

Abstract
Background
Anesthetics enhance gamma-aminobutyric acid (GABA)-mediated inhibition in the central nervous system. Different agents have been shown to act on tonic versus synaptic GABA receptors to different degrees, but it remains unknown whether different forms of synaptic inhibition are also differentially engaged. With this in mind, we tested the hypothesis that different types of GABA-mediated synapses exhibit different anesthetic sensitivities. The present study compared effects produced by isoflurane, halothane, pentobarbital, thiopental and propofol on paired pulse GABAA receptor-mediated synaptic inhibition. Effects on glutamate-mediated facilitation were also studied.
Methods
Synaptic responses were measured in rat hippocampal brain slices. Orthodromic paired pulse stimulation was used to assess anesthetic effects on either glutamate-mediated excitatory inputs or GABA-mediated inhibitory inputs to CA1 neurons. Antidromic stimulation was used to assess anesthetic effects on CA1 background excitability. Agents were studied at equi-effective concentrations for population spike depression to compare their relative degree of effect on synaptic inhibition.
Results
Differing degrees of anesthetic effect on paired pulse facilitation at excitatory glutamate synapses were evident, and blocking GABA inhibition revealed a previously unseen presynaptic action for pentobarbital. Although all five anesthetics depressed synaptically evoked excitation of CA1 neurons, the involvement of enhanced GABA-mediated inhibition differed considerably among agents. Single pulse inhibition was enhanced by propofol, thiopental and pentobarbital, but only marginally by halothane and isoflurane. In contrast, isoflurane enhanced paired pulse inhibition strongly, as did thiopental, but propofol, pentobarbital and halothane were less effective.
Conclusions
These observations support the idea that different GABA synapses use receptors with differing subunit compositions, and that anesthetics exhibit differing degrees of selectivity for these receptors. The differing anesthetic sensitivities seen in the present study, at glutamate and GABA synapses, help explain the unique behavioral/clinical profiles produced by different classes of anesthetics, and indicate that there are selective targets for new agent development.
Introduction
General anesthetics have long been known to enhance gamma-aminobutyric acid (GABA)A-mediated inhibition, particularly in hippocampal cortex.(1-4) There is no doubt that this effect contributes to anesthesia because GABA is the most important neurotransmitter mediating synaptic inhibition in all higher brain regions. It has been estimated that almost 1/3 of all synapses in hippocampus, thalamus and neocortex use GABA, and GABA-mediated inhibitory postsynaptic currents (IPSCs) have been observed in virtually all types of subcortical,(5) hippocampal and cortical neurons, including inhibitory interneurons.(6-8) In vivo studies using knock-in mice with a single point mutation in GABAA receptors that makes them insensitive to some anesthetics have shown that behavioral responses to the anesthetics propofol and etomidate are markedly attenuated.(9) That said, there remains a good deal of controversy about the relative contribution of enhanced GABA-mediated inhibition to various anesthetic endpoints, especially for immobility and/or amnesia.(10,11)
Several forms of inhibition mediated by GABAA receptors have been described.(12,13) Hippocampal CA1 neurons, for example, exhibit at least two forms of GABAA synaptic currents, fast IPSCs with decay time constants of 3 to 10 ms, and slow GABAA IPSCs with decay times of 30 to 70 ms,(14) not to be confused with GABAB synaptic currents that last much longer, > 100 to 1000 ms. Synapses exhibiting fast kinetics appear to be localized to the cell body (stratum pyramidal) region, while slow IPSCs appear to occur preferentially in dendritic regions of CA1 neurons. In addition, CA1 neurons also exhibit tonic GABAA-mediated currents, thought to be generated by extrasynaptic receptors.(15,16) Tonic GABAA receptors appear to differ from synaptic receptors by incorporating alpha5 and possibly beta3 subunits (17) that impart a high sensitivity to GABA (in the μM range), and relatively nondesensitizing responses to GABA, and also by their extrasynaptic localization.(18) It is also likely that the different forms of synaptic inhibition (fast vs slow) are also mediated by GABA receptors made up of different subunit compositions.(19,20)
Little is known about the anesthetic sensitivity of different forms of GABAA-mediated inhibition. Several studies suggest that tonic receptors on CA1 neurons may be particularly sensitive to some agents, for example, ethanol,(21,22) propofol and thiopental,(16) but not to isoflurane.(23) Differential anesthetic effects on different types of GABAA receptors could help explain the diverse profiles of effects produced by different anesthetic classes in vivo.(24) GABAA subtype selective anesthetic effects could also lead to the development of more selectively targeted agents for anesthesia. The purpose of the present study was to investigate the effects of five anesthetics on GABAA-mediated synaptic inhibition associated with paired pulse responses in the hippocampal CA1 circuit. Agents from four distinct chemical classes were chosen, as well as two barbiturates with distinctly different potencies and kinetic profiles, for comparison.
