

Background: Genetic variants in the FADS cluster are determinants of arachidonate synthesis, but their role in eicosanoid generation remains unclear.
Results: FADS SNP, rs174537 is associated with leukotriene B4 and 5-HETE production from stimulated human blood.
Conclusion: A FADS SNP affects the synthesis of 5-lipoxygenase products.
Significance: FADS variation may influence inflammation via eicosanoid biosynthesis.
Keywords: Arachidonic Acid (AA) (ARA), Eicosanoid, Inflammation, Nutrition, Polyunsaturated Fatty Acid (PUFA), Single-nucleotide Polymorphism (SNP), Fatty Acid Desaturase
Abstract
Dramatic shifts in the Western diet have led to a marked increase in the dietary intake of the n-6 polyunsaturated fatty acid (PUFA), linoleic acid (LA). Dietary LA can then be converted to arachidonic acid (ARA) utilizing three enzymatic steps. Two of these steps are encoded for by the fatty acid desaturase (FADS) cluster (chromosome 11, 11q12.2-q13) and certain genetic variants within the cluster are highly associated with ARA levels. However, no study to date has examined whether these variants further influence pro-inflammatory, cyclooxygenase and lipoxygenase eicosanoid products. This study examined the impact of a highly influential FADS SNP, rs174537 on leukotriene, HETE, prostaglandin, and thromboxane biosynthesis in stimulated whole blood. Thirty subjects were genotyped at rs174537 (GG, n = 11; GT, n = 13; TT, n = 6), a panel of fatty acids from whole serum was analyzed, and precursor-to-product PUFA ratios were calculated as a marker of the capacity of tissues (particularly the liver) to synthesize long chain PUFAs. Eicosanoids produced by stimulated human blood were measured by LC-MS/MS. We observed an association between rs174537 and the ratio of ARA/LA, leukotriene B4, and 5-HETE but no effect on levels of cyclooxygenase products. Our results suggest that variation at rs174537 not only impacts the synthesis of ARA but the overall capacity of whole blood to synthesize 5-lipoxygenase products; these genotype-related changes in eicosanoid levels could have important implications in a variety of inflammatory diseases.
Introduction
There have been dramatic changes in the Western diet for the past 50 years. Perhaps nowhere has this been seen more than the dramatic increase in dietary n-6 PUFA with the addition of vegetable oil products (soybean, corn, palm, and canola oils as well as margarine and shortenings) (1–5). For example, the n-6 PUFA LA2 has increased 3–4-fold, now making up 6–8% of energy consumed (2). Once ingested, humans have the capacity for LA to be converted to ARA utilizing three (two desaturation and one elongation) enzymatic steps (6–9). The two desaturation steps are encoded by two genes (FADS2 and FADS1) found in a region of chromosome 11 known as the FADS cluster (11q12.2-q13), and have long been recognized as the rate-limiting steps in ARA biosynthesis (see Fig. 1) (6, 10–12).
FIGURE 1.

Biochemical pathway representing the metabolism of LA into arachidonic acid and subsequently into proinflammatory eicosanoids. Gene names are in pink with green (FADS1 and FADS2), and common enzyme names are in blue. Abbreviations: LA, linoleic acid; GLA, γ-linolenic acid; DGLA, dihomo-γ-linolenic acid; MGLL, monoacylglycerol lipase; PC, phosphatidylcholine; PE, phosphatidylethanolamine.
Once ARA is synthesized in a tissue such as the liver, it can be transported to other cells and tissues in circulation as free fatty acids bound to albumin or esterified to complex lipids such as phospholipids, cholesterol esters, and triglycerides in lipoprotein particles (see Fig. 1) (13, 14). Once taken up, ARA is acted upon by specific ARA-CoA synthetase(s) that converts free ARA into ARA-CoA to be utilized by ARA-CoA: l-acyl-2-lysophosphoglyceride acyltransferase(s) to yield 1-acyl-2-ARA phospholipids (15, 16). ARA is then remodeled into the sn-2 position of 1-alkyl and 1-alk-1-enyl phospholipids utilizing CoA-dependent and -independent acyl transferases.
ARA can then be liberated from membrane phospholipids (typically after cellular activation) as a free fatty acid by a family of phospholipases (17–21). ARA itself is a potent cellular signal, and it can be converted into a large family of eicosanoid products (including prostaglandins, thromboxanes, leukotrienes, and lipoxins) via cyclooxygenase, lipoxygenase, and cytochrome P450 enzymes (Fig. 1) (22–24). In general, these ARA-derived eicosanoids act like local hormones to promote acute and chronic inflammation in numerous human diseases. In addition, radical-based oxidation of ARA is known to generate complex mixtures of additional products, and some of these oxidations products are important biomarkers of chronic diseases such as coronary artery disease (25, 26).
Initial candidate gene studies by Schaeffer and colleagues (27), followed by almost 20 other candidate gene and gene wide association studies, have showed that numerous SNPs in and around FADS2 and FADS1 have dramatic effects on ARA and ARA-containing phospholipid levels (12, 27–33). rs174537 is the most strongly associated SNP with ARA levels (p = 5.95 × 10−46) (33) in a large genome-wide association study is just upstream of FADS1. A few studies have addressed the mechanism of action by which FADS variation impacts PUFA levels and shown that it may alter the expression of FADS cluster genes, through promoter usage or stability of transcript (34, 35) Our laboratory recently demonstrated that many of the peak-associated SNPs (described in both candidate gene and genome-wide association studies) are strongly associated with methylation status a few CpG sites within regulatory region with an “enhancer signature” (36). This region sits between the promoters for FADS1 and FADS2, and this observation raises the question of whether the methylation status of these CpG sites may play a role in regulating the transcription of FADS1 and FADS2. There are striking differences in the minor allele frequency at rs174537 between populations (37–39); 79–82% of African Americans carry two copies of the G allele compared with only 42–45% of European Americans. Importantly, the allelic effect of the G allele, which is associated with enhanced conversion of dihomo-γ-linolenic acid to ARA on enzymatic efficiency was similar in both groups.
