

Abstract
The human X and Y chromosomes evolved from an ordinary pair of autosomes, but millions of years ago genetic decay ravaged the Y chromosome, and only three percent of its ancestral genes survived. We reconstructed the evolution of the Y chromosome across eight mammals to identify biases in gene content and the selective pressures that preserved the surviving ancestral genes. Our findings indicate that survival was non-random, and in two cases, convergent across placental and marsupial mammals. We conclude that the Y chromosome's gene content became specialized through selection to maintain the ancestral dosage of homologous X-Y gene pairs that function as broadly expressed regulators of transcription, translation and protein stability. We propose that beyond its roles in testis determination and spermatogenesis, the Y chromosome is essential for male viability, and plays unappreciated roles in Turner syndrome and in phenotypic differences between the sexes in health and disease.
The human X and Y chromosomes evolved from autosomes over the past 300 million years1. Only 3% of ancestral genes survive on the human Y chromosome2,3, compared to 98% on the X chromosome4. Y-chromosome decay was initially rapid but has virtually halted over the last 25 million years, leaving a stable set of ancestral genes5-7. Mathematical models of Y-chromosome decay assume all ancestral genes are equally likely to survive. However, our initial studies of the human Y chromosome suggested that its gene content is functionally coherent8, leading us to ask whether mammalian Y chromosomes preferentially retained a subset of ancestral genes, and, if so, what qualities these survivors share.
Our earlier analyses8 of the human Y chromosome were hampered by limited knowledge of the gene content of the ancestral autosomes. Our recent cross-species comparisons enabled us to reconstruct their gene content and identify acquired genes on the X and Y chromosomes. The human X chromosome acquired and amplified testis-expressed gene families2,4. Similarly, our comparisons of the human, chimpanzee and rhesus Y chromosomes indicated recent acquisition and amplification of testis-specific genes3,5,6. Thus, both the human X and Y chromosomes gained a specialization for male reproduction by acquiring genes that were not present on the ancestral autosomes2-4.
We excluded acquired genes to independently examine ancestral Y-linked genes for characteristics that distinguished survivors from genes lost to decay. Because the human, chimpanzee and rhesus Y chromosomes share nearly identical ancestral gene content, we analyzed five additional mammals to enhance our ability to detect biases in the decay and survival of ancestral genes. We produced finished sequence of the ancestral portions of the Y chromosomes of marmoset, mouse, rat, bull and opossum and compared them to the published sequences of the human, chimpanzee and rhesus Y chromosomes, all eight corresponding X chromosomes and the orthologous chicken autosomes. Using this expanded tree of species, we reconstructed the evolution of mammalian Y chromosomes from their origin to the present. We concluded that surviving Y-linked genes form a functionally coherent group enriched for dosage-sensitive, broadly expressed regulators of transcription, translation and protein stability.
We produced finished sequence using the SHIMS (Single-Haplotype Iterative Mapping and Sequencing) strategy we employed on primate Y, human X and chicken Z chromosomes (Methods)2-7. These sequences comprise 17 megabases (Mb) and are accurate to about 1 nucleotide per 0.3 Mb (Supplementary Table 1, Extended Data Fig. 1, Methods). To identify ancestral X-Y gene pairs, we searched for Y-homologs of protein-coding genes we had identified as ancestral (Supplementary Tables 2 and 3)2,5. We validated each putative gene by verifying transcriptional activity (Extended Data Fig. 2) and comparing its open reading frame to its chicken ortholog (Supplementary Data 1& 2). We identified 36 different ancestral X-Y gene pairs across all eight species, adding 18 ancestral X-Y gene pairs to the 18 known to be present on the human, chimpanzee and rhesus Y chromosomes (Fig. 1).
Figure 1. Y-linked genes by species and human X homolog location.
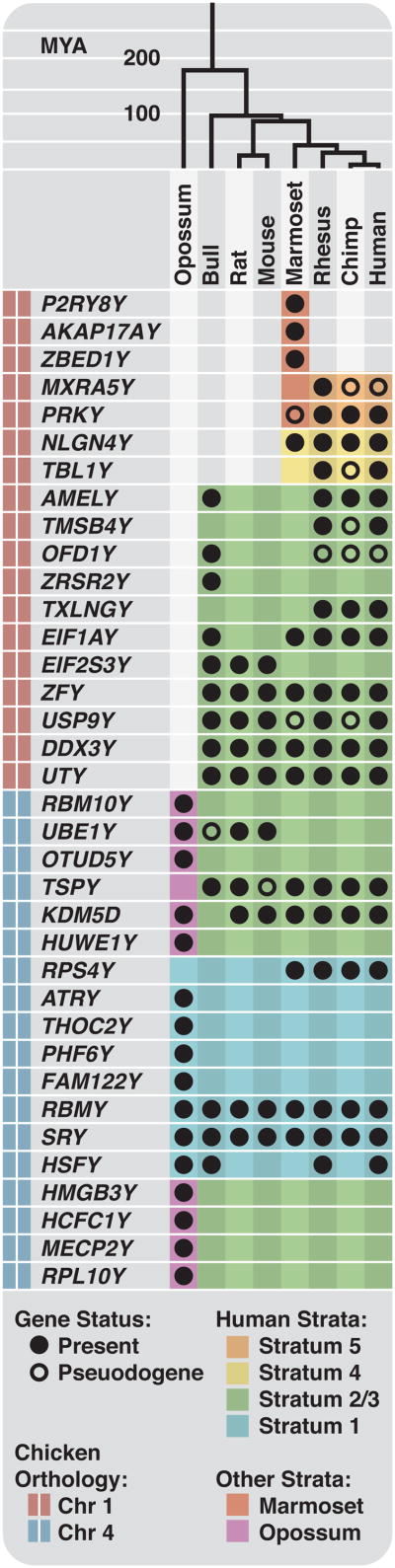
Y-linked genes (filled circles) and pseuodgenes (open circles) listed in order of position of human X chromosome homolog. Added (red bar) and conserved (blue bar) regions of the sex chromosomes are indicated on the left. Human sex chromosome evolution was punctuated by formation of at least 4 evolutionary strata (light blue, green, yellow and orange); other strata formed independently in opossum (purple) and marmoset (red).
Regulatory functions of X-Y gene pairs
Seventeen years ago, we characterized human X-Y gene pairs as specialized in cellular housekeeping functions8. Since then, annotation of the human genome has increased in detail and completeness. We therefore revisited the question of functional coherence and found evidence that X-Y pair genes perform an array of regulatory functions (Fig. 2). Based on annotations of their X homologs, ancestral Y-linked genes appear to regulate each stage of the central dogma: histone lysine demethylases KDM5D (H3K4) and UTY (H3K27); the transcription factor ZFY, regulating stem-cell self-renewal; spliceosomal component RBMY; translation initiation factors DDX3Y and EIF1AY; and the deubiquitinase USP9Y (Fig. 2). Compared to other ancestral genes that survive on the X chromosome, X-Y pair genes are enriched for annotations such as nucleic-acid binding, transcription and translation (Extended Data Table 1, Methods, Supplementary Table 4), suggesting that X-Y pair genes can govern expression of targets throughout the genome.
Figure 2. Regulatory annotations of X-Y pair genes.

Venn diagram depicting regulatory functions predicted for selected X-Y pair genes on basis of UniProt annotations of human X-homolog. Common alternatives to official gene symbols in parentheses.
