

Abstract
The mechanism by which Haemophilus influenzae causes meningitis is unclear. Previously, we established murine meningitis by intranasal instillation of H. influenzae as a cell-bound organism (CBO). In this study, we aimed to identify the molecules associated with inhibiting the transmigration of cells across the blood–brain barrier (BBB). Two-dimensional difference gel electrophoresis and protein identification by mass spectrometry were used for proteomic analysis. Analysis of the membranous extract from a tumor necrosis factor (TNF)-α-treated human brain microvascular endothelial cell (HBMEC) monolayer revealed 41 differentially expressed proteins. Zyxin, which is thought to be essential for tight cell-to-cell junctions, decreased 1.8-fold in TNF-α-treated HBMECs. In addition, zyxin transcript levels decreased 1.5-fold in cells derived from TNF-α-treated HBMECs. Intranasal instillation of CBOs in zyxin-deficient mice resulted in a significant higher mortality rate than in wild-type mice. Transmigration of CBOs across a HBMEC monolayer pretreated with TNF-α (1 ng/mL), interleukin (IL)-1β (10 ng/mL), or lipopolysaccharide (LPS; 10 ng/mL) was assayed by counting CBOs that migrated from an upper chamber into a lower chamber. HBMEC pretreated with TNF-α exhibited significantly greater migration (P < 0.01) than did control cells or cells treated with IL-1β or LPS. Our findings highlight that zyxin is an important protein protecting the tight junction of the BBB against cell transmigration across the BBB. Finally, TNF-α produced in respiratory infection when the primary infection reached the BBB caused decreased zyxin levels in BBB cell membranes. Furthermore, H. influenzae reaching the BBB as CBOs could transmigrate into cerebrospinal fluid across the zyxin-decreased BBB.
Keywords: TNF-α, blood-brain barrier, cell-bound organism, intracellular parasite, meningitis, secondary infection, zyxin
Introduction
The blood–brain barrier (BBB) is a structural and functional interface between the circulation and the central nervous system (CNS); it regulates the passage of blood-borne substances and cells into the brain, and thus maintains homeostasis in the neural microenvironment, which is crucial for normal neuronal activity and function.1 One of the unique structural characteristics of the BBB is its minimal permeability to water and solutes, a feature that is essential for normal CNS function. Several proinflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, IL-8, and interferon (IFN)-γ have been implicated in the regulation of BBB permeability.2 Various pathogens transmigrate across the BBB and infect the CNS through transcellular or paracellular routes, and/or via infected phagocytes.3
The mechanism by which Haemophilus influenzae causes meningitis is unclear. Based on the fact that H. influenzae is an intracellular parasite, we previously established a murine pneumonia model based on the inoculation of cell-bound organisms (CBOs) with H. influenzae as the intracellular parasite.4 Furthermore, we used the same method to establish a murine meningitis model by H. influenzae.5 In this model, the appearance of TNF-α, IL-6, and IL-1β in serum derived from infected mice commenced after 3 d, 5 d, and 1 d post-infection, respectively.6 The appearance of TNF-α in mouse serum coincided with the commencement of mortality in the mice due to meningitis.5,6
In this study, we investigated the factor(s) associated with meningitis caused by H. influenzae and sought to identify molecules associated with the inhibition of cell transmigration across the BBB using a proteomics-based approach to analyze specialized membrane-associated molecules of the BBB. Our results identified the protein zyxin as a tight junction candidate in both human and mouse CNS microvascular endothelia.
Results
Proteomic analysis of a membrane extract from human brain microvascular endothelial cells (HBMECs) exposed to TNF-α treatment
The tight junction in the BBB is composed of the transmembrane proteins occludin and claudin.7,8 The increase in BBB permeability during acute liver failure paralleled the elevated serum level of TNF-α, and could be blocked by an IgG antibody against TNF-α.9 Based on these observations, we performed proteomic analyses of membranous extracts of a HBMEC monolayer exposed to TNF-α or tissue culture medium alone, in order to identify proteins whose expression was altered by TNF-α. Forty-one proteins were induced or repressed upon exposure to TNF-α but not to the culture medium alone; we defined changes of greater or less than 1.5-fold as significant (Table 1). Thirty-seven of the proteins were identified based on their homology to known eukaryotic proteins in the NCBInr database. Proteins of the reticulum family were prominent among the most highly induced proteins (1.9-fold increase), while moesin production was reduced 1.8-fold by TNF-α exposure. These proteins are generally thought to control cellular function and morphology. Zyxin content decreased 1.8-fold. Zyxin is thought to be essential for tight cell-to-cell junctions.10 We measured zyxin transcript expression by reverse transcription polymerase chain reaction (RT-PCR) in HBMECs treated with TNF-α and found a 1.5-fold decrease vs. the control, consistent with the decrease in protein content. These results suggest that zyxin is required in the tight junctions of the BBB.