Interestingly, the same SNPs that are associated ARA levels are also strongly associated with traditional markers of cardiovascular disease and inflammation, including LDL cholesterol, triglycerides, HDL cholesterol, total cholesterol levels, and C-reactive protein as well as coronary artery disease (CAD) (30, 40–42). FADS variation has also been associated with urinary excretion of 8-epi-prostaglandin F(2α) in both controls and CAD patients, and this has been widely recognized as a sensitive and independent risk factor for CAD (25, 26, 43).
A critical question that remains is whether these associations with ARA and ARA-containing phospholipids levels are also observed with enzymatic eicosanoid products. This is a complicated question due to the fact that eicosanoids are typically local signaling molecules that are rapidly metabolized (44–47). Consequently, it is difficult to get accurate in vivo measurements in blood and tissues from humans. Fradin and colleagues (48) described a whole blood stimulation technique combined with LC-MS/MS that could monitor the capacity of whole blood to synthesize a wide variety of eicosanoids but primarily leukotriene B4 made as a result of polymorphonuclear leukocyte phagocytosis of zymosan particles. Ex vivo whole blood stimulation has been used as a pharmacodynamics end point to monitor the impact of eicosanoid blockers such as 5-lipoxygenase inhibitors on leukotriene levels (49). In the current study, we have examined the relationship between genotypes at an important FADS variant rs174537 and leukotriene, HETE, prostaglandin, and thromboxane levels after whole blood stimulation.
EXPERIMENTAL PROCEDURES
Subjects
This study was reviewed and approved for human subjects by the Institutional Review Board at Wake Forest University Baptist Health. Subjects were recruited by telephone, letter, or E-mail from September 2011 until January 2014. These subjects were asked to come to the Clinical Research Unit at Wake Forest Baptist Hospital. Written consent was obtained from all subjects prior to enrollment. To be included in this study, subjects had to be considered healthy Caucasian females, between the ages of 21 and 65. Subjects were excluded from the study if they carried diagnoses of diabetes, cancer, heart attack or vascular surgery within the past year, heart disease, high blood pressure, history of stroke, atherosclerosis, asthma, multiple sclerosis, or chronic joint disease. They were also excluded for gallbladder disease or history of cholecystectomy, use of tobacco products within the last six months, current pregnancy, fasting triglycerides >150 mg/dl, fasting glucose >125 mg/dl, blood pressure >130/90 mmHg, a body mass index of ≥30 kg/m2 or <19 kg/m2, taking >100 mg of aspirin per day, taking NSAIDs or oral corticosteroids, or taking a monoleukast-type of medicine.
Biochemical Measurements
Potential subjects were brought to the clinic for an initial screening visit and to provide informed consent. At that time, they donated fasting blood and urine samples. Heparinized blood was used for whole blood zymosan stimulation (48), and the remainder was saved for red blood cell and plasma aliquots. An EDTA tube was also drawn for DNA isolation in order to genotype at the rs174537 SNP locus. A final tube was drawn for serum and biochemical measurements. All subjects were screened for total cholesterol, triglycerides, HDL, VLDL, LDL, serum glucose, and high sensitivity C-reactive protein. Anthropometric measurements were also taken at this encounter, including systolic and diastolic blood pressure, resting heart rate, weight, percent body fat, body mass index, and waist and hip circumference. All values were determined by Lab Corp (Burlington, NC).
Fatty Acid Analysis
Each subject had a tube of blood drawn for serum for determination of fatty acid content. A standard protocol was used and levels were determined in the presence of an internal standard (triheptadecanoin; Nuchek Prep, Elysian, MN) as described previously (50). Briefly, fatty acid methyl esters were prepared (51) after saponification of duplicate serum samples (100 μl). A panel of 23 different fatty acids was quantified by gas chromatography with flame ionization detection.
DNA and Genotype Analysis
Each subject had an EDTA tube drawn that was reserved specifically for DNA isolation, whereas early patients also gave saliva samples for DNA isolation. Each pellet of DNA was isolated using a standard protocol, washed, and rehydrated in Tris-EDTA for analysis. Concentration of DNA per sample was determined using Nanodrop technology. All samples were then diluted to a concentration of 10 nl/μl and 20 μl of each sample was plated into a 96-deep well plate for analysis via a Sequenome platform. A total of 61 SNPs were evaluated via this platform, including the gene locus rs174537.
Eicosanoid Stimulation and Analysis
A single heparinized tube of blood was taken from each patient and part was utilized for zymosan (Sigma-Aldrich), stimulation of whole blood as described previously (48). All samples were processed within 30 min of the blood draw. For each subject, the assay was performed in duplicate for control (950 μl of whole blood plus 50 μl of PBS) and stimulated 950 μl of whole blood plus 50 μl of 50 mg/ml zymosan) conditions. Tubes were incubated in a 37 °C shaking water bath for 30 min, and the reaction was stopped on ice for 5 min. Tubes were then centrifuged at 2000 rpm for 10 min at 4 °C. The supernatant was aliquoted and stored under argon at −20 °C until analysis. Supernatants were analyzed for eicosanoids by LC-MS/MS at the University of Colorado at Denver.