Convergent survival of X-Y gene pairs
To gain insight into the decay and survival of ancestral genes, we reconstructed Y chromosome evolution, taking advantage of our earlier discovery that synonymous nucleotide divergence between the X and Y sequences of each gene pair increases in stepwise fashion along the human X chromosome1,3,9. This suggested a series of discrete events, most likely inversions on the Y chromosome, that suppressed X-Y crossing over in a single region, or “stratum,” without disturbing gene order on the X chromosome1,9. We used the 36X-Y gene pairs to recalibrate previous reconstructions of evolutionary strata (Extended Data Table 2, Extended Data Figs. 3-5, Methods, Supplementary Tables 2 and 5). In broad agreement with previous reconstructions1-3,9,10, we concluded that the human X and Y chromosomes evolved from ordinary autosomes through chromosomal fusion and formation of at least four strata (Fig. 3, Methods).
Figure 3. Reconstruction of human sex chromosome evolution.

Major events in the evolution of the human sex chromsomes are labeled with approximate dates. After SRY evolved, at least 4 evolutionary strata (light blue, green, yellow and orange) formed in the lineage leading to the human Y chromosome. Each stratum expanded the MSY (male-specific region of the Y, deep blue) at the expense of the PAR (pseudoautosomal region, grey). Genetic decay eliminated most genes from MSY. A chromosomal fusion extended the PAR, generating conserved (XCR/YCR) and added (XAR/YAR) regions.
Our results indicate that the stratum containing UBE1Y and KDM5D formed independently in the placental and marsupial lineages (Extended Data Fig. 4). The same set of ancestral genes became subject to genetic decay in each lineage, forming replicates of the same natural experiment. Out of the 184 ancestral genes shared between these strata, nine survived on the Y chromosome in marsupials, and three survived in placental mammals, but both lineages retained UBE1Y and KDM5D (Fig. 1, Supplementary Table 2, Methods). The convergent survival of two ancestral genes is unlikely to occur under a model where genes survive genetic decay at random (one-tailed Fisher Exact test, P <6.25×10-3).
Remarkable longevity of X-Y gene pairs
Using these recalibrated evolutionary strata, we reexamined the kinetics of genetic decay among ancestral Y-chromosome genes. Analysis of primate Y chromosomes had led us to conclude that, within a stratum, rapid gene loss was followed by stabilization at a baseline set of genes5. With five more divergent mammals, we doubled the constraints on the kinetics of gene loss during human Y chromosome evolution (Fig. 4, Methods) and traced the stability of human Y-chromosome genes to the origin of mammals (Fig. 4). We infer that 97 million years ago, the Y chromosome of the common ancestor of placental mammals carried 18 ancestral genes from stratum 1 and stratum 2/3 (Fig. 1). Of those 18 genes, 14 survive in the human lineage (Fig. 1), and none have been lost in the last 44 million years (Fig. 4). We also examined whether ancestral Y-linked genes were stable in marsupials. Recent analyses of the tammar wallaby Y chromosome identified ten genes shared with the Tasmanian devil11; we observe that all are ancestral and survive in the opossum. This suggests the opossum lineage maintained these genes over the last 78 million years12. We conclude that in both placental and marsupial lineages, some ancestral X-Y gene pairs were remarkably long lived despite rapid decay of surrounding genes.
Figure 4. Decay of Y-linked genes to a baseline level.

Gene numbers (on a log scale on y axis) plotted versus time (in Myr before present on × axis). Filled circles show inferred or observed gene numbers in (from left to right) Ancestral X-Y genes (before stratum formation), the MSY of common ancestor of human and opossum (176 Myr ago), bull (97 Myr ago), mouse and rat (91 Myr ago), marmoset (44 Myr ago), rhesus (30 Myr ago) and chimpanzee (6 Myr ago), and modern human MSY. Lines represent best-fit curves to data points using alternate models of decay. Exponential decay to a constant baseline provides the best fit; shaded regions represent parameters producing an equally good fit.
Two strategies preserved Y-linked genes
In light of the regulatory annotations of X-Y gene pairs, convergent survival of X-Y gene pairs in the placental and marsupial lineages, and the longevity of ancestral X-Y gene pairs across mammals, we sought the evolutionary pressures that drove their survival. We had previously hypothesized that biases in the gene content of the human Y chromosome could arise through two evolutionary strategies: retention and amplification of testis-specific gene families, and conservation of ancestral X-Y gene pairs to maintain comparable expression between males and females8. Using the set of 639 ancestral genes reconstructed through cross-species comparisons of the human X chromosome and orthologous chicken autosomes2,4,5, we tested whether these hypotheses account for the 36 ancestral X-Y pair genes found on eight present-day Y chromosomes.
The Y chromosome was predicted to accumulate genes that enhance male reproductive fitness13, which depends upon sperm production in the adult testis. In each species we studied, ancestral genes that are amplified into multi-copy families are expressed exclusively or predominantly in the testis (Extended Data Fig. 2). However, many such genes have broadly expressed single-copy homologs on orthologous chicken autosomes, on mammalian X chromosomes, and in cases like DDX3Y, EIF1AY, UBE1Y and ZFY, on other Y chromosomes (Extended Data Fig. 2, Supplementary Table 2). This suggests that adoption of testis-specific function preceded gene amplification.
In light of evidence that intrachromosomal gene conversion preserved testis-specific gene families in primate Y-chromosome palindromes14, we speculated that gene amplification contributed to longevity. We ranked surviving Y-linked genes by total branch length across our tree of eight species (Fig. 5a)12. Genes that are amplified in at least one species have a significantly greater branch length than those that are single copy in every species (one-tailed Mann-Whitney U-test: P <4.27 × 10-5) (Fig. 5a). This correlation remains robust when the opossum lineage, with a large number of unique single-copy genes, is excluded (one-tailed Mann-Whitney U-test: P < 5.54 × 10-4). Gene families in tandem arrays show high intraspecies identity and interspecies divergence, a sign that gene conversion is more frequent than mutation in these structures (Extended Data Fig. 6). Two pairs of Y-linked genes – RPS4Y1 and RPS4Y2 in primates and Zfy1 and Zfy2 in mouse – are exceptions; both are physically dispersed and show no sign of recent Y-Y gene conversion (Extended Data Fig. 6). We conclude that genes specialized for male reproduction avoided genetic decay through intrachromosomal gene conversion among members of a Y-linked, multi-copy gene family.
Figure 5. Factors in the survival of Y-linked genes.

Violin plots, white bar - interquartile range, circle - median value; asterisk - significant difference in one-tailed Mann-Whitney U-test. a, Multicopy genes (n = 9) have greater longevity than single copy genes (n = 27) (P < 4.28 × 10-5). b, X-Y pair genes (n = 32) have higher haploinsufficiency probability than other ancestral X genes (n = 478) (P < 6.59 × 10-3). c, X-Y pair genes (n = 28) are have broader expression across human tissues than other ancestral X genes (n = 383) (P < 2.20 × 10-3). d, XY-pair genes (n = 27) have lower dN/dS ratio than other ancestral X genes (n = 489) (P < 3.39 × 10-4).
Next, we examined whether single-copy genes on the Y chromosome survived due to selection to preserve the correct dosage of broadly expressed genes critical to both sexes3,8,15. Most genes on the Y chromosome were lost to genetic decay, and the X chromosome evolved mechanisms to compensate for the lost dosage of Y-linked genes in males8,16,17. The Y chromosome might preferentially retain genes for which the transition state of this process, with a non-functional Y-linked gene and a functional but non-dosage-compensated X-linked homolog, was disadvantageous. Dosage-sensitive genes functioning in many tissues and cell types might be particularly sensitive to these pressures15. We reanalyzed published datasets for evidence that our set of 36 X-Y pair genes systematically differ from the 613 other ancestral genes on the X chromosome with regard to dosage sensitivity18-20, breadth of expression21,22, and intensity of purifying selection23.
