

Background: MYPT1 is a regulatory subunit of myosin phosphatase.
Results: Deleting MYPT1 in vascular smooth muscle enhances myosin phosphorylation, contractility, and blood pressure.
Conclusion: Genetic evidence shows MYPT1 plays a role in modulating vascular smooth muscle contractility.
Significance: Although MYPT1 is not essential for vascular smooth muscle contractility, it contributes to blood pressure maintenance in vivo through signaling to myosin phosphorylation.
Keywords: Contractile Protein; Myosin; Phosphatase; Smooth Muscle; Vascular Biology; Myosin Light Chain Phosphatase, Myosin Phosphorylation, Vascular Smooth Muscle
Abstract
Myosin light chain phosphatase with its regulatory subunit, myosin phosphatase target subunit 1 (MYPT1) modulates Ca2+-dependent phosphorylation of myosin light chain by myosin light chain kinase, which is essential for smooth muscle contraction. The role of MYPT1 in vascular smooth muscle was investigated in adult MYPT1 smooth muscle specific knock-out mice. MYPT1 deletion enhanced phosphorylation of myosin regulatory light chain and contractile force in isolated mesenteric arteries treated with KCl and various vascular agonists. The contractile responses of arteries from knock-out mice to norepinephrine were inhibited by Rho-associated kinase (ROCK) and protein kinase C inhibitors and were associated with inhibition of phosphorylation of the myosin light chain phosphatase inhibitor CPI-17. Additionally, stimulation of the NO/cGMP/protein kinase G (PKG) signaling pathway still resulted in relaxation of MYPT1-deficient mesenteric arteries, indicating phosphorylation of MYPT1 by PKG is not a major contributor to the relaxation response. Thus, MYPT1 enhances myosin light chain phosphatase activity sufficient for blood pressure maintenance. Rho-associated kinase phosphorylation of CPI-17 plays a significant role in enhancing vascular contractile responses, whereas phosphorylation of MYPT1 in the NO/cGMP/PKG signaling module is not necessary for relaxation.
Introduction
Blood pressure is influenced by multiple factors, including the pumping action of the heart, vascular resistance, and blood volume. Vascular resistance depends on a balance between contraction and relaxation of vascular smooth muscle cells to establish arterial tone (1–4). Depolarization of the cell membrane by external stimuli activates voltage-gated Ca2+ channels, resulting in Ca2+ influx (5, 6). Vascular tone is regulated via G-protein-coupled receptors (GPCRs) acting on two major signaling modules involving the heterotrimeric G proteins Gq/G11 and G12/G13. Gq/G11 mediates the activation of phospholipase C to generate inositol 1,4,5-trisphosphate, which releases Ca2+ from the sarcoplasmic reticulum, leading to Ca2+/calmodulin-dependent activation of myosin light chain kinase (7–9). The G12/G13 proteins couple to Rho guanine nucleotide exchange factor (RhoGEF) proteins to activate RhoA and thereby enhance the Ca2+-dependent contraction via RhoA kinase-dependent inhibition of myosin light chain phosphatase (MLCP)3 (Ca2+-sensitization). Gq/G11-mediated signaling is required for basal vascular tone induced by vasoactive mediators, whereas Gq/G11 and G12/G13 are needed for pathological increases in vascular tone in hypertension (9).
Ca2+/calmodulin-dependent myosin light chain kinase (MLCK) phosphorylates myosin regulatory light chain (RLC) leading to displacement of myosin cross-bridges from the thick filament to cycle on actin filaments for force development and cell shortening (10–13). RLC is dephosphorylated by myosin light chain phosphatase (MLCP), thereby resulting in relaxation. Thus, the extent of RLC phosphorylation depends on the relative activities of MLCK and MLCP.
MLCP is a heterotrimer that consists of a catalytic type 1 phosphatase subunit (PP1cδ), a regulatory subunit, MYPT1, and a 20-KDa subunit (M20) with unknown function (14, 15). MYPT1 enhances the catalytic activity and specificity of PP1cδ toward phosphorylated RLC in its binding to myosin (16). MYPT1 and PP1cδ also bind other proteins. which allow MLCP activity to be regulated through multiple signaling pathways (17–20). Moreover, multiple phosphorylation sites within MYPT1 that regulate PP1cδ activity toward RLC serve as an important platform for relaying signals for contraction or relaxation (21). Phosphorylation of MYPT1 Thr-696 or Thr-853 inhibits PP1cδ activity by an intramolecular mechanism (22). Phosphorylation of both sites is catalyzed by a RhoA-associated kinase, ROCK, whereas other Ca2+-independent kinases phosphorylate Thr-696 constitutively in vascular smooth muscle (23). Phosphorylation at these Thr sites prevents phosphorylation at the adjacent Ser residues (Ser-695 and Ser-852) by cyclic nucleotide-dependent kinase and hence exerts its inhibitory effect on MLCP activity (24), thereby causing Ca2+-sensitization. If Ser-695 and Ser-852 are phosphorylated, phosphorylation of Thr-696 and Thr-853 are inhibited. In addition, MYPT1 changes the conformation of PP1cδ to increase its sensitivity for binding CPI-17 phosphorylated by PKC, thereby further inhibiting MLCP activity (22). Finally, MYPT1 acts as a scaffold for other proteins including Par-4, HSP27, and M-RIP (25). Thus, MYPT1 appears to play a central role in regulating MLCP activity through direct interactions with PP1cδ, its phosphorylation by different kinases, and its actions as a scaffold for other proteins that may affect RLC phosphorylation (8, 11, 14, 25).
Although an important role for modulation of RLC phosphorylation by MYPT1 is implicated by numerous biochemical investigations and studies in intact and permeable smooth muscle fibers, insights into its role in vivo are lacking. A conventional total body knock-out of MYPT1 is embryonic-lethal in mice (26). Using conditional knock-out mice with a specific deletion of MYPT1 in smooth muscle cells, we previously found that the loss of MYPT1 in smooth muscle was not lethal but did alter the contractile phenotypes of gut phasic smooth muscle (27). We have now investigated the role of MYPT1 in tonic vascular smooth muscle contraction and blood pressure maintenance in vivo. The changes in vascular contractile response resulting from MYPT1 deficiency appear to be sufficient to elevate blood pressure in vivo.
EXPERIMENTAL PROCEDURES
Establishment of MYPT1SMKO Animals
The establishment of MYPT1 conditional knock-out mice is described in our previous report (27). Briefly, two loxP sites were inserted at each end of the first exon of the Mypt1 gene, and the resultant Mypt1flox/flox mice were crossed with transgenic mice with Cre expression driven by a smooth muscle α-actin promoter (Mypt1flox/flox: SMA-Cre mice, MYPT1SMKO) (28). The littermates of the Mypt1flox/flox/+:SMA-Cre mice were used as a control (both male and female mice of 4∼60 weeks old). All mice were specific pathogen-free and maintained in compliance with the Guidelines of the Care and Use of Laboratory Animals in the Model Animal Research Center of Nanjing University.
Blood Pressure Measurements
The systolic, diastolic, and mean blood pressures of conscious adult mice were recorded using a tail-cuff system (ALC-Non-invasive Blood Pressure System, Shanghai Alcott Biotech, China) as previously reported (13). Mice were trained by repeated blood measurement for 7 days before measurements were recorded. Mice were kept in a prewarmed box at 32 °C and pressure-measured for 20 min at the same time every day. Data were calculated as the mean value of three sequential days.