Eicosanoid Analysis by LC-MS/MS
After addition of stable isotope-labeled internal standards to the zymosan-stimulated blood, the eicosanoids were extracted and analyzed essentially as described previously (52), with some modifications: 1 ng of [d4]PGE2, [d4]thromboxane B2, [d4]LTB4, [d5]LTC4, [d8]5-HETE, 5 ng of [d4]6-keto PGF1α. Samples were diluted with water to a final methanol concentration of <10% and then extracted using solid phase cartridges, Strata-X, 33 Polymeric Reversed Phase (Phenomenex). The eluate (1 ml of methanol) was dried down and solubilized in 75 μl of HPLC solvent A (8.3 mm acetic acid buffered to pH 5.7 with ammonium hydroxide) plus 25 μl of HPLC solvent B (acetonitrile-methanol, 65:35, v/v). An aliquot of each sample (30 μl) was injected onto a Kinetex C18 5 μm 50 × 3.0 mm column (Phenomenex), at 250 μl/min, with a linear gradient from 25% solvent B to 75% in 12 min, 75 to 98% in 2 min, 5 min hold and re-equilibration for 5 min. The HPLC system was directly interfaced into the electrospray source of an AB Sciex Q Trap 5500 mass spectrometer (PE-Sciex, Thornhill, Ontario, Canada) for mass spectrometric analysis in the negative ion mode using multiple reaction monitoring of the specific transitions, and the following m/z transitions monitored were as follows: m/z 335 → 195 for LTB4 and Δ6-trans-LTB4; m/z 351 → 195 for 20-OH-LTB4; m/z 365 → 195 for 20-COOH-LTB4; m/z 624 → 272 for LTC4; m/z 495 → 177 for LTD4; m/z 438 → 333 for LTE4; m/z 319 → 115 for 5-HETE; m/z 319 → 179 for 12-HETE; m/z 319 → 219 for 15-HETE; m/z 351 → 271 for PGE2; m/z 351 → 333 PGD2; m/z 353 → 309 for PGF2α; m/z 369 → 163, 6-keto PGF1α; m/z 369 → 169 TXB2; 629 → 272 [d5]LTC4; 327 → 116 [d8]5-HETE; m/z 339 → 197 for[d4]LTB4; m/z 373 → 173 for [d4]TXB2; m/z 373 → 167 [d4]6-keto PGF1α, and m/z 355 → 275 for [d4]PGE2 (Fig. 2). The stereochemistry of 5-and 15-HETE was not determined in this study.
FIGURE 2.

Analysis of eicosanoids released into whole blood by specific LC-MS/MS assay of unstimulated (A) and stimulated (B) by the addition of zymosan in an ex vivo assay. The eicosanoids are separated by reverse phase HPLC and specific eicosanoids detected as negative ions transitions. The scales are normalized to the most abundant ion transition observed (B). The inset to B has the same normalization factor as that of A to show the internal standard abundance in the two samples. Abbreviations: TXB2, thromboxane B2; PGE2, prostaglandin E2.
Data Analysis
Quantitative trait data were examined for the presence of outliers and for skewness; tests for association are performed under a linear model assuming an additive effect of the alleles as the SNP and thereby sensitive to both outliers and non-normality. Outlier observations were dropped and data were log transformed if necessary. (LTB4 and 5-HETE were log10 transformed and, to accommodate zero values for PGD2, 12-HETE, 15-HETE, LTC4, and LTD4, these variables were analyzed as log10(1+ value), i.e. a constant of 1 was added to each value prior to transformation because log(0) is undefined.) To accommodate the additive effect at rs174537, the SNP genotype was coded as 0/1/2 for GG/GT/TT reflecting 0, 1, or two copies of the minor (T) allele. Pearson correlation coefficients were generated for all quantitative variables against each other. Once again, transformed variables were used as necessary.
RESULTS
Characteristics of the Study Population
Thirty healthy Caucasian females with an average age of 41.2 ± 12.7 years and ranging from 22 years to 62 years were recruited into the study. This study population had triglycerides, total cholesterol, fasting glucose, and body mass indexes of 70.9 ± 25.7 mg/dl, 171.2 ± 32.7 mg/dl, 87.5 ± 8.2 mg/dl, and 23.6 ± 2.8, respectively. There were 11 subjects with the GG genotype, 13 with the GT genotype, and six with the TT genotype within the study.
Stimulation of Whole Blood
Fig. 2 illustrates the LC-MS/MS analysis of eicosanoids from a typical sample of unstimulated (A) and zymosan-stimulated whole blood (B). Examination of unstimulated samples showed primarily the deuterated internal standards and low levels of most eicosanoids including TxB2, PGE2, and 5-HETE. Zymosan stimulation of whole blood induced a dramatic increase in 5-lipoxygenase products such as LTB4 and 5-HETE. There were also more modest increases (3–4-fold) in TxB2 and other eicosanoids. In Fig. 2B, the scales were normalized to the most abundant ion transition observed. The inset to B has the same normalization factor as that of A to show the internal standard abundance in the two samples.
Effect of Genotype of rs174537 on Fatty Acid Profile
An additive model was used to examine the effect of rs174537 on the phenotypic expression of serum fatty acids and eicosanoids in subjects (Table 1). As expected, there was a negative allelic effect between the minor allele at rs174537 and ARA levels (p = 0.067) indicating decreasing levels of ARA with increasing copies of the T allele, consistent with all prior publications; the lack of statistical significance is likely due to the small sample size (Table 1). There was a statistically significant genotypic effect (p = 0.035) when ARA is expressed as a ratio of ARA to its metabolic precursor, LA, once again replicating numerous candidate gene and genome-wide association studies (12, 27–33). Fig. 3 shows genotype at rs174537 as a function of ARA and ARA/LA. In support of prior studies, GG at rs174537 had the highest levels of ARA and ratio of ARA to LA with progressively lower levels and ratios for subjects with the GT and TT genotypes, respectively.
TABLE 1.
Summary of tests for association between rs174537 and PUFA levels, PUFA ratios, and eicosanoid production
| Phenotype | Estimate of the effect of rs174537 under an additive model β (95% CI) | p value |
|---|---|---|
| LA | 1.056 (−0.708, 2.819) | 0.251 |
| ALA | 0.023 (−0.061, 0.107) | 0.591 |
| DGLA | −0.03 (−0.251, 0.192) | 0.796 |
| ARA | −0.956 (−1.939, 0.027) | 0.067a |
| EPA | −0.005 (−0.201, 0.191) | 0.958 |
| DHA | 0.102 (−0.145, 0.35) | 0.426 |
| ARA/LA | −0.034 (−0.063, −0.004) | 0.035b |
| ARA/DGLA | −0.255 (−0.956, 0.446) | 0.481 |
| EPA/ALA | −0.161 (−0.567, 0.244) | 0.442 |
| DHA/ALA | 0.136 (−0.201, 0.473) | 0.437 |
| PGE2 | 0.017 (−0.15, 0.183) | 0.847 |
| PGD2c | 0.02 (−0.029, 0.069) | 0.426 |
| LTB4c | −0.195 (−0.303, −0.087) | 0.001d |
| LTE4 | −1.667 (−6.989, 3.655) | 0.544 |
| 5-HETEc | −0.122 (−0.237, −0.007) | 0.048b |
| 12-HETEc | −0.028 (−0.223, 0.166) | 0.778 |
| 15-HETEc | −0.094 (−0.191, 0.003) | 0.068a |
| LTC4c | 0.003 (−0.118, 0.123) | 0.964 |
| LTD4c | 0.064 (−0.101, 0.23) | 0.451 |
| TXB2 | 0.014 (−0.826, 0.853) | 0.975 |
a p < 0.1.
b p < 0.05.
c Trait was transformed for analysis.
d p < 0.01.