Methods
Brain Slice Preparation
Experimental protocols were approved by the animal use committee at Stanford University and adhered to published guidelines of the National Institutes of Health as well as the Society for Neuroscience. Brain slices were prepared from male Sprague-Dawley rats that had been deeply anesthetized with isoflurane. The brain was quickly removed and placed in ice cold artificial cerebrospinal fluid (ACSF) that was saturated with 95% O2 and 5% CO2 (carbogen) to achieve a pH of 7.4. The ACSF was composed of the following ions dissolved in spectrophotometry grade water (EMD Chemicals Inc., N.J.) and stated in mM concentrations: Na 151.25, Cl 131.5, HCO3 26, K 2.5, Ca 2, Mg 2, SO4 2, H2PO4 1.25, and glucose 10. Coronal brain slices were prepared using a vibratome (Model 1500, Technical Products International Inc.) and 450-micron thick slices were hemisected and placed on cellulose filter papers. They were stored at a gas (carbogen) - liquid (ACSF) interface in a holding chamber for at least one hour before use.
Single slices were transferred to a recording chamber and submerged in room temperature (22 °C) ACSF flowing at a rate of 3.0 ml/min. ACSF was saturated with carbogen by bubbling the solution for at least 15 minutes before use. Room temperature slices exhibit stable synaptic responses for several hours and appear to remain viable for much longer time periods compared to slices maintained at physiological temperatures.(25,26) Anesthetic solubility, especially for volatile agents, increases at room temperature, relative to physiological temperatures, so care was taken to ensure concentrations were measured and appropriately compensated.(27,28) Teflon storage vessels and tubing were used throughout the perfusion system to ensure minimal loss of anesthetic agents due to diffusion or binding.
Electrophysiology
Evoked field potentials were measured using thin walled glass pipettes (1.0 ID, 1.5 OD; Garner Glass Co., CA) filled with ACSF. Electrodes were pulled on a two stage patch pipette puller (Model PP-830, Narishige, Japan) and had a final tip resistance of 2 to 3 MOhm. Electrodes were placed in stratum radiatum to record field excitatory postsynaptic potentials (EPSPs) or placed in stratum oriens, close to the CA1 cell body layer, to record population spikes (Figure 1). Bipolar tungsten microelectrodes (10 MOhm, Frederick Haer & Co., ME) were placed in stratum radiatum and used to stimulate Schaffer-collateral fibers to synaptically drive CA1 neurons. Single or paired stimulus pulses (0.01 to 0.03 ms duration; unipolar, 10 to 80 μA at 1.0 to 8.0 V) were generated from constant current stimulus isolation units (SIU 6D, Grass Instruments, MA) driven by a Grass S8800 stimulator. Stimulus rates of 0.05 Hz were used and paired pulses had interstimulus intervals of 60 ms. Paired pulse stimulation was used to measure anesthetic effects on tonic and fast (1st pulse) and slow (2nd pulse) inhibitory pathways in the CA1 area neural circuit (Figure 1), since 60 ms is too late to involve fast synapses, but ideal for the time-course of slow synapses. Effects on tonic inhibition would contribute equally to both responses. Antidromic activation of CA1 neurons was produced by stimulating output axons with electrodes placed in the alveus (Figure 1) to test for nonsynaptic depression of CA1 neurons. Recorded signals were conditioned using an Axoclamp 2B preamplifier (Axon Instruments, CA) with a bandpass from DC to 30 KHz and gain of 10×. Signals were further amplified (100×) and conditioned (DC offset, DC to 30KHz) using a differential instrumentation amplifier in single-ended mode (Model 440, BrownLee Precision, CA). Signals were digitized at 10 KHz using a National Instruments analogue to digital converter (USB 6009) and Igor Pro software (Wavemetrics Inc., OR) on a MacBook Pro computer. Responses were measured and plotted in real time to ensure that stable baselines (< ± 2.0% variation) were recorded for at least 10 minutes before experiments began. Preparations showing any upward or downward drift in baseline were not used for experiments. Responses were analyzed off line and graphed for publication figures using IgorPro software on MacBook Pro computers.