Western Blot Analysis
Proteins were extracted from isolated arteries fixed in trichloroacetic acid. Briefly, the arteries were frozen quickly by immersion in 10% trichloroacetic acid and 10 mm dithiothreitol in acetone cooled in liquid nitrogen to −78 °C. After homogenizing in 10% trichloroacetic acid and 10 mm dithiothreitol in H2O, the protein pellets were collected by centrifugation and then washed 3 times with ether. The resultant dried protein was dissolved in sample buffer, boiled for 4 min, and then resolved by SDS-polyacrylamide gel electrophoresis. The blotted membrane was probed with specific primary antibodies (anti-MYPT1 (1:1,000), anti-MLCK (1:10,000), anti-CPI-17 (1:5,000), anti-ROCKII (1:1,000), or anti-PP1cδ (1:500)), and the appropriate horseradish peroxidase-conjugated secondary antibodies and then visualized with enhanced chemiluminescence (ECL, Pierce).
RNA Extraction and RT-PCR
Total RNA was extracted from vessels and reverse-transcribed with PrimeScript RT reagent kit (Takara). The following primers were used for RT-PCR: MBS85 forward (ATCTGGACGAGGCTCGCCTGAT) and reverse (TCCCCGTCACTGTTGACAGCAG), PCR product size 322 bp; MYPT2 forward (AGACGGTGCGGTCTTTCTAGCT) and reverse (CCTCTGCAAGGTCAGAAGGAAC), PCR product size 326 bp; MYPT3 forward (AGCAAGAAGGGCCATGGGGA) and reverse (AATCCAGCGTCTGTGCGTCC), PCR product size 418 bp; β-actin forward (5-CACCCTGTGCTGCTCACC-3) and reverse (5-GCACGATTTCCCTCTCAG-3), PCR product size 328 bp.
Histological Analysis and Immunofluorescent Staining
Tissues were fixed with 4% paraformaldehyde overnight and then embedded in paraffin for histological analyses. Transverse sections (5 μm) were stained with hematoxylin/eosin and examined under a microscope. For the immunofluorescence analysis, fresh arteries were embedded with OCT (Leica), and sections were blocked with phosphate-buffered saline containing 0.1% Triton X-100, 0.1% Tween 20, 1% bovine serum albumin, and 5% non-immune goat serum for 1 h at room temperature. Primary antibodies (anti-MYPT1 (1:200), Upstate Biotechnology; anti-smooth muscle α-actin (1:200), Thermo Scientific) and fluorescent secondary antibodies (Invitrogen) were used. The fluorescent staining was examined with a confocal microscope (Olympus).
Echocardiography
Echocardiographic parameters were collected using a Micro-Echocardiography system (Vevo 770TM, Visual Sonics, Toronto, ON, Canada). Mice were anesthetized with avertin (250 mg/kg, intraperitoneal injection) and maintained on a platform at 37 °C. Transthoracic M-mode echocardiography was performed with a 30-MHz RMV-707B scanning head.
Measurement of Plasma Renin, Angiotensin II, and Aldosterone
Plasma concentrations of renin, angiotensin II, and aldosterone were assayed with an ELISA kit (angiotensin II and aldosterone ELISA kits were from Enzo Life Sciences; mouse renin ELISA kit were from Xinqidi Biological Technology) and performed according to the protocols provided by the manufacturers. Plasma samples were collected, and EDTA was used as an anticoagulant. After centrifugation at 1000 × g for 15 min at 4 °C, the supernatant was aliquoted and stored at −80 °C before the measurements were performed.
Myography Assay for Vessel Contractility
The secondary branch of the mesenteric artery was isolated from adventitial tissues and mounted for isometric tension measurements (610-M, Danish Myo Technology) as previously reported (13). After equilibrating in pre-oxygenated HEPES-Tyrode buffer (137 mm NaCl, 2.7 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5.6 mm glucose, and 10 mm HEPES, pH 7.4) at 37 °C for 30 min, the resting tension was adjusted to a value comparable to 100 mm Hg pressure in vivo. The artery segments were then equilibrated for 20 min before testing contractile capacity by exposure to 124 mm KCl-containing buffer (15.7 mm NaCl, 124 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5.6 mm glucose, and 10 mm HEPES, pH 7.4). The contractile ability of the vessels was then determined by exposure to KCl and norepinephrine.
Measurement of Myosin Light Chain Phosphorylation
Arterial strips were frozen by immersion in 10% trichloroacetic acid in acetone chilled to a slurry in liquid nitrogen at the indicated time points. After thawing in trichloroacetic acid/acetone to room temperature, the samples were transferred to 10% trichloroacetic acid in H2O and homogenized. The extracted proteins were resolved with urea-glycerol PAGE followed by membrane transfer. Western blotting analysis was performed with a monoclonal antibody against myosin light chain (29). The percentage of phosphorylated myosin light chain relative to the total amount of myosin light chain was calculated as described previously (13).
Chemicals and Antibodies
Norepinephrine, angiotensin II, U46619, Arg-vasopressin, sodium nitroprusside (SNP), HA1077, acetylcholine, and Gö6976 were purchased from Sigma; endothelin-1, Y27632, and H1152 were purchased from Calbiochem. Calyculin A and GF109203X were purchased from Tocris. ECL was purchased from Thermo Scientific. Antibodies against MYPT1 and PP1cδ were obtained from Millipore, p-CPI-17 (phosphorylated CPI-17) was from Bio-world, MLCK (K36), β-actin was from Sigma, ROCKII was from Santa Cruz Biotechnology, PKGIα was from Cayman Chemical, and PKGIβ was from Stressgen Bioregeagents. Antibody against CPI-17 was previously described (30).
Statistics
Data are presented as the mean ± S.E. Statistics was analyzed with GraphPad Prism 5.0. Student's t test was used between two groups data analysis. For multiple groups, significance was analyzed with two-way analysis of variance analysis followed by the Bonferroni post test. Significant differences between groups were considered when p < 0.05.
RESULTS
Ablated Expression of MYPT1 in Vascular Smooth Muscle
We disrupted the Mypt1 gene specifically in smooth muscle cells by crossing Mypt1-floxed mice (Mypt1flox/flox) with SMA-Cre transgenic mice (28). To examine the knock-out efficiency of Mypt1 in vascular smooth muscle from the resultant mice (Mypt1flox/flox: SMA-Cre, MYPT1SMKO), we estimated the amount of MYPT1 protein via Western blot analysis and immunofluorescence. Western blotting showed either a trace or undetectable amounts of MYPT1 protein in mesenteric, femoral, and aortic smooth muscle tissues from MYPT1SMKO mice (Fig. 1A). Immunofluorescence analyses showed no obvious staining for MYPT1 within the smooth muscle layer of tissues from knock-out animals (Fig. 1B). We also analyzed the expression of MLCK, ROCKII, CPI-17, PKGIα/β, and PP1cδ in smooth muscles from MYPT1SMKO and control mice. For MLCK, ROCKII CPI-17, and PKGIα/β no significant differences in expressed amounts were observed between the MYPT1SMKO and control groups (Fig. 1, C and E). We also measured the expression pattern of structural proteins by SDS-PAGE as we reported previously (27), and no difference among the proteins was observed (data not shown). However, PP1cδ expression in the MYPT1-deficient arteries was reduced ∼50% compared with the control vessels (Fig. 1, D and E). The reduction in PP1cδ is likely due to the cellular degradation of unbound PP1cδ compared with PP1cδ in complex with MYPT1 (31). We examined the expression of MYPT2, MYPT3, and MBS85 in the knock-out vascular smooth muscle by RT-PCR and found no significant MYPT2 and MYPT3 expression in mutant or control smooth muscles (Fig. 2). A weak expression of MBS85 could be detected but was similar in both mutant and control smooth muscles. Thus, these three MYPT family members are unlikely to compensate for the loss of MYPT1.