FIGURE 3.
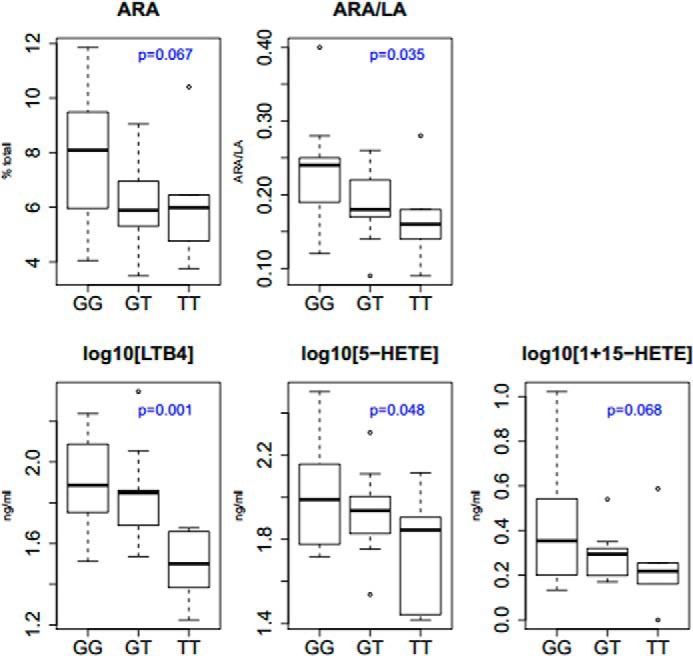
Distribution of fatty acid and eicosanoid levels by genotypes at rs174537. Serum fatty acid ARA and LA (as percent of total fatty acids) and eiconsanoids generated from zymonsan-stimulated whole blood were determined as described under “Experimental Procedures.” The data are grouped genotype at rs174537 and shown as the median. To accommodate zero values in the normalization of the data, a constant of 1 was added to each 15-HETE value generating the final trait for analysis as log10(1 + 15-HETE).
Effect of Genotype at rs174537 on Eicosanoid Production
Under an additive model at rs174537 on eicosanoid profiles of these subjects, it was determined there was a significant association between LTB4 and 5-HETE. Interestingly, the association with LTB4 (p = 0.001) was more significant that that with ARA. Fig. 3 shows levels of LTB4, 5-HETE, and 15-HETE, respectively, as a function of genotype; these quantitative phenotypes were transformed for analysis. The ARA/LA ratio subjects with the GG genotype had significantly higher levels of LTB4 and 5-HETE than those subjects of the GT genotype, and the GT genotype had higher levels than the TT genotype, supportive of the additive allelic effect at this locus. Although not quite reaching significance with a p value of 0.068, 15-HETE displayed a similar trend to LTB4 and 5-HETE. In contrast, none of the cyclooxygenase products examined showed any relationship to the rs174537 genotype (p > 0.1).
Relationship between ARA/LA ratio and Eicosanoid Production
To better understand whether there was a relationship between the capacity to synthesize ARA from LA and the capacity to produce eicosanoids, Pearson correlation coefficients were generated between ARA/LA and levels of LTB4, 5-HETE, and 15-HETE (Fig. 4). A strong positive correlation was observed between ARA/LA and LTB4 (r = 0.441, p = 0.0148), suggesting that the ratio is a strong independent predictor. In contrast, the correlations between the ARA/LA ratio and 5-HETE and 15-HETE levels were not statistically significant (r = 0.267, p = 0.153 and 0.127, p = 0.505, respectively).
FIGURE 4.
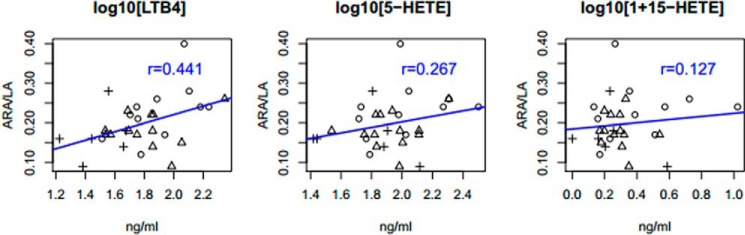
Correlation between ARA/LA ratio and eicosanoid levels. Sample labels indicate genotype of the individual sample at rs174537 (plus sign, TT; triangle, GT; circle, GG). To accommodate zero values in the normalization of the data, a constant of 1 was added to each 15-HETE value generating the final trait for analysis as log10(1 + 15-HETE).
DISCUSSION
There have been numerous studies showing that genetic variation in the FADS cluster is associated with LC-PUFA biosynthesis (12, 27–33). The role of FADS variants in complex lipid and inflammatory phenotypes is also well documented. Genome-wide association studies have identified associations between FADS variants and cardiovascular disease risk factors, including LDL, triglyceride, HDL, and total cholesterol levels (30, 40–42). Recently, five African American cohorts (n ∼ 8,000) were meta-analyzed and confirmed the association of FADS SNPs with lipid phenotypes and CAD (53).