We examined whether X-Y pair genes show signs of dosage sensitivity. In humans, gene-by-gene estimates predict a greater likelihood of haploinsufficiency18 for ancestral X-linked genes with surviving Y homologs compared to those lacking Y homologs (one-tailed Mann-Whitney U-test: P <6.59 × 10-3) (Fig. 5b). If surviving X-Y gene pairs maintain ancestral gene dosage, then X-linked genes with surviving Y-linked homologs should escape X inactivation. In human19, mouse20, and opossum24, data on allele-specific expression in females is informative for a subset of ancestral genes (Supplementary Table 2). In each species, a higher proportion of X-linked genes with surviving Y-linked homologs escape X-inactivation compared to those without surviving Y-linked homologs (Supplementary Table 2), and X-Y gene pairs where the X-homolog is subject to X-inactivation have Y-homologs that show signs of functional differentiation. In humans, 12 of 14 informative X-Y pair genes escape X inactivation, but only 168 of 385 remaining ancestral X genes escape (one-tailed Fisher Exact Test P < 1.89 × 10-3) (Supplementary Table 2). The two exceptions, TSPY and RBMY, are amplified into testis-specific gene families (Extended Data Figs. 2 and 6). In mouse, where X chromosome inactivation is more complete20, four of nine informative X-Y pair genes escape X inactivation, while only five of 344 remaining ancestral genes escape (one-tailed Fisher Exact Test P < 2.36 × 10-5)(Supplementary Table 2). All five exceptions (Sry, Rbmy, Ube1y, Usp9y and Zfy) evolved testis-specific expression in mouse (Extended Data Fig. 2). Lastly, despite differences in the mechanisms of X inactivation between placental and marsupial mammals, all eight informative opossum X-Y pair genes escape X inactivation, but only 15 of 138 remaining ancestral genes escape (one-tailed Fisher Exact Test P < 1.17 × 10-7) (Supplementary Table 2).
Additionally, the Turner syndrome phenotype (classically associated with a 45,X karyotype, or monosomy X) suggests a strict dosage requirement for one or more sex-linked genes in humans. If dosage of X-Y pair genes is partially responsible for the Turner syndrome phenotype, it could explain the differing features of monosomy X in humans and mice. Monosomy X in humans results in poor in utero viability. Less than 1 in 100 45,X conceptuses survive to term25,26. Those that do survive are often mosaic for all or part of a second sex chromosome26,27, so that variability in the Turner phenotype may reflect variability in dosage of X-Y pair genes among tissues as well as individuals. The mouse phenotype of monosomy X is less severe; animals are small but viable and have reduced fertility28-30. This milder phenotype may reflect a dearth of genes on the mouse X chromosome that require two doses: only nine ancestral genes survive on the mouse Y chromosome (compared to 17 in human), and fewer X-linked genes escape inactivation.
Finally, human X-linked intellectual disability syndromes provide evidence for the dosage sensitivity of specific X-Y pair genes. UTX, KDM5C and NLGN4X all have Y homologs, escape X inactivation, and appear to be haploinsufficient (Supplementary Table 2). Mutations in UTX cause Kabuki syndrome; both duplications and deletions result in multiple congenital anomalies and intellectual disability in males and females31. KDM5C is associated with X-linked intellectual disability in hemizygous males, and heterozygous females with mild intellectual disability have been reported in several families32. In both hemizygous males and heterozygous females, NLGN4X mutations are associated with autism spectrum disorders and learning disabilities reminiscent of the cognitive and behavioral phenotypes of Turner Syndrome33. Even the human X-homologs of X-Y gene pairs identified only in the opossum (HCFC1, HUWE1 and MECP2) still display acute sensitivity to gene dosage. In humans each of these X-linked genes has no Y homolog and is subject to X inactivation19 (Supplementary Table 2). Nevertheless, a non-coding mutation causing overexpression of HCFC1, as well as duplications of HUWE1 and MECP2, have been implicated in X-linked intellectual disability in human males34-36. Thus, even though the human Y-homologs of HCFC1, HUWE1 and MECP2 were lost their Y-homologs and the surviving X-homologs have evolved dosage compensation, their gene dosage remains tightly constrained.
X-Y pair genes functioning across many tissues and cell types may face additional selective constraints that prevent both loss of the Y-linked gene and evolution of a dosage-compensated gene on the X chromosome. In all eight species, single-copy Y-linked genes are broadly expressed across adult tissues (Extended Data Fig. 2), with two major exceptions, in which both members of the X-Y pair share ancestrally restricted expression: AMELY, whose ortholog disappeared in the toothless avian lineage, is expressed only in developing tooth buds37; and HSFY, which is testis specific, and has a chicken ortholog that is predominantly expressed in testis. In chicken, the autosomal orthologs of mammalian X-Y pair genes have significantly broader expression across adult tissues than do the orthologs of ancestral genes that survive only on the X chromosome, and X-Y pair genes maintain this broader expression across mammals (one-tailed Mann-Whitney U-test: chicken P < 3.38 × 10-3, human P < 2.20 × 10-3, rhesus P < 1.39 × 10-7, mouse P < 4.74 × 10-8, rat P < 4.63 × 10-6, bull P < 1.20 × 10-5) (Fig. 5c, Supplementary Table 2). This breadth of expression extends to the earliest stages of development. Relative to other X-linked ancestral genes on the X chromosome, X-Y pair genes are enriched for genes up-regulated after the onset of zygotic gene activation in a time course of human, mouse and bovine pre-implantation development (one-tailed Fisher Exact Test, human P < 2.13 × 10-2, mouse P < 5.93 × 10-4, bull P < 1.37 × 10-2) (Supplementary Table 2). X-Y pair genes are more broadly expressed than other ancestral genes that survive on the X chromosome, across multiple tissues and developmental time.
Unlike the testis-expressed multi-copy gene families, the broadly expressed, dosage-sensitive single-copy genes of the Y chromosome cannot avoid genetic decay through intrachromosomal gene conversion, and must rely on purifying selection. Our previous survey of human sequence variation among the Y chromosome's single-copy genes showed that natural selection operated effectively to preserve the amino acid sequences of Y-linked genes in the human lineage38. If X-Y gene-pairs are haploinsufficient, alleles that alter the function of the X-linked homologs should be detrimental in both males and females. We examined Ensembl human-mouse ortholog alignment data for evidence that the X-linked homologs of X-Y gene pairs were subject to strong purifying selection23. Relative to other ancestral genes on the X chromosome, the X-linked homologs of X-Y gene pairs have a reduced ratio of non-synonymous to synonymous substitution rates (dN/dS) (one-tailed Mann-Whitney U-test: P < 3.39 × 10-4) (Fig. 5d). We conclude that these broadly expressed, dosage-sensitive X-Y pair genes are under more intense purifying selection than their neighbors on the X chromosome.
Human Y genes ensure male viability
We conclude that the longevity of many Y-linked genes is due to selection to maintain expression, in males, of dosage-sensitive, broadly expressed X-Y gene pairs at levels comparable to their autosomal ancestors. This model predicts that members of surviving single-copy X-Y gene pairs should be functionally interchangeable. Indeed, the human Y-linked genes RPS4Y and DDX3Y are functionally interchangeable with their X homologs in vitro39,40, and although the histone demethylase domain of the mouse Y-linked gene Uty appears to be inactive, mouse Utx and Uty are functionally redundant during mouse embryonic development41-43.