FIGURE 1.

MYPT1 was deleted in vascular smooth muscles from MYPT1SMKO mice. A, Western blot of MYPT1 in different vessels from MYPT1SMKO and CTR mice (8–10 weeks old), including the aorta, mesenteric artery, and femoral artery with β-actin loading control. B, immunofluorescence staining of mesenteric arteries. Smooth muscle α-actin (SMA, red) was stained as smooth muscle-specific marker. MYPT1 (green) was detected in the CTR smooth muscle layer and endothelium but only in the endothelium in MYPT1SMKO vessels. C, expression of other contractile proteins in MYPT1SMKO vascular smooth muscle. Western blots of MLCK, CPI-17, and ROCKII PKGIα and PKGIβ from mesenteric arteries showed no significant differences between MYPT1SMKO and CTR. D, measurement of PP1cδ expression in MYPT1SMKO and CTR mesenteric arteries by Western blotting. E, relative amounts of MLCK (n = 4), CPI-17 (n = 3), ROCKII (CTR n = 4, MYPT1SMKO n = 5) PKGIα (n = 3), PKGIβ (n = 4), and PP1cδ (CTR n = 4, MYPT1SMKO n = 5) in mesenteric arteries are shown.
FIGURE 2.
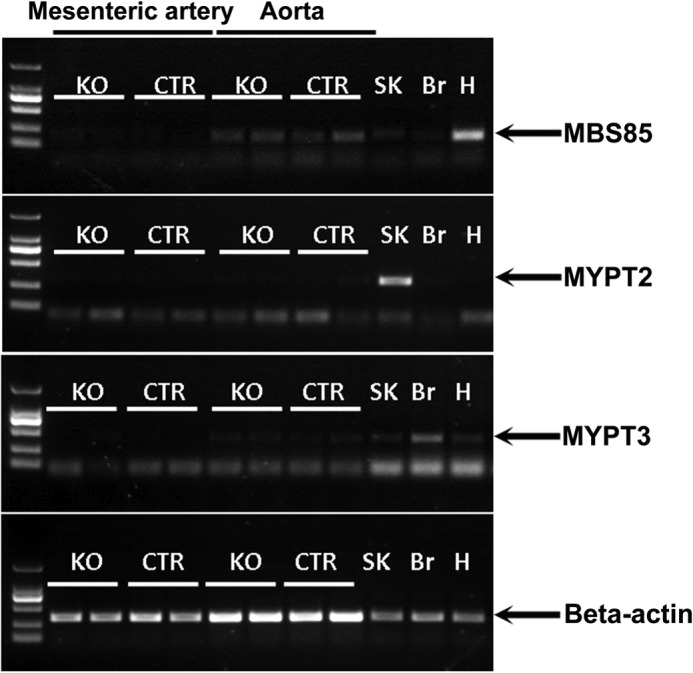
Expression of MYPT family members in vascular smooth muscles. Fresh mesenteric artery and aorta were isolated from MYPT1SMKO and CTR mice (8–10 weeks old). The total RNA extracted from these tissues was subjected to RT-PCR assay for 30 reaction cycles. β-Actin mRNA was used for internal control and mRNA from skeletal muscle (SK), brain (Br), heart (H) was used for a positive control. The MBS85 panel showed weak signals, whereas no signal showed in MYPT2 and MYPT3 panels of vascular smooth muscle. Nonspecific bands are present in MYPT2 and MYPT3 measurements, which migrate differently from expected products marked by arrows.
MYPT1 Knock-out Mice Displayed Permanent Hypertension
We measured the blood pressure of mice at different ages. The systolic arterial pressure of 8–48-week-old MYPT1SMKO mice was 138 ± 2.18 mm Hg (n = 14), which was significantly greater than that of control mice (114 ± 2.31 mm Hg, n = 14, p < 0.001, Fig. 3A); mean arterial pressure of the knock-out mice was also significantly greater than that of the control mice (115 ± 2.03 versus 95 ± 1.94 mm Hg; ***, p < 0.001, n = 14). Elevated blood pressures were detected as early as 1 month postnatal and lasted up to 15 months of age (Fig. 3B).
FIGURE 3.

Blood pressure of MYPT1SMKO mice was significantly increased without changes in vascular structure. A, blood pressure recordings of conscious MYPT1SMKO and control adult mice (8–48 weeks old). Systolic, diastolic, and mean blood pressures (SBP, DBP, MBP) were significantly elevated in MYPT1SMKO mice (***, p < 0.001, n = 14). B, systolic blood pressure in MYPT1SMKO and CTR mice at different ages. The systolic blood pressure of MYPT1SMKO mice was increased in 1-month-old mice (137 ± 2.9 mm Hg (MYPT1SMKO) versus 109.9 ± 7.3 mm Hg (CTR), ***, p < 0.001, n = 3) and maintained up to 15 months of age (128.4 mm Hg (MYPT1SMKO) versus 105.7 (CTR), **, p < 0.01, n = 3). C, histology of aortic and mesenteric arteries. Tissue slices of the arteries were stained with hematoxylin and eosin. The results showed apparently normal vascular morphology of the MYPT1SMKO blood vessels compared with CTR vessels. Scale bar, 100 μm.
Because the increased blood pressure might be affected by multiple factors, we examined cardiac and renal functions in addition to vascular properties of MYPT1SMKO mice. The heart rate of the KO mice (528 ± 11 beats per min) was comparable with the CTR heart rate (558 ± 21 beats per min, p > 0.05). Cardiac performance measures, including fractional shortening, ejection fraction, and ventricular volume, were also comparable in the MYPT1SMKO mice to values obtained with CTR mice (Table 1). The concentrations of renin, angiotensin II, and aldosterone in the peripheral blood were not altered in MYPT1SMKO mice (renin: CTR 19.41 ± 0.93 pg/ml, n = 6; MYPT1SMKO, 19.02 ± 1.91 pg/ml, n = 6, p > 0.01; angiotensin II (Ang II): CTR 79.61 ± 10.21 pg/ml; MYPT1SMKO, 86.33 ± 8.71 pg/ml, n = 12, p > 0.01; aldosterone: CTR 145.99 ± 20.54 pg/ml; MYPT1SMKO, 141.61 ± 24.14 pg/ml, n = 12, p > 0.01). Histological examination of MYPT1-deficient mesenteric vessels showed no gross changes in vessel diameter or thickness or in the organization of the smooth muscle layer (Fig. 3C). To assess arterial compliance in the MYPT1SMKO mice, we compared the circumference corresponding to a transmural pressure of 100-mm Hg using a previously described method (13). IC100 values for mesenteric arteries (n = 9) were comparable between the MYPT1SMKO and CTR vessels (362 ± 20 μm versus 361 ± 14 μm, p > 0.05). Collectively, our results suggest that hypertension in the MYPT1SMKO mice is unlikely attributable to alterations of cardiac and renal function or vascular structural properties.
TABLE 1.