However, there have been no studies to date that have examined potential association between common FADS variants and the enzymatic generation of eicosanoids from the lipoxygenase(s) and cyclooxygenases pathways. Examining such associations is extremely complex as leukotrienes, HETEs, prostaglandins, and thromboxanes are produced locally and rapidly metabolized (44–47). Analyzing eicosanoids after whole blood stimulation was established >20 years ago as a method to examine the capacity of cells in the blood (largely neutrophils) to be activated by zymosan phagocytosis (54). Upon stimulation, ARA is released from membrane phospholipids, and eicosanoids are formed. This assay integrates the mechanism of eicosanoid production, i.e. phospholipase A2 activation and release of free arachidonate, elevation of intracellular Ca2+, translocation of 5-lipoxygenase to the nuclear envelope, association with FLAP, and formation of leukotriene A4. Products measured by LC-MS/MS include TXA2 (measured as TXB2), PGE2, LTB4, and LTC4, along with 5-HETE, 12-HETE, and 15-HETE. Because this whole blood method of zymosan phagocytosis targets neutrophil eicosanoid production, the lack of a major effect on prostaglandin and thromboxane formation should not be overinterpreted. The neutrophil only poorly, if at all, expresses PGH synthase and those prostanoids observed (e.g. TXB2) were likely derived from other cells in the blood similar to the platelets that were activated as a secondary event to polymorphonuclear activation.
The current study demonstrated that there not only was the anticipated association between rs174537 and a key surrogate measure of the capacity to synthesize ARA from LA (ARA/LA ratio) but also the synthesis of 5-lipoxygenase products LTB4 and 5-HETE. The data also demonstrated a strong relationship between an individual's ARA/LA ratio and their capacity to synthesize LTB4, 5-HETE and to a lesser degree 15-HETE. Together, these studies have addressed the key question of whether associations between a key FADS variant and ARA levels extend to the enzymatic production of ARA products, eicosanoids.
In a metabolomics study, Geiger and colleagues demonstrated associations between rs174548 and phospholipids containing fatty acids with four double bonds (i.e. ARA), and this effect was observed for all major phospholipid species (phosphatidylcholine, phosphatidylethanolamine PI, including 1-acyl, 1-alkyl, and 1-alk-1-enyl phospholipids) (40). This SNP is in tight linkage disequilibrium with rs174537 (r2 = 0.81, and D′ = 1 based on the CEU (a cohort of Utah residents of northern and western European ancestry) samples within the Thousand Genomes Project). Moreover, the association with the SNP increased up to 14-fold (p < 10−21) when examining the ratios of putative PUFA-containing precursors and products. For example, the strongest effect size was observed with the ratio of ARA-containing phosphatidylcholine to dihomo-γ-linolenic acid-containing phosphatidylcholine (p = 2.4 × 10−22) with 28.6% of the total variance in the population being explained by one SNP, rs174548 in the FADS cluster. The authors point out that this effect is so strong that “if the molecular function of FADS1 had not been already known, the association between the SNP and the different glycerophospholipid concentrations per se would have allowed one to deduce its enzymatic activity of inserting a fourth double bond.” The aforementioned study extended the influence of FADS SNPs beyond ARA itself but to levels of the ARA-containing phospholipids that are substrates for phospholipases during cell activation. The current study suggests that the impact of loading this pool further impacts the synthesis of eicosanoids and particularly 5-lipoxygenase products in stimulated human blood.
The associations and the effect size between the genotype at rs174537 and LTB4 biosynthesis as well as the correlation between the ARA/LA ratio and LTB4 is strong despite the small sample size of the study. This suggests there may be a strong relationship between these variables. This study raises important questions regarding the potential impact of influential FADS variants in inflammatory responses mediated by 5-lipoxygenase products. These same variants have already been associated with C-reactive protein and enhanced levels of non-enzymatic oxidative products of ARA (urinary 8-epi-prostaglandin F(2α)). Both of these are recognized as independent CAD risk factors (25, 26).
Finally, we have recently demonstrated that African ancestry populations have significantly higher levels of circulating ARA (complexed to glycerolipids) compared with their European counterparts. This is due in large part to the increased frequency of the high ARA-converting genotype (GG) at rs174537 in the former (37, 38). We have also shown that there are identical estimated allelic effects of rs174537 on ARA levels in African Americans and European Americans (37); however, our studies also suggest that there are likely other factors that drive ARA levels even higher in African Americans. Given the strong correlation between ARA/LA ratios and LTB4 levels observed in this study, it will be important in future studies to determine whether African ancestry populations have greater capacity to produce eicosanoids and particularly 5-lipoxygenase products.
Limitations of this study include the small sample size and the fact that blood has to be stimulated ex vivo to measure eicosanoids. Here, we limit our investigation to a single SNP, rs174537. Nonetheless, this SNP is documented to have the strongest association with LC-PUFA levels in genome-wide association approaches and furthermore is within a block of extremely high linkage disequilibrium in European Americans, providing an excellent proxy for variation with the FADS gene cluster (37). However, the combination of a stimulation protocol that has been utilized for >20 years to monitor the capacity of individuals to produce leukotrienes revealed that those with the GG genotype at rs174537 would be more prone to synthesize proinflammatory leukotrienes.
This work was supported, in whole or in part, by National Institutes of Health Grant P50 AT002782. Dr. Chilton is an unpaid consultant for Gene Smart Health and receives no compensation in this role. This information has been disclosed to WFUHS and outside sponsors, as appropriate, and is institutionally managed.
- LA
- linoleic acid
- HETE
- hydroxyeicosatetraenoic acid
- LC-MS/MS
- liquid chromatography-tandem quadrupole mass spectrometry
- ARA
- arachidonic acid
- LTB4
- leukotriene B4
- CAD
- coronary artery disease.