Table 1. Properties of HBMC membrane proteins significantly expressed or repressed upon TNF-α exposure in comparison to non-treated cells.
| Ratioa | Gene name | MWb | pI | Cysteinec | P valued | Mascot scoree |
|---|---|---|---|---|---|---|
| 1.91 | Unnamed protein product | 24.7 | 8.66 | 3 | 3.20E−06 | 129 |
| Arginine-rich protein-human | 26.9 | 10.08 | 9 | 3.20E−06 | 51 | |
| Reticulon 3 isoform a | 25.6 | 8.67 | 3 | 3.20E−06 | 36 | |
| Reticulon 1 isoform A | 83.6 | 4.62 | 5 | 3.20E−06 | 36 | |
| Hypothetical protein | 66.3 | 7.68 | 10 | 3.20E−06 | 36 | |
| 1.75 | Unnamed protein product | 24.7 | 8.66 | 3 | 1.20E−05 | 91 |
| Enhancer protein | 22.1 | 8.16 | 6 | 1.20E−05 | 88 | |
| 1.74 | Lymphocyte specific 1 isoform 1 | 36.7 | 4.77 | 2 | 0.0043 | 81 |
| 1.7 | Dihydrolipoyl dehydrogenase | 54.1 | 7.59 | 10 | 0.017 | 59 |
| Structure of human glutamate dehydronase (Chain A) | 56.0 | 6.71 | 6 | 0.017 | 55 | |
| Glutamate dehydrogenase 1 variant | 61.3 | 8.05 | 6 | 0.017 | 51 | |
| 1.64 | Tyrosin3/tryptophan 5-mono-oxygenase activation protein | 29.3 | 4.63 | 2 | 0.0057 | 7 |
| Light polypeptide isoform a (Clathrin) | 23.2 | 4.63 | 2 | 0.0057 | 45 | |
| 1.62 | Manganese superoxidedismutase | 24.7 | 8.35 | 3 | 9.50E−05 | 49 |
| Enhancer protein | 22.1 | 8.16 | 6 | 9.50E−05 | 39 | |
| Ring finger protein 148 | 34.4 | 8.49 | 8 | 9.50E−05 | 34 | |
| 1.58 | Heterogeneous nuclear ribonucleo-protein A/B isoform a | 35.9 | 6.49 | 2 | 0.0037 | 84 |
| Protein tyrosin kinase | 40.3 | 6.48 | 3 | 0.0037 | 83 | |
| Ubiquitin fusion degradation1-like isoform A | 34.5 | 6.27 | 5 | 0.0037 | 70 | |
| Translation initiation factor eLF3 p40 40subunit eIF3p | 39.9 | 6.38 | 3 | 0.0037 | 69 | |
| Carboxyl terminal LIM domain protein | 36.1 | 6.80 | 8 | 0.0037 | 62 | |
| –1.5 | Hydroxysteroid (17-β) dehydronerase 10 isoform 1 | 26.9 | 7.66 | 4 | 6.90E−05 | 57 |
| Heterogeneous nuclear ribonucleo-protein A2/Ba isoform A2 | 36.0 | 8.67 | 1 | 6.90E−05 | 55 | |
| TPI1 protein | 27.4 | 8.48 | 6 | 6.90E−05 | 47 | |
| Triosephosphate isomerase (TIM) | 26.6 | 7.10 | 5 | 6.90E−05 | 46 | |
| –1.52 | Neuroleukin | 63.1 | 8.43 | 4 | 0.009 | 84 |
| Chaperonin containing TCP1, subunit 7 isoform a | 59.3 | 7.55 | 9 | 0.009 | 39 | |
| –1.55 | Unknown | 28.3 | 8.59 | 3 | 5.20E−05 | 46 |
| –1.63 | Heterongeneous nuclear ribonucleo-protein A1 | 38.8 | 9.26 | 2 | 5.20E−05 | 56 |
| –1.68 | Unknown | 28.3 | 8.59 | 3 | 0.0029 | 46 |
| Ribosomal protein small subunit structure of the karyopherin beta2-ran gppnhp | 30.0 | 9.75 | 4 | 0.0029 | 37 | |
| Nuclear transport complex (Chain C) | 24.3 | 7.01 | 3 | 0.0029 | 34 | |
| –1.72 | Cell cycle proteinp38–2G4 homolog | 43.8 | 6.13 | 6 | 0.0084 | 38 |
| –1.8 | Moesin | 67.8 | 6.08 | 2 | 0.0058 | 62 |
| ESP-2 (zyxin) | 53.1 | 5.89 | 21 | 0.0058 | 41 | |
| –1.83 | Adenylate kinase 2 isoform a | 26.5 | 7.67 | 4 | 0.0088 | 59 |
| High-mobility group box 2 | 24.0 | 7.62 | 3 | 0.0088 | 48 | |
| –1.84 | Heterogeneous nuclear ribonucleo-protein A1 | 38.8 | 9.26 | 2 | 0.00076 | 53 |
| Dermcidin preproprotein | 11.3 | 6.08 | 2 | 0.00076 | 40 | |
| Heterogeneous nuclear ribonucleo-protein A2/B1 isoform B1 | 37.4 | 8.91 | 1 | 0.00076 | 35 |
a Relative protein abundance TNF-α treated cells: non-treated cells; bMolecular weight; cNumber of cysteine residues (or is this number of predicted cysteine residues?); dP value (X test); eMascot score derived using Y method.
Establishment of a murine H. influenzae meningitis model using CBOs and comparison of mortality between wild and zyxin-deficient mice
We hypothesized that phagocytic dendritic cells (DCs) might gather in respiratory organs (primary infection site) and phagocytize H. influenzae organisms, which might thereby reach the BBB and then transmigrate across the BBB to cause meningitis. We, therefore, determined that DCs might be superior to mouse fetal lung cells for the induction and analysis of murine meningitis using CBOs. To test whether H. influenzae organisms could invade murine DCs, the bacteria were mixed with purified DCs as described previously.4,5 After 60 min of incubation, invading bacteria were observed within the cells by electron microscopy (data not shown) and the percentage of DCs receptive to H. influenzae organisms was more than 95%. These DCs, which contain intracellular H. influenzae, are referred to as CBOs.