Echocardiography parameters of MYPT1SMKO mice
Electrocardiography was performed as described under “Experimental Procedures.” Data are presented as the mean ± S.E. LV Vol;d, diastolic left ventricular volume; LV Vol,;s, systolic left ventricular volume; % EF, percent ejection fraction; % FS, percent fractional shortening; V mass, left ventricular mass. NS, no significant difference, p > 0.05; n, at least 11 values for each measurement.
| Cardiac parameters | KO | CTR | Summary |
|---|---|---|---|
| LV Vol;d (μl) | 45.3 ± 3.9 | 45.7 ± 2.5 | NS |
| LV Vol;s (μl) | 16.4 ± 2.5 | 14.8 ± 1.3 | NS |
| % EF (% ejection fraction) | 65.3 ± 2.9 | 67.8 ± 2.2 | NS |
| % FS (% fractional shortening) | 35.4 ± 2.1 | 37.3 ± 1.8 | NS |
| LV mass(mg) | 83.6 ± 4.8 | 73.4 ± 3.7 | NS |
Deletion of MYPT1 Enhanced Contractile Responses of Vascular Smooth Muscle
The contractile responses of mesenteric artery segments from MYPT1SMKO mice to different stimuli were modified compared with control tissues. Upon stimulation with 124 mm KCl, mesenteric smooth muscle from CTR mice produced a rapid and robust contraction followed by a lower sustained force (Fig. 4A). With deletion of MYPT1, segments developed a significantly greater initial force (4.27 ± 0.25 mN (KO) versus 3.33 ± 0.21 mN (CTR); p < 0.05) and a greater sustained force (3.98 ± 0.33 mN (KO) versus 1.92 ± 0.21 mN (CTR); p < 0.05; Fig. 4, A′ and H), indicating the functional involvement of MYPT1 in both phases of contraction. Similarly, upon stimulation with 10 μm norepinephrine, mesenteric arteries from MYPT1SMKO mice developed greater maximal force (4.47 ± 0.64 mN (nitric oxide (NO)) versus 2.38 ± 0.26 mN (CTR), p < 0.05; Fig. 4B, B′). We further examined the concentration-response effect of norepinephrine on force development (Fig. 4J). Both the control and mutant arteries initially developed force at 1 μm norepinephrine that increased up to 10 μm norepinephrine. At 10, 30, and 100 μm norepinephrine, the corresponding forces developed by MYPT1-deficient arteries were significantly greater than the control arteries (all p < 0.05; Fig. 4J). The norepinephrine concentration required for 50% of the maximal force (E50) was 1.97 ± 0.65 and 3.80 ± 0.76 μm for arteries from MYPT1SMKO and CTR mice, respectively. We also measured the contractile responses of mesenteric artery segments to other vasoconstrictors. Upon the addition of endothelin, Ang II, U46619 (a thromboxane analog) and Arg-vasopressin (Vas), all of the treated arteries from MYPT1SMKO mice developed significantly greater forces compared with the control arteries (endothelin 1: 4.55 ± 0.36 versus 2.25 ± 0.12 mN, p < 0.01; Ang II: 3.04 ± 0.38 versus 1.95 ± 0.12 mN, p < 0.05; U46619: 5.46 ± 0.62 versus 2.52 ± 0.31 mN, p < 0.01; Vas: 4.74 ± 0.37 versus 2.75 ± 0.40 mN, p < 0.01; Fig. 4, C–F and I). Therefore, we conclude that deletion of MYPT1 enhanced the contraction of vascular smooth muscle in response to depolarization and various vasoconstrictors. Signaling through MYPT1 appears to be shared by many vasoconstrictors in G-protein-coupled receptor-mediated contraction. The enhanced contractile response to vasoconstrictors might contribute to the development of hypertension in MYPT1SMKO mice.
FIGURE 4.

Vasoconstrictor responses of MYPT1SMKO mesenteric arteries. Representative force responses of CTR and MYPT1SMKO mesenteric arteries (8–10 weeks old) to 124 mm KCl (A and A′), 10 μm NE (B and B′), 0.1 μm endothelin 1 (ET1; C and C′), 10 μm Ang II (D and D′), 1 μm Vas (E and E′), and 0.1 μm U46619 are shown. H, quantified initial and sustained force responses to KCl stimulation of vessels from CTR and MYPT1SMKO mice (*, p < 0.05; ***, p < 0.001, n = 12). I, quantified peak force development responses for different vasoconstrictors showing significant differences between vessels from CTR and MYPT1SMKO mice (NE: n = 12; ***, p < 0.001; endothelin 1: n = 7; **, p < 0.01; Ang II: n = 7; Vas, n = 9; **, p < 0.01; U46619: n = 8, ***, p < 0.001). J, cumulative dose responses to NE relative to responses to 124 mm KCl for mesenteric arteries from CTR and MYPT1SMKO mice (*, p < 0.05, n = 7).
Deletion of MYPT1 Enhanced Myosin Light Chain Phosphorylation Responses of Vascular Smooth Muscle
Myosin light chain phosphorylation of vascular smooth muscle in response to depolarization and agonist stimulation was measured in mesenteric arteries (Fig. 5). With KCl stimulation, RLC phosphorylation in MYPT1-deficient arteries reached 49 ± 16% at 10 s and remained elevated (43 ± 6% at 60 s and 31 ± 6% at 300 s; Fig. 5, A and B). RLC phosphorylation in the control arteries also reached maximal values at 10 s (53 ± 9%) but rapidly declined subsequently (15 ± 5% at 60 s and 9 ± 5% at 300 s; Fig. 5, A and B). These results are consistent with the marked increase in sustained force development in mesenteric arteries from MYPT1SMKO mice. With norepinephrine stimulation for 10 s, RLC phosphorylation increased to 63 ± 9 and 49 ± 5% in MYPT1-deficient and CTR mesenteric arteries, respectively (Fig. 5, C and D). RLC phosphorylation in MYPT1-deficient vessels was greater than CTR values at 60 s, and both then decreased to 27 ± 8% at 300 s (Fig. 5, C and D). Thus, deletion of MYPT1 results in enhanced RLC phosphorylation with depolarization- and agonist-induced contractions.
FIGURE 5.

RLC phosphorylation is enhanced in mesenteric arteries from MYPT1SMKO mice. A and C, representative Western blots after urea/glycerol PAGE for vessels from MYPT1SMKO and CTR mice (8–10 weeks old) treated for 300 s with 124 mm KCl (A) or 10 μm NE (C). Nonphosphorylated (RLC) and phosphorylated (RLC-p) RLCs are identified. B and D, temporal responses for RLC phosphorylation to KCl (B) and NE (C) treatments. **, p < 0.01 compared with CTR tissue responses (KCl, n = 4; NE, n = 6).
Signaling to MLCP in the Vascular Smooth Muscle Lacking MYPT1
Inactivation of G12-G13/Rho/ROCK signaling by ROCK inhibitors causes disinhibition of MLCP activity, resulting in reduced RLC phosphorylation and force development (23). Y27632, an inhibitor of ROCK, inhibits either the KCl- or norepinephrine-induced contraction of control arteries (Fig. 6). Upon pretreatment with Y27632, MYPT1-deficient mesenteric arteries developed a normal initial response induced by KCl, but the sustained force was greatly inhibited in a manner similar to that which affected the control muscle (Fig. 6, A–C). Upon treatment with Y27632 during the sustained contraction, the control arteries almost completely relaxed when contracted in response to KCl (Fig. 6D) or norepinephrine (Fig. 6G) treatment. Adding Y27632 to MYPT1-deficient arteries also inhibited sustained contractile responses induced by KCl (Fig. 6E) or norepinephrine (Fig. 6H). The norepinephrine-induced force response of MYPT1-deficient arteries was as sensitive to Y27632 as the responses in control muscles (IC50 of 2.51 ± 0.91 μm (KO) versus 1.81 ± 0.62 μm (CTR), p > 0.05; Fig. 6I). The KCl-induced force responses for the MYPT1-deficient muscles also appeared similarly sensitive to Y27632 compared with the control at the higher concentration of Y27632 (Fig. 6F). To further analyze the effect of ROCK on MYPT1-deficient mesenteric arteries, we examined the effects of other ROCK-specific inhibitors, HA1077 and H1152 (11). No differences in sensitivity to these inhibitors were observed between arteries from CTR and MYPT1SMKO mice (data not shown). These results collectively showed that the loss of MYPT1 was not sufficient to prevent inhibition of vascular smooth muscle contraction by inhibition of ROCK activity.