REFERENCES
- 1. Cordain L., Eaton S. B., Sebastian A., Mann N., Lindeberg S., Watkins B. A., O'Keefe J. H., Brand-Miller J. (2005) Origins and evolution of the Western diet: health implications for the 21st century. Am. J. Clin. Nutr. 81, 341–354 [DOI] [PubMed] [Google Scholar]
- 2. Blasbalg T. L., Hibbeln J. R., Ramsden C. E., Majchrzak S. F., Rawlings R. R. (2011) Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am. J. Clin. Nutr. 93, 950–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gerrior S., Bente L., Hiza H. (2004) Nutrient Content of the U. S. Food Supply, 1909–2000 (Home Economics Research Report No. 56), pp. 14–31, U. S. Department of Agriculture, Center for Nutrition Policy and Promotion [Google Scholar]
- 4. Eaton S. (1992) Humans, lipids and evolution. Lipids 27, 814–820 [DOI] [PubMed] [Google Scholar]
- 5. Hibbeln J. R., Nieminen L. R., Blasbalg T. L., Riggs J. A., Lands W. E. (2006) Healthy intakes of n-3 and n-6 fatty acids: estimations considering worldwide diversity. Am. J. Clin. Nutr. 83, 1483S–1493S [DOI] [PubMed] [Google Scholar]
- 6. Park W. J., Kothapalli K. S., Lawrence P., Tyburczy C., Brenna J. T. (2009) An alternate pathway to long-chain polyunsaturates: the FADS2 gene product Delta8-desaturates 20:2n-6 and 20:3n-3. J. Lipid Res. 50, 1195–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Los D. A., Murata N. (1998) Structure and expression of fatty acid desaturases. Biochim. Biophys. Acta 1394, 3–15 [DOI] [PubMed] [Google Scholar]
- 8. Nakamura M. T., Nara T. Y. (2003) Essential fatty acid synthesis and its regulation in mammals. Prostaglandins Leukot. Essent. Fatty Acids 68, 145–150 [DOI] [PubMed] [Google Scholar]
- 9. Sprecher H. (1981) Biochemistry of essential fatty acids. Prog. Lipid Res. 20, 13–22 [DOI] [PubMed] [Google Scholar]
- 10. Cho H. P., Nakamura M. T., Clarke S. D. (1999) Cloning, expression, and nutritional regulation of the mammalian δ-6 desaturase. J. Biol. Chem. 274, 471–477 [DOI] [PubMed] [Google Scholar]
- 11. Cho H. P., Nakamura M., Clarke S. D. (1999) Cloning, expression, and fatty acid regulation of the human δ-5 desaturase. J. Biol. Chem. 274, 37335–37339 [DOI] [PubMed] [Google Scholar]
- 12. Koletzko B., Lattka E., Zeilinger S., Illig T., Steer C. (2011) Genetic variants of the fatty acid desaturase gene cluster predict amounts of red blood cell docosahexaenoic and other polyunsaturated fatty acids in pregnant women: findings from the Avon Longitudinal Study of Parents and Children. Am. J. Clin. Nutr. 93, 211–219 [DOI] [PubMed] [Google Scholar]
- 13. Spector A. A. (2001) Plasma free fatty acid and lipoproteins as sources of polyunsaturated fatty acid for the brain. J. Mol. Neurosci. 16, 159–165 [DOI] [PubMed] [Google Scholar]
- 14. Lands W. E., Libelt B., Morris A., Kramer N. C., Prewitt T. E., Bowen P., Schmeisser D., Davidson M. H., Burns J. H. (1992) Maintenance of lower proportions of (n-6) eicosanoid precursors in phospholipids of human plasma in response to added dietary (n-3) fatty acids. Biochim. Biophys. Acta 1180, 147–162 [DOI] [PubMed] [Google Scholar]
- 15. Lands W., Hart P. (1965) Metabolism of glycerolipids. VI. Specificities of acyl coenzyme A: phospolipid acyltransferases. J. Biol. Chem. 240, 1905–1911 [PubMed] [Google Scholar]
- 16. Chilton F. H., Murphy R. C. (1986) Remodeling of arachidonate-containing phosphoglycerides within the human neutrophil. J. Biol. Chem. 261, 7771–7777 [PubMed] [Google Scholar]
- 17. Astudillo A. M., Balgoma D., Balboa M. A., Balsinde J. (2012) Dynamics of arachidonic acid mobilization by inflammatory cells. Biochim. Biophys. Acta 1821, 249–256 [DOI] [PubMed] [Google Scholar]
- 18. Clark J. D., Lin L. L., Kriz R. W., Ramesha C. S., Sultzman L. A., Lin A. Y., Milona N., Knopf J. L. (1991) A novel arachidonic acid-selective cytosolic PLA2 contains a Ca2+-dependent translocation domain with homology to PKC and GAP. Cell 65, 1043–1051 [DOI] [PubMed] [Google Scholar]
- 19. Chilton F. H., Fonteh A. N., Surette M. E., Triggiani M., Winkler J. D. (1996) Control of arachidonate levels within inflammatory cells. Biochim. Biophys. Acta 1299, 1–15 [DOI] [PubMed] [Google Scholar]
- 20. Balsinde J., Balboa M. A., Insel P. A., Dennis E. A. (1999) Regulation and inhibition of phospholipase A2. Annu. Rev. Pharmacol. Toxicol. 39, 175–189 [DOI] [PubMed] [Google Scholar]
- 21. Balsinde J., Winstead M. V., Dennis E. A. (2002) Phospholipase A(2) regulation of arachidonic acid mobilization. FEBS Lett. 531, 2–6 [DOI] [PubMed] [Google Scholar]
- 22. Spector A. A. (1999) Essentiality of fatty acids. Lipids 34, S1–3 [DOI] [PubMed] [Google Scholar]
- 23. Smith W. L., DeWitt D. L., Garavito R. M. (2000) Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69, 145–182 [DOI] [PubMed] [Google Scholar]
- 24. Buczynski M. W., Dumlao D. S., Dennis E. A. (2009) An integrated omics analysis of eicosanoid biology. J. Lipid Res. 50, 1015–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schwedhelm E., Bartling A., Lenzen H., Tsikas D., Maas R., Brümmer J., Gutzki F. M., Berger J., Frölich J. C., Böger R. H. (2004) Urinary 8-iso-prostaglandin F2α as a risk marker in patients with coronary heart disease: a matched case-control study. Circulation 109, 843–848 [DOI] [PubMed] [Google Scholar]