Previous observations suggest that the selective pressures that maintained these Y-linked genes remain strong in the human lineage; about 99% of human 45,X conceptuses are inviable, and those that survive to term are often mosaic for all or part of a second sex chromosome25-27. Therefore, we also conclude that the broadly expressed, dosage-sensitive genes of the human Y chromosome – along with their X-homologs, which escape X chromosome inactivation – are collectively haplolethal. We propose that, as a set, these dozen Y-linked genes are essential for the viability of 46,XY fetuses (Methods, Extended Data Fig. 7). Thus we propose a third organismal function of the human Y chromosome: that it carries single-copy genes that ensure male viability. This is distinct from the human Y chromosome's more widely appreciated roles in testis determination through SRY and sperm production through ampliconic gene families.
Sex differences in health and disease
The myriad differences between human males and females – from anatomy to disease susceptibility – arise from differences in the genes of the X and Y chromosomes that appeared as these chromosomes diverged in gene content from their autosomal ancestors. Of the 17 surviving ancestral genes on the human Y chromosome, four (SRY, RBMY, TSPY, and HSFY) have clearly diverged in function from their X homologs (SOX3, RBMX, TSPX, and HSFX) to play male-specific roles in reproductive development or gametogenesis. Since all genes on the Y chromosome were exposed to selection only in males, even widely expressed ancestral genes may exhibit subtle functional differences from their X-linked homologs. Particularly worthy of consideration are eight global regulators of gene activity that exist as X-encoded and Y-encoded (male-specific) protein isoforms in diverse human tissues: UTX/Y, EIF1AX/Y, ZFX/Y, RPS4X/Y, KDM5C/D, DDX3X/Y, USP9X/Y, and TBL1X/Y. These exemplify a fundamental sexual dimorphism, at a biochemical level, throughout the human body, that derives directly from genetic differences between the X and Y chromosomes. It will surely be of interest to determine whether this dimorphism plays a role in diseases, outside the reproductive tract, that occur with greater frequency or severity in males or females.
Online Only Methods
1. Single-Haplotype Iterative Mapping and Sequencing (SHIMS) strategy
The single-haplotype iterative mapping and sequencing (SHIMS) strategy was used to assemble partial male-specific region of the Y (MSY) sequences for marmoset, mouse, rat, bull, and opossum. We previously employed the SHIMS strategy to obtain the full-length MSY sequences of human, chimpanzee, and rhesus macaque5,6,51. The major steps in the SHIMS strategy are outlined below:
Initial BAC selection and sequencing. MSY-derived bacterial artificial chromosome (BAC) clones are identified and organized into contigs of overlapping BACs using one or more of the following methods based on resource availability: i. high-density filter hybridization using pools of overgo probes, ii. electronic mapping of BAC-end sequences to female genomic sequence, and iii. BAC fingerprint contig analysis. Assembled MSY contigs are verified by PCR using MSY-specific STS markers. Tiling paths of clones are selected for sequencing.
Distinguishing repeat copies and finding true tiling paths. Overlaps between BACs within repetitive regions are scrutinized for sequence differences or sequence family variants (SFVs). If SFVs are found, this indicates that the BACs belong to distinct copies of the same repeat unit. SFV patterns are then used to identify true overlapping BACs. New tiling paths are produced, and the process is reiterated until all overlaps are consistent.
Extension and joining of BAC contigs. Identify clones that extend outward from or link existing contigs using high-density filter hybridization.
Clone selection
We designed overgo probes from male-specific sequences identified by electronic subtraction of female genomic sequences from male (or mixed male and female) genomic sequences. Because of this approach, our clone selection was not biased towards gene-containing regions. We selected clones from existing male BAC libraries CHORI-259, RPCI-24, CHORI-240, and VMRC-6 (http://bacpac.chori.org), as well as custom BAC libraries MARMAEX, RNAEX, RNECO, BTDAEX, and MDAEX constructed by Amplicon Express (http://www.genomex.com).
Sequencing error rate
The sequencing error rate for the partial MSY sequences for marmoset, mouse, rat, bull, and opossum is approximately one nucleotide per 0.3Mb.
Order and orientation of contigs
We ordered and oriented our clone-based contigs using both radiation hybrid mapping and fluorescence in-situ hybridization (FISH). We used a previously published 10,000-rad rhesus macaque radiation hybrid panel52, and a set of new 25,000-rad radiation hybrid panels from marmoset, mouse, bull, and opossum, constructed by William J. Murphy, James E. Womack, and Elaine Owens. For bull FISH, we used a primary fibroblast cell line derived from the sequenced animal, L1 Domino (JEW 85), received from James E. Womack and Elaine Owens of Texas A&M University. For marmoset FISH, we used cell lines WHT5952 (father of sequenced animal) and WHT5955 (brother of sequenced animal) received from Suzette Tardif and Peter Hornsby in the Sam and Ann Barshop Institute for Longevity and Aging Studies at the University of Texas Health Science Center. For rat FISH, we created cell line WHT5890, embryonic fibroblasts derived from non-phenotypic SHR rat line from Charles River Labs. For mouse FISH, we established embryonic fibroblast cell lines from the C57BL/6 strain from Jackson Laboratories. For opossum FISH, we used primary fibroblast cell line WHT6354 derived from opossum A0067 from Paul Samollow of Texas A&M University.
Gap closure
Regions composed of repeats with units less than 10kb and greater than 99% identity frustrate the assembly of individual BAC clones and are not well-represented in our assemblies. These regions include both gene-poor regions like centromeres, telomeres, and heterochromatin, as well as gene-rich regions, such as the TSPY arrays on the human and bull Y-chromosomes. No current technology is able to access these regions. Wherever possible we attempted to find the boundaries of these arrays, obtain a representative repeat unit, and verify the contiguity of the array by FISH.
The gaps in both bull and opossum assemblies (Extended Data Figure 1) are the result of arrays of short, highly-identical repeats of this type. The bull Y-chromosome assembly is interrupted by extremely long tandem arrays of a ∼3kb repeat unit, but all contigs are ordered and oriented, and the homogeneity of these arrays was confirmed by FISH. The opossum Y-chromosome assembly is interrupted by stretches of several different heterochromatic repeat units. The opossum Y chromosome is too small to resolve these regions by FISH. However, we are confident that our assembly is not biased towards gene-rich regions due to our almost exclusive use of electronic subtraction to generate probes.
2. PANTHER Statistical Overrepresentation Test
We employed the PANTHER statistical overrepresentation test to identify functional coherence among the 36 ancestral X-Y pair genes relative to the remaining ancestral X genes. For each functional category, the PANTHER software employs a binomial test to identify statistically significant overrepresentation (or underrepresentation) of the genes in an input list relative to the genes in a reference list53. This test makes no assumptions about the processes that generated either the input or reference gene lists, aside from the null hypothesis that both the input and reference list are drawn from the same population, such that each functional category is equally well represented in the two lists53.
We manually curated our gene lists to ensure that any overrepresentation we identified was the result of processes that favored the survival of ancestral genes on the Y chromosome, rather than the processes that drove gene acquisition and amplification. First, we restricted our analyses to X-Y gene pairs that included one of the 639 ancestral X-linked genes we identified in our reconstruction of the ancestral autosomes from which the X and Y chromosomes evolved (Supplementary Table 2). Second, we excluded any X-Y gene pairs we could identify as arising from gene acquisition by the Y chromosome after the start of decay; for example, we excluded the X-Y pair genes resulting from the human-specific X-transposed region.
