) fibroblasts were grown in DMEM supplemented with l-glutamine and 10% fetal bovine serum. Cells were cultured under standard conditions (37 °C, 5% CO2, humidified air) and kept under 90% confluence. For a 6-well plate, 50,000 cells/well were plated and then the next day transfected with 20 nm siRNA according to the manufacturer's instructions using Oligofectamine (Invitrogen). After 48–72 h, media were changed 1 h prior to stimulation with either PMA (100 nm) or TNFα (20 ng/ml). For overexpression of ASM, cells were plated at 50,000 cells/well of a 6-well plate. The next day, cells were transfected with 1 μg of control vector (pEF6-V5/His6) or the ASM expression vector (pEF6-ASM-V5/His6)(9) according to the manufacturer's instructions using X-tremeGENE 9 (Roche Applied Science). After 24 h, medium was replaced, cells were incubated for an additional 24 h, and IL-6 expression was measured by qPCR as described.
Western Blotting
Cells were harvested by direct lysis in 1% SDS, 1 mm EDTA, 50 mm Tris-HCl, pH 7.4. Lysates were sonicated, and total protein was normalized by the BCA method. Laemmli buffer was added (6×), and then samples were boiled for 5 min. Equal volumes were loaded onto Criterion precast gels and run at 100 V. Proteins were transferred to nitrocellulose membranes, blocked with 5% nonfat milk in PBS plus 0.1% Tween 20 and were incubated overnight with primary antibodies in 5% nonfat milk in PBS plus 0.1% Tween 20. Membranes were washed in PBS plus 0.1% Tween 20 at least three times, incubated with secondary antibodies (1:5,000), washed again, and then developed in ECL chemiluminescence reagent (Pierce).
Reverse Transcriptase Reaction and PCR
Equal amounts of RNA were isolated (Qiagen, RNeasy kit) and used in a reverse transcriptase reaction according to the manufacturer's protocol (Superscript II). The final reaction was diluted to 2 mg of initial RNA/ml. For qPCR analysis, 5 μl of cDNA was used per 25-μl reaction in duplicate or triplicate using SYBR Green SuperMix (Bio-Rad). Real-time PCR was performed using an iCycler (Bio-Rad) consisting of a program of 95 °C for 3 min followed by 40 cycles of 95 °C for 15 s, 60 °C for 45 s, and 68 °C for 30 s. Threshold cycle (Ct) values of target genes were normalized to actin Ct values generating mean normalized expression using Q-gene software (16) and expressed as -fold change compared with control. Primers for IL-6 (Table 1) were designed using Primer-blast (NCBI) in order to specifically amplify mature mRNA, referred to as mRNA (reverse primer aligning to contiguous regions of exon 1 and 2), hnRNA (reverse primer completely within intron), or both (reverse primer aligning to adjacent exon, exon 2, to the forward primer in exon 1) with a common forward primer for these reverse primers (see Scheme 2). IL-6 primer specificity was validated with melt curves and by DNA electrophoresis. The primer set, which would amplify both species, was only used for semiquantitative PCR, whereas the others were used for qRT-PCR. ASM, CCL5/RANTES, and β-actin primer sequences were described previously (12, 17).
TABLE 1.
IL-6 primers
| Primer | Sequence (5′–3′) |
|---|---|
| Forward (F) | CGA GCC CAC CGG GAA CGA AA |
| hnRNA reverse (Rhn) | CCA GGG CTA AGG ATT TCC TGC ACT T |
| mRNA reverse (Rm) | TGG ACC GAA GGC GCT TGT GGA |
| hn/mRNA reverse (Rboth) | CAG CCC CAG GGA GAA GGC AAC T |
SCHEME 2.

IL-6 hnRNA and mRNA showing primers designed specifically for each species (Rhn or Rm) or both (Rboth) with a common forward primer (F). Approximate, relative size of exons is shown (E1–E5).
IL-6 ELISA
Cultured media were harvested, and IL-6 was measured by human IL-6 ELISA according to the manufacturer's protocol (R&D, Quantikine). Cellular protein was harvested and measured by the BCA method. IL-6 concentrations were normalized to total cellular protein yielding pg of IL-6/mg of total protein unless otherwise indicated. In no case did normalization alter the results significantly, ruling out changes in total cell number or total protein content as the determinant.
Confocal Microscopy
Cells were washed with cold PBS, fixed in 3.7% paraformaldehyde, and permeabilized in 0.1% Triton X-100 for 10 min. Samples were blocked in 2% human serum for 1 h and then incubated with vimentin primary antibody in 2% human serum for 90 min at room temperature. Samples were washed and incubated with secondary antibodies conjugated to TRITC for 1 h. Samples were washed again and then incubated with DRAQ5 and phalloidin, and wash steps were repeated before imaging on a confocal laser microscope (LSM510, Zeiss).
Invasion Assay
HeLa cells were treated with siRNA for 48 h, trypsinized, and plated in transwells coated with Matrigel in serum-free medium with either serum-free or 10% serum in the bottom chamber. The cells were allowed to grow and invade for 24 h and then stained with fluorescein. Four representative images of each well were acquired, and the number of cells invaded through the Matrigel were counted. Cells were plated in triplicate.
ASM Activity Assay
Protein lysates from MCF-7, MDA-MB-231, or HeLa were assessed for ASM activity as described previously using radiolabeled SM (18).
Statistical Analysis
Statistical and kinetic analysis was performed with Prism/GraphPad using one phase exponential decay equation for calculating the half-life of IL-6 mRNA. Results displayed are mean ± S.E. or the range when n was <3 for a given experiment. Statistical tests were performed, depending on the experiment, with significance set at a p value of <0.05.
RESULTS
Differential Regulation of IL-6 Production and p38 Activation by Sphingolipid Salvage Enzymes
Stimulation of the breakdown of complex sphingolipids by PMA through the salvage pathway has been shown to be dependent on PKCδ, a novel PKC, in MCF-7 breast cancer cells (8). Moreover, activation of ASM, leading to ceramide accumulation, has been linked to PKCδ activity (9). Due to the interplay between PKC signaling and sphingolipids, it was of interest to define the roles of classical and novel PKCs in the context of IL-6 production. MCF-7 cells were chosen because they produce very low baseline levels of IL-6 but high levels in terms of -fold change upon stimulation with PMA, making this an ideal system to elucidate PKC-mediated mechanisms. MCF-7 breast cancer cells were pretreated with either Gö6976 or bisindoleilamide for 1 h and then stimulated with PMA, after which secreted IL-6 was measured. The classical PKC inhibitor, Gö6976, had no effect on PMA-induced IL-6 protein production, whereas the classical and novel PKC inhibitor, bisindoleilamide, ablated the PMA response, implicating novel PKCs in this process (Fig. 1A).