Intranasal inoculation of mice with CBOs produced meningitis in three of 20 tested wild-type mice after 3–5 d. Histopathological findings in infected wild-type mice showed the presence of neutrophils in the cerebrospinal fluid (CSF), but no neutrophils were observed in wild-type mice that received intranasal instillation of phosphate-buffered saline (PBS) (Fig. 1A and B). In addition, the bacilli associated with a monocyte could be observed in the CSF (Fig. 1C). The viable counts of H. influenzae in CSF derived from each mouse with meningitis were 6 × 104, 2 × 104, and 1.3 × 104 cfu/brain, respectively (Fig. 2A). Moreover, the histopathology of the brain and meninges in H. influenzae-infected mice demonstrated meningitis, epithelial cells were swollen (Fig. 1B). The same mice developed mild pneumonia as demonstrated by the presence of viable H. influenzae at 7 × 103, 1.9 × 103, and 1.6 × 103 cfu/lung (Fig. 2B).

Figure 1. Meningitis by intranasal inoculation of CBOs Histopathology of the brain and spinal marrow samples by hematoxylin-eosin staining (400×). (A) Control (without infection). (B) Epithelial cells were swollen with neutrophils and monocytes. The arrowheads indicate neutrophils. (C) Spinal fluid was obtained from a mouse by sub-occipital puncture using a 28-gauge butterfly needle 3 d post-infection, and stained with Giemsa stain. Representative findings in five mice, three of which were studied on day 3 post-infection, and two on day 5 post-infection. The arrowhead indicates an organism.
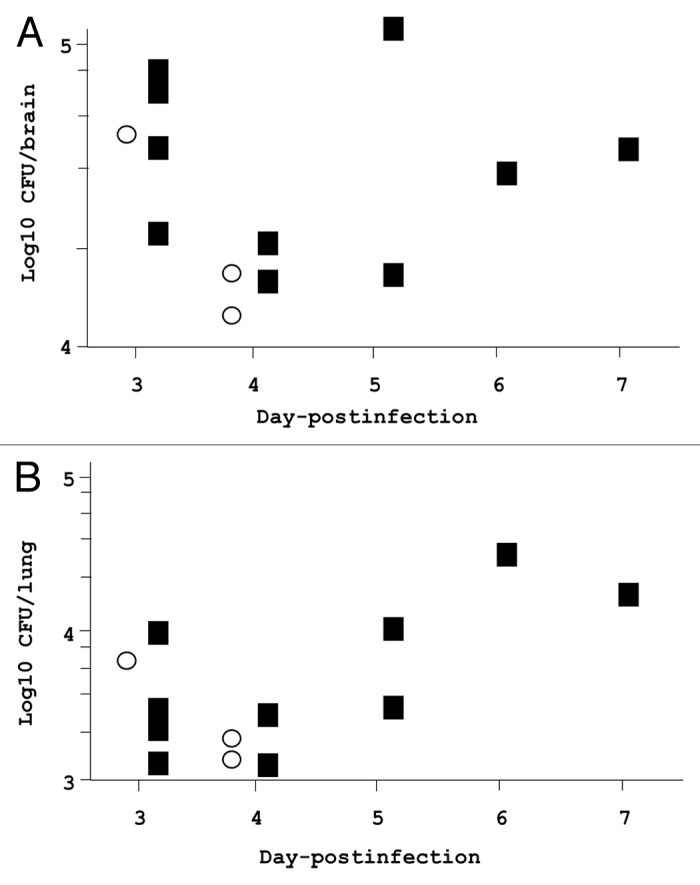
Figure 2. Viable organisms in the brain and lungs of zyxin-deficient and wild-type mice following intranasal inoculation of H. influenzae in CBOs. Brain and lung H. influenzae concentrations in zyxin-deficient and wild-type mice infected with intranasal CBO; the mice died due to infection. These data correspond to the experiment shown in Figure 3. (A) H. influenzae log10 cfu/brain as a function of time post-infection. (B) H. influenzae log10 cfu/lung as a function of time post-infection. Data are plotted by day of death. Open circles, wild type; closed squares, zyxin-deficient mice
To determine whether zyxin is one of the essential proteins that inhibit cell transmigration across the BBB, we created zyxin-deficient mice. Intranasal instillation of CBO into zyxin-deficient mice resulted in a significantly higher mortality fraction (50% vs. 15%, P = 0.03) and rate of death (inverse hazard ratio 3.8) than in wild-type mice (Fig. 3). These changes in zyxin-deficient mice did not appear to be due to higher bacterial concentrations in the brains or lungs (Fig. 2).