Out of the 639 ancestral X-linked genes, we identified 36 with Y homologs (Figure 1) that appear to have survived through the genetic decay of the Y chromosome in any one of our 8 species. All 36 of these genes mapped to a human identifier in PANTHER. Of the 613 remaining ancestral genes, 11 were lost in the human lineage, and 38 did not map to a human identifier in PANTHER, leaving 554 ancestral X genes without a surviving Y homolog in any of our 8 species (Supplementary Table 2).
We used the PANTHER statistical overrepresentation test to identify functional annotations that were enriched among the 36 ancestral X-Y pair genes that survive on the Y chromosome of one or more of the eight species we sequenced, relative to the reference list of 554 other ancestral X genes (Extended Data Table 1). We selected the 554 other ancestral X genes as a reference list, instead of all human genes, to control for any functional coherence among the ancestral genes that pre-dated the start of Y-chromosome decay, as well as the possibility that the annotation of the X chromosome is more complete than that of the autosomes.
We found that the annotation of the combined set of 590 ancestral X genes (36 ancestral X-Y pairs and 554 other ancestral X genes) is more complete than the rest of the human genome. Relative to all human genes, the 590 ancestral X genes are significantly underrepresented for genes that are “Unclassified” in the GO Biological Process (P < 1.96 × 10-7), GO Molecular Function (P < 1.52 × 10-2), and Panther Protein Class (P < 1.00 × 10-6) categories (Supplementary Table 4). On the other hand, the 590 ancestral X genes are overrepresented for three GO Biological Process annotations: “neurological system process” (P < 3.14 × 10-2), “cellular process” (P < 4.50 × 10-2), and “synaptic transmission” (P < 4.59 × 10-2) (Supplementary Table 4). We note that the “cellular process” annotation encompasses “synaptic transmission,” and that “cellular process” would not reach statistical significance if genes annotated as “synaptic transmission” were excluded. We obtained similar results when we excluded the 36 X-Y gene pairs and tested the 554 other ancestral X genes against all human genes, although the “Unclassified” annotation in the GO Molecular Function category failed to reach significance (Supplementary Table 4). We interpret these results as evidence that the intensive study of X-linked intellectual disability syndromes has produced a richer annotation of brain and cognitive functions on the X chromosome relative to the autosomes.
3. Identification and recalibration of evolutionary strata
We identified chromosomal fusions and evolutionary strata across our tree of species, using a combination of information: syntenic orthologs across species, synonymous nucleotide divergence between X-Y gene pairs, and phylogenetic analysis of X-Y gene pairs.
A chromosomal fusion in the ancestor of placental mammals
Previous comparisons between marsupial and placental sex chromosomes identified a conserved region shared between the sex chromosomes of placental and marsupial mammals, and an added region unique to the sex chromosomes of placental mammals10. Orthologs of genes from the added and conserved regions are found on separate autosomes in the chicken genome, the best assembled outgroup to placental and marsupial mammals, as well as in the genomes of 4 teleost fish2,9. These interspecies comparisons of X chromosomal and autosomal gene content established the model that the present day human X and Y chromosomes derived from the X-conserved region existed in the common ancestor of placental and marsupial mammals, and later, a chromosomal fusion brought the added and conserved regions together in the ancestor of placental mammals.
Our comparisons of Y-linked gene content support this model. Across all seven placental mammals, we identified 17 X-Y pairs that derive from the added region (Figure 1). As, expected, none of these pairs have an ortholog on the opossum Y chromosome (Figure 1). Additionally, we note that the opossum orthologs of placental added region genes reside on two autosomes in opossum, chromosome 4 and chromosome 7 (Supplementary Table 2, Extended Data Figure 3). Because the orthologs of placental X-added region genes are also syntenic in an outgroup, chicken2,9, we conclude that the ancestral autosome orthologous to the added region of the placental sex chromosomes broke apart in the opossum lineage (Figure 3).
Reconstruction of evolutionary strata
The chromosomal fusion event recorded in the placental added and conserved regions served as a palimpsest for the formation of evolutionary strata. Previous comparisons of the human X and Y chromosomes identified five evolutionary strata overlaid across the added and conserved regions on the X chromosome1,9. The oldest evolutionary strata, stratum one and stratum two, occupied the X-conserved region, while the X-added region contained strata three, four, and five, as well as the freely recombining pseudoautosomal region (PAR)1,9. We reexamined these findings across our expanded set of species and gene pairs. Within each species, we aligned single-copy X-Y gene pairs and calculated the nucleotide divergence (dS) between them (Supplementary Table 5). In the two oldest strata, uncertainty in the levels of divergence prevented us from distinguishing strata, in these cases we sought to distinguish strata by phylogenetic analysis (Extended Figure 4). The data from our broader comparison provides additional details that allow us to refine previous reconstructions of the evolutionary trajectory of the human sex chromosomes. In particular, we find no support of the distinction between strata two and three, and propose that a single combined stratum arose in the placental lineage after the fusion of the added and conserved regions.
Stratum two formed independently in placental and marsupial lineages
Based on the analysis of five X-Y gene pairs, previous reconstructions placed the two oldest strata before the divergence of placental and marsupial mammals1,3. We found that placental Y-linked genes from both stratum one and stratum two have orthologs in the opossum (Figure 1), as would be expected if both strata formed in the common ancestor of placental and marsupial mammals. Alternatively, the survival of Y-linked genes in both lineages could be the result of independent stratum formation and convergent survival of Y-linked genes after the divergence of marsupial and placental mammals. We examined both possibilities in light of our new data from the marsupial lineage. Sixteen opossum X-Y pairs are drawn from across the entire X-conserved region, encompassing both stratum one and stratum two. However, all opossum X-Y pairs (with the exception of SOX3/SRY) displayed a similarly high level of divergence (dS >= 1) (Supplementary Table 5).
Because saturation for synonymous substitutions prevented us from using nucleotide divergence to distinguish these ancient strata in the opossum, we sought to distinguish between them by phylogenetic analysis of X-Y gene pairs across all eight species, using autosomal orthologs in chicken as the outgroup. We found that across both placental and marsupial mammals, orthologs of the stratum one genes SRY, RBMY, and HSFY were more closely related to each other than to X-linked homologs (Extended Data Figure 4). Genes from stratum two showed a different pattern; as a group, placental orthologs of UBE1Y and KDM5D are more closely related to placental X-linked homologs than to their marsupial orthologs (Extended Data Figure 4). We conclude that stratum one, containing SRY, the male sex-determining gene54,55, evolved only once, before the divergence of marsupial and placental mammals, but that the formation of a second stratum proceeded independently in both lineages (Figure 1, Figure 3).
No support for the distinction between stratum two and stratum three
Previous reconstructions drew a distinction between stratum two and stratum three because stratum two had been dated before the divergence of placental and marsupial mammals and stratum three contained genes from the region added to the placental sex chromosomes. After finding that only the first and not the second stratum preceded the divergence of placental and marsupial lineages, we reexamined the distinction between stratum two and stratum three in placental mammals. We compared stratum two and stratum three gene pairs only from the four primate species; no single-copy gene pairs from stratum two survived on the bull Y chromosome, and single-copy gene pairs from both strata are saturated for synonymous substitutions in the rodent lineage (Figure 1, Supplementary Table 5). We also excluded AMELY and ZFY, which participated in interchromosomal gene conversion after stratum formation (Supplementary Table 5, Extended Data Figure 5)56,57. We found that within each of the four primate species, the divergence between KDM5C and KDM5D in stratum two is within the range of divergence of X-Y gene pairs from stratum three (Supplementary Table 5). Without phylogenetic or divergence data that distinguish stratum two from stratum three, we propose that together they represent a single stratum (Figure 1, Figure 3). This combined stratum formed in the ancestor of all placental mammals, after the chromosomal fusion event expanded the PAR of the X and Y chromosomes, but before bull diverged from the other six species, more than 97 million years ago (Figure 3)12.