- 26. Wolfram R., Oguogho A., Palumbo B., Sinzinger H. (2005) Enhanced oxidative stress in coronary heart disease and chronic heart failure as indicated by an increased 8-epi-PGF(2α). Eur. J Heart Fail. 7, 167–172 [DOI] [PubMed] [Google Scholar]
- 27. Schaeffer L., Gohlke H., Müller M., Heid I. M., Palmer L. J., Kompauer I., Demmelmair H., Illig T., Koletzko B., Heinrich J. (2006) Common genetic variants of the FADS1 FADS2 gene cluster and their reconstructed haplotypes are associated with the fatty acid composition in phospholipids. Hum. Mol. Genet. 15, 1745–1756 [DOI] [PubMed] [Google Scholar]
- 28. Baylin A., Ruiz-Narvaez E., Kraft P., Campos H. (2007) α-Linolenic acid, δ6-desaturase gene polymorphism, and the risk of nonfatal myocardial infarction. Am. J. Clin. Nutr. 85, 554–560 [DOI] [PubMed] [Google Scholar]
- 29. Malerba G., Schaeffer L., Xumerle L., Klopp N., Trabetti E., Biscuola M., Cavallari U., Galavotti R., Martinelli N., Guarini P., Girelli D., Olivieri O., Corrocher R., Heinrich J., Pignatti P. F., Illig T. (2008) SNPs of the FADS gene cluster are associated with polyunsaturated fatty acids in a cohort of patients with cardiovascular disease. Lipids 43, 289–299 [DOI] [PubMed] [Google Scholar]
- 30. Martinelli N., Girelli D., Malerba G., Guarini P., Illig T., Trabetti E., Sandri M., Friso S., Pizzolo F., Schaeffer L., Heinrich J., Pignatti P. F., Corrocher R., Olivieri O. (2008) FADS genotypes and desaturase activity estimated by the ratio of arachidonic acid to linoleic acid are associated with inflammation and coronary artery disease. Am. J. Clin. Nutr. 88, 941–949 [DOI] [PubMed] [Google Scholar]
- 31. Rzehak P., Heinrich J., Klopp N., Schaeffer L., Hoff S., Wolfram G., Illig T., Linseisen J. (2009) Evidence for an association between genetic variants of the fatty acid desaturase 1 fatty acid desaturase 2 (FADS1 FADS2) gene cluster and the fatty acid composition of erythrocyte membranes. Br. J. Nutr. 101, 20–26 [DOI] [PubMed] [Google Scholar]
- 32. Xie L., Innis S. M. (2008) Genetic variants of the FADS1 FADS2 gene cluster are associated with altered (n-6) and (n-3) essential fatty acids in plasma and erythrocyte phospholipids in women during pregnancy and in breast milk during lactation. J. Nutr. 138, 2222–2228 [DOI] [PubMed] [Google Scholar]
- 33. Tanaka T., Shen J., Abecasis G. R., Kisialiou A., Ordovas J. M., Guralnik J. M., Singleton A., Bandinelli S., Cherubini A., Arnett D., Tsai M. Y., Ferrucci L. (2009) Genome-wide association study of plasma polyunsaturated fatty acids in the InCHIANTI Study. PLoS Genet. 5, e1000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reardon H. T., Hsieh A. T., Park W. J., Kothapalli K. S., Anthony J. C., Nathanielsz P. W., Brenna J. T. (2013) Dietary long-chain polyunsaturated fatty acids upregulate expression of FADS3 transcripts. Prostaglandins Leukot. Essent. Fatty Acids 88, 15–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reardon H. T., Zhang J., Kothapalli K. S., Kim A. J., Park W. J., Brenna J. T. (2012) Insertion–deletions in a FADS2 intron 1 conserved regulatory locus control expression of fatty acid desaturases 1 and 2 and modulate response to simvastatin. Prostaglandins Leukot. Essent. Fatty Acids 87, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Howard T. D., Mathias R. A., Seeds M. C., Herrington D. M., Hixson J. E., Shimmin L. C., Hawkins G. A., Sellers M., Ainsworth H. C., Sergeant S., Miller L. R., Chilton F. H. (2014) DNA methylation in an enhancer region of the FADS cluster is associated with FADS activity in human liver. PLoS One 9, e97510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mathias R. A., Sergeant S., Ruczinski I., Torgerson D. G., Hugenschmidt C. E., Kubala M., Vaidya D., Suktitipat B., Ziegler J. T., Ivester P., Case D., Yanek L. R., Freedman B. I., Rudock M. E., Barnes K. C., Langefeld C. D., Becker L. C., Bowden D. W., Becker D. M., Chilton F. H. (2011) The impact of FADS genetic variants on ω6 polyunsaturated fatty acid metabolism in African Americans. BMC Genet. 12, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sergeant S., Hugenschmidt C. E., Rudock M. E., Ziegler J. T., Ivester P., Ainsworth H. C., Vaidya D., Case L. D., Langefeld C. D., Freedman B. I., Bowden D. W., Mathias R. A., Chilton F. H. (2012) Differences in arachidonic acid levels and fatty acid desaturase (FADS) gene variants in African Americans and European Americans with diabetes or the metabolic syndrome. Br. J. Nutr. 107, 547–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mathias R. A., Fu W., Akey J. M., Ainsworth H. C., Torgerson D. G., Ruczinski I., Sergeant S., Barnes K. C., Chilton F. H. (2012) Adaptive evolution of the FADS gene cluster within Africa. PLoS One 7, e44926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gieger C., Geistlinger L., Altmaier E., Hrabé de Angelis M., Kronenberg F., Meitinger T., Mewes H. W., Wichmann H. E., Weinberger K. M., Adamski J., Illig T., Suhre K. (2008) Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet. 4, e1000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kathiresan S., Willer C. J., Peloso G. M., Demissie S., Musunuru K., Schadt E. E., Kaplan L., Bennett D., Li Y., Tanaka T., Voight B. F., Bonnycastle L. L., Jackson A. U., Crawford G., Surti A., Guiducci C., Burtt N. P., Parish S., Clarke R., Zelenika