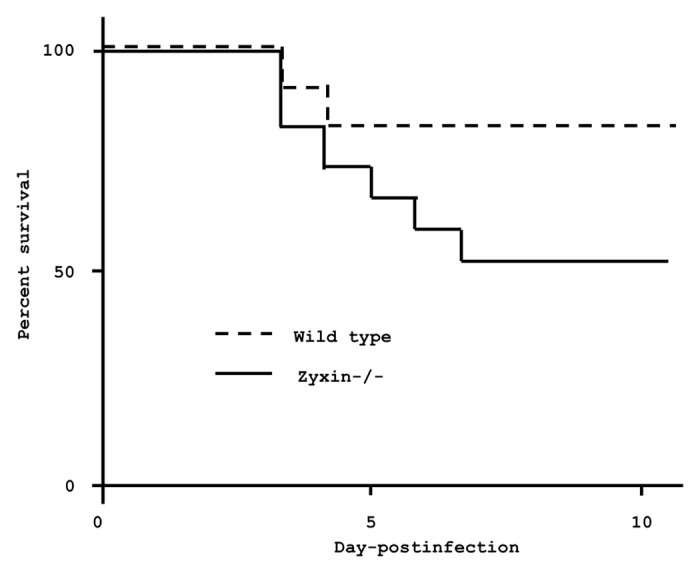
Figure 3. Survival of zyxin-deficient and wild-type mice after intranasal inoculation of H. influenzae in CBOs. Twenty mice of each genotype were infected with H. influenzae-containing CBOs delivered via the intranasal route; P = 0.03 by Mantel-Cox (log rank) test. Hazard ratio = 0.26; 95% confidence interval (CI) 0.08–0.82. A second independent experiment gave similar results, with P = 0.006 and a hazard ratio of 0.05 (0.007 to 0.43).
In contrast to the development of meningitis in zyxin-deficient mice challenged with intranasal CBOs, none of the 20 mice infected intravenously with CBOs (2 × 105 cfu/mouse) developed meningitis, as determined by autopsy findings 7 d post-infection. In addition, mice challenged with intravenous injection had no detectable H. influenzae organisms in blood, whole lung, or whole brain cultures, 24 h post-infection (data not shown). Respiratory infection, as a primary infection, occurred with intranasal instillation, but was not induced in intravenous injection. Collectively, these results show that the decrease of zyxin caused by TNF-α produced due to the primary infection might be necessary for the induction of meningitis.
The effects of cytokines and LPS on the transmigration of CBOs through the HBMEC monolayer
We previously reported that serum TNF-α and IL-1β levels were 375–421 and 3890–1810 pg/mL at 3 to 5 d post-infection.6 To confirm the involvement of these cytokines and LPS in BBB permeability, we determined the transmigration of CBOs through an HBMEC monolayer pretreated with TNF-α, IL-1β, and LPS. The number of CBOs transmigrating across the monolayer was significantly greater (P < 0.001, analysis of variance [ANOVA] and Dunnet post-hoc tests) after treatment with TNF-α than after treatment with IL-1β or LPS, or without treatment (Fig. 4). Moreover, transmigration of CBOs closely correlated with the concentration of TNF-α. In addition, the number of transmigrated CBOs was significantly greater with the addition of macrophage inhibitory protein (MIP)-1 into the lower chamber than without MIP-1 (P < 0.001, ANOVA). Collectively, those results confirm that TNF-α treatment accelerates the transmigration of CBOs across the BBB.

Figure 4. Treatment of HBMECs and the ability of CBO to transmigrate. Mean values of CBO transmigration in treated cells normalized to the null treatment control, with or without concomitant treatment with MIP-1 (10 ng/mL). TNF-α, 1 and 10 ng/mL; IL-1β, 10 ng/mL; LPS, 10 ng/mL. Error bars represent 95% confidence intervals. **P < 0.01 in comparison to non-MIP-1 treated control; ++P < 0.01 in comparison to MIP-1 treated control; NS, P > 0.05 in comparison to the controls. MIP-1 treated cells allowed significantly greater transmigration (P < 0.05) than did non-MIP-1 treated cells that were otherwise treated identically, with the exception of LPS treatment. Dotted horizontal lines represent the mean and 95% confidence intervals for control:control ratios. n = 5 for each data point.
Discussion
The incidence of meningitis due to Escherichia coli and Streptococcus pneumoniae, but not H. influenzae and Neisseria monocytogenes, is dependent on bacterial counts in the blood.11,12 Listeria monocytogenes-infected phagocytes are present in the bloodstream of experimentally infected mice, indicating that leukocytes transport intracellular bacteria to the brain and that cell-free bacteria are eliminated from the bloodstream during in vivo infections.13 In addition, in the present murine meningitis model, the detection of bacilli associated with monocytes in CSF indicate that H. influenzae organisms act as intracellular parasites, invading monocytes such as DCs in the primary infection site, the lungs. These data show that intracellular parasites reach the BBB and transmigrate across the BBB to induce meningitis. However, when viable organisms under CBO conditions were intravenously injected into both wild-type and zyxin-deficient mice, in the present study, unexpectedly no mice died during the observation period. In addition, infected organisms were not detected in blood, lungs, and brain tissue 24 h post-infection. This result indicates that there may be other pivotal factor(s) other than an intracellular parasite condition.
There is a close correlation between increased blood and CSF cytokine levels induced by injury, and breakdown of the BBB, as TNF-α is often a principal cause of changes in permeability.8 The detection of TNF-α in mouse serum coincided with the commencement of mortality due to meningitis after infection.9 This cytokine was produced at the primary infection site, such as the respiratory organs, because it was detected 3 d post-infection, and reached the BBB through the blood circulation.9 In addition, intravenous injection of TNF-α may increase the permeability of the BBB.14,15 Combined together, TNF-α might be a pivotal factor for the induction of meningitis. Furthermore, the present study indicated that neither influx nor efflux was mediated by LPS acting directly on BBB cells.16 On the other hand, upon cisternal puncture of IL-1β or TNF-α in rats, IL-1β is more effective than TNF-α as an independent inducer of meningitis.17 However, this method assumes that these cytokines are derived not from the blood but from the CSF. In essence, meningitis is caused by the transmigration of pathogens from the bloodstream into the CSF; such transmigration across the BBB should be due to TNF-α derived from blood vessels, not IL-1β in the CSF.
