

Abstract
Alzheimer's disease (AD) is a complex neurodegenerative disorder involving multiple cellular and molecular processes. The discovery of drug molecules capable of targeting multiple factors involved in AD pathogenesis would greatly facilitate in improving therapeutic strategies. The repositioning of existing non-toxic drugs could dramatically reduce the time and costs involved in developmental and clinical trial stages. In this study, preliminary screening of 140 FDA approved nervous system drugs by docking suggested the viability of the tricyclic group of antidepressants against three major AD targets, viz. Acetylcholinesterase (AChE), β-secretase (BACE-1), and amyloid β (Aβ) aggregation, with one member, protriptyline, showing highest inhibitory activity. Detailed biophysical assays, together with isothermal calorimetry, fluorescence quenching experiments, kinetic studies and atomic force microscopy established the strong inhibitory activity of protriptyline against all three major targets. The molecular basis of inhibition was supported with comprehensive molecular dynamics simulations. Further, the drug inhibited glycation induced amyloid aggregation, another important causal factor in AD progression. This study has led to the discovery of protriptyline as a potent multi target directed ligand and established its viability as a promising candidate for AD treatment.
Introduction
Alzheimer's disease (AD) is the foremost cause of dementia in the ageing population affecting over 35 million people worldwide. According to World Alzheimer Report 2013, this number is expected to increase by two fold in 2030. AD is a progressive neurodegenerative disorder that leads to the irreversible loss of neurons, intellectual abilities and eventually to death within a decade of diagnosis. Although the molecular bases of AD pathogenesis remains incompletely elucidated, the disease has been recognized as a multifactorial syndrome involving various molecular and cellular processes such as protein aggregation, oxidative stress, cell cycle deregulation and neuroinflammation [1].
There are currently several plausible hypotheses for AD pathogenesis. Cholinergic hypothesis states that the reduced cholinergic neurotransmission leads to the degeneration of cholinergic neurons and hence synaptic failure and cognitive dysfunction [2]. Following this, Acetylcholinesterase (AChE) was validated as a therapeutic target to reduce the degradation of acetylcholine in the synapse. AChE inhibitors (AChEIs) are effective in temporarily restoring cholinergic function, and constitute the majority of AD drugs currently available in the market [3]. However, they are incapable of delaying or preventing neurodegeneration [4], [5].
A variety of biochemical, genetic and pathological studies describe pivotal roles of the Amyloid β (Aβ) peptide in the pathogenesis of AD. “Amyloid hypothesis,” describes the altered synthesis, aggregation and accumulation of Aβ which results in amyloid plaque formation [6]. While extracellular deposits of amyloid plaques are highly neurotoxic, recent studies have also implicated soluble, oligomeric aggregates of Aβ in neurotoxicity [7]. Therefore, a key therapeutic strategy for the treatment of AD involves the development of drugs targeted at inhibiting Aβ production, aggregation, destabilization and clearance of preformed fibrils [8]. A crucial step in Aβ production is the specific N-terminal enzymatic cleavage of the membrane embedded Amyloid Precursor Protein (APP) by the transmembrane aspartyl protease, β-secretase (BACE-1) [9]–[12]. Therefore, inhibiting BACE-1 has been considered as another attractive approach to prevent Aβ neurotoxicity. It is noteworthy that no inhibitors of Aβ aggregation or BACE-1 activity have reached the market yet, despite strong evidence of the causative roles of Aβ in AD.
We further point out that Aβ is transported through a Receptor for Advanced Glycation End products (RAGE) and cause neuronal damage. Long-lived proteins are preferentially modified to form Advanced Glycation End products (AGE), and the stability of Aβ makes it an ideal substrate for non-enzymatic glycation and formation of AGEs [13]. In a recent study, it has been shown that Aβ-AGE formation may intensify the neurotoxicity whereas inhibition of this process significantly rescued the early cognitive deficit in mice [14]. Therefore, glycated Aβ has been considered as a more suitable ligand for RAGE, as it aggravates neuronal deterioration [14]. Hence inhibiting glycation of Aβ may be a valuable therapeutic strategy for AD. However, there has been no concerted effort to explore the inhibition of Aβ glycation as a therapeutic strategy.
Efforts to target AChE inhibition [15]; Aβ production [16]; Aβ aggregation [17]; tau phosphorylation and aggregation [18] have been investigated largely in isolation, despite the complex nature of AD etiology. Recently, drug discovery in AD has gradually inclined towards development of “multi-target-directed ligands” (MTDLs) [19]–[22] which are efficient in treating complex diseases because of their ability to target multiple modes of disease pathogenesis. Further, to evaluate MTDLs for the AD treatment, “drug repositioning” seems to be an appealing strategy, as this approach has several advantages, including reduced time and cost necessary for clinical trials23. Priority candidate drugs for hypertension, retinoid therapy, diabetes and antibiotics with sufficient supporting evidences have been considered for repositioning in AD [23]. However, to the best of our knowledge, repositioning drugs for multiple targets in AD is scarce. In this study, an in silico screening of 140 FDA approved drugs for neurological treatment was done against the primary targets of AD therapeutics, namely, acetylcholinesterase, BACE-1, and Aβ aggregation. Further in vitro studies showed that amongst selected molecules, the tricyclic antidepressant protriptyline exhibited significant inhibition of AChE, as well as inhibition of other targets of AD. In addition, protriptyline was also found to inhibit glycation mediated Aβ aggregation. Mechanistic insights into protriptyline binding and inhibition of AChE, Aβ, and BACE-1 activity were described in detail with molecular dynamics simulation studies.
Results and Discussion
Tricyclic Antidepressant Drugs Display Strong Binding against Various Targets of AD In silico
Multi-target-directed ligands (MTDLs) are likely to offer promising approaches for treatment of a disease as complex as AD [19]-[21]. The structures of 140 ligands were docked with the major targets of AD viz. AChE, BACE-1 and Aβ aggregation. Ligands were scored based on electrostatic and hydrophobic contributions to the binding energy [24]. Furthermore, polar interactions were considered by H-bonding interactions analysis. Docking scores were used to rank ligands, depending on presence of number of H-bond donors and acceptors within the active sites [25]. Binding energy scores represented in the Heatmap ( Figure 1A ) displayed variability in interactions of the ligands to the three targets of AD. There were several ligands that showed noteworthy interaction with at least two targets, but only few of them had strong interaction with all the targets. It was observed that five antidepressant drugs protriptyline, amytriptyline, maprotiline, doxepin and nortriptyline, which are tricyclic secondary amines showed strong binding affinity and broad specificity toward multiple targets of AD ( Figure 1B ).
Figure 1. Virtual screening by docking.

A. Heat map analysis of binding constants of 140 FDA approved nervous system drugs screened against Aβ, AChE and β-secretase by Autodock tool 4.2. In the gradient ruler, red colour indicated strong binding (ΔG<−6 kcal/mol), while green colour indicate weak binding (ΔG>−3 kcal/mol) and the five drugs showing higher affinity to all the above mentioned targets were zoomed. B. Chemical structures of the five drugs. All are tricyclic anti-depressant drugs.