FIGURE 1.

Regulation of IL-6 and p38 by sphingolipid salvage enzymes in MCF-7. A, PKC inhibitors Gö6976 (3 μm) and bisindoleilamide (Bis; 3 μm) were added to subconfluent MCF-7 cells for a 1-h pretreatment followed by PMA (100 nm) treatment for 18 h. IL-6 was measured by ELISA in cultured media as described under “Experimental Procedures” (n = 2). B, MCF-7 cells were treated with 20 nm siRNA for the indicated enzymes for 48 h and then stimulated with PMA for 18 h; IL-6 in cultured media was measured by ELISA (two-way analysis of variance, Bonferroni's post-test. **, p < 0.01; ***, p < 0.001 compared with control (Con) siRNA + PMA, n = 3). C, MCF-7 cells were treated with 20 nm siRNA for 48 h and stimulated with PMA (100 nm) for 30 min. Whole cell lysates were prepared, and equal amounts of protein were subjected to Western blotting for phospho-p38 and p38. Results are representative of two experiments. D, MCF-7 cells were cotreated with siRNA for the above enzymes for 48 h, followed by PMA treatment for 18 h. -Fold change was calculated to either control siRNA or ACD siRNA alone for each group (n = 2). E and F, MCF-7 cells were treated with 20 nm various siRNAs as labeled for 48 h. Cells were harvested either for qRT-PCR (E) or immunoblotting (F) as described under “Experimental Procedures.” Equal amounts of protein were loaded for siRNA validation for GBA and ACD (F). Error bars, S.E.
To delineate the role of distinct components of the sphingolipid salvage pathway (Scheme 1) for their specific contribution to IL-6 production, MCF-7 cells were treated with siRNA against ACD, ASM, or GBA1 and then stimulated with PMA for 18 h (Fig. 1B). These siRNAs were previously used and validated in our laboratory (8, 17) and were further validated either by Western blot or real-time PCR (Fig. 1, E and F). In accordance with our previous study (12), we found that silencing GBA potentiated production of IL-6 (Fig. 1B). Conversely, down-regulation of ASM significantly decreased the production of IL-6, and loss of ACD had no effect on IL-6 (Fig. 1B); however, only modest silencing was achieved for ACD. The stress kinase p38 is known to play a role in regulating both transcription and message stability of IL-6(19–22). Therefore, we tested the effect of siRNA against the sphingolipid catabolic enzymes on p38 activation. In a similar fashion, knockdown of ASM blunted PMA-induced p38 phosphorylation, whereas knockdown of GBA1 increased the p38 activation significantly (Fig. 1C). These results mirrored the effects of ASM, ACD, and GBA1 on IL-6 production, suggesting that p38 is probably a signaling pathway that is downstream of the sphingolipid enzymes upon PKC stimulation.
SCHEME 1.
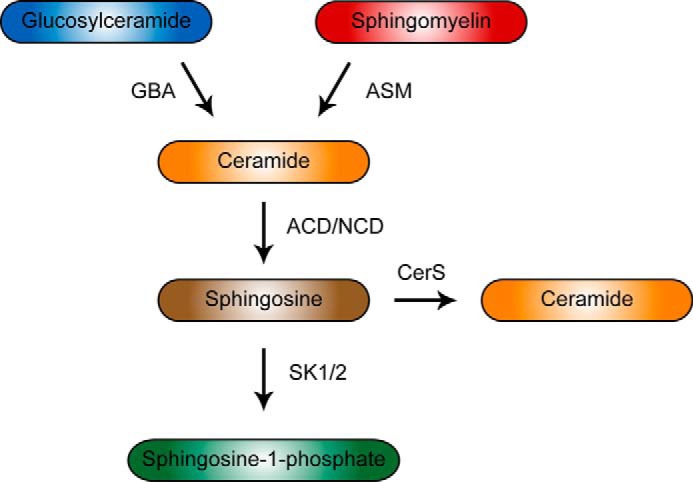
Sphingolipid salvage pathway
To test whether downstream metabolism of sphingolipids by ACD is required for the GBA1 or ASM response, co-knockdown of these enzymes was tested. The results demonstrated that in the presence or absence of ACD, there was a similar -fold change in IL-6 upon ASM and GBA1 silencing (Fig. 1D). Taken together, these results demonstrate that ASM is required for the induction of IL-6, but further metabolism of the ASM product ceramide to sphingosine via ACD may not be required in MCF-7 cells. Furthermore, these data reveal distinct and opposing roles for GBA1 and ASM in regulation of IL-6 production and p38 activation in response to PMA.
Involvement of ASM in IL-6 mRNA Induction in MCF-7 Cells
The mechanisms by which PKC and ASM regulate IL-6 RNA were next assessed in order to determine at which point of IL-6 synthesis these are involved. To achieve this goal, we utilized the standard actinomycin D time course and also a novel approach characterized by Zeisel et al. (23), which specifically measures mRNA or hnRNA, whereby inferences can be made whether changes seen are due to changes in transcription or mRNA stability. Therefore, specific primers for IL-6 mRNA and hnRNA were designed and validated (Scheme 2). Treatment of MCF-7 cells with ASM siRNA resulted in a decrease in PMA-induced IL-6 mRNA levels, probably ruling out mechanisms of translational or secretory regulation as the point of action but implicating transcription or message stability (Fig. 2A). To assess mRNA stabilization, an actinomycin D time course was performed in MCF-7 cells in the presence or absence of both PMA and ASM siRNA. PMA resulted in a doubling of IL-6 mRNA stability, and ASM silencing decreased message stability basally and with PMA (Fig. 2B). Additionally, IL-6 hnRNA was measured in a similar fashion as in Fig. 2A, which revealed no induction of IL-6 hnRNA upon treatment with PMA; however, ASM siRNA with or without PMA treatment yielded an approximately 40% decrease in IL-6 hnRNA (Fig. 2C). Taken together, these data show that ASM has effects on both transcription and stability of IL-6 mRNA. The results also show that PKC activation primarily results in increased IL-6 message stability.