The major molecular components of tight junctions in BBB include transmembrane and structural proteins, such as occludin, JAM, and claudin, and the sub-membranous peripheral ZO proteins.18 TNF-α inhibits occludin expression and increases BBB permeability.19 A direct link between Rho activation and tight junction disruption in the BBB was identified by treating monocytes with the brain endothelial proteins occludin and claudin-5.20 It has been reported that tight junction components in intact microvessels of the BBB are associated with α-actinin, vinculin, zyxin, cadherin, and A-CAM.19 We did not identify proteins other than zyxin because other proteins did not exhibit more than a 1.5-fold change in membranous extracts of a HBMEC monolayer compared with tissue culture medium alone. The present study confirmed that not only protein but also mRNA expression of zyxin dramatically decreased in the membranes of HBMECs treated with TNF-α. The murine meningitis experiment showed significantly higher mortality rate in zyxin-deficient mice than in wild-type mice. We conclude that zyxin is one of the important proteins protecting the tight junction of the BBB against the transmigration of phagocytes across the BBB.
In conclusion, the present study found that cells containing H. influenzae organisms, as well as the decrease in zyxin expression caused by TNF-α, both induce the occurrence of meningitis. However, the mortality rate of zyxin-deficient mice was not 100%; therefore, our future studies will aim to identify other factor(s) associated with meningitis.
Materials and Methods
Bacteria
H. influenzae strain 770235f+b+ was a gift from Dr L van Alphen. The bacteria were cultured overnight in Brain Heart Infusion broth (Difco) supplemented with NAD and hemin.
Generation of zyxin-deficient mice
Zyxin-deficient mice were generated by the inGenious Targeting Laboratory, Inc. as described previously.21 Briefly, F1 offspring were screened for heterozygotes (+/−) by PCR analysis using primer 1 (common reverse primer in Exon 8), 5′-AGGTGAAGCA CTGTGGGTGG TAG-3′; primer 2 (forward primer for the targeted allele in the neor gene), 5′-GCATCGCCTT CTATCGCCTT CTTG-3′; and primer 3 (forward primer for the wild-type allele in exon 6), 5′-TAGCCAGAGC TATGTAGTGA GATCCTGTCT C-3′. This allowed the simultaneous amplification of the wild-type and targeted alleles as 0.5-kb and 0.96-kb fragments, respectively. The heterozygous mice were backcrossed to C57BL/6N mice for eight generations. The zyxin-deficient mutant (−/−) strain was established by intercrossing the backcrossed heterozygous animals and was maintained by breeding −/− males and females.
Preparation of dendritic cells
Bone marrow was prepared as previously described from 4- to 6-wk-old female C57BL/6 mice (Charles River Japan) and zyxin−/−C57BL/6 mice anesthetized with pentobarbital sodium (50 mg/kg).22 Isolation of bone marrow cells was similar to the previously described method with the exception that on day 6, half of the culture supernatant was exchanged, and on day 8, non-adherent cells were collected by gentle pipetting. These non-adherent cells were centrifuged at 300× g for 5 min, and resuspended in 7 mL RPMI1460 (Life Technologies Japan).
Preparation of cell-bound organisms
The bacterial suspension (1 mL, 1 × 108 cfu/mL) was added to a 10-mL suspension of dendritic cells (2 × 106/mL), and gently shaken for 60 min at 35 °C. The suspension was centrifuged at 500 × g for 5 min; the pellet was suspended in 10 mL RPMI1640 containing gentamycin (100 mg/L), and then incubated for 30 min at room temperature to kill free bacteria. Finally, the precipitated CBOs were suspended in 6 mL RPMI 1640 containing 2 × 106 cfu/mL viable bacteria. To confirm that H. influenzae is an intracellular parasite, the viable organisms were assayed as described.4 The percentage of DCs receptive to H. influenzae organisms was calculated by the method described previously.23
Experimental infection in mice
The experimental protocol was approved by the Toho University Ethics Review Committee for Animal Experimentation on March 8, 2009 (permission no. 9-51-54). C57BL/6 and zyxin-deficient C57BL/6 mice were experimentally challenged with CBOs comprised of H. influenzae 770235f-b+ in dendritic cells via intranasal4 or intravenous routes. The inocula were 2 × 105 cfu/mouse (intravenous) and 1 × 105 cfu/mouse (intranasal). The intravenous inoculum was delivered via the caudal vein in a 0.1-mL volume. Twenty animals were used in each infection experiment, and each infection experiment was performed twice. Preliminary studies showed that 10% of the intranasal inoculum was retained in the lungs (unpublished data).
Giemsa staining
Cerebrospinal fluid (CSF) was drawn by sub-occipital puncture using a 28-gauge butterfly needle under anesthesia with pentobarbital from 1 d post-infection to 7 d post-infection, and the color and turbidity of the fluid was checked visually to confirm the absence of blood and debris. The specimen was Giemsa-stained. After cervical dislocation, brain and spinal marrow samples were fixed in phosphate-buffered 4% paraformaldehyde and processed with a conventional paraffin-embedding procedure for hematoxylin-eosin staining at 3 d post-infection.
Preparation of HBMECs
HBMECs were a gift from Prof KS Kim of Johns Hopkins University. HBMECs were cultured in RPMI 1640 medium (Life Technologies) supplemented with fetal calf serum (10%; Life Technologies), Nu serum IV (10%; Becton Dickinson), MEM vitamins (1%), non-essential amino acids (1%), sodium pyruvate (1 mM), and l-glutamine (2 mM) (all reagents from Pharmacia Biotech). The culture was incubated to confluence in T25 flasks (Corning Life Sciences), in a humid atmosphere at 37 °C with 5% CO2. Two days before use, the cells were treated with trypsin-EDTA (Life Technologies Japan) and seeded into gelatin-coated 24-well tissue culture plates (Corning Life Sciences) at a density of 5 × 104 cells/well. Cells were grown to about 2 × 106 cells/well before use.
