

Background: Several amino acids in proteins form hydroperoxides during oxidative stress.
Results: A fluorometric assay for real-time analyses of amino acid and protein hydroperoxides is developed.
Conclusion: The boronate-based assay is a convenient method for detection and absolute quantitation of amino acid/protein hydroperoxides.
Significance: The proposed strategy will enhance understanding of the mechanism(s) of oxidative and post-translational modification of proteins.
Keywords: Fluorescence, Free Radicals, Oxidative Stress, Oxygen Radicals, Protein Chemical Modification, Amino Acid Hydroperoxides, Boronates, Hydroperoxides, Singlet Oxygen, Tyrosine Hydroperoxide
Abstract
Hydroperoxides of amino acid and amino acid residues (tyrosine, cysteine, tryptophan, and histidine) in proteins are formed during oxidative modification induced by reactive oxygen species. Amino acid hydroperoxides are unstable intermediates that can further propagate oxidative damage in proteins. The existing assays (oxidation of ferrous cation and iodometric assays) cannot be used in real-time measurements. In this study, we show that the profluorescent coumarin boronic acid (CBA) probe reacts with amino acid and protein hydroperoxides to form the corresponding fluorescent product, 7-hydroxycoumarin. 7-Hydroxycoumarin formation was catalase-independent. Based on this observation, we have developed a fluorometric, real-time assay that is adapted to a multiwell plate format. This is the first report showing real-time monitoring of amino acid and protein hydroperoxides using the CBA-based assay. This approach was used to detect protein hydroperoxides in cell lysates obtained from macrophages exposed to visible light and photosensitizer (rose bengal). We also measured the rate constants for the reaction between amino acid hydroperoxides (tyrosyl, tryptophan, and histidine hydroperoxides) and CBA, and these values (7–23 m−1 s−1) were significantly higher than that measured for H2O2 (1.5 m−1 s−1). Using the CBA-based competition kinetics approach, the rate constants for amino acid hydroperoxides with ebselen, a glutathione peroxidase mimic, were also determined, and the values were within the range of 1.1–1.5 × 103 m−1 s−1. Both ebselen and boronates may be used as small molecule scavengers of amino acid and protein hydroperoxides. Here we also show formation of tryptophan hydroperoxide from tryptophan exposed to co-generated fluxes of nitric oxide and superoxide. This observation reveals a new mechanism for amino acid and protein hydroperoxide formation in biological systems.
Introduction
Several amino acids, peptides, and proteins undergo oxidation in the presence of reactive oxygen species (e.g. hydroxyl radical and singlet oxygen), forming the corresponding hydroperoxide as the major product (1, 2). Biological consequences of post-translational oxidative modification of amino acid residues in proteins are well documented (3). Amino acid hydroperoxides are unstable intermediates and can further propagate the oxidative damage. Protein hydroperoxides can induce secondary damage through inactivation of thiol-dependent enzymes that are essential to cellular function (2, 4). Protein hydroperoxides have also been reported to induce DNA damage (5–8). Despite their proposed involvement in multiple oxidative modifications, easy to use, sensitive, and reliable assays for measuring protein hydroperoxides in real time are still lacking, and there is an unmet need for developing such assays. Existing methods, including the oxidation of ferrous ion monitored with xylenol orange (FOX assay)3 and the iodometric assay (9, 10), have been widely used but have their own limitations. The requirement of a strict pH optimum, undefined stoichiometry of the reaction with protein hydroperoxides, and inhibition by metal ion chelators or ascorbate are a few of the shortcomings of the FOX assay (9–15). The iodometric assay involves a stoichiometric reaction with a broad range of hydroperoxides but requires strict anaerobic conditions, and this reaction is light-sensitive. Additionally, both assays cannot be used in real-time measurements.
Recently, a new class of boron-based profluorescent probes for detection of H2O2 and peroxynitrite was developed and successfully used in enzymatic and cellular systems (16–19). Upon reaction with H2O2 and peroxynitrite, the weakly fluorescent boronates are oxidatively converted into highly fluorescent phenolic products (Scheme 1). In this work, we show that a profluorescent coumarin boronic acid (CBA) probe also reacts with amino acid and protein-derived hydroperoxides to form the corresponding fluorescent product, 7-hydroxycoumarin (COH). Based on this finding, we have developed a fluorometric, real-time assay that is adapted to a multiwell plate format. We performed a direct comparison of the CBA-based assay with the FOX assay, using tyrosyl hydroperoxide (TyrOOH) as a model amino acid hydroperoxide. The pros and cons of using the CBA probe for the detection of protein hydroperoxides are discussed. We have applied this assay for detection of protein hydroperoxides in cell lysates derived from macrophages treated with visible light and photosensitizer. The CBA-based assay was also used to determine the kinetic parameters of the reactions between ebselen, a well known glutathione peroxidase (GPx) mimetic (20), and amino acid hydroperoxides.
SCHEME 1.
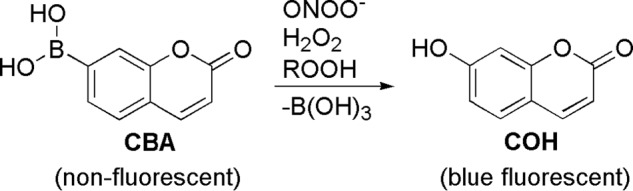
Oxidation of CBA probe.
EXPERIMENTAL PROCEDURES
Chemicals
Xanthine oxidase (cow milk) was purchased from Roche Applied Science. Superoxide dismutase (bovine erythrocytes), horseradish peroxidase, ferricytochrome c (equine heart), ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one), ebselen selenium oxide, tyrosine, 4-hydroxyphenylacetic acid, tryptophan, histidine, lysozyme (from chicken egg white), bovine serum albumin (BSA), reduced glutathione (GSH), oxidized glutathione (GSSG), cysteine, cystine, COH, xylenol orange disodium salt, sorbitol, ammonium ferrous sulfate, rose bengal, diethylenetriaminepentaacetic acid (DTPA), NaN3, H2O2, D2O, KH2PO4, K2HPO4, and H2SO4 were purchased at the highest purity available from Sigma-Aldrich. Catalase (beef liver) was from Roche Applied Science. PAPA-NONOate was from Cayman. CBA was synthesized according to the procedure described previously (17).
Hydroperoxide Generation
TyrOOH was generated in the horseradish peroxidase/xanthine oxidase (HRP/XO) system according to the reaction scheme shown (Scheme 2). Incubation mixtures consisted of tyrosine (1 mm), hypoxanthine (1 mm), and various concentrations of xanthine oxidase, horseradish peroxidase (2 μg/ml), phosphate buffer (50 mm; pH 7.4), and 0.1 mm DTPA (unless otherwise indicated). Catalase (250 units/ml) was present in incubation mixtures to inhibit oxidation of boronates by H2O2. Mixtures were incubated for 30–60 min at room temperature. SOD (0.1 mg/ml) was added to stop TyrOOH formation.
SCHEME 2.
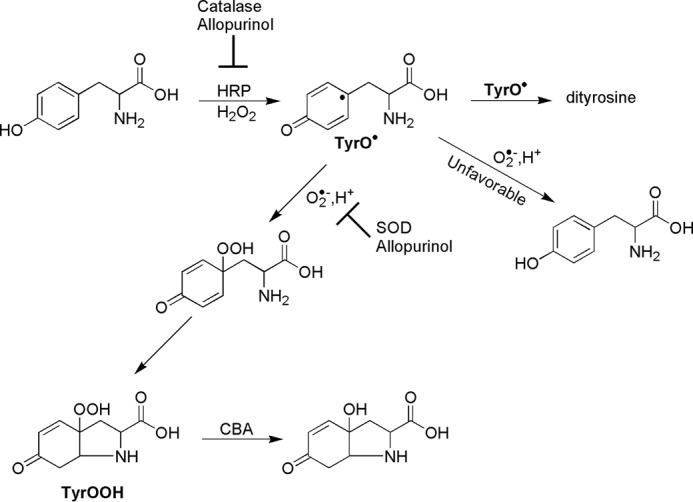
Mechanism of tyrosyl hydroperoxide synthesis in HRP/XO system.
Alternatively, TyrOOH and other hydroperoxides (Scheme 3) were generated by the reaction with singlet oxygen using rose bengal as the singlet oxygen generator. Photolysis experiments were carried out using a 300-watt xenon high pressure compact arc lamp (Eimac Model VIX 300 UV). Amino acid and protein hydroperoxides were generated by photolysis of solutions containing phosphate buffer (50 mm, pH 7.4), amino acid or protein, and DTPA (0.1 mm) in the presence of rose bengal (10 μm). Samples were placed in a fluorometer quartz cell and were photolyzed at room temperature with continuous oxygenation. The illuminating light was passed through a water filter and a 405-nm cut-off filter. Prior to the addition of CBA or FOX reagent, photolyzed samples were incubated with catalase (250 units/ml) for a period of 5 min to remove any H2O2 formed during the photolysis.
SCHEME 3.
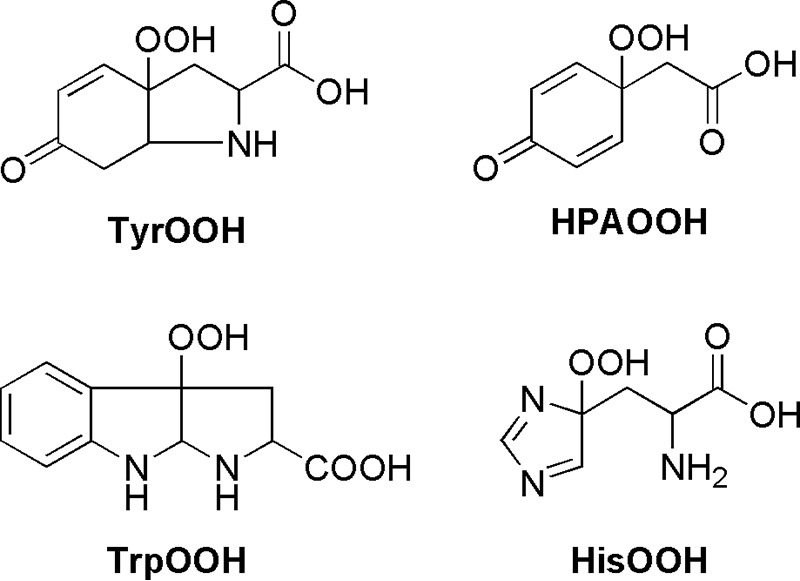
Structures of amino acid hydroperoxides.
Detection of Protein Hydroperoxides during Photosensitized Oxidation of Macrophages
RAW 264.7 murine macrophage-like cells (ATCC) were cultured in DMEM (Invitrogen), supplemented with 10% heat-inactivated FBS, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Prior to experimentation, cells were washed with HBSS and then incubated with 5 μm rose bengal in HBSS for 30 min at 37 °C in the dark. Next, cells were washed twice with HBSS to remove excess of rose bengal, and fresh HBSS was added. The rose bengal-loaded cells were placed on ice and exposed to visible light for up to 60 min. The 100-watt incandescent tungsten light bulb placed 15 cm above the samples was used for this purpose. The control cells loaded with rose bengal were kept in the dark on ice for the same period of time. Catalase (580 units/ml) was added immediately after cessation of photolysis, and the cells were incubated at room temperature for 2 min. Cells were then washed with Dulbecco's PBS, scraped with 1 ml of Dulbecco's PBS, transferred into a 1.5-ml tube, and centrifuged (1 min, 1000 × g). After centrifugation, the supernatant was discarded, and the cell pellets were frozen in liquid nitrogen and stored at −80 °C until needed or further processed as follows. The cell pellets were resuspended and lysed (10 syringe strokes) in water containing DTPA (0.1 mm) and catalase (100 units/ml). Such prepared lysates were transferred to 96-microwell plates and mixed with phosphate buffer (50 mm, pH 7.4) containing catalase (100 units/ml), DTPA (0.1 mm), and CBA (0.8 mm). Next, the samples were incubated for 2 h at room temperature, and fluorescence intensity of COH formed was measured by a plate reader (Beckman Coulter DTX 880). The protein concentration was determined using Bradford reagent and BSA standards.