Protriptyline Inhibits AChE Activity by Inducing Conformational Change in the Active Site
AChE currently remains the foremost therapeutic target for AD and the current treatment and management of AD mainly involves use of acetylcholinesterase inhibitors [4], [5]. Therefore, protriptyline, amitryptyline, maprotiline, doxepin and nortriptyline were assessed initially against AChE inhibition. While all the five drugs displayed inhibitory activity against this target ( Figure 2A ), protriptyline displayed highest inhibition, with the least IC50 of about 0.06 mM. In comparison, the other ligands displayed relatively higher IC50 values; the values corresponding to maprotiline, doxepin, nortriptyline and amitriptyline were 0.1 mM, 0.480 mM, 0.135 mM and 0.6 mM, respectively. Hence, the inhibitory activity of protriptyline against the other targets of AD was evaluated in detail in the remaining study.
Figure 2. Protriptyline inhibits AChE activity.

A. Determination of IC50 values of five drugs for AChE by using 0.05–0.8 mM concentration range of all the drugs B. Estimation of the kinetic constants by Lineweaver–Burk analysis. AChE inhibition by protriptyline showed competitive inhibition. C. Isothermal Titration Calorimetric analysis of protriptyline – AChE interactions. The upper panel shows the raw data in the form of heat effect during titration and the lower panel shows corresponding thermogram representing the best fit curve D. Snapshot of drug binding with catalytic subsite of AChE E. snapshot of drug binding with anionic subsite of AChE F. Distribution of Protriptyline –anionic subsite (solid line) and Protriptyline –esteratic subsite (broken line) nonbonded (nonb) interaction energy; data are averaged over last 20 ns G. Fluorescence quenching of AChE by protriptyline H. CD spectra of binding of protriptyline to AChE and I. CD pro analysis to study the conformational change J. Evolution of the backbone RMSD for the Protriptylline bound (solid line) and free (broken line) AChE active sites from MD trajectories K. SASA distributions of active sites for Pro-bound (solid line) and free (broken line) AChE active sites from MD trajectories L. Measurement of AChE activity after treatment of neuro2a cells with 25 µM and 60 µM protriptyline for 15 h.
The assessment of strong inhibitory activity of protriptyline was consolidated by enzyme kinetic studies, which suggested competitive inhibition of AChE by protriptyline. ( Figure 2B ). The apparent Km for AChE was determined to be ∼0.025 mM by Lineweaver-Burk plot and it was found to be increased in the presence of protriptyline. The inhibition constant Ki was determined from Cheng-Prusoff's equation and found to be ∼0.001 mM. Thermodynamic studies of protriptyline-AChE interaction was carried out using Isothermal titration calorimetry (ITC) as it is one of the most widely used quantitative technique for direct measurement of the enthalpy change when two species interact, allowing the determination of heat of association, stoichiometry, and binding affinity from a single experiment [26], [27]. The raw data and corresponding thermogram of the binding experiment is depicted in Figure 2C . Binding was strongly exothermic and showed 1∶1 stoichiometry for AChE and protriptyline. The spontaneity of the process is evidenced by a negative change in the enthalpy, ΔH, and a positive change in the entropy, ΔS. Kinetic and ITC data demonstrated the binding of single molecule of protriptyline to the active site of AChE. The active site of AChE comprises two subsites; the anionic subsite (Trp82, Glu198, Tyr333) and the esteratic subsite (Ser199, Glu330 and His443) [28]. Strong binding propensity of protriptyline to both the subsites of AChE was evidenced via the MD studies ( Figures 2D and E ). The mean interaction strengths of protriptyline with the anionic subsite is −31.3 (±9.1) kcal mol−1 and −19.4 (±5.0) kcal mol−1 with the esteratic subsite ( Figure 2F ). Breakup of the interactions into the non-bonded components showed that electrostatics plays a relatively stronger role than van der Waals interactions in protriptyline binding with active site. The mean interaction strengths of the active site residues with the ligand are provided in Table S1.
Protriptylline-AChE interaction was also studied by fluorescence spectrometry. A significant shift in the tryptophan fluorescence emission spectra was observed upon protriptyline binding suggested that the drug induces conformational change in AChE, thereby in its functional abilities. The fluorescence intensity of AChE exhibited an emission maximum at a wavelength, λmax, of 325 nm in the unbound state. However, titration of the native enzyme with increasing concentrations of inhibitor resulted in a concentration-dependent quenching of the tryptophanyl fluorescence ( Figure 2G ). A progressive red shift in the emission spectrum with increasing concentration of protriptyline suggested the conformation change in enzyme. Further, the conformational change was confirmed by CD analysis. CD analysis showed that the binding of protriptyline to AChE decreased the minima suggesting protriptyline induces conformational change in AChE. CDPro analysis revealed that binding of protriptyline increased β-sheets and decreased α-helices in AChE. These results are very well in accordance with the fluorescence quenching experiment ( Figure 2H and 2I ).
The evidence of structural distortion was supported by the MD simulation data. In Figure 2J , we compare the backbone root mean squared deviation (RMSD) of the active site residues in the free and protriptyline bound state for a sample MD simulation trajectory. The higher RMSD in the latter is a distinct demonstration of the perturbative effect of the ligand on the active site structure. This structural distortion is supported by changes in the inter-residue distances of the active site residues (see Table S2). Distinct increases in several of the inter-residue distances are noted, especially in the distances involving Tyr333 and His443. The sharpest increase in the mean inter-residue distance is found for two residues belong to the esteratic subsite, namely Ser199 and His443 that play critical roles in the hydrolysis of acetylcholine. The increase from 7.8 Å in the free state to 11.5 Å in the bound state strongly suggests that protriptyline binding directly affects the catalytic ability of AChE. Further, we found protriptyline binding at the active site to be commensurate with an increase in its solvent accessibility. The solvent accessible surface area, or SASA, was calculated by running a spherical probe of 1.8 Å radius around the surface and calculating the area covered by the probe / calculated in the standard manner. In Figure 2K , we have presented histograms of the SASA obtained from the MD trajectories of the free and protriptyline bound AChE. The active site SASA increased from 1533 (±38.9) Å2 to 1623 (±33.0) Å2 upon protriptyline binding.
Additionally, to confirm the in vitro results, the effect of protriptyline on AChE activity was assayed in neuro2a cell line. Protriptyline treated cells showed concentration dependent decrease in AChE activity ( Figure 2L ). These results demonstrated that the in vitro IC50 of protriptyline for AChE was sufficient to inhibit its activity in cell culture. Thus, this study suggested that protriptyline inhibits AChE activity in vitro as well as in neuro 2a cells, by binding to the active site and causing conformational change.