Transmigration assay
Transmigration assays were performed as described.24 Briefly, HBMECs were added (2.5 × 104 cells/well) to fibronectin-treated 12-µm pore, 12-mm diameter Costar transwells (Corning). The presence of a confluent monolayer was confirmed by microscopy. Prior to performing the transmigration assay, a pair of wells containing monolayer cells were pretreated with TNF-α (1 or 10 ng/mL) and IL-1β or LPS (10 ng/mL) for 24 h. CBOs (1–2 × 106 cells) were then added to each upper well, and monocyte chemotactic protein-1 (MCP-1, 10 ng/mL) was applied to the lower well of one of each pair of treated and control specimens. The transwells were kept for 4 h in a CO2 incubator at 37 °C. After incubation, the transmigrated CBOs were collected from the bottom of each well, centrifuged (10 min, 400 × g), resuspended in a defined volume, and counted with a hemocytometer.
Protein extraction from HBMEC membranes
HBMECs were cultured in two culture bottles containing 10 mL RPMI1460 for 5 d, and then one of the bottles was treated with TNF-α (10 ng/mL) for 16 h in a CO2 incubator at 37 °C. The monolayer cells in both bottles (treated and untreated) were independently harvested and suspended in 10 mL PBS, and washed three times with PBS. Proteins were extracted with ProteoExtract 71772-3 (Merck).
Two-dimensional difference gel electrophoresis
Two-dimensional difference gel electrophoresis (2D-DIGE) was performed with fluorescence labeling as described.25,26 In brief, a sample containing 5 μg protein was reduced with 2 nmol Tris(2-carboxyethyl)phosphine (TCEP) at 37 °C for 60 min. Subsequently, 4 nmol of Cy3 or Cy5 saturation dye in anhydrous dimethylformamide (DMF) was added, and the sample was incubated at 37 °C for 30 min. For preparative gel electrophoresis, a 250-μg protein sample was reduced with 100 nmol TCEP and labeled with 200 nmol Cy3 saturation dye. The labeling reaction was terminated by the addition of an equal volume of lysis buffer containing 130 mM dithiothreitol (DTT) and 2% IPG buffer, pH 3–11 NL. The labeled samples were stored at –80 °C.
First-dimension separation was performed using Immobiline Drystrips (24 cm, pH 3–11 NL). We loaded 5 μg of the Cy3-labeled protein, which was a mixture of equal amounts of all samples, as the internal control, and 5 μg of Cy5-labeled protein of the sample to be analyzed, on each gel. After rehydration of the IPG strips with the labeled samples diluted in rehydration buffer (lysis buffer containing 13 mM DTT and 1% IPG buffer, pH 3–11 NL) to 450 μL for 12 h at 20 °C, focusing was performed at 500 V and 1000 V for 1 h each, and 6000 V for a total of 90 KVh. The current was limited to 50 μA per strip. Prior to sodium dodecyl sulfate PAGE (SDS-PAGE) for the second dimensional separation, each strip was equilibrated twice with 10 mL equilibration buffer (100 mM TRIS-HCl, 6 M urea, 2% SDS, 30% glycerol, and 30 mM DTT, pH 8.8) for 15 min with gentle shaking. The IPG strips were then loaded and run on 12.5% polyacrylamide Laemmli gels27 using the Ettan DALT Twelve apparatus (GE Healthcare). The Laemmli gels were cast in 21 × 24 cm glass plates and the gels were run at 1 W per gel constant power at 20 °C. For preparative purposes, 500 μg Cy3-labeled protein was separated by two-dimensional difference gel electrophoresis in the same way, with the difference that one inner side of the Laemmli gel glass plate was precoated with bind-silane. All gel electrophoresis was performed in the dark, and three replicates of each sample were performed to minimize the effect of gel-to-gel variation. After separation by electrophoresis, gel images were collected with a Typhoon 9400 (GE Healthcare) fluorescence gel scanner. The Cy-dye images were scanned at 100-μm resolution and cropped with ImageQuant software (GE Healthcare), then imported into DeCyder software version 6.5 (GE Healthcare).
Expressed proteins, seen as spots, were detected with the differential in-gel analysis (DIA) module of the DeCyder software. These images were normalized to the Cy3 images as internal controls, and the biological variation analysis (BVA) module of DeCyder was used to calculate average abundance changes and analyze differential protein expression across all gels. Normalized spot volumes were analyzed by t test to compare average standardized abundances from triplicate samples. The spots that changed significantly were extracted by specifying thresholds for the average fold change and significance (P < 0.05).