FIGURE 2.

The role of ASM in the regulation of IL-6 RNA in MCF-7. A, MCF-7 cells were treated with 20 nm siRNA for 48 h and then stimulated with PMA for 90 min. Next, cDNA was synthesized, and qPCR was performed for IL-6 mRNA and normalized to actin as described under “Experimental Procedures.” -Fold change was calculated based on the untreated, control (Con) siRNA group (n = 3). B, MCF-7 cells were treated with siRNA as before, stimulated with PMA for 90 min, and then treated with 5 μg/ml actinomycin D for a 3-h time course, including time points of 0, 1, 2, and 3 h. RNA was harvested, cDNA was prepared, and IL-6 mRNA was measured by qPCR. The remaining amount of mRNA was calculated by normalizing each group to the values at t0 of actinomycin D. Values represent calculated t½ for each sample group (n = 2). C, MCF-7 cells were treated with 20 nm siRNA for 48 h and then stimulated with PMA for 90 min. As in A, cDNA was prepared, but qPCR was performed utilizing primers specific for IL-6 hnRNA (n = 3). D, MCF-7 cells were treated with either vehicle or PMA for 90 min, cDNA was prepared, qPCR was performed for IL-6, and CCL5/RANTES mRNA was expressed as -fold change from control group (n = 5). E, MCF-7 cells were treated with the indicated agents for 90 min, and RNA was harvested for qRT-PCR with primers specific to IL-6 (n = 3). F, MCF-7 cells were treated with BIRB796 and PMA for 90 min, and whole cell lysates were prepared. Equal protein was loaded in each lane of a 4–12% SDS-polyacrylamide gel and probed for total p38 or phospho-p38 (p-p38), as indicated. Error bars, S.E.
Previous work has demonstrated a role for ASM in the regulation of CCL5/RANTES in response to the cytokines, TNFα and IL-1β (17). In order to assess the possibility that PMA is also inducing RANTES through a novel mechanism, MCF-7 cells were treated with PMA and IL-6, and CCL5 mRNA was measured. PMA was found to be a specific inducer of IL-6 and not RANTES, suggesting a novel signaling function for ASM in response to PMA (Fig. 2D). Additionally, it was of interest to assess the contribution of p38 toward IL-6 expression in our system. Interestingly, there was only a modest decrease in IL-6 mRNA basally and with PMA treatment with the p38 inhibitor BIRB796 (24, 25) in MCF-7 cells (Fig. 2E) and HeLa cells (data not shown), although there was robust inhibition of p38 phosphorylation (Fig. 2F), suggesting that there could be other pathways by which ASM mediates its effect on IL-6. Therefore, in MCF-7 cells, ASM leads to p38 activation and eventual induction of IL-6 mediated by both changes in transcription and message stability.
Evaluation of IL-6 Protein and RNA in Normal and NPD Fibroblasts
Next, the role of ASM in IL-6 secretion was tested in fibroblasts, which are a significant source of IL-6 during wound healing (26) and can support tumor metastasis (27). Moreover, the availability of fibroblasts from patients with NPD, which have significantly decreased ASM activity (17), allows evaluation of the role of ASM in a genetic model. Here we found that NPD fibroblasts have profoundly defective IL-6 production basally and with TNFα when compared with healthy control fibroblasts (Fig. 3A).
FIGURE 3.

Altered expression and production of IL-6 in response to TNFα in human Niemann-Pick fibroblasts. A, equal numbers of control (Con) or Niemann-Pick type A disease fibroblasts were plated and treated the next day with TNFα for 18 h. Cultured media were assessed for IL-6 by ELISA (n = 2). B and C, WT and NPD fibroblasts were treated with TNF (20 ng/ml) or untreated for 90 min. RNA and cDNA were prepared, and IL-6 mRNA (B) and hnRNA (C) were measured as described under “Experimental Procedures” (n = 2). Error bars, S.E.
Next, we investigated the IL-6 RNA dynamics basally and in response to TNFα treatment in control and NPD fibroblasts. Loss of acid sphingomyelinase activity was found to decrease levels of IL-6 mRNA basally and with TNFα (Fig. 3B), as was found at the protein level. However, no difference was found basally in hnRNA levels between control and NPD fibroblasts, but TNFα-induced IL-6 hnRNA was significantly impaired in NPD fibroblasts (Fig. 3C), suggesting that ASM is required for TNFα-mediated transcriptional activation of the IL-6 gene in fibroblasts. Taken together, the results suggest that ASM plays a role in the basal stability of IL-6 mRNA due to the decrease of basal IL-6 mRNA (but not hnRNA) and then also in mediating TNFα-dependent transcription of IL-6 hnRNA.
The Role of ASM in IL-6 Production and p38 Activation in HeLa and MDA-MB-231 Cells
The contribution of ASM to IL-6 synthesis was assessed in invasive human cervical HeLa cancer cells as well as human breast MDA-MB-231 cancer cells, because invasive cell lines make significantly more IL-6 in absolute levels than non-invasive lines, and IL-6 plays a role in their malignant properties (3, 5, 28). First, we evaluated the role of ASM in these additional cell lines in regulating IL-6. Knockdown of ASM by siRNA in HeLa cells resulted in decreased IL-6 production in response to both PMA and TNFα (Fig. 4A). Further, treatment of MDA-231 and HeLa cells with desipramine (50 μm), a lysomotropic agent that induces the degradation of ASM (29), resulted in a dramatic decrease in IL-6 production in all cases (Fig. 4, B and C). Thus, these results show that ASM is involved in IL-6 production in response to TNFα and PMA in two invasive cancer cell lines. Likewise, inhibition of ASM with siRNA or desipramine abrogated p38 phosphorylation in HeLa cells (Fig. 4, D and E). Next, treatment of HeLa cells with desipramine (50 μm) was found to blunt PMA-induced IL-6 mRNA expression, consistent with the model that ASM and p38 induce IL-6 mRNA, leading ultimately to IL-6 protein secretion (Fig. 4F). The effectiveness of ASM knockdown in HeLa was demonstrated by a significant decrease in ASM mRNA and activity upon 48 h of ASM siRNA treatment (Fig. 4, G and H).
FIGURE 4.