CBA and FOX Assay
The FOX assay was used according to the procedure described elsewhere (21), using a 20-fold concentrated stock solution, with a slight modification as indicated below. The FOX assay consisted of 1.5 m H2SO4, 2 mm xylenol orange, 2 m sorbitol, and 5 mm ammonium ferrous sulfate. The concentration of sulfuric acid used was higher compared with the original protocol, in order to obtain the recommended pH in the range 1.7–1.9, after dilution of the FOX reagent with samples containing 50 mm phosphate buffer (11). Following hydroperoxide generation, 10 μl of the FOX reagent was mixed with 190 μl of a sample. Samples were left at room temperature for 30 min before reading the absorbance at 590 nm. DTPA was not added to the samples when FOX reagent was used (except when stated otherwise).
CBA was dissolved in DMSO at 0.1 m concentration, and this solution was added directly to the samples to obtain the desired concentration, usually 0.8 mm. For fluorescence detection of COH, which resulted from the oxidation of CBA, the excitation and emission wavelengths were set at 360 and 430 nm, respectively.
Absorbance and Fluorescence Measurements
Absorption and fluorescence measurements were performed on the Beckman Coulter DTX 880 multimode plate reader equipped with appropriate filters. Absorption spectra were recorded with an Agilent 8453 spectrophotometer. Fluorescence spectra were collected using the PerkinElmer Life Sciences LS 55 luminescence spectrometer. All measurements were carried out at room temperature.
HPLC Measurements
HPLC measurements were performed on an Agilent 1100 system equipped with UV-visible absorption and fluorescence detectors. HPLC analyses of the samples containing ebselen were carried out on the Kinetex PFP column (100 × 4.6 mm, 2.6 μm; Phenomenex). Prior to analyses, the column was equilibrated with a water/acetonitrile mobile phase (90:10, v/v) containing 0.1% trifluoroacetic acid (TFA). Compounds were separated by the linear increase of acetonitrile volume from 10 to 38% over 3.5 min. Next, the volume of organic phase was increased to 100% over 0.5 min and kept at this level for 2.5 min. For separating tyrosine and dityrosine, a Halo RP-amide column (100 × 4.6 mm, 2.6 μm; Advanced Materials Technology) was used. Prior to analyses, the column was equilibrated with an aqueous mobile phase containing 1% (v/v) of acetonitrile phase and 0.1% TFA. The organic phase during analysis was increased linearly in two stages: from 1 to 40% over 2.5 min and from 40 to 80% over the next 2 min. The samples containing tyrosine were acidified by adding TFA prior to injection. The injection volume and the flow rate used were 50 μl and 1.5 ml/min, respectively, in both HPLC methods.
LC-MS Analyses
The photolyzed samples of amino acids containing CBA were separated using an Acquity UPLC system (Waters) equipped with a photodiode array and coupled to an LCT Premier XE (Waters) mass spectrometer. MS spectra were recorded using an electrospray ionization probe in the positive ion mode. Separation of tyrosine photooxidation products and CBA was performed on a Waters UPLC column (Acquity UPLC CSH Phenyl-Hexyl, 1.7 μm, 2.1 × 50 mm) kept at 40 °C and equilibrated with 1% acetonitrile (containing 0.1% (v/v) TFA) in 0.1% TFA aqueous solution. The compounds were separated by the linear increase of the acetonitrile phase from 1 to 75% over 0.5 min and kept at this level for 1 min. Next, the concentration of organic phase was increased from 75 to 100% over 1.8 min. The samples were acidified by adding TFA directly before injection. Separation of tryptophan photooxidation products and CBA was performed on Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm). The column was equilibrated with acetonitrile/water mobile phase (10:90, v/v) containing 0.1% TFA. In order to separate the analytes, the concentration of organic phase was increased from 10 to 40% over 2 min. In both separation methods, a flow rate of 0.3 ml/min and injection volume of 1 μl were used. Alternatively, the Shimadzu Nexera HPLC system coupled with an LC-MS 8030 triple quadrupole mass detector was used for the tryptophan samples containing nitric oxide donor, xanthine oxidase, and hypoxanthine. The separation of analytes was performed on the Kinetex C18 column (1.3 μm, 50 × 2.1 mm) equilibrated with aqueous mobile phase containing 1% (v/v) acetonitrile and 0.1% TFA. The compounds were eluted by increasing the amount of acetonitrile from 1 to 21% over 4 min. Tryptophan-derived hydroperoxide (TrpOOH) and TrpOH were detected using multiple-reaction monitoring mode using the transitions 237.1 → 146.0 and 221.1 → 175.0, respectively.
Kinetic Measurements
All kinetic measurements were performed under pseudo-first order conditions, where the concentration of appropriate hydroperoxide was at least 10 times lower than that of CBA or ebselen. In the case of TyrOOH generated in the HRP/XO system, hydroperoxide was accumulated for 1 h in the presence of catalase. Next, SOD (final concentration of 0.1 mg/ml) and the appropriate volume of CBA solution were added. This mixture was quickly vortexed, and kinetic traces were collected using a luminescence spectrometer. In the case of 1O2-derived hydroperoxides, the solutions after photolysis were diluted with phosphate buffer and preincubated with catalase prior to the addition of CBA. Kinetic traces were recorded with a plate reader. In order to determine the rate constants of the reaction between ebselen and various hydroperoxides, the competition kinetics approach was applied. In this method, CBA and ebselen compete for hydroperoxide, which can be described by Reactions 1 and 2.
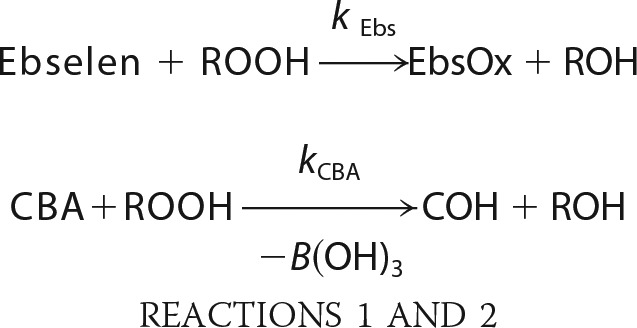 |
Because the concentration of both compounds remained relatively stable for the duration of the experiment, the competition model assuming two pseudo-first order reactions was applied. The equations that fit both the nonlinear (Equation 1) and linear relationship (Equation 2) were used.
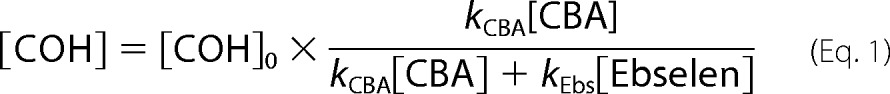 |
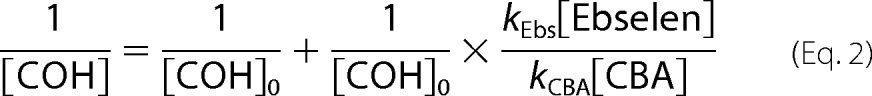 |
Evaluation of the Assay Quality by Z′-Factor
The Z′-factor is a simple statistical parameter that can be used to evaluate the quality of the assay (22). This dimensionless coefficient is defined by Equation 3,
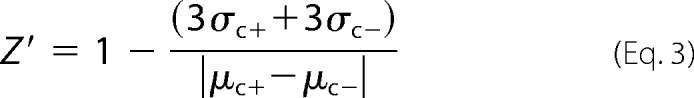 |
where σc+ and σc− are the S.D. values, and μc+ and μc− are the mean values for the positive and negative control, respectively (22).
With an increase in the assay quality, the value of the Z′-factor increases. The maximal value that the Z′ parameter can reach is 1. A Z′ value of 0.5 or higher indicates high quality of the assay.
RESULTS
Reaction between Tyrosyl Hydroperoxide and CBA
The TyrOOH was synthesized in the HRP/XO system (Scheme 2). Xanthine oxidase and hypoxanthine were used to generate superoxide radical anion (O2⨪) and hydrogen peroxide (H2O2) and horseradish peroxidase/H2O2-catalyzed oxidation of tyrosine to produce tyrosyl radicals. Tyrosyl radicals react with superoxide, forming TyrOOH. According to the published reports, this route is favored over the reduction of tyrosyl radical by O2⨪ back to tyrosine (23, 24). A similar reaction system was described by Winterbourn and co-workers (25–28). Previously, the detection and quantification of TyrOOH were carried out with the FOX assay (25). In the present work, the CBA-based assay was utilized to detect and quantitate TyrOOH (Scheme 1). The possible bicyclic structure of the TyrOOH formed shown in Scheme 2 was unambiguously identified in previous studies (29). The TyrOOH produced in the HRP/XO system converted the boronate probe into the fluorescent product. The fluorescence spectra collected upon oxidation of CBA were consistent with the formation of COH. The identity of CBA oxidation product was unequivocally confirmed by HPLC analysis, showing that the TyrOOH-mediated oxidation product of CBA is co-eluted with the authentic standard of COH with identical spectral properties under similar chromatographic conditions (Fig. 1). The presence of xanthine oxidase inhibitor, allopurinol, in the reaction mixture greatly inhibited COH formation.
FIGURE 1.