In-gel digestion
Protein spots of interest were excised with an automated spot collector (Ettan Spot Picker, GE Healthcare) from a preparative Cy3 2D-DIGE gel. The gel pieces were treated with a modified in-gel digestion method to prepare peptide samples for identification by mass spectrometry. The gel pieces were washed vigorously five times with 100 μL wash buffer (60% acetonitrile containing 100 mM ammonium bicarbonate) for 10 min; then the gel pieces were washed with 100 μL acetonitrile for 5 min and dried completely with a SpeedVac Concentrator (SPD2010, Thermo Electron Corp.) for 15 min. The dried gel pieces were swelled with 20 μL of 50 mM ammonium bicarbonate and incubated at 37 °C for 10 min. After the excess solution was removed, the gel pieces were dried in a SpeedVac Concentrator and re-swelled in 20 μL of 1 mM TRIS-HCl (pH 8.5) containing 200 ng Lys-C, and incubated on ice for 45 min. After the Lys-C solutions were removed and 20 μL of 1 mM TRIS-HCl (pH 8.5) was added, enzymatic digestions were performed overnight at 37 °C. The reaction solutions were transferred to new tubes and the peptide fragments in each gel piece were extracted by vortexing for 15 min with 50 μL of 60% acetonitrile. Each extract was combined with the reaction solution and evaporated to dryness with the SpeedVac Concentrator. The prepared in-gel digestion samples were stored at −30 °C.
Protein identification by mass spectrometry
Proteins from the spots of interest were identified with an Agilent 1200 series nanoLC-Chip MS system (Agilent Technologies) coupled with an HCT Ultra PTM Discovery mass spectrometer (Bruker Daltonics). In-gel digestion samples were desalted and separated with HPLC Chip (ProtID-Chip-150 II, 150 mm 300 Å C18 chip with 40 nL trap column, Agilent Technologies). The peptide fragments were eluted by an acetonitrile gradient and distilled water containing 0.1% formic acid at 200 nL/min, and injected directly into the HCT Ultra through an online ESI system of Chip-Cube MS interface (Agilent Technologies). The analytical conditions were as follows: m/z range; 350–1500, ionization mode; positive, voltage between ion spray tip and shield; 1800 V, dry gas temperature; 300 °C. MS spectra were repeatedly recorded and followed by two data-dependent CID MS/MS spectra generated from the two highest intensity precursor ions. Data acquisition and processing were performed with DataAnalysis 4.0 (Bruker Daltonics). Mascot 2.2 software (Matrix science) was used to search the mass spectrum data obtained from the nanoLC-ESI MS/MS analysis against the NCBI and UniprotKB databases for protein identification. Search parameters were set as follows: enzyme selected as Lys-C with two maximum missing cleavage sites; human species; mass tolerance of 0.3 Da for peptide tolerance and 0.6 Da for MS/MS tolerance; variable modifications of methionine oxidation; Cy3- and hydrolyzed Cy3-labeling of cysteine. Positive protein identifications were based on a significant MOWSE score.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by a grant of the strategic basis on research grounds for non-governmental schools at Heisei 20th from Ministry of Education, Culture, Sports, Science and Technology-Japan (S0801028). We thank Hiroyuki Okamoto, PhD (Diagnostics Division, Shionogi and Co., Ltd.), Atsushi Morita, PhD., Ms Yoshino Hirabaayashi-Ihsioka (Innovative Drug Discovery Research laboratories, Shionogi and Co., Ltd.), and Yoshinori Yamano, PhD (Medicinal Research laboratories, Shionogi and Co., Ltd.) for assistance with two-dimensional difference gel electrophoresis. Dr Paul Edelstein (Department of Pathology and Laboratory Medicine, University of Pennsylvania Perelman School of Medicine) provided editorial assistance.
Glossary
Abbreviations:
- CBO
cell-bound organism
- BBB
blood–brain barrier, TNF-α, tumor necrosis factor alpha
- HBMEC
human brain microvascular endothelial cell
- IL
interleukin
- LPS
lipopolysaccharide
- RT-PCR
reverse transcription polymerase chain reaction
- DC
dendritic cell
- CSF
cerebrospinal fluid
- CNS
central nervous system
References
- 1.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 2.Mayhan WG. Cellular mechanisms by which tumor necrosis factor-alpha produces disruption of the blood-brain barrier. Brain Res. 2002;927:144–52. doi: 10.1016/S0006-8993(01)03348-0. [DOI] [PubMed] [Google Scholar]
- 3.Pulzova L, Bhide MR, Andrej K. Pathogen translocation across the blood-brain barrier. FEMS Immunol Med Microbiol. 2009;57:203–13. doi: 10.1111/j.1574-695X.2009.00594.x. [DOI] [PubMed] [Google Scholar]
- 4.Miyazaki S, Nunoya T, Matsumoto T, Tateda K, Yamaguchi K. New murine model of bronchopneumonia due to cell-bound Haemophilus influenzae. J Infect Dis. 1997;175:205–9. doi: 10.1093/infdis/175.1.205. [DOI] [PubMed] [Google Scholar]
- 5.Miyazaki S, Matsumoto T, Furuya N, Tateda K, Yamaguchi K. The pathogenic role of fimbriae of Haemophilus influenzae type b in murine bacteraemia and meningitis. J Med Microbiol. 1999;48:383–8. doi: 10.1099/00222615-48-4-383. [DOI] [PubMed] [Google Scholar]
- 6.Miyazaki S, Nunoya T, Matsumoto T, Furuya N, Tateda K, Yamaguchi K. Lipoolygosaccharide indirectly enhances inflammatory lesions in lungs as a primary infection site by non-encapsulated and type B Haemophilus influenzae through production of cytokines. Cytokine. 2001;15:171–4. doi: 10.1006/cyto.2001.0908. [DOI] [PubMed] [Google Scholar]