FIGURE 6.

ROCK and PKC inhibitors decrease force responses of MYPT1SMKO mesenteric arteries. A and B, mesenteric arteries from the mice (8–10 weeks old) were pretreated with the ROCK inhibitor Y27632 and then stimulated with 124 mm KCl. C, quantified results of A and B (n = 4 ns: p > 0.05; *, p < 0.05). D and E, mesenteric arteries were precontracted with 124 mm KCl followed by cumulative increases in the concentration of Y27632 (0.1–10 μm) or vehicle (n = 7). F, quantified force responses for KCl contracted tissues treated with Y27632. Relative ratios of the relaxed force in steady state are expressed as the percent of the force development to KCl at the same time point of vehicle (relaxation % = (Fvehicle − Finhibitor)/Fvehicle, where F = force) (n = 7). G and H, mesenteric arteries from the mice (8–10 weeks old) were precontracted with 10 μm NE followed by cumulative increases in the concentration of ROCK inhibitor Y27632 (0.1–10 μm) or vehicle n = 7). I, quantified force responses for NE-contracted tissues treated with Y27632. Relative ratio of sustained force is expressed as the percent of the peak force to NE (n = 7). J and M, mesenteric arteries were precontracted with 124 mm KCl or 10 μm NE followed by cumulative increases in the concentration of the PKC inhibitors GF109203X (n = 3) and Gö6976 (n = 3).
Inactivation of PKC signaling by PKC inhibitors also results in disinhibition of MLCP activity by preventing CPI-17 phosphorylation (11). To assess the effect of PKC inhibitors on vascular smooth muscle force, we added Gö6976 and GF109203X to KCl- and NE-stimulated segments, respectively. The norepinephrine/KCl-induced contraction of MYPT1-deficient arteries was as sensitive as to Gö6976 and GF109203X as control muscle (Fig. 6, J–M).
To assess the role of MYPT1 in endothelium-dependent relaxation, a critical process in regulating vessel tone, we examined the effects of acetylcholine and nitric oxide on relaxing MYPT1-deficient arteries. In the control arteries, acetylcholine relaxed KCl-induced force with a similar reduction in force in the knock-out arteries (Fig. 7, A and B), showing a similar sensitivity to acetylcholine (ACh; Fig. 7C). Thus, MYPT1-deficient mesenteric segments were as sensitive to relaxation to acetylcholine as control segments. Because NO is a downstream product of acetylcholine signaling, we also examined the effect of the NO donor, SNP, on the relaxation of KCl-induced contraction, and a similar conclusion was obtained (Fig. 7, G–I). We next examined the relaxation of NE-induced contraction in mesenteric arteries after treatment with ACh and SNP. The norepinephrine-induced forces in arteries from MYPT1SMKO and control mice were relaxed by acetylcholine with similar concentration dependence (Fig. 7, D–F). Surprisingly, the deletion of MYPT1 did affect SNP-mediated relaxation of norepinephrine-induced force.
FIGURE 7.

Nitric oxide signaling relaxes mesenteric arteries from MYPT1SMKO mice. A and B, relaxation responses to the cumulative addition of ACh or vehicle for mesenteric arteries from 8–10 week-old-mice with intact endothelium contracted with 124 mm KCl (n = 4) (A and B). C, concentration dependence of relaxation responses to ACh with KCl-induced contraction is shown (n = 4). (Relaxation % = (relaxation % = (Fvehicle − Finhibitor)/Fvehicle, where F = force). D and E, relaxation responses to the cumulative addition of ACh or vehicle for mesenteric arteries with intact endothelium contracted with 10 μm NE (n = 6). F, concentration dependence of relaxation responses to ACh with NE-induced contractions are shown (n = 6). G and H, representative concentration-dependent relaxation responses to the cumulative addition of SNP or vehicle for mesenteric arteries contracted with 124 mm KCl (n = 8). I, concentration dependence of relaxation responses to SNP with KCl-induced contraction is shown. J and K, relaxation responses to the cumulative addition of SNP or vehicle for mesenteric arteries contracted with 10 μm NE. L, concentration dependence of relaxation responses to SNP with NE-induced contraction are shown (n = 4).
MLCP Activity Is Regulated in Mesenteric Segments from MYPT1SMKO Mice
The minimal effect of MYPT1 deletion on inhibition of norepinephrine contractile responses to ROCK inhibitors and NO generators led us to consider the existence of redundant regulation of MLCP activity. Because vascular smooth muscles predominantly express CPI-17, which inhibits PP1cδ activity after phosphorylation by PKC or ROCK, we hypothesized that CPI-17 phosphorylation might inhibit PP1cδ activity and, hence, relax vessels in the absence of MYPT1. To test this hypothesis, we pretreated intact mesenteric segments with the PP1c inhibitor calyculin A and measured the relaxation effect of H1152. In control segments, pretreatment with calyculin A attenuated H1152-mediated inhibition of the contractile response (Fig. 8A). In MYPT1-deficient segments pretreated with calyculin A, the attenuation of the H1152-mediated relaxation effect was greater compared with CTR (Fig. 8, B and C). Upon stimulation with norepinephrine, CPI-17 phosphorylation in the MYPT1-deficient smooth muscle was reduced compared with the control vessels, whereas the total amount of CPI-17 was unchanged (Fig. 8, D–F). Y27632 inhibited CPI-17 phosphorylation in response to norepinephrine treatment in both MYPT1-deficient and CTR mesenteric arteries. Collectively, the results show that ROCK acting on CPI-17 may regulate MLCP activity in vascular smooth muscle.
FIGURE 8.

Regulation of CPI-17 phosphorylation in mesenteric arteries from MYPT1SMKO mice. A and B, relaxation effects of the ROCK inhibitor H1152 before and after inhibition of PP1cδ by calyculin A treatment. After force development with 10 μm NE, mesenteric arteries from CTR (A) and MYPT1SMKO (B) mice from 8–10-week-old mice were treated with cumulative concentrations of H1152 (0.01–0.10 μm) in the absence of calyculin A (CLA). The vessels were washed and then incubated with 10 nm CLA for 30 min. The tissues were treated again with 10 μm NE + 10 nm CLA to induce force development followed by treatment with increasing concentrations of H1152 (right). The inhibitory effect of H1152 was attenuated after CLA treatment. C, quantified results for responses shown in A and B (MYPT1SMKO, n = 3; CTR, n = 4; MYPT1SMKO CLA, n = 3; CTR CLA, n = 5). D, measurement of p-CPI-17 before and after treating mesenteric arteries with 10 μm NE for 60 s in MYPT1SMKO and CTR mesenteric arteries. E, inhibition of CPI-17 phosphorylation in mesenteric arteries by 10 μm Y27632. Note that this panel shows enhanced signal of p-CPI-17 by longer exposure so as to make a weak band clear. F, quantified results for responses shown in C and D. p-CPI-17 was expressed as fraction of p-CPI-17 level in CTR mesenteric arteries treated with NE for 60 s. CPI-17 phosphorylation of NE-treated MYPT1SMKO arteries (KO+NE) was 48 ± 17% of that of CTR arteries (CTR+NE) (n = 8). Y27632 decreased p-CPI-17 in both KO and CTR arteries (KO+NE+Y27632, 3 ± 1% (n = 3); CTR+NE+Y27632, 27 ± 4%, n = 3).