Protriptyline Inhibits Aβ Self-Assembly
Recent studies suggest that the K16LVFF20 segment in the Aβ sequence is crucial for the peptide's oligomeric properties as well as fibrillogenetic behavior [29]–[33]. The sequence HHQKLVFFAE corresponding to Aβ13–22 was used for our in vitro amyloid aggregation inhibition studies. β-sheet rich structures are a common feature of amyloid aggregates [34] and bind to the molecule Thioflavin T (ThT). Therefore, ThT fluorescence assays are frequently used to monitor aggregation of amyloidogenic peptides [35]. In Figure 3A , we present ThT fluorescence as a function of time for Aβ13–22 in the absence or presence of 10 µM protriptyline. It is observed that the lag time corresponding to the nucleation phase is increased from ∼25 to about ∼50 hours in the presence of protriptyline. The data further shows a dramatic inhibition in the growth phase in the presence of protriptyline. The fluorescence intensity corresponding to the saturation phase was further lowered in the presence of protriptyline, demonstrating the ability of protriptyline to reduce Aβ aggregation. Concentration dependent decrease in Thioflavin T fluorescence was observed in presence of protriptyline at 7th day (Figure S1). Static light scattering experiments were performed to investigate the relative decrease in average molecular mass of Aβ aggregates in the presence of protriptyline ( Figure 3B ). As expected, protriptyline reduced the light scattering in a concentration dependent manner. Furthermore, Far-UV CD spectra of the Aβ13–22 were recorded with and without protriptyline to monitor any possible alterations in secondary structural propensities ( Figure 3C ). CDPro analysis comparing the spectra obtained at the two conditions showed that protriptyline treatment reduced β-sheet formation and increased overall helicity. The reduced β-sheet formation by CD analysis corroborated the Thioflavin T results. We further performed atomic force microscopy (AFM) experiments on the peptide aggregates formed with and without protriptyline treatment. The results of an AFM scans for Aβ13–22 incubated for 7 days with and without protriptyline over scanning areas of 10 µm×10 µm and 20 µm×20 µm are compared in Figure 3D .
Figure 3. Inhibition of Aβ aggregation by protriptyline.

Aβ13–22 aggregation in the absence and presence of protriptyline was investigated by A. Thioflavin T assay B. Light scattering C. CD analysis D. Atomic Force Microscopy images [i and iv] of aggregated amyloid and Amyloid + protriptyline (0.010 mM) in 10×10 µm2 and 20×20 µm2 surface area respectively. It is also represented in Line profile [ii and v] and 3D images [iii and vi].
First, the AFM profiles suggested a distinct reduction in the fibrillar density and a high degree of size dispersion resulting from protriptyline treatment. Further, the line profile and 3D AFM images showed that the average height of the Aβ13–22 aggregates was ∼400 nm, with the maximum height reaching ∼600 nm. The height of the aggregates reduced to ∼100 nm in case of protriptyline treated samples. It was quite convincing from the AFM images that the amyloid protein is aggregated and protriptyline treated sample protein showed decreased aggregation.
For a molecular level insight into the inhibitory action of protriptyline on Aβ self-assembly, we performed MD simulations of Aβ dimerisation in the absence and presence of protriptyline molecules. A β-sheet rich full-length monomer was considered for these studies. A single protriptyline molecule was found to bind to the Aβ monomer with a mean binding strength of −84.0 (±34.0) kcal mol−1 (Figure S2A). The mean monomer-monomer interaction strength obtained at the end of multiple independent trajectories over a combined total simulation time of 60 ns was −249.1 (±87.1) kcal mol−1. In the presence of protriptyline, however, the monomer-monomer interaction weakened significantly, with a mean value of only −69.7 (±31.5) kcal mol−1. In Figure 4A , we have depicted evolution of monomer-monomer interaction over simulation time for sample simulations in the absence and in the presence of protriptyline; the distributions of these interactions, along with distribution of the protriptyline-dimer interaction is depicted as an inset. The weakening of the inter-monomer interactions due to protriptyline binding results in the formation of complexes that are less compact compared to the pure dimeric forms. In Figure 4B , we compare distributions of asphericities Iγ, of the pure and protriptyline-bound dimer complexes; Iγ value of 0.0 denotes perfect sphericity while increasing values denote increased asphericity. The protriptyline bound dimer complexes have a wider distribution and a higher mean value of Iγ; the mean asphericity values of the free and protriptyline -bound complexes are 0.2 and 0.5, respectively. Further, the mean radii of gyration (Rg) of these complexes were found to be 13.0 (±0.1) and 16.5 (1.0) Å, respectively. The difference in dimeric compactness upon protriptyline binding is evident from representative snapshots depicted in Figures 4C and 4D . Additionally, clustering analysis was done to identify the key residues of Aβ interacting with the protriptyline during molecular dynamics simulation. A representative snapshot from most populated cluster is shown in Figure 4D . In two of the most populated clusters (70% of total snapshots used in the clustering analysis), protriptyline interacts most strongly with the KLVFF region.
Figure 4. Destabilization of amyloid dimer by protriptyline.

A. Evolution of monomer-monomer interaction strength over time for free dimer (broken line) and Protriptylline-bound dimer (solid line). Inset. Distributions of the interactions from multiple trajectories, and the dimer interactions with Protriptylline (in brown) B. Distributions of the asphericity for free (in broken line) and Protriptylline-bound (solid line) dimer C. Representative snapshot of most populated cluster of free, and D. Protriptylline-bound dimer [16–20 region in blue colour with 19–20 showed in line representation; protriptyline in red colour and two Aβ peptides are in cyan and limon colour respectively] E. Residue-residue contact probabilities for free dimer, and F. Protriptylline-bound dimer G. Residue-wise Beta sheet percentages for free dimer (in red) and Protriptylline-bound dimer (in blue) H. Residue-wise helical percentages for free dimer (in red) and Pro-bound dimer (in blue).
In order to evaluate the effects of protriptyline binding on the inter-monomer associations, we have analyzed the nature of inter-residue contacts. The average numbers of inter-residue side-chain cross-contacts were reduced up to 46% upon protriptyline binding. Here, as in previous studies [36], we have defined two residues to be in contact if the maximum separating distance of their side-chains does not exceed 7 Å. In Figures 4E and 4F , we compare the inter-residue contact probabilities for the free and protriptyline-bound complexes from the MD data. The largest numbers of contacts between the KLVFF regions are lost upon protriptyline binding. However, we note the emergence of a small extent of extraneous contacts, particularly involving the N- and C-terminal regions of the different monomeric units.
Further the effect on the secondary structural propensities of the Aβ units due to protriptyline binding was investigated. Protriptyline was found to reduce β-sheet and induce higher helical propensities in the monomeric form of Aβ (see Figure S2 B). Figure 4G and 4H present residue-wise β-sheet and helical propensities of the free and protriptyline-bound dimeric complexes, respectively, from the MD data. A sharp decrease in β-sheet propensity is found uniformly along the Aβ sequence, including in the residue span H13HQKLVFFAE22. The decrease in β-sheet propensity is accompanied with an overall increase in helical conformations. Sharp increase in helicity was observed near the N-terminal and KLVFF regions. The alterations to secondary structural propensities thus observed from MD analysis are an excellent corroboration of the CD and ThT binding data.