Identification of the product formed from the reaction between CBA and TyrOOH. Insets show the online absorption spectra collected by a diode array detector for the eluted analytes. TyrOOH was generated in the reaction mixture containing tyrosine (1 mm), hypoxanthine (0.5 mm), catalase (0.25 kilounits/ml), HRP (2 μg/ml), XO (0.6 milliunits/ml), phosphate buffer (50 mm, pH 7.4), and DTPA (0.1 mm) with and without allopurinol (0.3 mm), as indicated. After 1 h of incubation, the reaction was stopped by the addition of allopurinol to the allopurinol-free sample, and CBA (0.1 mm) was added to both samples. The concentration of CBA and COH standards was 0.1 mm.
The product detected, COH, is also formed from oxidation of CBA by H2O2 and peroxynitrite (ONOO−), as described previously (17). The reported rate constants for the reaction of CBA with H2O2 and ONOO− at pH 7.4 are 1.5 ± 0.2 m−1 s−1 and (1.1 ± 0.2) × 106 m−1 s−1, respectively. We measured the rate constant of TyrOOH/CBA reaction at the same pH by following the build-up of COH fluorescence (Fig. 2), and from the plot of the observed pseudo-first order rate constants of COH formation on the CBA concentration, the second order rate constant of 15.4 ± 0.3 m−1 s−1 was determined (Fig. 2, inset). The TyrOOH/CBA rate constant is 10 times higher than for the H2O2/CBA reaction and nearly 5 orders of magnitude lower than that reported for the ONOO−/CBA reaction.
FIGURE 2.

Kinetic traces of COH formation during oxidation of CBA by TyrOOH. Inset, dependence of pseudo-first order rate constants of COH formation on the concentration of CBA. Reaction mixtures containing tyrosine (1 mm), hypoxanthine (1 mm), catalase (0.25 kilounits/ml), HRP (0.5 units/ml), and XO (0.6 milliunits/ml) in phosphate buffer (50 mm, pH 7.4) and DTPA (0.1 mm) were incubated for 1 h. After incubation, SOD (0.1 mg/ml) and CBA (0.05–0.8 mm) were added, and fluorescence measurements were started.
To investigate whether the CBA assay can be used for real-time monitoring of TyrOOH formation in the HRP/XO system, the following experiment was performed (Fig. 3). We monitored oxidation of CBA to COH in the mixture containing hypoxanthine and xanthine oxidase in the presence or absence of several components, including tyrosine, HRP, two different concentrations of catalase, SOD, and ebselen. The reaction was monitored in a plate reader (Fig. 3A). The rate of fluorescence signal increase was maximal for incubations containing HRP in addition to HX/XO and tyrosine. In this system, most of the H2O2 is expected to be consumed by HRP with the production of tyrosyl radicals. In order to exclude the contribution of H2O2 to the overall rate of CBA oxidation, catalase was added (250 units/ml) to the incubation mixture. Catalase partly diminished the rate of CBA oxidation. The observed inhibition is not only due to exclusion of the CBA/H2O2 reaction; the COH rate formation is also lowered, as expected, due to the competition between HRP and catalase for H2O2. The formation of COH was completely inhibited in the presence of both SOD and catalase in the reaction mixture. In the absence of catalase, the COH signal was strongly inhibited by SOD, but the residual oxidation of CBA was still observed. This indicates that H2O2 is the oxidant contributing to the observed fluorescence increase. The COH signal was completely mitigated in the presence of a high concentration of catalase (25 kilounits/ml), suggesting that all H2O2 was consumed by catalase, not by HRP, resulting in the inhibition of tyrosyl radical formation. In the absence of any component crucial for TyrOOH generation, such as tyrosine or HRP enzyme, the build-up of COH fluorescence was not observed even at a low concentration of catalase. The fluorescence signal was also abolished if the antioxidant drug ebselen (a glutathione peroxidase mimetic) (20) was present during TyrOOH synthesis. The amounts of COH produced in various incubations were also measured by HPLC. The HPLC chromatograms are shown in Fig. 3B and are consistent with the results obtained by monitoring fluorescence intensity using the plate reader.
FIGURE 3.

A, time course of CBA oxidation measured in the reaction mixtures containing CBA (0.8 mm), tyrosine (1 mm), HX (1 mm), XO (1 milliunits/ml), and DTPA (0.1 mm) in phosphate buffer (50 mm, pH 7.4) with and without HRP (0.5 units/ml), catalase (0.25 kilounits/ml for CATLow or 25 kilounits/ml for CATHigh), SOD (0.1 mg/ml), and ebselen (0.1 mm). B, comparison of HPLC traces showing COH peaks detected after 30 min of incubation. HPLC traces were collected using a fluorescence detector with an emission wavelength set at 430 nm and an excitation wavelength set at 320 nm.
Tyrosyl radical formed in HRP/H2O2 oxidation of tyrosine undergoes dimerization in the absence of superoxide to form dityrosine, which is detectable by fluorescence (28). We used the HPLC method with fluorescence detection for simultaneous monitoring of COH and dityrosine to investigate the influence of SOD on dityrosine formation and on the conversion of CBA into COH. The authentic standards of COH and dityrosine were used to identify the observed peaks. The product profiles and HPLC traces obtained from the real-time monitoring experiment are summarized in Fig. 4, A–C. As shown, the presence of SOD greatly enhanced the amount of dityrosine produced over the time of incubation (Fig. 4C), with concomitant inhibition of the conversion of CBA into COH (Fig. 4B). This is reasonable in light of the proposed role of O2⨪ in the conversion of tyrosyl radical into TyrOOH (Scheme 2). Other peaks that can be ascribed to trityrosine or higher oligomer formation were also observed by UV-visible detection during HPLC analyses (data not shown). These results indicate that the CBA probe is especially suitable for real-time monitoring of tyrosine hydroperoxide formation in enzymatic systems.
FIGURE 4.

Simultaneous monitoring of dityrosine and COH formation by HPLC with fluorescence detection. Reaction mixtures contained tyrosine (1 mm), hypoxanthine (1 mm), catalase (0.25 kilounits/ml), HRP (0.5 units/ml), XO (0.6 milliunits/ml), SOD (where indicated, 0.1 mg/ml), 0.8 mm CBA, phosphate buffer (50 mm, pH 7.4), and DTPA (0.1 mm) and were incubated for 1 h before analysis. A, collected HPLC traces; B, COH peak areas; C, dityrosine peak areas.
Comparison between CBA and FOX Assays
Next, we compared the assay developed here with the FOX assay, commonly used for determination of amino acid hydroperoxides (29). Both assays are based on the different detection modes, and the detection principles are presented in Fig. 5 (A and E). In the case of the FOX assay, tyrosyl hydroperoxide oxidizes the ferrous ion to the ferric ion selectively in dilute acid, and the resultant ferric ions are determined with Fe3+-sensitive dye, xylenol orange (21). Xylenol orange binds ferric ions, forming a colored (blue-purple) complex. Under the experimental conditions used, the absorption maximum of this complex was detected at 590 nm (Fig. 5A). The spectra obtained at different ferric ion concentrations and the calibration curve used are shown (Fig. 5A, inset).
FIGURE 5.

Comparison between FOX and CBA assays. A, absorption spectra recorded during detection of ferric ions by the FOX assay. Inset, calibration curve for ferric ions. B, build-up of absorbance signal at 590 nm upon TyrOOH detection by the FOX assay. C, end point measurement of TyrOOH using the FOX assay. The amount of TyrOOH is given as ferric ion concentration (left axis) and as H2O2 equivalents (right axis). D, accumulation of TyrOOH with time, shown as ferric ion concentration (left axis) and as H2O2 equivalents (right axis), measured by the FOX assay ([XO] = 1.94 milliunits/ml). E, excitation and emission spectra of COH at different concentrations. Inset, calibration curve for COH measured with the plate reader. F, build-up of COH fluorescence signal upon TyrOOH detection using the CBA assay. G, end point measurement and real-time monitoring of TyrOOH by the CBA assay. H, accumulation of TyrOOH with time measured by the CBA assay ([XO] = 1.94 milliunits/ml). Reaction mixtures containing tyrosine (1 mm), hypoxanthine (1 mm), catalase (0.25 kilounits/ml), HRP (0.5 units/ml), XO (0.1–5 milliunits/ml), phosphate buffer (50 mm, pH 7.4), DTPA (0.1 mm, except if FOX reagent was used), and SOD (0.1 mg/ml) were incubated for 1 h (except in D and H). Error bars, S.D.
CBA is a profluorescent probe that, upon oxidation by TyrOOH, is converted to COH, as discussed above. The fluorescence excitation and emission spectra of COH as well as the calibration curve used for the quantification of TyrOOH produced in HRP/XO system are shown in Fig. 5E. The time courses of signal formation for both assays upon reaction with TyrOOH are also presented (Fig. 5, B and F). As shown, the signal of the Fe3+-xylenol orange complex is developed within 15 min of incubation (Fig. 5B) and is relatively stable over the time scale shown. For the CBA assay, the time needed to obtain a stable COH fluorescence signal is about 5 times shorter at 0.8 mm CBA concentration (Fig. 5F).
To estimate the relative sensitivity of both assays for detection of TyrOOH, we varied H2O2/O2⨪ fluxes and consequently the amounts of TyrOOH generated in the HRP/XO system. The fluxes of H2O2 and O2⨪ species were regulated by changing the XO concentration. The time and H2O2/O2⨪ flux dependence of signal detected by FOX and CBA assays are shown (Fig. 5, C, D, G, and H). Because the authentic standard for TyrOOH is not available, we decided to express the quantity of TyrOOH in the FOX assay as ferric ion concentration as well as H2O2 equivalents. The concentration of TyrOOH measured by the CBA assay can be given directly, assuming a CBA/TyrOOH stoichiometry of 1:1, the same as in the case of the CBA/H2O2 reaction. Comparing the end point measurements of TyrOOH by CBA and FOX assays in the HRP/XO system (Fig. 5, C and G), the amount of TyrOOH measured by the FOX assay (expressed as H2O2 equivalents) is about 3-fold lower than the amount of TyrOOH measured by the CBA assay. In turn, the amounts of TyrOOH measured in situ by the CBA assay were 3–5 times higher than by the FOX assay. This is consistent with the data reported previously by Winterbourn and co-workers (26, 28). They reported that the amount of TyrOOH assessed by the FOX assay and expressed as H2O2 equivalents was much lower (about 6-fold) than the superoxide-dependent loss of tyrosine quantified by HPLC (26, 28). The difference between the TyrOOH amounts measured in real time and in end point modes by the CBA assay (Fig. 5G) must be due to the instability of hydroperoxide, which can be expected to be converted to secondary products during incubation.
Fig. 5, C and G, was also used to calculate the Z′-factor. We used the Z′-factor, a simple statistical parameter, to directly compare the quality of both assays (22). According to the Z′-factor definition (see “Experimental Procedures”), the values of calculated coefficient for the highest concentration of TyrOOH (or XO) are equal to 0.533 and 0.946 for the FOX and CBA assay, respectively. This indicates that the CBA assay quality is superior to that of the FOX assay, mainly because of the higher variability of the data obtained by the FOX assay (see calculated S.D. values). Comparing Fig. 5, C and G, it is evident that the curve shape of negative control containing SOD for the CBA assay is close to a straight line, and the values are close to zero. With the FOX assay, the shape of the SOD control is more complex. This further confirms the better quality of the CBA assay.
The kinetics of TyrOOH accumulation during incubation has been determined by FOX and CBA assays in the end point measurement mode, and the data are shown in Fig. 5 (D and H) for the concentration of XO equal to 1.94 milliunits/ml. Although both assays show the accumulation of the peroxide, the CBA assay yields higher values of the absolute concentration of TyrOOH with a significantly lower background signal.
Both assays tested above were used for the detection of TyrOOH generated in the HRP/XO system. To confirm whether the FOX reagent and the CBA probe react with the same species, we performed a competition experiment, as illustrated in Fig. 6. The addition of a low concentration of CBA probe (0.1 mm) to the samples containing TyrOOH caused a build-up of fluorescence signal (solid squares). Simultaneously, aliquots of the same CBA/TyrOOH mixture were transferred into the wells containing the FOX reagent in 5-min intervals. The reaction between CBA and TyrOOH is pH-dependent and at low pH values is very slow. Thus, after the addition of FOX reagent, when pH of the solution decreased to ∼1.8, the residual TyrOOH reacted with the FOX assay but not with the CBA probe. The FOX signal measured for control sample (open circles) and for sample containing CBA (open squares) is shown (Fig. 6). Clearly, the presence of CBA accelerates the decomposition of FOX-reactive product. Both fluorescence (CBA assay) and absorbance (FOX assay) signals were abolished if SOD was present during generation of TyrOOH. The results show that CBA time-dependently diminished the FOX assay signal, with concomitant formation of COH product, indicating that both assays detect the same species. Also, a slow decrease of FOX signal in the absence of CBA (open circles) over the first 30 min, followed by signal stabilization, indicates that at least two species (hydroperoxides) of different stability are detected in the FOX assay. There are some limitations in the use of the FOX reagent (e.g. compounds that bind ferric ions interfere with the FOX assay through competition with xylenol orange for ferric ions) (21). Fig. 7A shows the influence of DTPA, a metal ion chelator, on the FOX and CBA assay signal upon detection of TyrOOH. TyrOOH was produced in HRP/XO system, as described above. These results clearly demonstrate that DTPA, at the concentration of 100 μm, commonly used in buffers, completely inhibited the formation of the colored Fe3+-xylenol orange complex, whereas it did not interfere with the CBA assay. The amount of CBA converted into COH upon reaction with TyrOOH was independent of the concentration of DTPA.
FIGURE 6.