Regulation of p38/IL-6 by ASM in HeLa cells. A, HeLa cells were treated with control or ASM siRNA for 48 h, followed by 18 h of PMA treatment. IL-6 in cultured media was measured by ELISA. B and C, HeLa (B) or MDA-MB-231 cells (C) were treated with 50 μm desipramine or PBS control (Con) for 1 h and then stimulated with PMA and TNFα for 3 h. IL-6 in cultured media was measured as above (n = 2). D, HeLa cells were treated with the indicated siRNA for 72 h and then stimulated with TNFα (20 ng/ml) for 15 min. Whole cell lysates were subjected to Western blotting for phospho-p38 (p-p38) and p38. Equal amounts of protein were loaded (representative of two experiments). E, HeLa cells were pretreated with 25 μm desipramine for 1 h and treated with TNFα (20 ng/ml) for 15 min. Whole cell lysates were prepared, and equal amounts of protein were subjected to Western blotting for phospho-p38 and p38 (representative of three experiments). F, HeLa cells were pretreated with PBS or desipramine (50 μm) for 1 h and then stimulated with PMA for 90 min. Next, cDNA was synthesized, and qPCR was performed for IL-6 mRNA and normalized to actin as described under “Experimental Procedures” (n = 3). G, HeLa cells were treated with ASM siRNA for 48 h. RNA and cDNA were harvested and prepared, and qPCR was performed for ASM mRNA and normalized to actin (n = 3). H, HeLa cells were transfected as previously for 48 h, and then lysates were harvested, and acid sphingomyelinase activity was measured as described under “Experimental Procedures” (n = 3). Error bars, S.E.
Differential IL-6 RNA Dynamics in MCF-7 Compared with HeLa and MDA-MB-231 Cells in Response to PMA
Synthesis of IL-6 is a dynamic process with multiple regulated steps, including transcription, message stability, and translation. It is known that protein and mRNA levels of IL-6 are increased in cell lines such as MDA-MB-231 compared with MCF-7, but it is not known whether this is due to stability or transcription (5). To investigate potential differences and mechanisms in IL-6 RNA dynamics in response to PKC signaling between MCF-7, MDA-MB-231, and HeLa cells, a time course of PMA was performed, and levels of mRNA and hnRNA were determined, utilizing primers with either exonic or intronic reverse primers (23). In MCF-7, PMA treatment resulted in a 35-fold induction of IL-6 mRNA, yet no detectable change in hnRNA was observed, suggesting that IL-6 induction is primarily due to increased message stability (Fig. 5A). To further assess the process of transcription, a luciferase-based promoter assay was utilized. Using a human IL-6 promoter assay, PMA stimulated promoter activity by 2.3-fold in MCF7 cells (data not shown). This moderate change does not explain the dramatic induction at the mRNA and protein level in response to PMA (Fig. 1), yet it is difficult to reconcile with the unchanged hnRNA levels (Fig. 5A). Taken together, PKC-mediated induction of IL-6 mRNA in MCF-7 is mediated primarily by message stability and a possible minor contribution of transcription.
FIGURE 5.

IL-6 RNA dynamics in MCF-7, MDA-MB-231, and HeLa; up-regulation of ASM and IL-6 in invasive cancer cells. A–C, MCF-7 (A), MDA-MB-231 (B), and HeLa (C) cells were treated with PMA for 45 or 90 min. Total RNA was harvested, and cDNA was synthesized according to “Experimental Procedures.” IL-6 hnRNA and mRNA were measured by qPCR using specific primers for each species, as described under “Experimental Procedures.” -Fold change was calculated from untreated controls, and t0 was represented by the average of untreated samples at 45 and 90 min, being nearly identical (n = 2). D, basal IL-6 hnRNA and mRNA levels from the various cell lines were calculated after normalizing to actin, representing relative amounts between cell lines and between hnRNA versus mRNA. E, ASM mRNA levels were measured by RT-PCR as described under “Experimental Procedures” in MCF-7, MDA-MB-231, and HeLa cells under basal conditions (n = 2). F, HeLa cells were transfected for 48 h with either control or ASM siRNA. RNA was harvested, and RT-PCR was performed for IL-6 mRNA as described above (n = 2). G, MCF-7 cells were transfected for 48 h with either pEF6-EV or pEF6-ASM-V5-His6 as described under “Experimental Procedures,” after which cDNA was prepared, and IL-6 mRNA was measured by RT-PCR and expressed as -fold change (unpaired t test; **, p < 0.01, n = 3). Error bars, S.E.
Next, IL-6 RNA dynamics were assessed in MDA-MB-231 and HeLa cells. PMA treatment resulted in IL-6 mRNA induction, and further, there was a 2- and 6-fold induction of hnRNA at 45 min in MDA-MB-231 and HeLa, respectively, which preceded the time-dependent increase in mRNA at 90 min (Fig. 5, B and C). Additionally, IL-6 promoter activity was assessed in HeLa, and the results showed a 2.8-fold increase in promoter activity in response to PMA (data not shown). Therefore, based on increased IL-6 luciferase promoter activity and increased endogenous hnRNA, PMA results in transcriptional activation of IL-6 expression in HeLa and MDA-MB-231. Conversely, we can infer that PMA also results in increased message stability because, for example, in HeLa cells, a 6-fold increase in transcription is not sufficient to explain the 25-fold increase in IL-6 mRNA. Based on the RNA profiles of MDA-MB-231 and HeLa and the promoter activity assay, IL-6 RNA induction is explained by a combination of transcriptional activation and message stabilization.
Basal Induction of the ASM/IL-6 Pathway in Invasive Cancer Cells
The most striking difference between the cell lines, in the context of IL-6, is the basal level of IL-6 mRNA despite having approximately equivalent basal hnRNA (Fig. 5D). We wondered if this difference could be due to ASM. To investigate this possibility, we hypothesized that there may be differences in ASM expression between the three cell lines. ASM mRNA was measured by real-time PCR, and the results showed a 2–3-fold up-regulation of ASM message in HeLa and MDA-MB-231 compared with MCF-7 (Fig. 5E). Furthermore, we determined the effects of silencing ASM in HeLa on basal levels of IL-6 mRNA, and the results showed that this caused a 40% reduction in basal IL-6 mRNA (Fig. 5F). Additionally, overexpression of ASM in MCF-7 led to a 5.3-fold up-regulation of basal IL-6 mRNA (Fig. 5G). In summary, up-regulation of ASM in invasive cancer cells may play a role in the stabilization of IL-6 mRNA, thus explaining the previous findings that MDA-MB-231 and HeLa cells have higher levels of IL-6 mRNA than MCF7 cells.