Protriptyline Inhibits BACE-1 Activity
BACE-1 is a key enzyme required for Aβ production. Hence, BACE-1 inhibition is an attractive target for countering AD [10]–[12]. BACE-1 assay demonstrated decreased activity with increasing concentrations of protriptyline having IC50 ∼0.025 mM ( Figure 5A ). Protriptyline inhibited BACE-1 by competitive inhibition as depicted by Lineweaver-Burk plot ( Figure 5B ). Apparent Km and Ki of BACE-1 were calculated as mentioned above for AChE and found to be 0.0025 mM and 0.005 mM respectively. Apparent Km of BACE-1 was increased in the presence of protriptyline Competitive inhibition was evidenced by MD simulation analysis that illustrated protriptyline binds strongly at the active site of BACE-1 comprised of Asp32 and Asp228 ( Figure 5C ). The mean binding strength required for protriptyline to bind to the active site is −29.5 (±7.0) kcal mol−1. The center of mass distance between the two residues increases from 5 Å to 8 Å as a result of protriptyline binding ( Figure 5D ). The comparison of the root mean squared deviation (RMSD) of Cα atoms of the active site in the unbound state with the protriptyline bound state indicated structural distortion of the arrangement of the active site (see Figure S3A). The binding was also found to induce significant alterations to the local secondary structural propensity around the active site (see Figure S3B). Therefore, inhibition of BACE-1 by protriptyline is an extra benefit as it prevents Aβ generation. And even if there is some production, protriptyline will obstruct it to get aggregate.
Figure 5. BACE-1 inhibition by protriptyline.

A. Determination of IC50 of BACE-1 by using various concentrations of protriptyline. The sigmoidal curve indicates the best fit for the percentage inhibition data obtained B. Lineweaver-Burk analysis to estimate the kinetic constants. It showed competitive inhibition. C. Snapshot of drug binding with active site of BACE-1. Active site residues in BACE-1 are in line representation. D. Active site of BACE-1. The structures from unbound (green) and ligand bound (orange) simulations are shown after all - atom superimposition. Snapshots are generated using PyMol.
Protriptyline Inhibits Glycation Associated Aggregation of Aβ
AD is also referred as type III diabetes [37] and its pathogenesis has been correlated with the extent of glycation [38]. Recent studies suggested glycated Aβ is more neurotoxic than native Aβ [13]; therefore the effect of protriptyline on glycation of Aβ was investigated. Glycated proteins emit fluorescence at 440 nm upon excitation at 370 nm. Fluorescence assay illustrated that Aβ undergoes glycation. The increase in glycation associated fluorescence was reduced by protriptyline in a concentration dependent manner ( Figure 6A ). Glycation enhances the aggregation and also alters the secondary structure of proteins. Static light scattering was used to study glycation induced protein aggregation. Concentration dependent decrease in light scattering was observed ( Figure 6B ). Further, it was studied by Thioflavin T fluorescence assay. Kinetics of aggregation displayed increased Thioflavin T fluorescence during glycation reaction. Lag phase for Aβ and glycated Aβ aggregation was increased in the presence of protriptyline with decreased Thioflavin T fluorescence ( Figure 6C ). In addition to inhibition of Aβ glycation, the drug also inhibited glycation of insulin and BSA(Bovine Serum Albumin) evidenced by MALDI, AGE fluorescence and Thioflavin T fluorescence assay (See Figure S4 and S5).
Figure 6. Protriptyline inhibits glycation.

A. Fluorescence emmission of Aβ and glycated Aβ in presence of various concentration of protriptyline B. Light scattering and C. Kinetics of amyloid aggregation by Thioflavin T of Aβ13–22 and glycated Aβ13–22 in the absence and presence of protriptyline.
Protriptyline Does Not Affect Other Proteases
Protriptyline inhibited multiple targets of AD; therefore we further studied the influence of this drug on other enzymes such as trypsin and α-secretase. It was interesting to observe that it was not able to inhibit trypsin activity even at 0.1 and 0.5 mM ( Figure 7A ). For ADAM 17, protriptyline was found to be a weak inhibitor. It was showing ∼3% inhibition at 0.1 mM and ∼25% inhibition at 0.5 mM ( Figure 7B ). It suggested that protriptyline is not a non-specific inhibitor as it was not able to inhibit other proteases.
Figure 7. Protriptyline Does Not Affect Other Proteases.
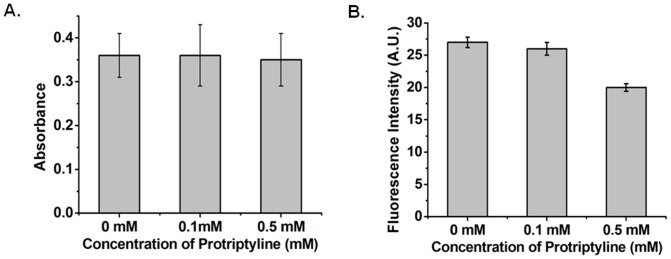
Effect of protriptyline on A. Trypsin B. ADAM 17activity. Specific synthetic substrate BApNA and fluorogenic peptide was used for analyzing activity on trypsin and ADAM 17, respectively. Trypsin activity was unaffected, while ADMA 17 showed weak inhibition in presence of protriptyline.
Viability of neuro2a cells in the presence of protriptyline
The effect of various concentrations of protriptyline (25–500 µM) on cell viability was evaluated by MTT assay in neuro2a neuroblastoma cells. It was observed that there was more than 90% of cells were viable upto 150 µM of protriptyline, and a drastic reduction in cell viability was observed at 200–500 µM protriptyline concentrations ( Figure 8 ). In this study the IC50 of protriptyline for all the three targets AChE, Aβ, BACE1 was less than 150 µM, this concentration was found to be non toxic to cells.
Figure 8. Cell viability in neuro2a cells.
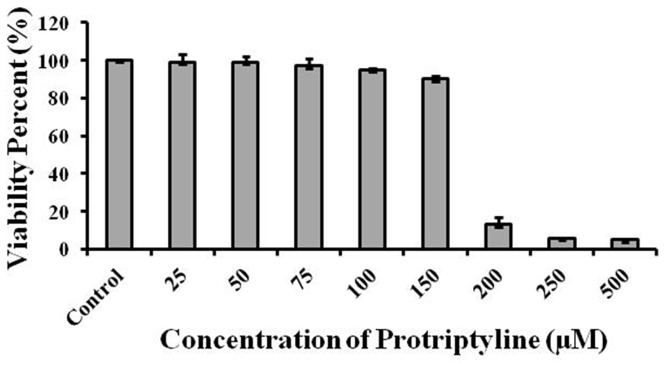
Effect of various concentrations of protriptyline (25–500 µM) on cell viability was assessed by MTT assay. Cells were 90% viable up to 150 µM protriptyline concentration.
Conclusion
The multifactorial nature of AD makes its treatment complex and unmanageable. The discovery of molecules that can inhibit multiple pathways of the disease should significantly advance therapeutic strategies. In this study, we investigated the efficacy of the tricyclic antidepressant, protriptyline, against important AD targets. Our in vitro and in silico investigations established the inhibitory effects of the drug on AChE, amyloid aggregation, BACE-1 and glycation ( Figure 9 ). Protriptyline was able to inhibit AChE and β-secretase by binding at the active site and causing conformational changes. In addition, it strongly prevented self-assembly of Aβ and glycated Aβ. It is a FDA approved drug for the treatment of depression, narcolepsy, Attention Deficit Hyperactivity Disorder (ADHD) and headaches and its ability to cross blood brain barrier (BBB) [39] is an additional advantage, which is a crucial requirement of molecules used for intra-cranial diseases. Furthermore, as there is high prevalence rate (30–50%) of AD and depression co-morbidity, the use of antidepressants can be a rational complementary therapy for AD treatment [40]. Therefore, antidepressant activity of this drug could be an added advantage when dealing with AD complications related to depression. To the best of our knowledge, this is the first study in which an anti-depressant drug has been shown to inhibit multiple targets of AD. Our results strongly ratify protriptyline as a promising candidate for AD therapy, and its further evaluation in animal and clinical studies.