Competition between CBA and FOX reagent for TyrOOH generated in XO/HRP system. Reaction mixtures containing tyrosine (1 mm), hypoxanthine (1 mm), catalase (0.25 kilounits/ml), HRP (0.5 units/ml), XO (2.8 milliunits/ml), phosphate buffer (50 mm, pH 7.4), DTPA (0.1 mm), and SOD (0.1 mg/ml, where indicated) were incubated for 1 h, after which SOD was added to all samples to stop the production of TyrOOH. Next, CBA (final concentration, 0.1 mm) or FOX or both reagents were added (see “Results”). Open symbols are related to absorbance scale (left axis); solid symbols are related to fluorescence scale (right axis). Error bars, S.D.
FIGURE 7.

A, influence of iron chelator DTPA on the observed signal for FOX (circles, absorbance scale, left axis) and CBA (squares, fluorescence scale, right axis) assays at constant concentration of TyrOOH. B, effect of DTPA on the yield of TyrOOH generated in the HRP/XO system at different concentrations of ferric ions (0, 10, and 50 μm). Error bars, S.D.
We also tested the influence of ferric ions on the yield of TyrOOH formed in the HRP/XO system (Fig. 7B) with and without DTPA. In the presence of 10 or 50 μm ferric ions, the amount of TyrOOH formed in the incubation mixture that did not contain DTPA was distinctly lower than the TyrOOH concentration measured in the absence of redox-active iron. The presence of DTPA increased the yield of TyrOOH formed in the samples containing ferric ions. The effect of ferric ions on the TyrOOH yields was abolished by 20 μm DTPA for the samples containing 10 μm ferric ions and by 100 μm DTPA for the samples containing 50 μm ferric ions (Fig. 7B).
Detection of 1O2-derived Hydroperoxides by CBA Assay
In order to test the applicability of the CBA probe for detecting hydroperoxides generated during the reaction between amino acids/proteins and the singlet oxygen, we photolyzed solutions containing tyrosine, 4-hydroxyphenylacetic acid (HPA), tryptophan, histidine, lysozyme, and BSA, at pH 7.4 with visible light (λ > 405 nm) in the presence of 10 μm rose bengal as a photosensitizer. Photolyzed samples were pretreated with catalase to remove any photogenerated H2O2. Next, CBA was added, and an increase in the fluorescence signal with time was monitored (Fig. 8). No peroxides were detected in nonphotolyzed solutions containing all reactants. Experiments carried out in 60% D2O resulted in an increase of peroxide yield, whereas the inclusion of azide led to a marked decrease in peroxide formation. Because D2O significantly increases the lifetime of 1O2 and the azide anion is an efficient scavenger of 1O2, these results strongly suggest that the conversion of CBA to COH is mediated by peroxides formed in 1O2-dependent processes (2). Additionally, the presence of tyrosyl and tryptophan hydroperoxides was confirmed by LC-MS analysis, which also provided additional evidence for the reaction of tyrosyl and tryptophan hydroperoxides with the CBA probe (Figs. 9 and 10). Direct LC-MS analysis of the mixture containing tyrosine and rose bengal subjected to the visible light resulted in the detection of ions consistent with the presence of tyrosine, [M + H]+ = 182.1, and tyrosyl hydroperoxide, [M + H]+ = 214.1. The incubation of photogenerated tyrosyl hydroperoxide with CBA led to the decrease of tyrosyl hydroperoxide peak with concomitant build-up of the peak corresponding to the appropriate alcohol, [M + H]+ = 198.1 (Fig. 9A). The conversion of tyrosyl hydroperoxide to the alcohol was accompanied by consumption of CBA (eluting at 2 min) and formation of COH (eluting at 2.2 min; Fig. 9B). The reaction profiles are shown in Fig. 9C, where open symbols represent the samples containing both CBA and TyrOOH, and closed symbols represent the control samples where CBA or TyrOOH were present alone. The reaction profiles indicate that both CBA and TyrOOH were relatively stable over a 1-h incubation time when present alone but underwent almost complete consumption within 1 h when mixed together. This confirms that the observed changes are due to the direct reaction between CBA and TyrOOH. Identical results were obtained for the reaction between CBA and TrpOOH (Fig. 10). In this case, besides the mass of tryptophan [M + H]+ = 205.1, the two peaks eluting at 0.9 min and 1.1 nm corresponding to the mass [M + H]+ = 237.1 were detected (Fig. 10, A and B). These peaks can be attributed to the cis and trans isomers of tryptophan hydroperoxide (32). The addition of CBA to the sample containing tryptophan hydroperoxide caused the decay of the observed hydroperoxides with concomitant formation of isomeric alcohols, appearing as peaks at 0.7 min and 0.9 min corresponding to the mass [M + H]+ = 221.1. Again, the reaction profiles confirm the direct reaction of CBA with tryptophan hydroperoxide and the relative stability of CBA and TrpOOH during the experiment when present alone (Fig. 10C).
FIGURE 8.

Detection of 1O2-derived hydroperoxides with CBA. Reaction mixtures contained tyrosine (1 mm) (A), 4-hydroxyphenylacetic acid (1 mm) (B), tryptophan (1 mm) (C), histidine (1 mm) (D), lysozyme (10 mg/ml) (E), bovine serum albumin (5 mg/ml) (F), and rose bengal (10 μm). Mixtures were photolyzed for 1–15 min in the presence or absence of azide ion (10 mm) and deuterated water (50–70% (v/v)). After photolysis, samples were incubated with catalase (250 units/ml) for 5 min and for an additional 1 min with ebselen, where indicated. Next, samples were diluted 10-fold (A–D) with buffer containing CBA (0.8 mm), or CBA was added directly to the samples (E and F). All samples were prepared in phosphate buffer (50 mm, pH 7.4) containing DTPA (0.1 mm) and were continuously oxygenated during photolysis.
FIGURE 9.

LC/MS analysis of products formed from TyrOOH-mediated oxidation of CBA. A, MS traces of incubation mixtures containing CBA (0.1 mm) and TyrOOH. Data represent extracted ion chromatograms of species with parent ions with m/z [M + H]+ 214.07 and 198.08. B, UPLC traces of mixtures of CBA (0.1 mm) with TyrOOH (0.1 mm) collected using the absorption detector set at 300 nm. Prior to the addition of CBA, the sample of tyrosine (1 mm) was photolyzed in the presence of rose bengal (10 μm), phosphate buffer (50 mm, pH 7.4) over 30 min and, next, preincubated with catalase (100 units/ml) for 5 min. C, profiles of the reactants (i.e. CBA, COH, TyrOOH, and TyrOH, as indicated). Open symbols, reaction profiles when both CBA and TyrOOH were present in the incubation mixtures; solid symbols, reaction profiles upon the absence of CBA or TyrOOH, as indicated.
FIGURE 10.