Role of ASM in Cancer Cell Invasion
Due to the abundant evidence linking IL-6 to proinvasive biological processes, it became important to assess the role of ASM in cancer cell invasion. HeLa cells were treated with either control or ASM siRNA for 48 h and subsequently transferred to precoated Matrigel invasion wells. Knockdown of ASM resulted in a dramatic decrease in serum-induced invasion through Matrigel (Fig. 6A). ASM was also required for invasion in MDA-MB-231, suggesting that ASM could be playing a fundamental role in the pathological process of invasion (data not shown). Furthermore, we assessed the roles of PKC and p38 in invasion by adding PMA or BIRB796 to the upper chamber during invasion (Fig. 6B). PMA treatment enhanced invasion over serum-replete medium alone, whereas BIRB796 largely blocked PMA-induced invasion. Pretreatment of BIRB796 with HeLa cells effectively blocks PMA-induced p38 phosphorylation, showing that it is active at the used concentration (Fig. 6C). Additionally, to assess for morphologic changes, immunofluorescent microscopy was performed, which revealed significant loss of typical F-actin stress fibers with a rearrangement of cellular architecture upon ASM knockdown (Fig. 6D). This striking change was mirrored by decreased levels of phosphorylation of myosin light chain in HeLa (data not shown). Taken together, the results demonstrate a role for ASM in sustaining cytoskeletal changes toward invasion in cancer cells, which is consistent with its role in regulating p38 and IL-6.
FIGURE 6.

Role of ASM in invasion and cytoskeletal changes in HeLa cells. A, HeLa cells were treated with control (Con) or ASM siRNA for 48 h and trypsinized, and equal cells were plated in precoated Matrigel wells. After 48 h of incubation with or without serum in the lower chamber, invaded cells were stained and counted in 4 fields/well (n = 2). B, HeLa cells were treated with the indicated agents for 90 min, trypsinized, resuspended in serum-free medium, and seeded at 2.5 × 104 cells/well in the upper well of a Matrigel transwell invasion assay plate. The indicated agents were then added to the upper wells of the transwell plate, and either serum-replete or serum-free medium was added to the lower chamber (two-way analysis of variance, Bonferroni's post-test; ****, p < 0.0001, control compared with PMA and PMA compared with PMA + BIRB796, n = 3). C, HeLa cells were treated with the indicated agents for 90 min, and total protein was harvested. Equal protein was loaded in each lane of a 4–12% SDS-polyacrylamide gel and probed for total p38 or phospho-p38 as indicated. D, HeLa cells transfected with control or ASM siRNA for 48 h were visualized by immunofluorescence for vimentin (green), F-actin (red; phalloidin), and nuclei (blue; DRAQ5). E, scheme depicting the role of p38 and ASM in IL-6 production and malignant progression of tumor cells.
DISCUSSION
In this study, a role for ASM was demonstrated in the regulation of p38 signaling and the proinvasive cytokine IL-6 with potential implications for a role for ASM in cancer progression. The biochemical and signaling functions of ASM were specific in that they could be distinguished from those of GBA1, the other key enzyme involved in generation of lysosomal ceramide. ASM was demonstrated to play an important role in IL-6 production at the protein and message level with effects on transcription and message stability. Further, ASM was shown to be critically involved in the pathobiological process of invasion, a possible consequence of its regulation of IL-6.
Regulation of IL-6 by PKC and Sphingolipid Metabolism
This work reveals a new role for the novel PKC family in up-regulating IL-6. This relates to previous work from our laboratory, which showed that the novel isoform PKCδ is required for ASM activation leading to ceramide accumulation (9). Moreover, ASM and GBA1 both contribute to PKCδ-dependent ceramide accumulation, which is predominantly C16-ceramide (8). However, unique biological roles for ASM- and GBA1-derived ceramide had not been defined in the same context. Here we show that the novel isoforms of PKC are required for PMA-induced IL-6 production in MCF-7 (Fig. 1A). Thus, the data provide further evidence linking PKCs to sphingolipid-mediated pathways.
Our results reveal distinct roles for ASM and GBA1 in the regulation of IL-6, suggesting that ASM- and GBA1-derived ceramides have opposing biological functions despite being predominantly structurally identical (mostly C16-ceramide) (Fig. 1B)(8). These findings raise the possibility of a novel paradigm that not only the specific identity of a bioactive sphingolipid is important, but the enzyme from which it is produced within a subcellular location may dictate its biologic function. Based on our studies, the subcellular topology was not defined, but we speculate that ASM and GBA1 are acting on their respective substrates at somewhat distinct points in the endolysosomal system, which is involved in recycling SM and glucosylceramide from the plasma membrane to the lysosome (30, 31). In terms of transducing a signal, the plasma membrane or early endosomes appear to be a more likely candidate than the lysosome, which itself is encased by the limiting membrane, where ASM and GBA1 are in the lumen, bound to and degrading internal membranes within the lysosome. Alternatively, or in addition, the levels of substrates of these two enzymes could play a key role in dictating the specific functions attributed to ASM versus those attributed to GBA1.
Regulation of Cytokine Production and p38 by ASM
Previous reports have studied cytokine regulation by ASM with varying results. This is probably due to the use of various approaches and distinct regulation of different cytokines. Previous studies have used ASM−/− peritoneal macrophages, showing no requirement of ASM for the production of various inflammatory mediators, including nitric oxide, TNFα, and IL-1β (13, 32). Conversely, a transcriptional inhibitor of ASM, SMA-7, was shown to reduce cytokine production, although the specificity of this inhibitor has not been well established (14). Additionally, while this work was in progress, Kumagai et al. (15) showed involvement of ASM in the synergistic production of IL-6 in response to carcinogenic electrophiles with TNFα but not TNFα alone in bladder cancer cells, where these electrophiles resulted in ASM up-regulation. Further, recent work by Jin et al. (34) showed ASM involvement in palmitic acid-induced IL-6 up-regulation in macrophages, suggesting a common role for ASM in IL-6 production.