Figure 9. Protriptyline as MTDL.

The scheme represents that protriptyline (at the center) is able to inhibit key targets of AD pathogenesis such as AChE, BACE-1, Amyloid aggregation and glycation induced amyloid aggregation.
Experimental Section
Materials
All chemicals were procured from sigma unless and otherwise stated.
Virtual Screening for Multi-target Drug Ligand for AD
Structures of 140 FDA approved antiepileptics, psycoleptics, analgesics, psycoanaleptics, anti-Parkinson and other nervous system drugs were obtained from DrugBank (The DrugBank database website. Available: http://www.drugbank.ca/. Accessed 2013 Feb 19) database and optimized for their 3D coordinates using Marvin Sketch Tool (ChemAxon website. Available: http://www.chemaxon.com. Accessed 2013 Feb 21). Three dimensional structures of Human acetylcholinesterse (PDB ID: 1B41), β-secretase (PDB ID: 2HM1) and Aβ peptide (PDB ID: 1ZOQ) were accessed from RCSB PDB. Protein structures were energy minimized using Swiss PDB viewer (Swiss-PdbViewer application. Available: http://spdbv.vital-it.ch/. Accessed 2013 Feb 21). AutoDock tool 4.2 [41] was used to convert receptor and ligand from *.pdb to *.pdbqt format and to set other docking parameters. Grid map was set around the active site of acetylcholinesterase (Ser199, Glu330 and His443), β-secretase (Asp32 and Asp228) and KLVFF (residue 17 to 21) region of Aβ protein involved in aggregation. Virtual screening was carried out using AutoDock Vina software and the Lamarckian genetic algorithm as a searching procedure [42]. Binding energy obtained for each complex was represented in heat map format using MeV software packages (MeV: MultiExperiment Viewer. Available: http://www.tm4.org/mev/. Accessed 2013 Mar 5) [43]. The gradient ruler is an indicator of interaction strength. Molecules showing strong binding against all the selected targets were selected for further in vitro and molecular simulation studies.
Acetylcholinesterase Inhibition Assay
The modified method of Ellman et al. [44] was adopted to measure AChE activity. Briefly, 25 µl (0.3 U/ml) AChE from Electric eel fish (Electrophorus electricus) was incubated with and without different concentrations of drugs selected from Molecular Docking studies. The reaction was carried out for 15 min at 25°C. 500 µl of 5, 5-dithiobis (2-nitrobenzioc) acid DTNB (3 mM) was then added and reaction was initiated by the addition of 100 µl substrate acetyl thiocholine iodide (ATCI) (15 mM). Total volume of the reaction was made up to 1 ml by Tris buffer, pH 8.0. ATCI hydrolysis was measured by colored product formation, 5-thio-2-nitrobenzoate anion by reaction between DTNB and thiocholine, a hydrolysis product of ATCI. The formation of the colored product was measured at 405 nm wavelength after 30 min. The background was determined from negative controls (omission of AChE enzyme).
Isothermal Titration Calorimetry
ITC was performed using Microcal Auto-iTC instrument (GE Healthcare). 40 injections of 2 µl protriptyline (Stock = 2.2 mM) was titrated against 0.3 U/µl solution of AChE. Experiments were carried out at 25°C in a Tris buffer, pH 8.0. Reference titration was carried out by injecting the same concentration of protriptyline into buffer. Reference titration was subtracted from experimental titration. Origin 6.0 software was used to derive affinity constants (Kd), the molar reaction enthalpy (ΔH) and the stoichiometry of binding (N), by fitting the integrated titration peaks.
Fluorescence Analysis of AchE-Protriptyline Interaction
AchE-protriptyline interaction was also studied by measuring tryptophan fluorescence using Varioscan plate reader. AChE was excited at 280 nm and emission was scanned from 300 nm to 500 nm. Titration of enzyme with protriptyline was performed by the addition of different concentrations of inhibitor (10 µM–100 µM) to a fixed concentration (0.2 U/µl) of enzyme solution. Background buffer and inhibitor spectra were subtracted and graphs were smoothed.
BACE-1 Inhibition Assay
BACE-1 activity was studied in accordance with the manufacturer's instructions (Sigma). To test the effect of protriptyline on BACE-1 activity, 1.8 U of BACE-1 enzyme and 10 µM of β-secretase specific peptides conjugated to fluorogenic reporter molecules were incubated with or without various concentrations of protriptyline for 2 h at 37°C. Negative control included all the reactants except BACE-1 enzyme. After 2 h, fluorescence emission was measured at 405 nm upon excitation at 320 nm.
Inhibition Kinetics for AChE and BACE-1
Michaelis–Menten constant (Km) was determined by measuring the activities of AChE and BACE-1 using various concentrations of ATCI (100 µM–1000 µM) and BACE-1 substrate (0.25 µM–6 µM) respectively. Lineweaver-Burk double reciprocal plot was plotted in order to determine the Km. The protriptyline inhibition kinetics was analyzed over a range of concentration (50 µM–1500 µM) and (10 µM–150 µM) for AChE and BACE-1 respectively. The IC50 of protriptyline for both the enzymes was calculated by determining the inhibitor concentration at which the enzyme activity is 50% inhibited. The Ki was calculated directly from IC50 value using Cheng-Prussoffs classical equation [45].
In order to determine the type of inhibition, AChE and BACE-1 were incubated with 60 µM and 15 µM protriptyline concentrations respectively and assayed at increasing concentrations of AChE substrate (ATCI, 100 µM–1000 µM) and BACE-1 substrate (1 µM–10 µM) respectively. The reciprocals of reaction rate (1/v) for each inhibitor concentration were plotted against the reciprocals of the substrate concentrations (1/S). Mode of inhibition by protriptyline was determined from the graphical representation.
Inhibition of Aβ Aggregation
Aβ13–22 peptide (HHQKLVFFAE), the aggregation prone region of Aβ, was synthesized from Thermo Fisher Scientific. Synthetic Aβ13–22 peptide was dissolved in 10% ammonium hydroxide and sonicated for 5 min. It was then diluted in 10 mM PBS, pH 7.0 to a final concentration of 200 µM. 100 µM of Aβ13–22 was incubated with and without various concentrations of protriptyline in 10 mM PBS, pH 7.0 at 37°C for a week and these samples were further used for aggregation inhibition assays.
Glycation Inhibition Assay
Anti-glycation activity of protriptyline was studied using insulin, BSA and Aβ. The details of insulin and BSA glycation are described in Method S1. In Aβ glycation studies, 100 µM Aβ13–22 and 0.1 M glucose were incubated with and without various concentrations of protriptyline in 10 mM PBS, pH 7.0 for 7 days. These samples were further used to study glycation mediated Aβ aggregation by different assays.