Location of the ancestral placental PAR boundary
The formation of this combined stratum defined the PAR boundary in the placental ancestor, but subsequent X-Y gene conversion events in AMELY have made it difficult to establish the location of this boundary using only data from the human X and Y chromosomes, with proposed boundaries ranging in location from as distal as between KAL1 and TBL1X and as proximal as between AMELX and TMSB4X1,3,9,58. The 4.2 megabases between KAL1 and TMSB4X comprise almost 3% of the human X chromosome. Our expanded dataset provides additional constraints that narrow this region by a factor of 10. We find that AMELY is present on the human, chimpanzee, rhesus macaque, and bull Y chromosomes, while TBL1Y is present only in human, rhesus macaque and, as a pseudogene, in chimpanzee (Figure 1). The bovine ortholog of TBL1X is located in the PAR, and furthermore, MID1, which is located between TBL1X and AMELX on the human X chromosome, has an ortholog in the mouse PAR (Extended Data Table 2)59. We conclude that the ancestral placental PAR boundary was proximal to both TBL1X and MID1, but distal to AMELX. This places TBL1Y in stratum four, and AMELY in the combined stratum two/three. The low divergence between AMELX and AMELY is likely the result of lineage-specific X-Y gene conversion events after stratum formation, similar to what has been observed for ZFY (Supplementary Table 5, Extended Data Figure 5)56,57.
Lineage-specific evolutionary strata in primates
After the formation of the stratum that established the ancestral placental PAR boundary, lineage-specific evolutionary strata continued to form. Previous reconstructions identified two additional strata in the human lineage with a boundary between PRKX and NLGN4X9. We recalculated the age of human strata 4 and 5 following previously published methods9, using the updated figure of 29.6 MYA for the divergence between old world monkeys and hominoids12.
NLGN4Y, from stratum four, is present in all four primate species, while TBL1Y is present in human and rhesus macaque, with a pseudogene in chimpanzee. The X-Y divergence in human stratum four is compatible with an origin in the simian ancestor, over 44 million years ago, close to the time of divergence of platyrrhine and catarrhine primates (Figure 3)9,12.
In contrast, human stratum five dates to 32-34 million years ago, prior to the divergence of rhesus macaque from human and chimpanzee9,12. All three species share the PRKY gene, as well as a common PAR boundary5. We conclude that stratum five was already established in the catarrhine ancestor, and afterwards, no further strata formed in the human, chimpanzee, and rhesus lineages (Figure 3), although subsequent insertions, deletions, and rearrangements generated different configurations of the male-specific region of the Y chromosome in each species5.
Independently, the marmoset lineage also formed a fifth stratum with a more distal PAR boundary than the human, chimpanzee, and rhesus (Figure 1, Supplementary Figure 7). Because the marmoset whole genome shotgun sequence is a mixture of male and female sequence, and this marmoset-specific stratum formed relatively recently, it is not possible to differentiate between X and Y derived contigs in the marmoset whole genome shotgun sequence. P2RY8Y, SFRS17AY, and ZBED1Y are the only survivors out of 24 ancestral genes in this stratum (Figure 1, Supplementary Table 2), demonstrating that, at least while strata are young, genetic decay is both swift and extensive5,60.
4. Modeling kinetics of Y-chromosome decay
We modeled the numbers of ancestral genes within individual MSY strata as a function of time in millions of years before the present by fitting a one-phase exponential decay model with a baseline constant (below) to our data using nonlinear regression analysis in GraphPad Prism 5.0. Parameters for each stratum are given in the Source Data for Figure 4. This one-phase exponential decay model gives the number of genes at time t, N(t):
Where N0 is the number of genes within given stratum in ancestral autosomal/pseudoautosomal portion of genome at the start of stratum formation, K is the decay constant, and b is the baseline (approximated by the number of active ancestral genes within that stratum on the human Y chromosome).
5. Haplolethality of broadly expressed, dosage-sensitive X-Y pair genes
We propose that the broadly expressed, dosage-sensitive genes of the human Y chromosome, along with their X homologs that escape X chromosome inactivation, are collectively haplolethal. Twelve human XY-gene pairs meet this criterion: RPS4X/RPS4Y, ZFX/ZFY, TBL1X/TBL1Y, PRKX/PRKY, USP9X/USP9Y, DDX3X/DDX3Y, UTX/UTY, TMSB4X/TMSB4Y, NLGN4X/NLGN4Y, TXLNG/CYORF15, KDM5C/KDM5D, and EIF1AX/EIF1AY.
We compiled a list of cases with non-mosaic partial-Y deletions removing one or more of these genes to determine if any single gene was haplolethal. We found that the Y-homolog of each X-Y gene pair was deleted in one or more cases (Extended Data Figure 7, Extended Data Table 3). Thus, we attribute the inviability of 45,X conceptuses to a collective haplolethality for several X-Y gene pairs, and not to any single gene pair. Supporting the notion that these gene pairs are dosage-sensitive, TBL1Y and PRKY, two genes deleted in the rare J2e1*/M241 Y chromosome haplotype61, are the only 2 of these 12 gene pairs with X-linked homologs that do not always escape X-inactivation19.
We also searched the literature for reports of structurally variant X chromosomes in females, where one X chromosome was deleted for one or more of these 12 genes (Extended Data Figure 7, Extended Data Table 3). These reports are not inconsistent with a collective haplolethality for X-Y gene pairs, but the interpretation of these cases is complicated by viability effects mediated by the X-inactivation center (XIC), and a possible critical region for ovarian failure near USP9X62.
We found cases where a variant X chromosome has been transmitted from mother to daughter, and which are therefore unlikely to be mosaic, that delete as many as 7 genes (PRKX, NLGN4X, TBL1X, TMSB4X, TXLNG, EIF1AX, and ZFX)63-69.
We also found reports of extensive de novo deletions that eliminate 11 of these 12 genes, leaving only RPS4X on the long arm66,69. However, we cannot exclude the possibility that these cases are mosaic for 46,XX cells in a cell lineage other than the blood. The absence of familial cases of deletions of this type may because of a critical region for ovarian failure on the short arm of the X chromosome; both ZFX and USP9X have been proposed as candidate genes62.
We could not find any reports of deletions of RPS4X. RPS4X is located on the long arm, between the centromere and the XIC. We believe that the absence of reports of X chromosome variants deleted for RPS4X reflects the proximity of RPS4X to the XIC rather than haplolethality of RPS4X.
Supplementary Material
Acknowledgments
We thank W.J. Murphy, E. Owens and J. E. Womak for generating radiation hybrid panels and for assistance in mapping; L. Lyons and W.J.M. for providing the rhesus radiation hybrid panel; A. Kaur for a rhesus cell line; S. Austad, P. Hornsby and S. Tardif for marmoset cell lines; M. Brown for rat cell lines; J.E.W. for bull fibroblasts; W. Johnson and S. O'Neil for rhesus tissues samples; W.J., S.O. and S.T. for marmoset tissue samples; M. Turner for rat tissue samples; J.E.W. for bull tissue samples; P. Samollow for opossum tissue samples; E. Vallender for Tamarin DNA; B. Chowdhary and T. Raudsepp for FISH experiments in the bull; C. Friedman and B. Trask for flow-sorted marmoset Y chromosomes; B.T. for sizing rat Y chromosomes; C. Burge for permission to assemble transcriptome data from SRR594455, SRR594463, and SRR594508; J. Alföldi for permission to assemble transcriptome data from SRR500909; R.B. Norgren for permission to assemble transcriptome data from SRR544870; and A. Godfrey, Y. Hu and B. Lesch for comments on the manuscript. Supported by National Institutes of Health and Howard Hughes Medical Institute.