D., Kubalanza K. A., Morken M. A., Scott L. J., Stringham H. M., Galan P., Swift A. J., Kuusisto J., Bergman R. N., Sundvall J., Laakso M., Ferrucci L., Scheet P., Sanna S., Uda M., Yang Q., Lunetta K. L., Dupuis J., de Bakker P. I., O'Donnell C. J., Chambers J. C., Kooner J. S., Hercberg S., Meneton P., Lakatta E. G., Scuteri A., Schlessinger D., Tuomilehto J., Collins F. S., Groop L., Altshuler D., Collins R., Lathrop G. M., Melander O., Salomaa V., Peltonen L., Orho-Melander M., Ordovas J. M., Boehnke M., Abecasis G. R., Mohlke K. L., Cupples L. A. (2009) Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 41, 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aulchenko Y. S., Ripatti S., Lindqvist I., Boomsma D., Heid I. M., Pramstaller P. P., Penninx B. W., Janssens A. C., Wilson J. F., Spector T., Martin N. G., Pedersen N. L., Kyvik K. O., Kaprio J., Hofman A., Freimer N. B., Jarvelin M. R., Gyllensten U., Campbell H., Rudan I., Johansson A., Marroni F., Hayward C., Vitart V., Jonasson I., Pattaro C., Wright A., Hastie N., Pichler I., Hicks A. A., Falchi M., Willemsen G., Hottenga J. J., de Geus E. J., Montgomery G. W., Whitfield J., Magnusson P., Saharinen J., Perola M., Silander K., Isaacs A., Sijbrands E. J., Uitterlinden A. G., Witteman J. C., Oostra B. A., Elliott P., Ruokonen A., Sabatti C., Gieger C., Meitinger T., Kronenberg F., Döring A., Wichmann H. E., Smit J. H., McCarthy M. I., van Duijn C. M., Peltonen L. (2009) Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet. 41, 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Park J. Y., Paik J. K., Kim O. Y., Chae J. S., Jang Y., Lee J. H. (2010) Interactions between the APOA5–1131T>C and the FEN1 10154G>T polymorphisms on ω6 polyunsaturated fatty acids in serum phospholipids and coronary artery disease. J. Lipid Res. 51, 3281–3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kumlin M., Granström E. (1986) Radioimmunoassay for 11-dehydro-TXB2: a method for monitoring thromboxane production in vivo. Prostaglandins 32, 741–767 [DOI] [PubMed] [Google Scholar]
- 45. Catella F., Healy D., Lawson J. A., FitzGerald G. A. (1986) 11-Dehydrothromboxane B2: a quantitative index of thromboxane A2 formation in the human circulation. Proc. Natl. Acad. Sci. U.S.A. 83, 5861–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cho H., Huang L., Hamza A., Gao D., Zhan C. G., Tai H. H. (2006) Role of glutamine 148 of human 15-hydroxyprostaglandin dehydrogenase in catalytic oxidation of prostaglandin E2. Bioorg. Med. Chem. 14, 6486–6491 [DOI] [PubMed] [Google Scholar]
- 47. Ensor C. M., Zhang H., Tai H. H. (1998) Purification, cDNA cloning and expression of 15-oxoprostaglandin 13-reductase from pig lung. Biochem. J. 330, 103–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fradin A., Zirrolli J. A., Maclouf J., Vausbinder L., Henson P. M., Murphy R. C. (1989) Platelet-activating factor and leukotriene biosynthesis in whole blood: a model for the study of transcellular arachidonate metabolism. J. Immunol. 143, 3680–3685 [PubMed] [Google Scholar]
- 49. Hui K. P., Taylor I. K., Taylor G. W., Rubin P., Kesterson J., Barnes N. C., Barnes P. J. (1991) Effect of a 5-lipoxygenase inhibitor on leukotriene generation and airway responses after allergen challenge in asthmatic patients. Thorax 46, 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weaver K. L., Ivester P., Seeds M., Case L. D., Arm J. P., Chilton F. H. (2009) Effect of dietary fatty acids on inflammatory gene expression in healthy humans. J. Biol. Chem. 284, 15400–15407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Metcalfe L. D., Schmitz A. A., Pelka J. R. (1966) Rapid preparation of fatty acid esters from lipids for gas chromatographic analysis. Anal. Chem. 38, 514–515 [Google Scholar]
- 52. Gijón M. A., Zarini S., Murphy R. C. (2007) Biosynthesis of eicosanoids and transcellular metabolism of leukotrienes in murine bone marrow cells. J. Lipid Res. 48, 716–725 [DOI] [PubMed] [Google Scholar]
- 53. Lettre G., Palmer C. D., Young T., Ejebe K. G., Allayee H., Benjamin E. J., Bennett F., Bowden D. W., Chakravarti A., Dreisbach A., Farlow D. N., Folsom A. R., Fornage M., Forrester T., Fox E., Haiman C. A., Hartiala J., Harris T. B., Hazen S. L., Heckbert S. R., Henderson B. E., Hirschhorn J. N., Keating B. J., Kritchevsky S. B., Larkin E., Li M., Rudock M. E., McKenzie C. A., Meigs J. B., Meng Y. A., Mosley T. H., Newman A. B., Newton-Cheh C. H., Paltoo D. N., Papanicolaou G. J., Patterson N., Post W. S., Psaty B. M., Qasim A. N., Qu L., Rader D. J., Redline S., Reilly M. P., Reiner A. P., Rich S. S., Rotter J. I., Liu Y., Shrader P., Siscovick D. S., Tang W. H., Taylor H. A., Tracy R. P., Vasan R. S., Waters K. M., Wilks R., Wilson J. G., Fabsitz R. R., Gabriel S. B., Kathiresan S., Boerwinkle E. (2011) Genome-wide association study of coronary heart disease and its risk factors in 8,090 African Americans: the NHLBI CARe Project. PLoS Genet. 7, e1001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bellavite P., Dri P., Della Bianca V., Serra M.C. (1983) The measurement of superoxide anion production by granulocytes In whole blood. A clinical test for the evaluation of phagocyte function and serum opsonic capacity. Eur. J. Clin. Invest. 13, 363–368 [DOI] [PubMed] [Google Scholar]