Thioflavin T Assay
Thioflavin T assay was performed on 7th day of incubation. 50 µl (25 µM) of Aβ13–22 was mixed with 150 µl of Thioflavin T (ThT) stock solution (50 µM ThT in PBS pH 7.0) and placed in 96-well plate (black with flat bottom, Cornings). Fluorescence emission was measured at 460–550 nm upon excitation at 440 nm. To account for background fluorescence, the fluorescence intensity measured from each control solution without Aβ was subtracted from each solution containing Aβ13–22. Similarly, Thoflavin T assay was performed for aggregation kinetics of Aβ13–22 and Aβ13–22 glycation with and without protriptyline.
Light Scattering Analysis
Inhibition of aggregation/ glycation mediated aggregation of Aβ was detected by static light scattering method using a Perkin-Elmer Luminescence spectrometer LS50B. Both excitation and emission wavelengths were set at 400 nm. Excitation and emission slit width was set to 10 nm and 2.5 nm, respectively. Scattering was recorded for 60 sec.
Circular Dichroism Spectroscopy
The far UV CD spectra (in wavelength range of 190–250 nm) of Aβ13–22 (20 µg/ml) with and without protriptyline was recorded on a Jasco-J815 spectropolarimeter at ambient temperature. In case of AChE, the enzyme was incubated with protriptyline and CD spectra were recorded at 0 min and 30 min. Each CD spectrum was accumulated from three scans at 50 nm/min with cell path length of 0.1 cm. Contribution due to buffer was corrected in all spectra and observed values were converted to mean residual ellipticity (MRE) in deg cm2 dmol−1 defined as
Where M is the molecular weight of the protein, θλ is CD in millidegree, d is the path length in cm, c is the protein concentration in mg/ml and r is the number of amino acid residues in the protein. Secondary structure content of the amyloid with and without protriptyline was calculated using the CDPro software (CDPro software package. Available: http://lamar.colostate.edu/~sreeram/CDPro/main.html. Accessed 2013 Oct 18).
Atomic Force Microscopy
For atomic force microscopy (AFM) analysis, 10 µl of each sample was deposited on a piece of freshly cleaved mica disk. The disk was washed with water and dried overnight. The sample was mounted onto a Multimode scanning probe microscope equipped with a Nanoscope IV controller from Veeco Instrument Inc., Santa Barbara, CA. All the AFM measurements were done under ambient conditions using the tapping-mode AFM probes model - Tap190Al purchased from Budget Sensors. The radii of tips used in this study were less than 10 nm, and their height was ∼17 µm. The cantilever used had a resonant frequency of ca. 162 kHz and nominal spring constant of ca. 48 N/m with a 30 nm thick aluminium reflex coating on the back side of the cantilever of the length 225 µm. For each sample, three locations with a surface area of 20×20 µm2 and 10×10 µm2 for amyloid and protriptyline treated amyloid were imaged with a frequency of 1 Hz and at a resolution of 512×512 dpi. Representative images were selected for comparative studies.
Measurement of Glycation Associated Fluorescence
Glycation associated fluorescence of Aβ was measured in amyloid, glycated amyloid treated with or without protryptiline at 370 nm excitation and emission was scanned from 400–550 nm.
BApNA Assay
Activity of Bovine trypsin was estimated using enzyme-specific chromogenic substrate BApNA [46]. In brief, 10 µg Bovine trypsin was incubated with and without 100 µM, 200 µM, 500 µM protriptyline at 37°C for 15 min and volume was made upto 150 µl with 0.1 M Tris-HCl pH 7.8. Further, 1 ml BApNA was added to the reaction mixture and incubated for 10 min at 37°C. The reaction was terminated by addition of 200 µl of 30% acetic acid and absorbance was measured at 410 nm.
ADAM17 Assay
ADAM17 assay kit (Enzo Life Sciences) was used to study the effect of protriptyline on ADAM17. Briefly, ADAM17 and fluorogenic peptide (substrate) was incubated with and without 100 µM protriptyline at 37°C for 10 min. The negative controls had the reaction mixture without enzyme. Fluorescence was measured at 328 nm and 420 nm for excitation and emission respectively.
Cell culture
Murine neuro2a neuroblastoma cells were obtained from National Center for Cell Science, Pune, India. The cells were maintained in Dulbecco's modified Eagle's medium (DMEM) and 10% Fetal Bovine Serum (FBS). Cells were maintained at 37°C in humidified air containing 5% CO2 and were grown in monolayer cultures.
Determination of AChE inhibition in cultured cells
Equal numbers of cells were seeded in 25 cm2 flasks (1.5 million). Cells were allowed to adhere and attain their morphology. Cells were serum starved for 24 h and treated with or without 25 µM or 60 µM of protriptyline concentration for 16 h. Cells were trypsinised and given two washes with ice cold PBS. Protein was extracted by sonicating the cells for 20 min. The cell lysate was centrifuged for 60 min at 16000 rpm. Supernatant was collected and protein concentration was determined by Bradford's method. Acetylcholinesterase activity was assayed as described above by Ellman's assay.
Cell viability
Cell viability following exposure to protriptyline was measured by MTT reduction assay. Neuro 2a (N2a) neuroblastoma cells were seeded at a cell density of 1×104 cells per well in a 96 well plate. After the cells adhered and attained their morphology, they were serum starved for 24 h prior to treatment with various concentrations (25–500 µM) of protriptyline in triplicate for 15 h. After incubation, cells were given one wash with PBS and 100 µl fresh serum free media was added. 20 µl of 5 mg/ml MTT (dissolved in PBS) was added to each well and incubated in dark at 37°C until violet formazan crystals were observed. Media from each well was discarded and crystals were dissolved in 100 µl DMSO. Absorbance was measured at 555 nm using Biorad iMark microplate reader.
Molecular Dynamics Simulations
All simulations in this study were carried out with the NAMD2.9 package [47], using the CHARMM22 all-atom force field with CMAP correction for the proteins [48], [49]. Force field parameters for protriptyline were generated using the SwissPARAM tool [50], and refined via electronic structure calculations using Gaussian03. This strategy has been used in several recent studies [51]–[53]. Simulations were carried using a time step of 2 fs in the isothermal-isobaric (NPT) ensemble at a temperature of 310 K and a pressure of 1 atmosphere. Each system was sampled for a total duration of 60 ns with multiple trajectories. The SHAKE algorithm [54] was used to constrain bond lengths involving hydrogen atoms. Constant temperature was maintained with Langevin dynamics with a collision frequency of 1 ps−1, and constant pressure was maintained using the Langevin piston Nose-Hoover method [55]. Three-dimensional orthorhombic periodic boundary conditions were employed and full electrostatics calculated with the particle-mesh Ewald method [56]. A non-bonded cutoff distance of 12 Å was employed, which were smoothened at a distance of 10.5 Å. Details of system setups and trajectory analysis are provided in Method S2.
Statistical Analysis
All the experiments performed independently three times. Student's t-test was used for statistical analysis. Data were expressed as mean ±SD. A p-value <0.05 was considered as statistically significant.
Supporting Information
Thioflavin T Assay. Protriptyline causes concentration dependent decrease in Aβ aggregation.
(DOCX)
A. Snapshot of drug binding with Aβ monomer. Central hydrophobic core are in line representation B. Comparison of residue wise percentage of helix from unbound and ligand bound Aβ monomer simulated trajectory.