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions: D.W.B., J.F.H., H.S., S. Rozen, W.C.W., R.A.G., R.K.W. and D.C.P. planned the project. J.F.H., H.S., L.G.B., T.-J.C., N.K., S.Z., and J.A. performed BAC mapping, radiation hybrid mapping and real-time polymerase chain reaction analyses. T.G., S. Rock, C.K., R.S.F, S.D., Y.D., D.M., Z.K., L.L., C.B., Q.W., J.W., M.H., S.L., L.N. and D.M.M. were responsible for BAC sequencing. D.W.B., J.F.H. and H.S. performed comparative sequence analyses. T.P. performed FISH analyses. D.W.B. and D.C.P. wrote the paper.
Author Information cDNA sequences of Y-linked genes and their X-linked homologs have been deposited in GenBank (http://www.ncbi.nlm.nih.gov) under accession numbers FJ526999-FJ527008, FJ627275, FJ627276, FJ627278, FJ659845, FJ959389, GQ253467-GQ253475, GQ338825, GU304599-GU304603, GU304606-GU304607, JF487792-JF487795, JF827151, JF827152, JN086997, JN585955, JN585956, JQ313990-JQ313992 and bioproject PRJNA221163. 454 and Illumina testis cDNA sequences have been deposited in GenBank under accession numbers SRX335333, SRX335335, SRX335470, SRX335472, SRX335475-SRX335477, SRX358238, and SRX359414.
The authors declare no competing financial interests.
Literature Cited
- 1.Lahn BT, Page DC. Four evolutionary strata on the human X chromosome. Science. 1999;286:964–967. doi: 10.1126/science.286.5441.964. [DOI] [PubMed] [Google Scholar]
- 2.Bellott DW, et al. Convergent evolution of chicken Z and human X chromosomes by expansion and gene acquisition. Nature. 2010;466:612–616. doi: 10.1038/nature09172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skaletsky H, et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–837. doi: 10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- 4.Mueller JL, et al. Independent specialization of the human and mouse X chromosomes for the male germline. Nature Genet. 2013;45:1083–1087. doi: 10.1038/ng.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hughes JF, et al. Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature. 2012;483:82–86. doi: 10.1038/nature10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hughes JF, et al. Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature. 2010;463:536–539. doi: 10.1038/nature08700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes JF, et al. Conservation of Y-linked genes during human evolution revealed by comparative sequencing in chimpanzee. Nature. 2005;437:100–103. doi: 10.1038/nature04101. [DOI] [PubMed] [Google Scholar]
- 8.Lahn BT, Page DC. Functional coherence of the human Y chromosome. Science. 1997;278:675–680. doi: 10.1126/science.278.5338.675. [DOI] [PubMed] [Google Scholar]
- 9.Ross MT, et al. The DNA sequence of the human X chromosome. Nature. 2005;434:325–337. doi: 10.1038/nature03440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watson JM, Spencer JA, Riggs AD, Graves JA. The X chromosome of monotremes shares a highly conserved region with the eutherian and marsupial X chromosomes despite the absence of X chromosome inactivation. Proc Natl Acad Sci USA. 1990;87:7125–7129. doi: 10.1073/pnas.87.18.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murtagh VJ, et al. Evolutionary history of novel genes on the tammar wallaby Y chromosome: Implications for sex chromosome evolution. Genome Res. 2012;22:498–507. doi: 10.1101/gr.120790.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hedges SB, Dudley J, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22:2971–2972. doi: 10.1093/bioinformatics/btl505. [DOI] [PubMed] [Google Scholar]
- 13.Fisher RA. The Evolution of Dominance. Biol Rev. 1931;6:345–368. [Google Scholar]
- 14.Rozen S, et al. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature. 2003;423:873–876. doi: 10.1038/nature01723. [DOI] [PubMed] [Google Scholar]
- 15.Kaiser VB, Zhou Q, Bachtrog D. Nonrandom gene loss from the Drosophila miranda neo-Y chromosome. Genome Biol Evol. 2011;3:1329–1337. doi: 10.1093/gbe/evr103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jegalian K, Page DC. A proposed path by which genes common to mammalian X and Y chromosomes evolve to become X inactivated. Nature. 1998;394:776–780. doi: 10.1038/29522. [DOI] [PubMed] [Google Scholar]
- 17.Ohno S. Sex Chromosomes and Sex-linked Genes. Springer-Verlag; Berlin: 1967. [Google Scholar]
- 18.Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- 20.Yang F, Babak T, Shendure J, Disteche CM. Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res. 2010;20:614–622. doi: 10.1101/gr.103200.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science. 2012;338:1593–1599. doi: 10.1126/science.1228186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie D, et al. Rewirable gene regulatory networks in the preimplantation embryonic development of three mammalian species. Genome Res. 2010;20:804–815. doi: 10.1101/gr.100594.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flicek P, et al. Ensembl 2014. Nucleic Acids Res. 2014;42:D749–755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Douglas KC, Vandeberg JL, Clark AG, Samollow PB. Chromosome-wide profiling of X-chromosome inactivation and epigenetic states in fetal brain and placenta of the opossum, Monodelphis domestica. Genome Res. 2014;24:70–83. doi: 10.1101/gr.161919.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cockwell A, MacKenzie M, Youings S, Jacobs P. A cytogenetic and molecular study of a series of 45,X fetuses and their parents. J Med Genet. 1991;28:151–155. doi: 10.1136/jmg.28.3.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hook EB, Warburton D. The distribution of chromosomal genotypes associated with Turner's syndrome: livebirth prevalence rates and evidence for diminished fetal mortality and severity in genotypes associated with structural X abnormalities or mosaicism. Hum Genet. 1983;64:24–27. doi: 10.1007/BF00289473. [DOI] [PubMed] [Google Scholar]
- 27.Hassold T, Benham F, Leppert M. Cytogenetic and molecular analysis of sex-chromosome monosomy. Am J Hum Genet. 1988;42:534–541. [PMC free article] [PubMed] [Google Scholar]
- 28.Burgoyne PS, Tam PP, Evans EP. Retarded development of XO conceptuses during early pregnancy in the mouse. J Reprod Fertil. 1983;68:387–393. doi: 10.1530/jrf.0.0680387. [DOI] [PubMed] [Google Scholar]
- 29.Burgoyne PS, Baker TG. Oocyte depletion in XO mice and their XX sibs from 12 to 200 days post partum. J Reprod Fertil. 1981;61:207–212. doi: 10.1530/jrf.0.0610207. [DOI] [PubMed] [Google Scholar]
- 30.Burgoyne PS, Evans EP, Holland K. XO monosomy is associated with reduced birthweight and lowered weight gain in the mouse. J Reprod Fertil. 1983;68:381–385. doi: 10.1530/jrf.0.0680381. [DOI] [PubMed] [Google Scholar]
- 31.Lindgren AM, et al. Haploinsufficiency of KDM6A is associated with severe psychomotor retardation, global growth restriction, seizures and cleft palate. Hum Genet. 2013;132:537–552. doi: 10.1007/s00439-013-1263-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rujirabanjerd S, et al. Identification and characterization of two novel JARID1C mutations: suggestion of an emerging genotype-phenotype correlation. Eur J Hum Genet. 2010;18:330–335. doi: 10.1038/ejhg.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawson-Yuen A, Saldivar JS, Sommer S, Picker J. Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur J Hum Genet. 2008;16:614–618. doi: 10.1038/sj.ejhg.5202006. [DOI] [PubMed] [Google Scholar]