DISCUSSION
Fundamental to blood pressure maintenance are cardiac output, circulating volume, and vascular tone. The latter is actively regulated by contraction and relaxation of vascular smooth muscle cells in response to mechanical stresses and vasoactive signals from sympathetic nerves, endocrine organs, and parenchymal cells. As a focal point for smooth muscle contractile signaling, RLC phosphorylation is regulated by multiple pathways involving MLCK activity regulated by Ca2+/calmodulin and phosphorylation (10). MLCP activity is regulated by MYPT1 and CPI-17 phosphorylation (11, 14, 15, 25). Interacting temporal and spatial signaling responses basically control the ratio of MLCK to MLCP activities, thereby physiologically fine-tuning the extent of RLC phosphorylation and arterial tone.
Smooth muscle-specific knock-out of MLCK is lethal and results in general smooth muscle failure with markedly impaired contractile responses in ileal (32), airway, vascular (13, 33), and bladder (34) (30) smooth muscle tissues. In contrast smooth muscle-specific knock-out of MYPT1 is not lethal (27) but does lead to measurable changes in contractile properties of smooth muscle tissues with in vivo consequences as with hypertension described herein. These results support the conclusion that whereas MYPT1 is not an essential regulatory effector for myosin dephosphorylation, it serves a modulatory role.
Biochemically, MYPT1 is a primary PP1cδ-binding protein in smooth muscle where it regulates the activity of MLCP holoenzyme by localizing the catalytic subunit to myosin filaments (35). In MYPT1-deficient smooth muscles from mesenteric artery or intestine, 50 or 100% PP1cδ protein remained, respectively, and RLC was dephosphorylated after treatments that increase its phosphorylation (27). Myosin targeting subunits normally expressed in other non-smooth muscle cells and tissues were not abundant and did not compensate by increased expression in MYPT1-deficient smooth muscles. Thus, MYPT1 is not essential for phosphatase activity directed toward phosphorylated RLC. We, therefore, suggest that the remaining PP1cδ is sufficient to dephosphorylate phosphorylated RLC, although the activity is less than in the holoenzyme (36).
Using a smooth muscle-specific knock-out of MYPT1 in mice, we found that its deletion enhanced RLC phosphorylation and contractility of isolated vascular smooth muscle in response to various vasoactive stimuli and resulted in hypertension. These results are consistent with a decrease in MLCP activity due to the loss of MYPT1 and the partial decline in PP1cδ protein (15, 25, 37). The smooth muscle knock-out of MYPT1 in mice led to no apparent changes in cardiac performance or blood volume regulating hormones so the increased blood pressure may result from the enhanced arterial contractile responses.
In response to KCl depolarization and norepinephrine, vascular smooth muscle produces an initial rapid and then a sustained contraction (38–40). The signaling mechanisms for this contraction involve a rapid activation of MLCK by Ca2+/calmodulin for rapid RLC phosphorylation followed by recruitment of Ca2+-sensitization mechanisms for RLC phosphorylation. In general, Ca2+-sensitization mechanisms may be mediated by G12-G13/RhoA/ROCK inhibition of MLCP activity by MYPT1 and CPI-17 phosphorylation (9, 22, 23). The relative physiological significance of these two distinct regulatory pathways in smooth muscle is not completely clear, although published reports indicate that phosphorylation of these proteins fine-tune contractile responses in different smooth muscle tissues in which MYPT1 and CPI-17 are differentially expressed (23, 41). The inhibition of MLCP activity by ROCK-mediated MYPT1 phosphorylation is thought to play a key role in Ca2+-sensitized contractions in different kinds of smooth muscles, including different vascular smooth muscles. In MYPT1-deficient mesenteric arteries, force enhancement in the initial and sustained contraction phases was associated with enhanced RLC phosphorylation. Considering the proposed central role MYPT1 plays in regulating MLCP activity, it was surprising that MYPT1 deletion in vascular smooth muscle was not more consequential. The apparent decrease in MLCP activity in tissues was associated with diminished amounts of PP1cδ. Because MYPT1 is absent in MYPT1SMKO mice, the enhanced response to norepinephrine may result from CPI-17 phosphorylation with inhibition of the reduced amount of PP1cδ. Vascular smooth muscles have an abundance of CPI-17 protein relative to other kinds of smooth muscles (42). Both ROCK and PKC inhibitors inhibited agonist-induced sustained contractions in MYPT1-deficient vascular smooth muscle similar to control smooth muscles, suggesting a contribution of CPI-17 phosphorylation by ROCK in regulating PP1cδ activity. Contractile responses were not inhibited by a ROCK inhibitor in MYPT1-deficient intestinal smooth muscle, which has much less CPI-17 compared with vascular smooth muscle (27). Rock inhibition also resulted in inhibition of CPI-17 phosphorylation in MYPT1-deficient mesenteric arteries. Additionally, our results are consistent with a recent report suggesting α-adrenergic receptor activation acted through CPI-17, not MYPT1 phosphorylation, in rat mesenteric arteries to affect Ca2+ sensitization (43). Thus, PKC and ROCK pathways may both serve as important Ca2+-sensitization mechanisms involving CPI-17 phosphorylation in mesenteric arteries.
In contracting smooth muscles, relaxation may result from a decrease in [Ca2+]i and/or reduction in Ca2+-sensitization of RLC phosphorylation and force development. NO initiates vascular smooth muscle relaxation by stimulating cGMP formation and activating PKG (44–46). PKG activation results in a decrease in [Ca2+]i by inhibiting PLC and inositol 1,4,5-trisphosphate formation (47, 48), a desensitization of RLC phosphorylation to Ca2+ by phosphorylating MYPT1, resulting in MLCP activation (49), an activation of the large conductance Ca2+-activated K+ channel (BK) (50), and RhoA phosphorylation that inhibits activation and membrane translocation (51). The latter inhibits activation of ROCK by RhoA (52). Mice containing selective mutations in the amino-terminal leucine zipper domain of PKG show both decreased MYPT1 phosphorylation and increased activation of RhoA in vascular smooth muscles (4). This disruption of PKG regulation contributes to abnormal relaxation responses and increased systemic blood pressure. The NO/cGMP/PKG signaling module is an important physiological system modulating vascular tone that has multiple cell targets affecting contractility. We found that activation of the NO/cGMP/PKG signaling pathway relaxed evoked force in MYPT1-deficient vascular smooth muscle similar to results obtained with control tissues. This finding suggests that NO/cGMP/PKG signaling mechanisms effecting reduction in [Ca2+]i are sufficient to relax contraction of mesenteric vessels. In summary, deletion of MYPT1 in vascular smooth muscle enhanced RLC phosphorylation and contractile activity, which may contribute to the development of hypertension in vivo.