LC/MS analysis of products formed from TrpOOH-mediated oxidation of CBA. A, MS traces of mixture of CBA (0.1 mm) with TrpOOH. Data represent extracted ion chromatograms of species with parent ions with m/z [M + H]+ 237.09 and 221.09. B, UPLC traces of a mixture of CBA (0.1 mm) with TrpOOH (0.1 mm) collected using the absorption detector set at 300 nm. Prior to the addition of CBA, the sample of tryptophan (1 mm) was photolyzed in the presence of rose bengal (10 μm), phosphate buffer (50 mm, pH 7.4) over 5 min and, next, preincubated with catalase (100 units/ml) for 5 min. C, profiles of the reactants (i.e. CBA, COH, TrpOOH, and TrpOH) (as indicated). Open symbols, reaction profiles when both CBA and TrpOOH were present in the incubation mixture; solid symbols, reaction profiles upon the absence of CBA or TrpOOH, as indicated.
The photolytically generated hydroperoxides were also assayed using the FOX reagent (Fig. 11). The addition of the FOX reagent to the photolyzed samples preincubated with catalase caused an increase in the absorbance signal at 590 nm, whereas only background signal (absorbance of ∼0.03) was detected in the samples pretreated with CBA (0.8 mm) or ebselen (0.1 mm). However, there was an exception related to a lysozyme sample (Fig. 11E); even incubation of photolyzed sample with CBA for 60 min did not remove completely the species responsible for oxidation of FOX reagent, and a noticeable increase of absorbance signal compared with the non-photolyzed sample was seen. This may be due to the location of hydroperoxide in lysozyme protein not accessible to CBA probe and/or a high value of pKa of hydroperoxide, resulting in a much slower reaction with boronic compounds. It is known that, due to high pKa, aliphatic hydroperoxides are practically unreactive toward boronate probes at neutral pH. Some oxidation of the FOX reagent was also observed for unirradiated BSA sample, but the magnitude of absorbance signal change was small.
FIGURE 11.

Detection of singlet oxygen-derived hydroperoxides using the FOX assay. Reaction mixtures contained tyrosine (1 mm) (A), 4-hydroxyphenylacetic acid (1 mm) (B), tryptophan (1 mm) (C), histidine (1 mm) (D), lysozyme (10 mg/ml) (E), or bovine serum albumin (5 mg/ml) (F). Mixtures were photolyzed from 1 to 5 min in the presence of rose bengal (10 μm) in phosphate buffer (50 mm). After photolysis, samples were incubated with catalase (250 units/ml) for 5 min and diluted 10-fold (A–D) with phosphate buffer or with phosphate buffer containing CBA (0.8 mm) or ebselen (0.1 mm). To sample E and F, CBA and ebselen were added directly. Prior to the addition of FOX reagent, samples were incubated for another 5 min (samples A–D) or 30–60 min (samples E and F). Absorbance readings were performed 30 min after adding the FOX reagent in a plate reader using a 590-nm filter. Error bars, S.D.
Next, the rate constants of the reaction between CBA and tyrosine, HPA, tryptophan, and histidine hydroperoxides were determined under pseudo-first order conditions at pH 7.4. The rate constants obtained from the exponential fittings of the kinetic traces acquired for COH formation (Fig. 12, A–D) showed the linear dependence on the CBA concentration (Fig. 12, E–H). The determined second order rate constants are listed in Table 1. Because the oxidation of CBA by lysozyme or BSA hydroperoxides did not follow the pseudo-first order rate law, their rate constants were not determined. In general, we can conclude that amino acid hydroperoxides react with CBA about 10 times faster than does H2O2.
FIGURE 12.

Kinetics of the reaction between 1O2-derived amino acid hydroperoxides and CBA. A–D, kinetic traces with fitted curves for tyrosine (A), 4-hydroxyphenylacetic acid (B), tryptophan (C), and histidine (D). E–H, dependence of the pseudo-first order rate constants of COH formation on the CBA concentration for the reaction with hydroperoxides derived from tyrosine (E), 4-hydroxyphenylacetic acid (F), tryptophan (G), and histidine (H). Error bars, S.D.
TABLE 1.
Rate constants of the reactions of amino acid hydroperoxides with CBA and ebselen
| Hydroperoxide | kROOH + CBA | kROOH + ebselen |
|---|---|---|
| m−1 s−1 | m−1 s−1 | |
| TyrOOH | 23.4 ± 0.5 | (1.11 ± 0.06) × 103 |
| HPAOOHa | 7.0 ± 0.1 | (1.07 ± 0.08) × 103 |
| TrpOOH | 11.4 ± 0.2 | (1.54 ± 0.05) × 103 |
| HisOOHb | 23.3 ± 0.7 | (1.05 ± 0.07) × 103 |
| H2O2 | 1.5 ± 0.2 | 35 ± 1 |
a HPA-derived hydroperoxide.
b Histidine-derived hydroperoxide.
Detection of Protein Hydroperoxides Generated in a Cellular System
In order to extend the applicability of the CBA assay to complex biological systems, we decided to test the possibility to detect hydroperoxides in cell lysates from macrophages exposed to visible light in the presence of a singlet oxygen-generating photosensitizer. It has been shown that exposure of rose bengal-loaded cells to visible light generates intracellular protein peroxides (33, 34). The addition of a cell lysate originating from photolyzed cells to a buffered solution (pH 7.4) containing CBA resulted in a formation of highly fluorescent product, COH. The identity of the product was confirmed on the basis of fluorescence spectra. The signal intensity was linearly dependent on the amount of lysate mixed with CBA solution (Fig 13A). The analogous treatment of cell lysates originating from cells incubated with rose bengal but not exposed to visible light did not lead to an increase of COH fluorescence signal. This indicates that oxidative conversion of CBA into COH was due to the presence of photolytically generated oxidant(s). Due to the presence of catalase in the lysis buffer and assay buffer, we can exclude the possibility of H2O2 being the oxidant detected. The signal intensity was also proportional to the time of exposure of rose bengal-loaded cells to visible light (Fig 13B). Because prolonged exposure of cells to a visible light led to a decrease of the cell viability, the protein concentration was taken into account during calculations. The addition of the hydroperoxide scavenger ebselen to the cell lysates prior to mixing with CBA solution greatly inhibited the increase of COH fluorescence signal (Fig 13C).
FIGURE 13.

Detection of protein hydroperoxides in RAW 264.7 cell lysates. A, dependence of the concentration of COH formed on the amount of cell lysate added, as expressed by final cellular protein concentration in the reaction mixture. B, dependence of protein hydroperoxides concentration (expressed as COH concentration) in the cell lysates on the time of photolysis. C, effect of ebselen (50 μm) on the concentration of protein hydroperoxides detected in cell lysates by CBA probe. Macrophages RAW 264.7 were preincubated with 5 μm rose bengal, washed with HBSS, and exposed to visible light (100-watt incandescent tungsten light bulb) from 0 to 60 min; next, the cell lysates were prepared and used for further experimentation. The amount of COH produced was normalized to the total amount of protein in the cell lysates. See “Experimental Procedures” for further details. Error bars, S.D.
Reaction of Glutathione with 1O2
Because GSH is one of the most abundant low molecular weight antioxidants in cells, we tested whether CBA can be used to directly probe the formation of GSH-derived peroxy species formed from the reaction of GSH with 1O2. Incubation mixtures containing glutathione (1 mm) were photolyzed with the visible light in the presence of rose bengal (10 μm) and CBA (0.8 mm) at pH 7.4. The intensity of the fluorescence signal from COH formed was measured at different time points, as shown in Fig. 14A. Photolysis of this mixture in the presence of catalase led to an increase in fluorescence intensity (due to CBA to COH conversion) over the duration of illumination. In the absence of catalase the rate of increase in the fluorescence intensity was higher due to the reaction between CBA and H2O2 (probably produced upon illumination from a type 1 mechanism). The presence of the azide ions during photolysis decreased the yield of COH formed. The highest fluorescence increase with time was observed from incubations in D2O. No change in the fluorescence intensity was observed for the control sample photolyzed in the absence of glutathione. The same was true when the control sample containing glutathione and other components was incubated in the dark. Additionally, if CBA was added after sample photolysis, no further increase of fluorescence signal was observed (results not shown). This indicates that the peroxy species formed from the GSH are not stable, consistent with the lack of positive signal in the FOX assay (Fig. 14A, inset).
FIGURE 14.

Detection of peroxy species from the reaction between singlet oxygen and GSH (A), GSSG (B), cysteine (C), and cystine (D). Incubation mixtures contained rose bengal (10 μm) and CBA (0.8 mm) and were photolyzed in the presence or absence of an appropriate amino acid or peptide (1 mm), catalase (250 units/ml), deuterated water (∼60%), and azide ion (10 mm) (as indicated). Insets, photolyzed mixtures (photolysis time 10 min; 10 μm rose bengal) of GSH (1 mm) (A) or cysteine (C) assayed with FOX reagent. Samples contained phosphate buffer (50 mm, pH 7.4) and DTPA (0.1 mm) (except when FOX reagent was used) and were continuously oxygenated during photolysis. Error bars, S.D.
Upon prolonged illumination (up to 10 min) of the solution of CBA probe, rose bengal, and catalase at pH 7.4, in the presence or absence of GSSG (1 mm), negligible changes in the fluorescence intensity were noticed (Fig. 14B). A similar set of experiments was carried out for cysteine and cystine (Fig. 14, C and D). These results are analogous to those obtained for glutathione and GSSG. Insets in Fig. 14, A and C, show the results for the photolyzed samples of glutathione and cysteine assayed with FOX reagent. As shown, there was no increase of absorbance at 590 nm for these samples compared with the nonphotolyzed samples.
Reactivity of Ebselen toward Amino Acid Hydroperoxides
Because ebselen has been found to compete with CBA for amino acid hydroperoxides (Fig. 3), we decided to perform HPLC analysis to characterize the product formed during the reaction of ebselen with tyrosine hydroperoxide. TyrOOH was preformed in mixtures containing rose bengal, tyrosine, catalase, and DTPA in phosphate buffer (pH 7.4) subjected to photolysis with visible light. Immediately after turning off the light, ebselen (0.1 mm) was added, and the oxidation products were analyzed using the reversed phase HPLC. Oxidation of ebselen by TyrOOH led to a major product eluting at a retention time of 2.9 min (Fig. 15A), and the concentration of this product increased with the time of photolysis (Fig. 15A). The product derived from the ebselen reaction with TyrOOH eluted at the same time as the authentic standard, ebselen selenium oxide, and the product of the reaction of ebselen with H2O2. As shown in Fig. 15B, catalase slightly diminished the yield of ebselen selenium oxide. For reactions in D2O, the amount of ebselen selenium oxide produced was significantly increased, whereas the azide anion (1O2 scavenger) almost completely abolished its formation, consistent with 1O2-derived TyrOOH being the oxidant.
FIGURE 15.