(DOCX)
A. Root mean square deviation (RMSD) of active site regions of β-secretase in unbound (red) and ligand bound (blue) simulated trajectory. B. Comparison of residue wise percentage of beta from unbound (red) and ligand bound (blue) simulated trajectory.
(DOCX)
A. Control insulin B. Glycated insulin C. glycation inhibition in presence of 500 µM and D. 1000 µM protriptyline. These spectra were acquired on a positive reflector mode by MALDI-TOF-MS. Glycated peaks are shown by black arrow.
(DOCX)
BSA (Bovine Serum Albumin) glycation inhibition assay. Glycation inhibition of BSA was studied by A. Measurement of AGE fluorescence, % glycation inhibition was plotted and B. Thioflavin T fluorescence assay, concentration dependent decrease in thioflavin T fluorescence was observed.
(DOCX)
Acetylcholinesterase-protriptyline interaction energy calculations. Interaction energy of ligand with active site residues averaged over last 20 ns of all simulated trajectories. Interaction strengths are in kcal mol-1 unit. Standard deviations are provided within braces.
(DOCX)
Inter-residue distance between active site of AChE residues averaged over last 20 ns of free (vertical) and ligand bound (horizontal) simulated trajectories. Distances are in Å unit. Standard deviations are provided within braces.
(DOCX)
Inhibition of Insulin and BSA glycation.
(DOCX)
System Setup for Molecular Dynamics Simulation.
(DOCX)
Acknowledgments
This work was supported by CSIR network project BSC0115. SBB, KBB acknowledge CSIR and AKJ, RSJ acknowledge UGC for the research fellowships. We thank Dr. Sourav Pal (Director) and Dr. Vidya Gupta (Head, Biochemical Sciences) for their support. We thank Dr. Amitava Das, Dr. Thulasiram, Dr. Sushama Gaikwad for allowing their lab facility to be used. CSIR-NCL for providing ITC facility.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by CSIR Network project, BSC0115. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Roberson E, Mucke L (2006) 100 Years and Counting: Prospects for Defeating Alzheimer's Disease. Science 314: 781–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bartus RT, Dean III RL, Beer B, Lippa AS (1982) The Cholinergic Hypothesis of Geriatric Memory Dysfunction. Science 217: 408–414. [DOI] [PubMed] [Google Scholar]
- 3. Alzheimer's Association: FDA-approved treatments for Alzheimers. Available: https://www.alz.org/national/documents/topicsheet_treatments.pdf [Google Scholar]
- 4. Small DH (2005) Acetylcholinesterase inhibitors for the treatment of dementia in Alzheimer's disease: do we need new inhibitors? Expert Opin Emerg Drugs 10: 817–825. [DOI] [PubMed] [Google Scholar]
- 5. Terry AV Jr, Buccafusco JJ (2003) The cholinergic hypothesis of age and Alzheimer's disease-related cognitive deficits: recent challenges and their implications for novel drug development. J Pharmacol Exp Ther 306: 821–827. [DOI] [PubMed] [Google Scholar]
- 6. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's dis-ease: progress and problems on the road to therapeutics. Science 297: 353–356. [DOI] [PubMed] [Google Scholar]
- 7. Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat Rev Mol Cell Biol 8: 101–112. [DOI] [PubMed] [Google Scholar]
- 8. Golde TE (2006) Disease modifying therapy for AD? J Neurochem 99: 689–707. [DOI] [PubMed] [Google Scholar]
- 9. Selkoe DJ (1999) Translating cell biology into therapeutic advances in Alzheimer's disease. Nature 399: A23–A31. [DOI] [PubMed] [Google Scholar]
- 10. Citron M (2002) Beta-secretase as a target for the treatment of Alzheimer's disease. J Neurosci Res 70: 373–379. [DOI] [PubMed] [Google Scholar]
- 11. Hussain I (2004) The potential for BACE-1 inhibitors in the treatment of Alzheimer's disease. IDrugs 7: 653–658. [PubMed] [Google Scholar]
- 12. Hills ID, Vacca JP (2007) Progress toward a practical BACE-1 inhibitor. Curr Opin Drug Discov Devel 10: 383–91. [PubMed] [Google Scholar]
- 13. Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, et al. (1994) Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Nat Acad Sci USA 91: 4766–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li XH, Du LL, Cheng XS, Jiang X, Zhang Y, et al. (2013) Glycation exacerbates the neuronal toxicity of β-amyloid. Cell Death and Disease 4: e673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anand P, Singh B (2013) A review on cholinesterase inhibitors for Alzheimer's disease. Arch Pharm Res 36: 375–99. [DOI] [PubMed] [Google Scholar]
- 16. Zhang C (2012) Natural compounds that modulate BACE1-processing of amyloid-beta precursor protein in Alzheimer's disease. Discov Med 14: 189–197. [PubMed] [Google Scholar]
- 17. Schenk D, Basi GS, Pangalos MN (2012) Treatment strategies targeting amyloid β-protein. Cold Spring Harb Perspect Med 2: a006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Götz J, Ittner A (2012) Tau-targeted treatment strategies in Alzheimer's disease. Br J Pharmacol 165: 1246–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Youdim MB, Buccafusco JJ (2005) Multi-functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol Sci 26: 27–35. [DOI] [PubMed] [Google Scholar]
- 20. Zhang HY (2005) One-compound-multiple-targets strategy to combat Alzheimer's disease. FEBS Lett 579: 5260–5264. [DOI] [PubMed] [Google Scholar]
- 21. Cappelli A, Gallelli A, Manini M, Anzini M, Mennuni L, et al. (2005) Further studies on the interaction of the 5-hydroxytryptamine3 (5-HT3) receptor with arylpiperazine ligands. Development of a new 5-HT3 receptor ligand showing potent acetylcholinesterase inhibitor properties. J Med Chem 48: 3564–3575. [DOI] [PubMed] [Google Scholar]
- 22. Lee S, Zheng X, Krishnamoorthy J, Savelieff MG, Park HM, et al. (2014) Rational design of a structural framework with potential use to develop chemical reagents that target and modulate multiple facets of Alzheimer's disease. J Am Chem Soc 136(1): 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Corbett A, Pickett J, Burns A, Corcoran J, Dunnett SB, et al. (2012) Drug repositioning for Alzheimer's disease. Nat Rev Drug Discov 11: 833–846. [DOI] [PubMed] [Google Scholar]
- 24. Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3: 935–949. [DOI] [PubMed] [Google Scholar]
- 25. Kellenberger E, Rodrigo J, Muller P, Rognan D (2004) Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins 57: 225–242. [DOI] [PubMed] [Google Scholar]
- 26. Campoy AV, Freire E (2005) ITC in the post-genomic era…? Priceless. Biophys Chem 115: 115–124. [DOI] [PubMed] [Google Scholar]
- 27. Shoemaker BA, Panchenko AR (2007) Deciphering Protein–Protein Interactions. Part I. Experimental Techniques and Databases. PLoS Comput Biol 3: 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tai K, Shen T, Henchman RH, Bourne Y, Marchot P, et al. (2002) Mechanism of Acetylcholinesterase Inhibition by Fasciculin: A 5-ns Molecular Dynamics Simulation. J Am Chem Soc 124: 6153–6161. [DOI] [PubMed] [Google Scholar]