This work was supported, in whole or in part, by National Institutes of Health Grants 1R01HL112778 and 5P01HL110869. This work was also supported by the National Key Scientific Research Program of China (2014CB964701), National Natural Science Funding of China (31272311 and 31330034; to M.-S. Z.), the Moss Heart Fund, the Fouad A. and Val Imm Bashour Distinguished Chair in Physiology (to J. T. S.), and the Fundamental Research Funds for the Central Universities (GK201402057 to Y.-N. Q.).
- MLCP
- myosin light chain phosphatase
- MYPT1
- myosin phosphatase target subunit 1
- MLCK
- myosin light chain kinase
- PP1cδ
- myosin phosphatase catalytic subunit
- RLC
- myosin regulatory light chain
- ROCK
- Rho-associated kinase
- NE
- norepinephrine
- Ang II
- angiotensin II
- Vas
- Arg-vasopressin
- SNP
- sodium nitroprusside
- CTR
- control group
- MYPT1SMKO
- MYPT1 smooth muscle-specific knockout group
- CPI-17
- myosin phosphatase inhibitory protein of 17 kDa
- p-CPI-17
- phospho-CPI-17 (Thr-38)
- PKG
- protein kinase G
- CLA
- calyculin A
- MBS85
- myosin phosphatase binding subunit of 85 kDa
- mN
- millinewtons.
REFERENCES
- 1. Crowley S. D., Gurley S. B., Oliverio M. I., Pazmino A. K., Griffiths R., Flannery P. J., Spurney R. F., Kim H.-S., Smithies O., Le T. H., Coffman T. M. (2005) Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J. Clin. Invest. 115, 1092–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cowley A. W. (2006) The genetic dissection of essential hypertension. Nat. Rev. Genet. 7, 829–840 [DOI] [PubMed] [Google Scholar]
- 3. Kobori H., Nangaku M., Navar L. G., Nishiyama A. (2007) The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol. Rev. 59, 251–287 [DOI] [PubMed] [Google Scholar]
- 4. Michael S. K., Surks H. K., Wang Y., Zhu Y., Blanton R., Jamnongjit M., Aronovitz M., Baur W., Ohtani K., Wilkerson M. K., Bonev A. D., Nelson M. T., Karas R. H., Mendelsohn M. E. (2008) High blood pressure arising from a defect in vascular function. Proc. Natl. Acad. Sci. U.S.A. 105, 6702–6707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davis M. J., Hill M. A. (1999) Signaling mechanisms underlying the vascular myogenic response. Physiol. Rev. 79, 387–423 [DOI] [PubMed] [Google Scholar]
- 6. Moosmang S., Lenhardt P., Haider N., Hofmann F., Wegener J. W. (2005) Mouse models to study L-type calcium channel function. Pharmacol. Ther. 106, 347–355 [DOI] [PubMed] [Google Scholar]
- 7. Somlyo A. P., Somlyo A. V. (2000) Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J. Physiol. 522, 177–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cole W. C., Welsh D. G. (2011) Role of myosin light chain kinase and myosin light chain phosphatase in the resistance arterial myogenic response to intravascular pressure. Arch. Biochem. Biophys. 510, 160–173 [DOI] [PubMed] [Google Scholar]
- 9. Wirth A., Benyó Z., Lukasova M., Leutgeb B., Wettschureck N., Gorbey S., Orsy P., Horváth B., Maser-Gluth C., Greiner E., Lemmer B., Schütz G., Gutkind J. S., Offermanns S. (2008) G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 14, 64–68 [DOI] [PubMed] [Google Scholar]
- 10. Kamm K. E., Stull J. T. (2001) Dedicated myosin light chain kinases with diverse cellular functions. J. Biol. Chem. 276, 4527–4530 [DOI] [PubMed] [Google Scholar]
- 11. Somlyo A. P., Somlyo A. V. (2003) Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325–1358 [DOI] [PubMed] [Google Scholar]
- 12. Lowey S., Trybus K. M. (2010) Common structural motifs for the regulation of divergent class II myosins. J. Biol. Chem. 285, 16403–16407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. He W.-Q., Qiao Y.-N., Zhang C.-H., Peng Y.-J., Chen C., Wang P., Gao Y.-Q., Chen C., Chen X., Tao T., Su X.-H., Li C.-J., Kamm K. E., Stull J. T., Zhu M.-S. (2011) Role of myosin light chain kinase in regulation of basal blood pressure and maintenance of salt-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 301, H584–H591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsumura F., Hartshorne D. J. (2008) Myosin phosphatase target subunit: many roles in cell function. Biochem. Biophys. Res. Commun. 369, 149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hartshorne D. J., Ito M., Erdödi F. (2004) Role of protein phosphatase type 1 in contractile functions: myosin phosphatase. J. Biol. Chem. 279, 37211–37214 [DOI] [PubMed] [Google Scholar]
- 16. Velasco G., Armstrong C., Morrice N., Frame S., Cohen P. (2002) Phosphorylation of the regulatory subunit of smooth muscle protein phosphatase 1M at Thr-850 induces its dissociation from myosin. FEBS Lett. 527, 101–104 [DOI] [PubMed] [Google Scholar]
- 17. Strålfors P., Hemmings H. C., Jr., Greengard P. (1989) Inhibitors of protein phosphatase-1. Eur. J. Biochem. 180, 143–148 [DOI] [PubMed] [Google Scholar]
- 18. Greengard P., Allen P. B., Nairn A. C. (1999) Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron 23, 435–447 [DOI] [PubMed] [Google Scholar]
- 19. Endo S., Suzuki M., Sumi M., Nairn A. C., Morita R., Yamakawa K., Greengard P., Ito M. (1999) Molecular identification of human G-substrate, a possible downstream component of the cGMP-dependent protein kinase cascade in cerebellar Purkinje cells. Proc. Natl. Acad. Sci. U.S.A. 96, 2467–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dey N. B., Lincoln T. M. (2012) Possible involvement of Cyclic-GMP-dependent protein kinase on matrix metalloproteinase-2 expression in rat aortic smooth muscle cells. Mol. Cell. Biochem. 368, 27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Puetz S., Lubomirov L. T., Pfitzer G. (2009) Regulation of smooth muscle contraction by small GTPases. Physiology 24, 342–356 [DOI] [PubMed] [Google Scholar]
- 22. Khromov A., Choudhury N., Stevenson A. S., Somlyo A. V., Eto M. (2009) Phosphorylation-dependent autoinhibition of myosin light chain phosphatase accounts for Ca2+ sensitization force of smooth muscle contraction. J. Biol. Chem. 284, 21569–21579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dimopoulos G. J., Semba S., Kitazawa K., Eto M., Kitazawa T. (2007) Ca2+-dependent rapid Ca2+ sensitization of contraction in arterial smooth muscle. Circ. Res. 100, 121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grassie M. E., Sutherland C., Ulke-Lemée A., Chappellaz M., Kiss E., Walsh M. P., MacDonald J. A. (2012) Cross-talk between Rho-associated kinase and cyclic nucleotide-dependent kinase signaling pathways in the regulation of smooth muscle myosin light chain phosphatase. J. Biol. Chem. 287, 36356–36369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grassie M. E., Moffat L. D., Walsh M. P., MacDonald J. A. (2011) The myosin phosphatase targeting protein (MYPT) family: a regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1δ. Arch. Biochem. Biophys. 510, 147–159 [DOI] [PubMed] [Google Scholar]