HPLC analysis of product formed from TyrOOH-mediated oxidation of ebselen. A, HPLC traces of ebselen (Ebs; 0.1 mm), ebselen oxide (EbsOx; 0.1 mm), a mixture of ebselen (0.1 mm) with H2O2 (1 mm), and photolyzed mixtures of tyrosine and ebselen (1 mm). Samples were photolyzed in the presence of rose bengal (10 μm), phosphate buffer (50 mm), and DTPA (0.1 mm) for the indicated periods of time. B, HPLC traces of tyrosine mixtures photolyzed in the presence or absence of deuterium oxide and azide ions. Before adding ebselen (final concentration, 0.1 mm), photolyzed samples were preincubated with catalase (250 units/ml) for 5 min (as indicated). All samples were prepared in phosphate buffer (50 mm, pH 7.4) containing DTPA (0.1 mm). HPLC traces were collected using the absorption detector set at 282 nm.
The rate constants of the reaction between amino acid hydroperoxides and ebselen were determined by a competition kinetics technique (as described under “Experimental Procedures”). As a competitor, CBA was used, and its previously determined rate constants with hydroperoxides (Table 1) were utilized in the calculations. Fig. 16A shows the dependence of COH peak area, obtained for the incubations containing photolytically generated TyrOOH, CBA, and different concentrations of ebselen, as a function of ebselen concentration. With increasing ebselen concentration, the HPLC peak areas due to COH decreased. A rate constant of 2k = 1.1 ± 0.07 × 103 m−1 s−1 due to the ebselen/TyrOOH reaction was determined from the nonlinear graph (Equation 1), as shown (Fig. 16A). The curve fitted using a linear equation (Equation 2) is also presented (Fig. 16A, inset). Fig. 16B shows the kinetic analysis data of the effect of ebselen on the amount of COH formed from incubations containing HPA-derived hydroperoxide. Similar experiments were performed for tryptophan and histidine hydroperoxides, and the results are shown in Fig. 16, C and D, respectively. The second order rate constants determined for the reaction between amino acid hydroperoxides and ebselen are summarized in Table 1.
FIGURE 16.

The competition kinetics analysis of the reaction between 1O2-derived hydroperoxides and ebselen. Shown is the dependence of COH peak area on the concentration of ebselen for the photogenerated hydroperoxides of tyrosine (A), 4-hydroxyphenylacetic acid (B), tryptophan (C), and histidine (D). The solid line represents the result of nonlinear fitting. Insets, results of fitting the experimental data using the linear relationship. Photolyzed samples were preincubated with catalase (250 units/ml) and diluted 10 times with solution containing CBA (0.8 mm) and ebselen (0–0.2 mm). All samples were prepared in phosphate buffer (50 mm, pH 7.4) containing DTPA (0.1 mm). Error bars, S.D.
Peroxynitrite-mediated Formation of Tryptophan Hydroperoxide
The diffusion-controlled reaction between nitric oxide and superoxide radical anion leads to the formation of an unstable intermediate, ONOO−. ONOO− is an oxidizing and nitrating agent able to traverse biological membranes. The reaction of ONOO− with amino acid residues of proteins may occur directly or through peroxynitrite-derived radicals. It has been shown that tryptophan reacts with ONOO− directly with a second order rate constant of 37 m−1 s−1 at pH 7.4 and 37 °C, but the free radicals derived from peroxynitrite can modify it as well (35, 36). Among the products and intermediates detected in peroxynitrite-damaged tryptophan residues, the tryptophanyl radical has been detected by EPR spin trapping (37). We reasoned that with co-generated fluxes of •NO and O2⨪, the tryptophan radical formed due to presence of peroxynitrite will react with O2⨪ to produce TrpOOH. Therefore, in this study we employed a continuous flux of O2⨪ and •NO with a ratio of 2:1 and tested whether hydroperoxidic species was formed (Fig 17A). We used PAPA-NONOate as the •NO donor (•NO flux of 1 μm/min) and the HX/XO system as a source of O2⨪ flux (2 μm/min). Because ONOO− reacts rapidly with boronates (38), we could not directly use the CBA probe to detect TrpOOH in such a system. We decided to use LC-MS detection and apply the boronate probe to confirm the identity of TrpOOH by monitoring boronate-induced transformation of TrpOOH into TrpOH, as shown above (Fig. 10). LC-MS analysis of the reaction mixture containing tryptophan under co-generated O2⨪ and •NO fluxes indicated the formation of TrpOOH species (retention time of 1.5 min) (Fig. 17B). Within 20 min after starting the incubation, the concentration of TrpOOH reached steady-state level (data not shown). The reaction was then stopped after 60 min by the addition of xanthine oxidase inhibitor, allopurinol, and it was tested whether the observed peak was sensitive to CBA. LC/MS analysis of the reaction mixture treated with allopurinol and CBA (0.2 mm) showed that the peak eluting at 1.5 min was eliminated (Fig. 17B). The decay of tryptophan hydroperoxide was accompanied by formation of the product eluting at a retention time of 1.3 min with the mass corresponding to the alcohol derivative (Fig. 17C). The LC/MS analyses unequivocally confirmed that tryptophan is transformed into hydroperoxide by co-generated O2⨪ and •NO according to the proposed mechanism (Fig. 17A).
FIGURE 17.