- 29. Jana AK, Sengupta N (2012) Adsorption mechanism and collapse propensities of the full-length, monomeric Aβ (1–42) on the surface of a single-walled carbon nanotube: a molecular dynamics simulation study. Biophys J 102: 1889–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Santini S, Wei G, Mousseau N, Derreumaux P (2004) Pathway Complexity of Alzheimer's β-Amyloid Aβ16–22 Peptide Assembly. Structure 12: 1245–1255. [DOI] [PubMed] [Google Scholar]
- 31. Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW, et al. (2000) Amyloid Fibril Formation by Aβ16–22, a Seven-Residue Fragment of the Alzheimer's β-Amyloid Peptide, and Structural Characterization by Solid State NMR. Biochemistry 39: 1374–1375. [DOI] [PubMed] [Google Scholar]
- 32. Bernstein SL, Wyttenbach T, Baumketner A, Shea JE, Bitan G, et al. (2005) Amyloid beta-protein: monomer structure and early aggregation states of Abeta42 and its Pro19 alloform. J Am Chem Soc 127(7): 2075–2084. [DOI] [PubMed] [Google Scholar]
- 33. Tjernberg LO, Näslund J, Lindqvist F, Johansson J, Karlström AR, et al. (1996) Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J Biol Chem 271: 8545–8548. [DOI] [PubMed] [Google Scholar]
- 34. Chimon S, Shaibat MA, Jones CR, Calero DC, Aizezi B, et al. (2007) Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer's β-amyloid. Nat Struct Mol Biol 14: 1157–1164. [DOI] [PubMed] [Google Scholar]
- 35. Levine H (1993) Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci 2: 404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee C, Ham S (2011) Characterizing amyloid-beta protein misfolding from molecular dynamics simulations with explicit water. J Comput Chem 32: 349–355. [DOI] [PubMed] [Google Scholar]
- 37. Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, et al. (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease-is this type 3 diabetes? J Alzheimers Dis 7: 63–80. [DOI] [PubMed] [Google Scholar]
- 38. Li J, Liu D, Sun L, Lu Y, Zhang Z (2012) Advanced glycation end products and neurodegenerative diseases: mechanisms and perspective. J Neurol Sci 317: 1–5. [DOI] [PubMed] [Google Scholar]
- 39. Gulyaeva N, Zaslavsky A, Lechner P, Chlenov M, McConnell O, et al. (2003) Relative hydrophobicity and lipophilicity of drugs measured by aqueous two-phase partitioning, octanol-buffer partitioning and HPLC. A simple model for predicting blood–brain distribution. Eur J Med Chem 38: 391–396. [DOI] [PubMed] [Google Scholar]
- 40. Aboukhatwa M, Dosanjh L, Luo Y (2010) Antidepressants are a rational complementary therapy for the treatment of Alzheimer's disease. . Mol Neurodegener 5: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, et al. (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 16: 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, et al. (2006) TM4 Microarray Software Suite. Methods Enzymol 411: 134–193. [DOI] [PubMed] [Google Scholar]
- 44. Ellman GL, Courtney D, Andres V, Featherston RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7: 88–95. [DOI] [PubMed] [Google Scholar]
- 45. Copeland RA, Lombardo D, Giannaras J, Decicco CP (1995) Estimating ki values for tight binding inhibitors from dose-response plots. Bioorganic & Medicinal Chemistry Letters 5: 1947–1952. [Google Scholar]
- 46. Tamhane VA, Chougule NP, Giri AP, Dixit AR, Sainani MN, et al. (2005) In vivo and in vitro effect of Capsicum annum proteinase inhibitors on Helicoverpa armigera guts proteinases. Biochim Biophys Acta 1722: 156–167. [DOI] [PubMed] [Google Scholar]
- 47. Kale L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, et al. (1999) NAMD2: Greater Scalability for Parallel Molecular Dynamics. Comput Phys 151: 283–312. [Google Scholar]
- 48. MacKerell AD Jr, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, et al. (1998) All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B 102: 3586–3616. [DOI] [PubMed] [Google Scholar]
- 49. Mackerell AD Jr, Feig M, Brooks CL 3rd (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 25: 1400–1415. [DOI] [PubMed] [Google Scholar]
- 50. Zoete V, Cuendet MA, Grosdidier A, Michielin O (2011) SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 32: 2359–2368. [DOI] [PubMed] [Google Scholar]
- 51. Hill EH, Stratton K, Whitten DG, Evans DG (2012) Molecular dynamics simulation study of the interaction of cationic biocides with lipid bilayers: aggregation effects and bilayer damage. Langmuir 28: 14849–14854. [DOI] [PubMed] [Google Scholar]
- 52. Mabanglo MF, Serohijos AW, Poulter CD (2012) The Streptomyces-produced antibiotic fosfomycin is a promiscuous substrate for Archaeal isopentenyl phosphate kinase. Biochemistry 51: 917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Caulfield T, Medina-Franco JL (2011) Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J Struct Biol. 176: 185–191. [DOI] [PubMed] [Google Scholar]
- 54. Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23: 327–341. [Google Scholar]
- 55. Feller SE, Zhang Y, Pastor RW, Brooks BR (1995) Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys 103: 4613–4621. [Google Scholar]
- 56. Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, et al. (1995) A smooth particle mesh Ewald method. J Chem Phys 103: 8577–8593. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Thioflavin T Assay. Protriptyline causes concentration dependent decrease in Aβ aggregation.
(DOCX)
A. Snapshot of drug binding with Aβ monomer. Central hydrophobic core are in line representation B. Comparison of residue wise percentage of helix from unbound and ligand bound Aβ monomer simulated trajectory.
(DOCX)
A. Root mean square deviation (RMSD) of active site regions of β-secretase in unbound (red) and ligand bound (blue) simulated trajectory. B. Comparison of residue wise percentage of beta from unbound (red) and ligand bound (blue) simulated trajectory.
(DOCX)
A. Control insulin B. Glycated insulin C. glycation inhibition in presence of 500 µM and D. 1000 µM protriptyline. These spectra were acquired on a positive reflector mode by MALDI-TOF-MS. Glycated peaks are shown by black arrow.
(DOCX)
BSA (Bovine Serum Albumin) glycation inhibition assay. Glycation inhibition of BSA was studied by A. Measurement of AGE fluorescence, % glycation inhibition was plotted and B. Thioflavin T fluorescence assay, concentration dependent decrease in thioflavin T fluorescence was observed.
(DOCX)
Acetylcholinesterase-protriptyline interaction energy calculations. Interaction energy of ligand with active site residues averaged over last 20 ns of all simulated trajectories. Interaction strengths are in kcal mol-1 unit. Standard deviations are provided within braces.
(DOCX)
Inter-residue distance between active site of AChE residues averaged over last 20 ns of free (vertical) and ligand bound (horizontal) simulated trajectories. Distances are in Å unit. Standard deviations are provided within braces.
(DOCX)
Inhibition of Insulin and BSA glycation.
(DOCX)
System Setup for Molecular Dynamics Simulation.
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.