Previous work from our laboratory has demonstrated a role for ASM in the production of the chemokine, CCL5/RANTES (17). Interestingly, this is probably a distinct pathway of regulation from that by which ASM regulates IL-6. This is supported by the requirement of ACD for induction of RANTES, whereas loss of ACD trended toward an increase in IL-6 production in MCF-7, thus distinguishing these two pathways. Additionally, PMA was found to be a specific inducer of IL-6 (Fig. 2D). One explanation of these data is that IL-6 and CCL5/RANTES are distinct in their expression pattern in response to cytokine stimulation, IL-6 being rapidly induced whereas RANTES displays slower kinetics (35). Consistent with this, IL-6 mRNA is basally unstable, containing multiple 3′-UTR AU-rich elements and is regulated substantially by increases in stability, whereas RANTES mRNA is intrinsically very stable (35). What is emerging, therefore, suggests coordinated regulation of these classes of cytokines by sphingolipids, such as ceramide, sphingosine, and sphingosine 1-phosphate. Ceramide, produced first metabolically, may be critical for IL-6, which is induced acutely, and then accumulation of sphingosine may trigger the transition to induction of late response genes, such as RANTES.
This work provides novel evidence for a role for ASM in p38 activation in cancer cells. Previous evidence showed that ASM is not required for p38 signaling in murine macrophages by using ASM−/− macrophages, a different context from that employed here and with inherent caveats concerning complete knock-out systems for sphingolipid enzymes (i.e. they recapitulate genetic disease but may not capture the biological role of the enzyme). More likely, the role of ASM in p38 signaling may be cell type-specific. There are some reports of exogenous ceramide activating p38 signaling, but there is no previous evidence demonstrating that a specific ceramide-producing enzyme is required for p38 activation (13, 36–38). Moreover, if the enzyme from which ceramide is derived dictates its function, then exogenous ceramide is unlikely to recapitulate the function of specific pools of endogenous ceramides. Despite a role for ASM in p38 activation shown here, the mechanism of this effect is still elusive. ASM may be directly involved in the signaling in response to outside signals to p38, or the loss of ASM may render cells unable to activate p38 by a gain of function upon inhibition of ASM. Preliminary results show activation of p38 upon overexpression of ASM in HeLa (data not shown), arguing against the possibility that loss of ASM results in a gain of function that blocks p38 activation. Additionally, recent work by Adada et al. has shown that sphingosine kinase 1 and sphingosine 1-phosphate are involved in p38 activation in HeLa (39), which raises the possibility that ASM, ACD and sphingosine kinase 1 may function in concert to produce sphingosine 1-phosphate, which could lead to activation of p38. However, this would be cell type-specific because, in MCF-7 cells, ACD is not required for p38 phosphorylation (Fig. 1C). In light of the body of literature showing activation of sphingomyelinase activity and ceramide accumulation in response to outside signals (8–10, 40), it is not far-fetched that ASM/ceramide participate in the acute activation of p38, and other ceramide-producing enzymes contribute to resolution of p38 activity.
Implications for Cancer Biology
The finding that ASM is involved in IL-6 production and invasion casts ASM in a novel role. Previous literature has implicated ASM in the accumulation of ceramide and downstream apoptosis in response to UV light (41, 42), ionizing radiation (43), and other stimuli (44–46). One could speculate that activation of ASM in normal cells is capable of inducing apoptosis, but once cancer cells are resistant to cell death, ASM activity could translate to increased inflammatory cytokines and invasiveness. In some ways, this is analogous to the dual nature of p38 signaling in apoptosis versus oncogenic properties (47). Additionally, due to the accumulating evidence for IL-6 as a critical player in cancer biology, targeting pathways that are involved in IL-6 synthesis or downstream effects may prove to be a powerful approach for attenuating invasion and metastasis (1, 33). Inhibition of ASM could be a therapeutic means to block IL-6 produced in the microenvironment by tumor cells, which is required for invasion. Future work is needed to evaluate the role of ASM in cancer progression or metastasis.
In summary, this work provides evidence for a clear role for ASM in the regulation of p38 signaling and IL-6 production. This regulation is specific in that other sphingolipid salvage pathway enzymes did not replicate this. ASM was found to be essential for p38 activation as well, defining a role in upstream signaling to IL-6. In line with this, ASM was required for invasion of HeLa cells. Moreover, ASM may play a critical role in mediating the increased levels of IL-6 found in invasive cell lines. The contribution of ASM to both malignant progression and IL-6 production is partially dependent on p38; however, a p38-independent mechanism may still directly link ASM to invasion and IL-6 production (Fig. 6E). This implicates ASM as a novel player in several malignant cancer properties and may indicate that ASM is an attractive target for therapeutic intervention in addition to p38. This work, for the first time, demonstrates a regulatory role for ASM in a signaling pathway and a biological outcome of cytokine induction, including mechanistic insights and disease implications. Further investigation is needed to understand how ASM is mediating its effects on p38 and IL-6.
Acknowledgments
We extend special thanks to Drs. Yan Huang and Junfei Jin for sharing IL-6 promoter constructs. We also thank Drs. Eytan Domany and Amit Zeisel for assistance in analysis with RNA dynamics. We thank Dr. Daniel Canals for careful review of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants CA 097132 (to Y. A. H. and L. M. O.) and GM097741 (to L. M. O.). This work was also supported by a Veterans Affairs merit award (to L. M. O.).
- SM
- sphingomyelin
- ASM
- acid sphingomyelinase
- GBA1
- acid β-glucosidase 1
- ACD
- acid ceramidase
- CCL5
- chemokine ligand 5
- RANTES
- regulated on activation normal T cell expressed and secreted
- PMA
- 4β-phorbol 12-myristate 13-acetate
- qPCR
- quantitative polymerase chain reaction
- NPD
- Niemann-Pick disease
- hnRNA
- heteronuclear RNA
- TRITC
- tetramethylrhodamine isothiocyanate.