- 7.Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141:1539–50. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–88. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lv S, Song HL, Zhou Y, Li LX, Cui W, Wang W, Liu P. Tumour necrosis factor-α affects blood-brain barrier permeability and tight junction-associated occludin in acute liver failure. Liver Int. 2010;30:1198–210. doi: 10.1111/j.1478-3231.2010.02211.x. [DOI] [PubMed] [Google Scholar]
- 10.Schulze C, Firth JA. Immunohistochemical localization of adherens junction components in blood-brain barrier microvessels of the rat. J Cell Sci. 1993;104:773–82. doi: 10.1242/jcs.104.3.773. [DOI] [PubMed] [Google Scholar]
- 11.Kim KS. Escherichia coli translocation at the blood-brain barrier. Infect Immun. 2001;69:5217–22. doi: 10.1128/IAI.69.9.5217-5222.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim KS. Strategy of Escherichia coli for crossing the blood-brain barrier. J Infect Dis. 2002;186(Suppl 2):S220–4. doi: 10.1086/344284. [DOI] [PubMed] [Google Scholar]
- 13.Drevets DA, Dillon MJ, Schawang JS, Van Rooijen N, Ehrchen J, Sunderkötter C, Leenen PJ. The Ly-6Chigh monocyte subpopulation transports Listeria monocytogenes into the brain during systemic infection of mice. J Immunol. 2004;172:4418–24. doi: 10.4049/jimmunol.172.7.4418. [DOI] [PubMed] [Google Scholar]
- 14.Takata F, Dohgu S, Matsumoto J, Takahashi H, Machida T, Wakigawa T, Harada E, Miyaji H, Koga M, Nishioku T, et al. Brain pericytes among cells constituting the blood-brain barrier are highly sensitive to tumor necrosis factor-α, releasing matrix metalloproteinase-9 and migrating in vitro. J Neuroinflammation. 2011;8:106–18. doi: 10.1186/1742-2094-8-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abraham CS, Deli MA, Joo F, Megyeri P, Torpier G. Intracarotid tumor necrosis factor-alpha administration increases the blood-brain barrier permeability in cerebral cortex of the newborn pig: quantitative aspects of double-labelling studies and confocal laser scanning analysis. Neurosci Lett. 1996;208:85–8. doi: 10.1016/0304-3940(96)12546-5. [DOI] [PubMed] [Google Scholar]
- 16.Jaeger LB, Dohgu S, Sultana R, Lynch JL, Owen JB, Erickson MA, Shah GN, Price TO, Fleegal-Demotta MA, Butterfield DA, et al. Lipopolysaccharide alters the blood-brain barrier transport of amyloid β protein: a mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav Immun. 2009;23:507–17. doi: 10.1016/j.bbi.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quagliarello VJ, Wispelwey B, Long WJ, Jr., Scheld WM. Recombinant human interleukin-1 induces meningitis and blood-brain barrier injury in the rat. Characterization and comparison with tumor necrosis factor. J Clin Invest. 1991;87:1360–6. doi: 10.1172/JCI115140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lagaraine C, Skipor J, Szczepkowska A, Dufourny L, Thiery JC. Tight junction proteins vary in the choroid plexus of ewes according to photoperiod. Brain Res. 2011;1393:44–51. doi: 10.1016/j.brainres.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Silwedel C, Förster C. Differential susceptibility of cerebral and cerebellar murine brain microvascular endothelial cells to loss of barrier properties in response to inflammatory stimuli. J Neuroimmunol. 2006;179:37–45. doi: 10.1016/j.jneuroim.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 20.Persidsky Y, Heilman D, Haorah J, Zelivyanskaya M, Persidsky R, Weber GA, Shimokawa H, Kaibuchi K, Ikezu T. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE) Blood. 2006;107:4770–80. doi: 10.1182/blood-2005-11-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffman LM, Nix DA, Benson B, Boot-Hanford R, Gustafsson E, Jamora C, Menzies AS, Goh KL, Jensen CC, Gertler FB, et al. Targeted disruption of the murine zyxin gene. Mol Cell Biol. 2003;23:70–9. doi: 10.1128/MCB.23.1.70-79.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lutz MB, Kukutsch N, Ogilvie AL, Rössner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/S0022-1759(98)00204-X. [DOI] [PubMed] [Google Scholar]
- 23.Miyazaki S, Matsunaga T, Kobayashi I, Yamaguchi K, Goto S. The other mediator for adherence of Haemophilus influenzae organisms without involvement of fimbriae. Microbiol Immunol. 1992;36:205–12. doi: 10.1111/j.1348-0421.1992.tb01658.x. [DOI] [PubMed] [Google Scholar]
- 24.O’Brien CD, Lim P, Sun J, Albelda SM. PECAM-1-dependent neutrophil transmigration is independent of monolayer PECAM-1 signaling or localization. Blood. 2003;101:2816–25. doi: 10.1182/blood-2002-08-2396. [DOI] [PubMed] [Google Scholar]
- 25.Shaw J, Rowlinson R, Nickson J, Stone T, Sweet A, Williams K, Tonge R. Evaluation of saturation labelling two-dimensional difference gel electrophoresis fluorescent dyes. Proteomics. 2003;3:1181–95. doi: 10.1002/pmic.200300439. [DOI] [PubMed] [Google Scholar]
- 26.Greengauz-Roberts O, Stöppler H, Nomura S, Yamaguchi H, Goldenring JR, Podolsky RH, Lee JR, Dynan WS. Saturation labeling with cysteine-reactive cyanine fluorescent dyes provides increased sensitivity for protein expression profiling of laser-microdissected clinical specimens. Proteomics. 2005;5:1746–57. doi: 10.1002/pmic.200401068. [DOI] [PubMed] [Google Scholar]
- 27.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]