A, the proposed mechanism of tryptophan hydroperoxide formation in the presence of co-generated •NO and O2⨪ fluxes. B, MRM chromatograms of TrpOOH (237.1 → 146.0) for the incubation mixture containing PAPA-NONOate (104 μm; •NO flux = 1 μm/min), xanthine oxidase (25 milliunits/ml; O2⨪ flux = 2 μm/min), hypoxanthine (1 mm), tryptophan (1 mm), catalase (100 units/ml), DTPA (100 μm), and phosphate buffer (50 mm), obtained by LC/MS after the indicated time of incubation. After 1 h, allopurinol and then CBA were added (100 μm). C, same as B, but the MRM chromatograms of TrpOH (221.1 → 175.0) are shown.
DISCUSSION
In this study, we show that the profluorescent probe CBA reacts with tyrosyl hydroperoxide and other amino acid and protein-derived hydroperoxides, forming a fluorescent product. We determined that the rate constant of the reaction between TyrOOH and CBA is about 10 times faster than between H2O2 and CBA. Catalase does not affect the stability of amino acid hydroperoxides (15), thus making it possible to monitor the formation of TyrOOH and other amino acid hydroperoxides in real time using the CBA probe even in the presence of H2O2. To our knowledge, this is the first report showing real-time measurements of amino acid/protein-derived hydroperoxides using the boronate-based profluorescent probe.
In this study, we have also compared the CBA assay described herein with the FOX assay that has been widely used to detect amino acid and protein hydroperoxides. Assuming 1:1 stoichiometry of the reaction of TyrOOH with CBA forming one molecule of COH, we were able to directly quantify the amount of TyrOOH formed in the HRP/XO system. The accuracy of our measurements is supported by the good agreement between our data and those reported in the literature (26, 28). Winterbourn and co-workers (26, 28) reported that the amount of TyrOOH measured by the FOX assay is 6 times lower than the superoxide-dependent loss of tyrosine quantified by HPLC. The amounts of TyrOOH measured in situ by the CBA assay were calculated to be 4–5 times higher than the amounts of TyrOOH measured by the FOX assay. We also showed that with unstable species, like TyrOOH, that readily convert into secondary products, it is essential to monitor in real time the formation of the primary hydroperoxide. The CBA assay enables real-time measurement of hydroperoxide formation in contrast to the FOX assay. The direct comparison of both assays using a simple assay quality parameter, Z′-factor, reveals that the quality of the CBA assay is better than that of the FOX assay, whereas both assays seem to be applicable to high throughput measurements. We also confirmed that both assays are sensitive to TyrOOH formed in the HRP/XO system and that despite different mechanisms of action, both assays detect the same hydroperoxide species.
Proteins are a major biological target for 1O2, due to their abundance and the high rate constants with this oxidant (2, 30). It has been reported that 1O2 reacts with proteins primarily at tyrosine, tryptophan, and histidine side chains, forming hydroperoxides of the structures shown in Scheme 3 (2, 30–32, 39). We generated hydroperoxides of these amino acids in a 1O2-mediated reaction employing rose bengal as the photosensitizer. We showed that CBA probe reacts with these species to form the fluorescent COH. The rate constants determined for the reaction between CBA and photolytically generated hydroperoxides are at least an order of magnitude higher than that of the CBA reaction with H2O2.
One of the most abundant antioxidants that can prevent the oxidation of proteins by 1O2 is GSH. The cysteinyl thiol group is responsible for the reaction of GSH with 1O2. The postulated intermediate formed in the reaction between cysteine and 1O2 is a zwitterionic peroxy species (e.g. RS(H)+-OO−) with disulfides and oxyacids as final products (2, 40, 41). We observed that the intermediate formed during the reaction of GSH (or CysSH) with 1O2 oxidizes the CBA probe to the fluorescent product, COH (Fig. 14). The probe oxidation was not observed when reduced thiols were replaced by corresponding disulfides. We conclude that the species involved in a conversion of CBA to COH is formed from the reaction between singlet oxygen and the sulfhydryl group of the thiol. The results also show that the intermediate species is unstable and should be detected in situ using the CBA probe because no signal was observed when the samples of GSH and cysteine were assayed with the CBA probe as well as FOX reagent directly after the photolysis. It can be concluded that the species formed in the GSH/1O2 and CysSH/1O2 systems react with the CBA probe. The structure of the proposed intermediate responsible for probe oxidation is similar to that of the postulated peroxy zwitterionic species (RS(H)+-OO−) and/or thiol hydroperoxide (RSOOH).
Peroxiredoxins (Prxs) are able to remove lysozyme and BSA hydroperoxides, showing that this enzyme family is a more efficient protein hydroperoxide scavenger than GPxs (15, 42). GPx1 was shown to be an efficient scavenger of low molecular weight hydroperoxides (15, 43). This is probably due to a more restricted access to the GPx1 active site by protein hydroperoxides compared with the Prxs active site. Low molecular weight catalytic scavengers of hydroperoxides, like ebselen, are superior to enzymatic systems in this regard. Selenium-containing amino acids have also been shown to act as catalytically efficient scavengers for amino acid and protein hydroperoxides (44, 45).
Ebselen, a selenoorganic compound, is capable of reducing a broad range of hydroperoxides using thiols as cosubstrates, thereby displaying GPx activity and acting as an antioxidant (20). There are, however, no kinetic data reported for the reaction between ebselen and protein hydroperoxides. Thus, no quantitative comparison could be made between non-enzymatic (GPx mimetics) and enzymatic scavengers of amino acid and protein hydroperoxides. We determined that amino acid hydroperoxides react with ebselen to form ebselen selenium oxide, which is in agreement with previous studies related to other hydroperoxides (20). Using the CBA assay and the competition kinetics, we were able to determine rate constants for the reaction of ebselen with tyrosine, tryptophan, and histidine hydroperoxides. We have also shown that ebselen removes lysozyme and BSA hydroperoxides. The kinetic data obtained in this study enabled us to compare its hydroperoxide scavenging properties with that of Prx enzymes. The bimolecular rate constants reported for the reaction of Prx2 and Prx3 with histidine hydroperoxide are equal to 2 × 103 and 3 × 103 m−1 s−1, respectively (42), whereas the measured rate constant for ebselen is (1.4 ± 0.1) × 103 m−1 s−1. Thus, we conclude that ebselen is a very efficient scavenger of amino acid and protein hydroperoxides and could contribute a pivotal role for ebselen's antioxidant mechanism.
A New Mechanistic Route for Protein Hydroperoxide Formation
Oxidation of proteins by reactive oxygen species leads to the formation of protein hydroperoxides via both radical and non-radical reactions. In vitro studies have shown that exposure of amino acids and proteins to highly reactive hydroxyl radical (•OH) in an oxygenated system results in the formation of their hydroperoxides with high yields (46). The other routes, also discussed in this paper, are related to the reactivity of singlet oxygen toward the tyrosine, tryptophane, and histidne residues as well as the reactivity of superoxide radical anion toward phenoxyl radicals formed on tyrosyl residues of proteins (25–28, 30, 47). The addition of superoxide radical anion to the tyrosyl radical was very well documented by Winterbourn et al. (25–28) and was considered to be physiologically plausible. Here we present the new peroxynitite-mediated route for the formation of amino acid and protein hydroperoxides, similar to the peroxidase-dependent generation of tyrosyl hydroperoxide described by Winterbourn et al. We have hypothesized that tryptophanyl radical formed in the reaction with peroxynitrite-derived radicals will react with O2⨪ to form hydroperoxide, and we have shown that during simultaneous generation of O2⨪ and •NO (2 μm/min of O2⨪ to 1 μm/min of •NO) in the presence of tryptophan, tryptophan is converted into hydroperoxide. The proposed mechanism (Fig. 17A), wherein tryptophan is oxidized to its radical by peroxynitrite derived oxidants and then the addition of O2⨪ occurs, is supported by the fact that among the intermediates detected in peroxynitrite-damaged tryptophan residues of proteins, the tryptophanyl radical has been detected. A similar mechanism can also be proposed for tyrosine residues. Overall, these studies provide new data on the mechanisms of peroxynitrite-mediated protein modification.
Measurement of Protein Hydroperoxides in Biological Samples
Measurements of lipid hydroperoxides in biological samples have been widely used as an indication of oxidative stress and associated with several diseases, including atherosclerosis, cancer, and neurodegenerative diseases. Nevertheless, in many reactive oxygen species-associated disorders, the peroxidation of proteins can be prevalent over the lipid peroxidation processes because proteins are the major non-water component of tissues or cellular or biological fluids and exhibit high reaction rates with many oxidants (46). For example, it has been shown that in cells subjected to peroxyl radicals, the hydroperoxide groups are generated on the cell proteins, and no peroxidation of lipids was observed (48). Furthermore, there is indirect evidence for the formation of hydroperoxides in a number of pathologies (49, 50).
Several methods have been employed for the detection of hydroperoxides in a wide variety of samples, including food, plasma, and tissues samples. The existing methods based on chemiluminescence, chromatographic, or spectroscopic detection of hydroperoxides demand complicated instrumentation and sample preparation procedures. Additionally, in many cases, the direct detection of hydroperoxides is not possible due to their instability, forcing the analysts to determine only hydroperoxide breakdown products (i.e. corresponding alcohols). Among the available methods, the titration and colorimetric methods based on iodide or iron oxidation have been widely used. Due to the lack of reliable assays for hydroperoxides, these methods, especially the FOX assay, were adapted for the detection of amino acid, peptide, and protein hydroperoxides. Although the FOX assay has been successfully applied for the detection of lipid and protein hydroperoxides in biological materials (e.g. cell lysates), the detection step has to be preceded by a relatively long sample preparation procedure involving multistep extraction, and this will lead to partial decomposition of cellular hydroperoxides (14). Here we show that the CBA-based assay can be performed directly in cell lysates. There is no need to separate protein hydroperoxides from lipid hydroperoxides because CBA is unreactive toward the latter. It is well established that hydroperoxides are generated on free amino acids and in proteins as well as in cell lysates upon exposure to a system that generates 1O2. The photochemical type II generation of 1O2 plays a crucial role in photodynamic therapy, inducing tumor cell death. The application of a CBA-based assay can facilitate the identification of an intracellular target of 1O2 involved in photodynamic therapy-induced cell death.
Photodynamic Mechanism in Mitochondria
The singlet oxygen and other reactive oxygen species generated from photodynamic activation of mitochondria-localized photosensitizer have been implicated in the selective inactivation of mitochondrial respiratory chain proteins (51). Most notable of the mitochondrial protein targets of Photofrin 11-mediated oxidant(s) were cytochrome c oxidase, F0F1-ATP synthase, and succinate dehydrogenase (51). Aminoleuvlinic acid is a heme precursor pro-drug that is preferentially taken up by tumor cells and converted to protoporphyrin IX in the mitochondria (52, 53). Selective inactivation of tumor cells during aminoleuvlinic acid/photodynamic therapy has been attributed to 1O2-mediated oxidative damage to mitochondrial proteins (53–55). Very little information with regard to oxidation of specific proteins or specific sites of oxidation is known, although the generally accepted mechanism includes peroxidation of lipids and proteins (51). It is conceivable that boronate probes targeted to mitochondria may be used to scavenge and quantitate mitochondrial protein hydroperoxides. In addition, post-treatment with boronates will convert protein hydroperoxides into the corresponding protein hydroxyl products that can be investigated using MALDI/MS techniques, thereby identifying the amino acid residue in specific proteins modified during photodynamic therapy.
Conclusions