REFERENCES
- 1. Grivennikov S. I., Karin M. (2011) Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann. Rheum. Dis. 70, i104–i108 [DOI] [PubMed] [Google Scholar]
- 2. Tawara K., Oxford J. T., Jorcyk C. L. (2011) Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer Manag. Res. 3, 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sullivan N. J., Sasser A. K., Axel A. E., Vesuna F., Raman V., Ramirez N., Oberyszyn T. M., Hall B. M. (2009) Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 28, 2940–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grivennikov S., Karin E., Terzic J., Mucida D., Yu G. Y., Vallabhapurapu S., Scheller J., Rose-John S., Cheroutre H., Eckmann L., Karin M. (2009) IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15, 103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Faggioli L., Costanzo C., Merola M., Bianchini E., Furia A., Carsana A., Palmieri M. (1996) Nuclear factor κB (NF-κB), nuclear factor interleukin-6 (NFIL-6 or C/EBPβ) and nuclear factor interleukin-6 β (NFIL6-β or C/EBP δ) are not sufficient to activate the endogenous interleukin-6 gene in the human breast carcinoma cell line MCF-7. Comparative analysis with MDA-MB-231 cells, an interleukin-6-expressing human breast carcinoma cell line. Eur. J. Biochem. 239, 624–631 [DOI] [PubMed] [Google Scholar]
- 6. Merrill A. H., Jr. (2011) Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem. Rev. 111, 6387–6422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kitatani K., Idkowiak-Baldys J., Hannun Y. A. (2008) The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 20, 1010–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kitatani K., Sheldon K., Rajagopalan V., Anelli V., Jenkins R. W., Sun Y., Grabowski G. A., Obeid L. M., Hannun Y. A. (2009) Involvement of acid β-glucosidase 1 in the salvage pathway of ceramide formation. J. Biol. Chem. 284, 12972–12978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zeidan Y. H., Hannun Y. A. (2007) Activation of acid sphingomyelinase by protein kinase Cδ-mediated phosphorylation. J. Biol. Chem. 282, 11549–11561 [DOI] [PubMed] [Google Scholar]
- 10. Becker K. P., Kitatani K., Idkowiak-Baldys J., Bielawski J., Hannun Y. A. (2005) Selective inhibition of juxtanuclear translocation of protein kinase C betaII by a negative feedback mechanism involving ceramide formed from the salvage pathway. J. Biol. Chem. 280, 2606–2612 [DOI] [PubMed] [Google Scholar]
- 11. Laulederkind S. J., Bielawska A., Raghow R., Hannun Y. A., Ballou L. R. (1995) Ceramide induces interleukin 6 gene expression in human fibroblasts. J. Exp. Med. 182, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kitatani K., Sheldon K., Anelli V., Jenkins R. W., Sun Y., Grabowski G. A., Obeid L. M., Hannun Y. A. (2009) Acid β-glucosidase 1 counteracts p38delta-dependent induction of interleukin-6: possible role for ceramide as an anti-inflammatory lipid. J. Biol. Chem. 284, 12979–12988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Manthey C. L., Schuchman E. H. (1998) Acid sphingomyelinase-derived ceramide is not required for inflammatory cytokine signalling in murine macrophages. Cytokine 10, 654–661 [DOI] [PubMed] [Google Scholar]
- 14. Sakata A., Ochiai T., Shimeno H., Hikishima S., Yokomatsu T., Shibuya S., Toda A., Eyanagi R., Soeda S. (2007) Acid sphingomyelinase inhibition suppresses lipopolysaccharide-mediated release of inflammatory cytokines from macrophages and protects against disease pathology in dextran sulphate sodium-induced colitis in mice. Immunology 122, 54–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kumagai T., Ishino T., Nakagawa Y. (2012) Acidic sphingomyelinase induced by electrophiles promotes proinflammatory cytokine production in human bladder carcinoma ECV-304 cells. Arch. Biochem. Biophys. 519, 8–16 [DOI] [PubMed] [Google Scholar]
- 16. Muller P. Y., Janovjak H., Miserez A. R., Dobbie Z. (2002) Processing of gene expression data generated by quantitative real-time RT-PCR. BioTechniques 32, 1372–1374, 1376,, 1378–1379 [PubMed] [Google Scholar]
- 17. Jenkins R. W., Clarke C. J., Canals D., Snider A. J., Gault C. R., Heffernan-Stroud L., Wu B. X., Simbari F., Roddy P., Kitatani K., Obeid L. M., Hannun Y. A. (2011) Regulation of CC ligand 5/RANTES by acid sphingomyelinase and acid ceramidase. J. Biol. Chem. 286, 13292–13303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jenkins R. W., Canals D., Idkowiak-Baldys J., Simbari F., Roddy P., Perry D. M., Kitatani K., Luberto C., Hannun Y. A. (2010) Regulated secretion of acid sphingomyelinase: implications for selectivity of ceramide formation. J. Biol. Chem. 285, 35706–35718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carter A. B., Monick M. M., Hunninghake G. W. (1999) Both Erk and p38 kinases are necessary for cytokine gene transcription. Am. J. Respir. Cell Mol. Biol. 20, 751–758 [DOI] [PubMed] [Google Scholar]
- 20. Kondo A., Koshihara Y., Togari A. (2001) Signal transduction system for interleukin-6 synthesis stimulated by lipopolysaccharide in human osteoblasts. J. Interferon Cytokine Res. 21, 943–950 [DOI] [PubMed] [Google Scholar]
- 21. Vanden Berghe W., Plaisance S., Boone E., De Bosscher K., Schmitz M. L., Fiers W., Haegeman G. (1998) p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-κB p65 transactivation mediated by tumor necrosis factor. J. Biol. Chem. 273, 3285–3290 [DOI] [PubMed] [Google Scholar]
- 22. Rousseau S., Morrice N., Peggie M., Campbell D. G., Gaestel M., Cohen P. (2002) Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP-K2 and its interaction with cytokine mRNAs. EMBO J. 21, 6505–6514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zeisel A., Köstler W. J., Molotski N., Tsai J. M., Krauthgamer R., Jacob-Hirsch J., Rechavi G., Soen Y., Jung S., Yarden Y., Domany E. (2011) Coupled pre-mRNA and mRNA dynamics unveil operational strategies underlying transcriptional responses to stimuli. Mol. Syst. Biol. 7, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuma Y., Sabio G., Bain J., Shpiro N., Márquez R., Cuenda A. (2005) BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J. Biol. Chem. 280, 19472–19479 [DOI] [PubMed] [Google Scholar]