- 26. Okamoto R., Ito M., Suzuki N., Kongo M., Moriki N., Saito H., Tsumura H., Imanaka-Yoshida K., Kimura K., Mizoguchi A., Hartshorne D. J., Nakano T. (2005) The targeted disruption of the MYPT1 gene results in embryonic lethality. Transgenic Res. 14, 337–340 [DOI] [PubMed] [Google Scholar]
- 27. He W. Q., Qiao Y. N., Peng Y. J., Zha J. M., Zhang C. H., Chen C., Chen C. P., Wang P., Yang X., Li C. J., Kamm K. E., Stull J. T., Zhu M. S. (2013) Altered contractile phenotypes of intestinal smooth muscle in mice deficient in myosin phosphatase target subunit 1. Gastroenterology 144, 1456–1465.e1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu Z., Yang L., Cai L., Zhang M., Cheng X., Yang X., Xu J. (2007) Detection of epithelial to mesenchymal transition in airways of a bleomycin induced pulmonary fibrosis model derived from an α-smooth muscle actin-Cre transgenic mouse. Respir. Res. 8, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Isotani E., Zhi G., Lau K. S., Huang J., Mizuno Y., Persechini A., Geguchadze R., Kamm K. E., Stull J. T. (2004) Real-time evaluation of myosin light chain kinase activation in smooth muscle tissues from a transgenic calmodulin-biosensor mouse. Proc. Natl. Acad. Sci. U.S.A. 101, 6279–6284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mizuno Y., Isotani E., Huang J., Ding H., Stull J. T., Kamm K. E. (2008) Myosin light chain kinase activation and calcium sensitization in smooth muscle in vivo. Am. J. Physiol. Cell Physiol. 295, C358–C364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scotto-Lavino E., Garcia-Diaz M., Du G., Frohman M. A. (2010) Basis for the isoform-specific interaction of myosin phosphatase subunits protein phosphatase 1c β and myosin phosphatase targeting subunit 1. J. Biol. Chem. 285, 6419–6424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He W.-Q., Peng Y.-J., Zhang W.-C., Lv N., Tang J., Chen C., Zhang C.-H., Gao S., Chen H.-Q., Zhi G., Feil R., Kamm K. E., Stull J. T., Gao X., Zhu M.-S. (2008) Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology 135, 610–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang W.-C., Peng Y.-J., Zhang G.-S., He W.-Q., Qiao Y.-N., Dong Y.-Y., Gao Y.-Q., Chen C., Zhang C.-H., Li W., Shen H.-H., Ning W., Kamm K. E., Stull J. T., Gao X., Zhu M.-S. (2010) Myosin light chain kinase is necessary for tonic airway smooth muscle contraction. J. Biol. Chem. 285, 5522–5531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gao N., Huang J., He W., Zhu M., Kamm K. E., Stull J. T. (2013) Signaling through myosin light chain kinase in smooth muscles. J. Biol. Chem. 288, 7596–7605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shin H.-M., Je H.-D., Gallant C., Tao T. C., Hartshorne D. J., Ito M., Morgan K. G. (2002) Differential association and localization of myosin phosphatase subunits during agonist-induced signal transduction in smooth muscle. Circ. Res. 90, 546–553 [DOI] [PubMed] [Google Scholar]
- 36. Eto M., Kitazawa T., Brautigan D. L. (2004) Phosphoprotein inhibitor CPI-17 specificity depends on allosteric regulation of protein phosphatase-1 by regulatory subunits. Proc. Natl. Acad. Sci. U.S.A. 101, 8888–8893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hartshorne D. J., Ito M., Erdödi F. (1998) Myosin light chain phosphatase: subunit composition, interactions and regulation. J. Muscle Res. Cell Motil. 19, 325–341 [DOI] [PubMed] [Google Scholar]
- 38. Masuo M., Reardon S., Ikebe M., Kitazawa T. (1994) A novel mechanism for the Ca2+-sensitizing effect of protein kinase C on vascular smooth muscle: inhibition of myosin light chain phosphatase. J. Gen. Physiol. 104, 265–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ogut O., Brozovich F. V. (2003) Regulation of force in vascular smooth muscle. J. Mol. Cell. Cardiol. 35, 347–355 [DOI] [PubMed] [Google Scholar]
- 40. Himpens B., Matthijs G., Somlyo A. P. (1989) Desensitization to cytoplasmic Ca2+ and Ca2+ sensitivities of guinea-pig ileum and rabbit pulmonary artery smooth muscle. J. Physiol. 413, 489–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lincoln T. M. (2007) Myosin phosphatase regulatory pathways. Circ. Res. 100, 10–12 [DOI] [PubMed] [Google Scholar]
- 42. Woodsome T. P., Eto M., Everett A., Brautigan D. L., Kitazawa T. (2001) Expression of CPI-17 and myosin phosphatase correlates with Ca2+ sensitivity of protein kinase C-induced contraction in rabbit smooth muscle. J. Physiol. 535, 553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kitazawa T., Kitazawa K. (2012) Size-dependent heterogeneity of contractile Ca2+ sensitization in rat arterial smooth muscle. J. Physiol. 590, 5401–5423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Murad F. (2006) Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N. Engl. J. Med. 355, 2003–2011 [DOI] [PubMed] [Google Scholar]
- 45. Hofmann F. (2005) The biology of cyclic GMP-dependent protein kinases. J. Biol. Chem. 280, 1–4 [DOI] [PubMed] [Google Scholar]
- 46. Morgado M., Cairrão E., Santos-Silva A. J., Verde I. (2012) Cyclic nucleotide-dependent relaxation pathways in vascular smooth muscle. Cell. Mol. Life Sci. 69, 247–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hirata M., Kohse K. P., Chang C. H., Ikebe T., Murad F. (1990) Mechanism of cyclic GMP inhibition of inositol phosphate formation in rat aorta segments and cultured bovine aortic smooth muscle cells. J. Biol. Chem. 265, 1268–1273 [PubMed] [Google Scholar]
- 48. Rapoport R. M. (1986) Cyclic guanosine monophosphate inhibition of contraction may be mediated through inhibition of phosphatidylinositol hydrolysis in rat aorta. Circ. Res. 58, 407–410 [DOI] [PubMed] [Google Scholar]
- 49. Surks H. K., Mochizuki N., Kasai Y., Georgescu S. P., Tang K. M., Ito M., Lincoln T. M., Mendelsohn M. E. (1999) Regulation of myosin phosphatase by a specific interaction with CGMP-dependent protein kinase Iα. Science 286, 1583–1587 [DOI] [PubMed] [Google Scholar]
- 50. Zhou X.-B., Arntz C., Kamm S., Motejlek K., Sausbier U., Wang G.-X., Ruth P., Korth M. (2001) A molecular switch for specific stimulation of the BKCa channel by cGMP and cAMP kinase. J. Biol. Chem. 276, 43239–43245 [DOI] [PubMed] [Google Scholar]
- 51. Kato M., Blanton R., Wang G. R., Judson T. J., Abe Y., Myoishi M., Karas R. H., Mendelsohn M. E. (2012) Direct binding and regulation of RhoA protein by cyclic GMP-dependent protein Kinase Iα. J. Biol. Chem. 287, 41342–41351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sauzeau V., Le Jeune H., Cario-Toumaniantz C., Smolenski A., Lohmann S. M., Bertoglio J., Chardin P., Pacaud P., Loirand G. (2000) Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J. Biol. Chem. 275, 21722–21729 [DOI] [PubMed] [Google Scholar]