We report a simple fluorometric assay for real-time measurement of amino acid and protein-derived hydroperoxides. The CBA-based assay is a convenient and sensitive method for detection and absolute quantification of amino acid and protein hydroperoxides. The proposed assay significantly simplifies the measurement of protein hydroperoxides in cells. Due to the possibility of real-time hydroperoxide monitoring using a CBA probe, the assay may be used to detect unstable peroxides, as exemplified by detection of glutathione- and cysteine-derived peroxide species. With the use of the CBA assay, we established the reactivity of ebselen, a versatile antioxidant molecule, with amino acid hydroperoxides. We conclude that ebselen reacts very effectively with amino acid-derived hydroperoxides, with rate constants comparable with those of peroxiredoxins, which indicates the possibility that scavenging of protein hydroperoxides contributes to antioxidative properties of ebselen. Boronates (aromatic and aliphatic), in general, could potentially serve as effective scavengers of amino acid/protein hydroperoxides, which may be used both in cytoprotection and for analytical purposes.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 HL073056 and R01 HL063119 (to B. K.). This work was also supported by a grant coordinated by Jagiellonian Centre for Experimental Therapeutics (JCET), POIG.01.01.02-00-069/09 (supported by the European Union from the resources of the European Regional Development Fund under the Innovative Economy Programme).
- FOX assay
- ferrous ion/xylenol orange assay
- CBA
- coumarin-7-boronic acid
- COH
- 7-hydroxycoumarin
- TyrOOH
- tyrosine hydroperoxide(s)
- GPx
- glutathione peroxidase
- DTPA
- diethylenetriaminepentaacetic acid
- XO
- xanthine oxidase
- SOD
- superoxide dismutase
- HPA
- 4-hydroxyphenylacetic acid
- CysSH
- cysteine, reduced form
- Prx
- peroxiredoxin
- CAT
- catalase
- TrpOOH
- tryptophan-derived hydroperoxide
- TrpOH
- tryptophan hydroperoxide-derived alcohol
- PAPA-NONOate
- propylamine propylamine NONOate
- HBSS
- Hanks' balanced salt solution
- DMSO
- dimethyl sulfoxide
- a.u.
- arbitrary units.
REFERENCES
- 1. Morgan P. E., Pattison D. I., Davies M. J. (2012) Quantification of hydroxyl radical-derived oxidation products in peptides containing glycine, alanine, valine, and proline. Free Radic. Biol. Med. 52, 328–339 [DOI] [PubMed] [Google Scholar]
- 2. Rahmanto A. S., Morgan P. E., Hawkins C. L., Davies M. J. (2010) Cellular effects of photogenerated oxidants and long-lived, reactive, hydroperoxide photoproducts. Free Radic. Biol. Med. 49, 1505–1515 [DOI] [PubMed] [Google Scholar]
- 3. Ryan K., Backos D. S., Reigan P., Patel M. (2012) Post-translational oxidative modification and inactivation of mitochondrial complex I in epileptogenesis. J. Neurosci. 32, 11250–11258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rahmanto A. S., Morgan P. E., Hawkins C. L., Davies M. J. (2010) Cellular effects of peptide and protein hydroperoxides. Free Radic. Biol. Med. 48, 1071–1078 [DOI] [PubMed] [Google Scholar]
- 5. Gebicki S., Gebicki J. M. (1999) Crosslinking of DNA and proteins induced by protein hydroperoxides. Biochem. J. 338, 629–636 [PMC free article] [PubMed] [Google Scholar]
- 6. Luxford C., Morin B., Dean R. T., Davies M. J. (1999) Histone H1- and other protein- and amino acid-hydroperoxides can give rise to free radicals which oxidize DNA. Biochem. J. 344, 125–134 [PMC free article] [PubMed] [Google Scholar]
- 7. Luxford C., Dean R. T., Davies M. J. (2000) Radicals derived from histone hydroperoxides damage nucleobases in RNA and DNA. Chem. Res. Toxicol. 13, 665–672 [DOI] [PubMed] [Google Scholar]
- 8. Luxford C., Dean R. T., Davies M. J. (2002) Induction of DNA damage by oxidised amino acids and proteins. Biogerontology 3, 95–102 [DOI] [PubMed] [Google Scholar]
- 9. Bou R., Codony R., Tres A., Decker E. A., Guardiola F. (2008) Determination of hydroperoxides in foods and biological samples by the ferrous oxidation-xylenol orange method: a review of the factors that influence the method's performance. Anal. Biochem. 377, 1–15 [DOI] [PubMed] [Google Scholar]
- 10. Gay C., Collins J., Gebicki J. M. (1999) Hydroperoxide assay with the ferric-xylenol orange complex. Anal. Biochem. 273, 149–155 [DOI] [PubMed] [Google Scholar]
- 11. Gay C., Collins J., Gebicki J. M. (1999) Determination of iron in solutions with the ferric-xylenol orange complex. Anal. Biochem. 273, 143–148 [DOI] [PubMed] [Google Scholar]
- 12. Gay C., Gebicki J. M. (2000) A critical evaluation of the effect of sorbitol on the ferric-xylenol orange hydroperoxide assay. Anal. Biochem. 284, 217–220 [DOI] [PubMed] [Google Scholar]
- 13. Gay C. A., Gebicki J. M. (2002) Perchloric acid enhances sensitivity and reproducibility of the ferric-xylenol orange peroxide assay. Anal. Biochem. 304, 42–46 [DOI] [PubMed] [Google Scholar]
- 14. Gay C. A., Gebicki J. M. (2003) Measurement of protein and lipid hydroperoxides in biological systems by the ferric-xylenol orange method. Anal. Biochem. 315, 29–35 [DOI] [PubMed] [Google Scholar]
- 15. Morgan P. E., Dean R. T., Davies M. J. (2004) Protective mechanisms against peptide and protein peroxides generated by singlet oxygen. Free Radic. Biol. Med. 36, 484–496 [DOI] [PubMed] [Google Scholar]
- 16. Lippert A. R., Van de Bittner G. C., Chang C. J. (2011) Boronate oxidation as a bioorthogonal reaction approach for studying the chemistry of hydrogen peroxide in living systems. Acc. Chem. Res. 44, 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zielonka J., Sikora A., Joseph J., Kalyanaraman B. (2010) Peroxynitrite is the major species formed from different flux ratios of co-generated nitric oxide and superoxide direct reaction with boronate-based fluorescent probe. J. Biol. Chem. 285, 14210–14216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zielonka J., Zielonka M., Sikora A., Adamus J., Joseph J., Hardy M., Ouari O., Dranka B. P., Kalyanaraman B. (2012) Global profiling of reactive oxygen and nitrogen species in biological systems: high-throughput real-time analyses. J. Biol. Chem. 287, 2984–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zielonka J., Sikora A., Hardy M., Joseph J., Dranka B. P., Kalyanaraman B. (2012) Boronate probes as diagnostic tools for real time monitoring of peroxynitrite and hydroperoxides. Chem. Res. Toxicol. 25, 1793–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sies H. (1993) Ebselen, a selenoorganic compound as glutathione-peroxidase mimic. Free Radic. Biol. Med. 14, 313–323 [DOI] [PubMed] [Google Scholar]
- 21. Wolff S. P. (1994) Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol. 233, 182–189 [Google Scholar]
- 22. Zhang J. H., Chung T. D., Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 [DOI] [PubMed] [Google Scholar]
- 23. d'Alessandro N., Bianchi G., Fang X. W., Jin F. M., Schuchmann H. P., von Sonntag C. (2000) Reaction of superoxide with phenoxyl-type radicals. J. Chem. Soc. Perkin Trans. 29, 1862–1867 [Google Scholar]
- 24. Jin F. M., Leitich J., von Sonntag C. (1993) The superoxide radical reacts with tyrosine-derived phenoxyl radicals by addition rather than by electron-transfer. J. Chem. Soc. Perkin Trans. 29, 1583–1588 [Google Scholar]
- 25. Nagy P., Kettle A. J., Winterbourn C. C. (2009) Superoxide-mediated formation of tyrosine hydroperoxides and methionine sulfoxide in peptides through radical addition and intramolecular oxygen transfer. J. Biol. Chem. 284, 14723–14733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pichorner H., Metodiewa D., Winterbourn C. C. (1995) Generation of superoxide and tyrosine peroxide as a result of tyrosyl radical scavenging by glutathione. Arch. Biochem. Biophys. 323, 429–437 [DOI] [PubMed] [Google Scholar]
- 27. Winterbourn C. C., Pichorner H., Kettle A. J. (1997) Myeloperoxidase-dependent generation of a tyrosine peroxide by neutrophils. Arch. Biochem. Biophys. 338, 15–21 [DOI] [PubMed] [Google Scholar]
- 28. Winterbourn C. C., Parsons-Mair H. N., Gebicki S., Gebicki J. M., Davies M. J. (2004) Requirements for superoxide-dependent tyrosine hydroperoxide formation in peptides. Biochem. J. 381, 241–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Möller M. N., Hatch D. M., Kim H. Y., Porter N. A. (2012) Superoxide reaction with tyrosyl radicals generates para-hydroperoxy and para-hydroxy derivatives of tyrosine. J. Am. Chem. Soc. 134, 16773–16780 [DOI] [PubMed] [Google Scholar]
- 30. Davies M. J. (2004) Reactive species formed on proteins exposed to singlet oxygen. Photochem. Photobiol. Sci. 3, 17–25 [DOI] [PubMed] [Google Scholar]
- 31. Agon V. V., Bubb W. A., Wright A., Hawkins C. L., Davies M. J. (2006) Sensitizer-mediated photooxidation of histidine residues: Evidence for the formation of reactive side-chain peroxides. Free Radic. Biol. Med. 40, 698–710 [DOI] [PubMed] [Google Scholar]
- 32. Gracanin M., Hawkins C. L., Pattison D. I., Davies M. J. (2009) Singlet-oxygen-mediated amino acid and protein oxidation: formation of tryptophan peroxides and decomposition products. Free Radic. Biol. Med. 47, 92–102 [DOI] [PubMed] [Google Scholar]
- 33. Wright A., Hawkins C. L., Davies M. J. (2003) Photo-oxidation of cells generates long-lived intracellular protein peroxides. Free Radic. Biol. Med. 34, 637–647 [DOI] [PubMed] [Google Scholar]
- 34. Morgan P. E., Pattison D. I., Hawkins C. L., Davies M. J. (2008) Separation, detection, and quantification of hydroperoxides formed at side-chain and backbone sites on amino acids, peptides, and proteins. Free Radic. Biol. Med. 45, 1279–1289 [DOI] [PubMed] [Google Scholar]
- 35. Alvarez B., Rubbo H., Kirk M., Barnes S., Freeman B. A., Radi R. (1996) Peroxynitrite-dependent tryptophan nitration. Chem. Res. Toxicol. 9, 390–396 [DOI] [PubMed] [Google Scholar]
- 36. Alvarez B., Radi R. (2003) Peroxynitrite reactivity with amino acid and proteins. Amino Acids 25, 295–311 [DOI] [PubMed] [Google Scholar]
- 37. Pietraforte D., Minetti M. (1997) One-electron oxidation pathway of peroxynitrite decomposition in human blood plasma: evidence for the formation of protein tryptophan-centered radicals. Biochem. J. 321, 743–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sikora A., Zielonka J., Lopez M., Joseph J., Kalyanaraman B. (2009) Direct oxidation of boronates by peroxynitrite: mechanism and implications in fluorescence imaging of peroxynitrite. Free Radic. Biol. Med. 47, 1401–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wright A., Hawkins C. L., Davies M. J. (2000) Singlet oxygen-mediated protein oxidation: evidence for the formation of reactive peroxides. Redox Rep. 5, 159–161 [DOI] [PubMed] [Google Scholar]
- 40. Foote C. S., Peters J. W. (1971) Chemistry of singlet oxygen. 14. Reactive intermediate in sulfide photooxidation. J. Am. Chem. Soc. 93, 3795–3796 [Google Scholar]
- 41. Rougee M., Bensasson R. V., Land E. J., Pariente R. (1988) Deactivation of singlet molecular-oxygen by thiols and related-compounds, possible protectors against skin photosensitivity. Photochem. Photobiol. 47, 485–489 [DOI] [PubMed] [Google Scholar]
- 42. Peskin A. V., Cox A. G., Nagy P., Morgan P. E., Hampton M. B., Davies M. J., Winterbourn C. C. (2010) Removal of amino acid, peptide and protein hydroperoxides by reaction with peroxiredoxins 2 and 3. Biochem. J. 432, 313–321 [DOI] [PubMed] [Google Scholar]
- 43. Gebicki S., Gill K. H., Dean R. T., Gebicki J. M. (2002) Action of peroxidases on protein hydroperoxides. Redox Rep. 7, 235–242 [DOI] [PubMed] [Google Scholar]
- 44. Suryo Rahmanto A., Davies M. J. (2011) Catalytic activity of selenomethionine in removing amino acid, peptide, and protein hydroperoxides. Free Radic. Biol. Med. 51, 2288–2299 [DOI] [PubMed] [Google Scholar]
- 45. Rahmanto A. S., Davies M. J. (2012) Selenium-containing amino acids as direct and indirect antioxidants. IUBMB Life 64, 863–871 [DOI] [PubMed] [Google Scholar]
- 46. Davies M. J. (2005) The oxidative environment and protein damage. Biochim. Biophys. Acta 1703, 93–109 [DOI] [PubMed] [Google Scholar]
- 47. Pattison D. I., Rahmanto A. S., Davies M. J. (2012) Photo-oxidation of proteins. Photochem. Photobiol. Sci. 11, 38–53 [DOI] [PubMed] [Google Scholar]
- 48. Gieseg S., Duggan S., Gebicki J. M. (2000) Peroxidation of proteins before lipids in U937 cells exposed to peroxyl padicals. Biochem. J. 350, 215–218 [PMC free article] [PubMed] [Google Scholar]
- 49. Fu S., Davies M. J., Stocker R., Dean R. T. (1998) Evidence for roles of radicals in protein oxidation in advanced human atherosclerotic plaque. Biochem. J. 333, 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fu S., Dean R., Southan M., Truscott R. (1998) The hydroxyl radical in lens nuclear cataractogenesis. J. Biol. Chem. 273, 28603–28609 [DOI] [PubMed] [Google Scholar]
- 51. Hilf R. (2007) Mitochondria are targets of photodynamic therapy. J. Bioenerg. Biomembr. 39, 85–89 [DOI] [PubMed] [Google Scholar]
- 52. Hunter G. A., Ferreira G. C. (2011) Molecular enzymology of 5-aminolevulinate synthase, the gatekeeper of heme biosynthesis. Biochim. Biophys. Acta 1814, 1467–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nokes B, Apel M., Jones C., Brown G., Lang J. E. (2013) Aminolevulinic acid (ALA): photodynamic detection and potential therapeutic applications. J. Surg. Res. 181, 262–271 [DOI] [PubMed] [Google Scholar]
- 54. Kim C. H., Chung C. W., Choi K. H., Yoo J. J., Kim do H., Jeong Y. I., Kang D. H. (2011) Effect of 5-aminolevulinic acid-based photodynamic therapy via reactive oxygen species in human cholangiocarcinoma cells. Int. J. Nanomedicine 6, 1357–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Golding J. P., Wardhaugh T., Patrick L., Turner M., Phillips J. B., Bruce J. I., Kimani S. G. (2013) Targeting tumour energy metabolism potentiates the cytotoxicity of 5-aminolevulinic acid photodynamic therapy. Br. J. Cancer 109, 976–982 [DOI] [PMC free article] [PubMed] [Google Scholar]