- 25. Pargellis C., Tong L., Churchill L., Cirillo P. F., Gilmore T., Graham A. G., Grob P. M., Hickey E. R., Moss N., Pav S., Regan J. (2002) Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 9, 268–272 [DOI] [PubMed] [Google Scholar]
- 26. Gallucci R. M., Sloan D. K., Heck J. M., Murray A. R., O'Dell S. J. (2004) Interleukin 6 indirectly induces keratinocyte migration. J. Invest. Dermatol. 122, 764–772 [DOI] [PubMed] [Google Scholar]
- 27. Studebaker A. W., Storci G., Werbeck J. L., Sansone P., Sasser A. K., Tavolari S., Huang T., Chan M. W., Marini F. C., Rosol T. J., Bonafé M., Hall B. M. (2008) Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. 68, 9087–9095 [DOI] [PubMed] [Google Scholar]
- 28. Oh K., Ko E., Kim H. S., Park A. K., Moon H. G., Noh D. Y., Lee D. S. (2011) Transglutaminase 2 facilitates the distant hematogenous metastasis of breast cancer by modulating interleukin-6 in cancer cells. Breast Cancer Res. 13, R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hurwitz R., Ferlinz K., Sandhoff K. (1994) The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol. Chem. Hoppe Seyler 375, 447–450 [DOI] [PubMed] [Google Scholar]
- 30. Aureli M., Masilamani A. P., Illuzzi G., Loberto N., Scandroglio F., Prinetti A., Chigorno V., Sonnino S. (2009) Activity of plasma membrane beta-galactosidase and β-glucosidase. FEBS Lett. 583, 2469–2473 [DOI] [PubMed] [Google Scholar]
- 31. Schissel S. L., Keesler G. A., Schuchman E. H., Williams K. J., Tabas I. (1998) The cellular trafficking and zinc dependence of secretory and lysosomal sphingomyelinase, two products of the acid sphingomyelinase gene. J. Biol. Chem. 273, 18250–18259 [DOI] [PubMed] [Google Scholar]
- 32. Rozenova K. A., Deevska G. M., Karakashian A. A., Nikolova-Karakashian M. N. (2010) Studies on the role of acid sphingomyelinase and ceramide in the regulation of tumor necrosis factor α (TNFα)-converting enzyme activity and TNFα secretion in macrophages. J. Biol. Chem. 285, 21103–21113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oh K., Ko E., Kim H. S., Park A. K., Moon H. G., Noh D. Y., Lee D. S. (2011) Transglutaminase 2 facilitates the distant hematogenous metastasis of breast cancer by modulating interleukin-6 in cancer cells. Breast Cancer Res. 13, R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jin J., Zhang X., Lu Z., Perry D. M., Li Y., Russo S. B., Cowart L. A., Hannun Y. A., Huang Y. (2013) Acid sphingomyelinase plays a key role in palmitic acid-amplified inflammatory signaling triggered by lipopolysaccharide at low concentration in macrophages. Am. J. Physiol. Endocrinol. Metab. 305, E853–E867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hao S., Baltimore D. (2009) The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 10, 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bauer J., Huy C., Brenmoehl J., Obermeier F., Bock J. (2009) Matrix metalloproteinase-1 expression induced by IL-1β requires acid sphingomyelinase. FEBS Lett. 583, 915–920 [DOI] [PubMed] [Google Scholar]
- 37. Kato Y., Ozawa S., Tsukuda M., Kubota E., Miyazaki K., St-Pierre Y., Hata R. (2007) Acidic extracellular pH increases calcium influx-triggered phospholipase D activity along with acidic sphingomyelinase activation to induce matrix metalloproteinase-9 expression in mouse metastatic melanoma. FEBS J. 274, 3171–3183 [DOI] [PubMed] [Google Scholar]
- 38. Liangpunsakul S., Rahmini Y., Ross R. A., Zhao Z., Xu Y., Crabb D. W. (2012) Imipramine blocks ethanol-induced ASMase activation, ceramide generation, and PP2A activation, and ameliorates hepatic steatosis in ethanol-fed mice. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G515–G523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adada M. M., Orr-Gandy K. A., Snider A. J., Canals D., Hannun Y. A., Obeid L. M., Clarke C. J. (2013) Sphingosine kinase 1 regulates tumor necrosis factor-mediated RANTES induction through p38 mitogen-activated protein kinase but independently of nuclear factor κB activation. J. Biol. Chem. 288, 27667–27679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Modur V., Zimmerman G. A., Prescott S. M., McIntyre T. M. (1996) Endothelial cell inflammatory responses to tumor necrosis factor alpha. Ceramide-dependent and -independent mitogen-activated protein kinase cascades. J. Biol. Chem. 271, 13094–13102 [DOI] [PubMed] [Google Scholar]
- 41. Zeidan Y. H., Wu B. X., Jenkins R. W., Obeid L. M., Hannun Y. A. (2008) A novel role for protein kinase Cdelta-mediated phosphorylation of acid sphingomyelinase in UV light-induced mitochondrial injury. FASEB J. 22, 183–193 [DOI] [PubMed] [Google Scholar]
- 42. Charruyer A., Jean C., Colomba A., Jaffrézou J. P., Quillet-Mary A., Laurent G., Bezombes C. (2007) PKCζ protects against UV-C-induced apoptosis by inhibiting acid sphingomyelinase-dependent ceramide production. Biochem. J. 405, 77–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peña L. A., Fuks Z., Kolesnick R. N. (2000) Radiation-induced apoptosis of endothelial cells in the murine central nervous system: protection by fibroblast growth factor and sphingomyelinase deficiency. Cancer Res. 60, 321–327 [PubMed] [Google Scholar]
- 44. Ion G., Fajka-Boja R., Kovács F., Szebeni G., Gombos I., Czibula A., Matkó J., Monostori E. (2006) Acid sphingomyelinase mediated release of ceramide is essential to trigger the mitochondrial pathway of apoptosis by galectin-1. Cell. Signal. 18, 1887–1896 [DOI] [PubMed] [Google Scholar]
- 45. Falcone S., Perrotta C., De Palma C., Pisconti A., Sciorati C., Capobianco A., Rovere-Querini P., Manfredi A. A., Clementi E. (2004) Activation of acid sphingomyelinase and its inhibition by the nitric oxide/cyclic guanosine 3′,5′-monophosphate pathway: key events in Escherichia coli-elicited apoptosis of dendritic cells. J. Immunol. 173, 4452–4463 [DOI] [PubMed] [Google Scholar]
- 46. Zhang Y., Li X., Carpinteiro A., Gulbins E. (2008) Acid sphingomyelinase amplifies redox signaling in Pseudomonas aeruginosa-induced macrophage apoptosis. J. Immunol. 181, 4247–4254 [DOI] [PubMed] [Google Scholar]
- 47. Feng Y., Wen J., Chang C. C. (2009) p38 Mitogen-activated protein kinase and hematologic malignancies. Arch. Pathol. Lab. Med. 133, 1850–1856 [DOI] [PubMed] [Google Scholar]