- 34.Huang L, et al. A noncoding, regulatory mutation implicates HCFC1 in nonsyndromic intellectual disability. Am J Hum Genet. 2012;91:694–702. doi: 10.1016/j.ajhg.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A. 2010;152A:1079–1088. doi: 10.1002/ajmg.a.33184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Froyen G, et al. Copy-number gains of HUWE1 due to replication- and recombination-based rearrangements. Am J Hum Genet. 2012;91:252–264. doi: 10.1016/j.ajhg.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lau EC, Mohandas TK, Shapiro LJ, Slavkin HC, Snead ML. Human and mouse amelogenin gene loci are on the sex chromosomes. Genomics. 1989;4:162–168. doi: 10.1016/0888-7543(89)90295-4. [DOI] [PubMed] [Google Scholar]
- 38.Rozen S, Marszalek JD, Alagappan RK, Skaletsky H, Page DC. Remarkably little variation in proteins encoded by the Y chromosome's single-copy genes, implying effective purifying selection. Am J Hum Genet. 2009;85:923–928. doi: 10.1016/j.ajhg.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watanabe M, Zinn AR, Page DC, Nishimoto T. Functional equivalence of human X- and Y-encoded isoforms of ribosomal protein S4 consistent with a role in Turner syndrome. Nature Genet. 1993;4:268–271. doi: 10.1038/ng0793-268. [DOI] [PubMed] [Google Scholar]
- 40.Sekiguchi T, Iida H, Fukumura J, Nishimoto T. Human DDX3Y, the Y-encoded isoform of RNA helicase DDX3, rescues a hamster temperature-sensitive ET24 mutant cell line with a DDX3X mutation. Exp Cell Res. 2004;300:213–222. doi: 10.1016/j.yexcr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 41.Welstead GG, et al. X-linked H3K27me3 demethylase Utx is required for embryonic development in a sex-specific manner. Proc Natl Acad Sci USA. 2012;109:13004–13009. doi: 10.1073/pnas.1210787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shpargel KB, Sengoku T, Yokoyama S, Magnuson T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 2012;8:e1002964. doi: 10.1371/journal.pgen.1002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee S, Lee JW, Lee SK. UTX, a histone H3-lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program. Dev Cell. 2012;22:25–37. doi: 10.1016/j.devcel.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slonim D, Kruglyak L, Stein L, Lander E. Building human genome maps with radiation hybrids. J Comput Biol. 1997;4:487–504. doi: 10.1089/cmb.1997.4.487. [DOI] [PubMed] [Google Scholar]
- 45.Saxena R, et al. The DAZ gene cluster on the human Y chromosome arose from an autosomal gene that was transposed, repeatedly amplified and pruned. Nature Genet. 1996;14:292–299. doi: 10.1038/ng1196-292. [DOI] [PubMed] [Google Scholar]
- 46.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 47.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Felsenstein J. Distributed by the author. Department of Genome Sciences, University of Washington; Seattle: 2005. [Google Scholar]
- 49.Thomas PD, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuroda-Kawaguchi T, et al. The AZFc region of the Y chromosome features massive palindromes and uniform recurrent deletions in infertile men. Nature Genet. 2001;29:279–286. doi: 10.1038/ng757. [DOI] [PubMed] [Google Scholar]
- 52.Karere GM, Froenicke L, Millon L, Womack JE, Lyons LA. A high-resolution radiation hybrid map of rhesus macaque chromosome 5 identifies rearrangements in the genome assembly. Genomics. 2008;92:210–218. doi: 10.1016/j.ygeno.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gubbay J, et al. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature. 1990;346:245–250. doi: 10.1038/346245a0. [DOI] [PubMed] [Google Scholar]
- 55.Sinclair AH, et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. 1990;346:240–244. doi: 10.1038/346240a0. [DOI] [PubMed] [Google Scholar]
- 56.Hayashida H, Kuma K, Miyata T. Interchromosomal gene conversion as a possible mechanism for explaining divergence patterns of ZFY-related genes. J Mol Evol. 1992;35:181–183. doi: 10.1007/BF00183228. [DOI] [PubMed] [Google Scholar]
- 57.Marais G, Galtier N. Sex chromosomes: how X-Y recombination stops. Curr Biol. 2003;13:R641–R643. doi: 10.1016/s0960-9822(03)00570-0. [DOI] [PubMed] [Google Scholar]
- 58.Iwase M, et al. The amelogenin loci span an ancient pseudoautosomal boundary in diverse mammalian species. Proc Natl Acad Sci USA. 2003;100:5258–5263. doi: 10.1073/pnas.0635848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dal Zotto L, et al. The mouse Mid1 gene: implications for the pathogenesis of Opitz syndrome and the evolution of the mammalian pseudoautosomal region. Hum Mol Genet. 1998;7:489–499. doi: 10.1093/hmg/7.3.489. [DOI] [PubMed] [Google Scholar]
- 60.Bachtrog D. The temporal dynamics of processes underlying Y chromosome degeneration. Genetics. 2008;179:1513–1525. doi: 10.1534/genetics.107.084012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jobling MA, et al. Structural variation on the short arm of the human Y chromosome: recurrent multigene deletions encompassing Amelogenin Y. Hum Mol Genet. 2007;16:307–316. doi: 10.1093/hmg/ddl465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jones MH, et al. The Drosophila developmental gene fat facets has a human homologue in Xp11.4 which escapes X-inactivation and has related sequences on Yq11.2. Hum Mol Genet. 1996;5:1695–1701. doi: 10.1093/hmg/5.11.1695. [DOI] [PubMed] [Google Scholar]
- 63.Adachi M, Tachibana K, Asakura Y, Muroya K, Ogata T. Del(X)(p21.1) in a mother and two daughters: genotype-phenotype correlation of Turner features. Hum Genet. 2000;106:306–310. doi: 10.1007/s004390051042. [DOI] [PubMed] [Google Scholar]
- 64.Chocholska S, Rossier E, Barbi G, Kehrer-Sawatzki H. Molecular cytogenetic analysis of a familial interstitial deletion Xp22.2-22.3 with a highly variable phenotype in female carriers. Am J Med Genet A. 2006;140:604–610. doi: 10.1002/ajmg.a.31145. [DOI] [PubMed] [Google Scholar]
- 65.Good CD, et al. Dosage-sensitive X-linked locus influences the development of amygdala and orbitofrontal cortex, and fear recognition in humans. Brain. 2003;126:2431–2446. doi: 10.1093/brain/awg242. [DOI] [PubMed] [Google Scholar]
- 66.James RS, et al. A study of females with deletions of the short arm of the X chromosome. Hum Genet. 1998;102:507–516. doi: 10.1007/s004390050733. [DOI] [PubMed] [Google Scholar]
- 67.Massa G, Vanderschueren-Lodeweyckx M, Fryns JP. Deletion of the short arm of the X chromosome: a hereditary form of Turner syndrome. Eur J Pediatr. 1992;151:893–894. doi: 10.1007/BF01954124. [DOI] [PubMed] [Google Scholar]
- 68.Zinn AR, et al. Del (X)(p21.2) in a mother and two daughters with variable ovarian function. Clin Genet. 1997;52:235–239. doi: 10.1111/j.1399-0004.1997.tb02554.x. [DOI] [PubMed] [Google Scholar]
- 69.Zinn AR, et al. Evidence for a Turner syndrome locus or loci at Xp11.2-p22.1. Am J Hum Genet. 1998;63:1757–1766. doi: 10.1086/302152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.