Figure 1.
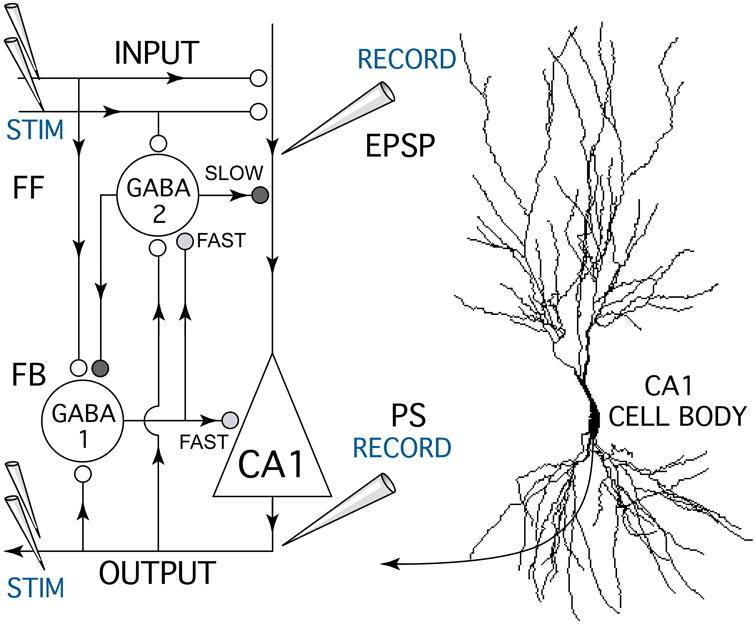
Schematic diagram of the hippocampal CA1 circuit showing the experimental setup used to test anesthetic effects on excitatory glutamate synapses and GABA-mediated inhibitory feedforward (FF) and feedback (FB) synapses. A bipolar stimulating electrode (STIM) was placed on Schaffer-collateral fiber excitatory inputs (INPUT) which make glutamate synapses (white circles) onto CA1 pyramidal neuron apical dendrites. Excitatory postsynaptic potentials (EPSPs) resulting from input stimulation were recorded using a field potential electrode (RECORD) placed in the apical dendrite zone where synaptic currents were generated. A second recording electrode was placed near CA1 neuron cell bodies to measure population spike discharges (PS, OUTPUT) generated in response to Schaffer-collateral synaptic activation. Paired pulse stimuli were used measure anesthetic effects on different forms of GABA-mediated synaptic inhibition (grey and black symbols) generated by inhibitory neurons (GABA 1-FAST & 2-SLOW). Antidromic population spike responses were generated by stimulating CA1 neuron axons (STIM OUTPUT) with a bipolar electrode placed in the alveus. Arrowheads indicate the direction of information flow in the CA1 circuit.
Drug Solutions
The volatile anesthetics (isoflurane or halothane) were applied in the ACSF perfusate after at least 15 minutes of equilibration, by bubbling into the ACSF with a carbogen carrier gas. Concentrations were determined using calibrated vaporizers (Cyprane, UK) and gas phase concentrations were measured using an anesthetic airway gas monitor (Datex Model 254, Puritan-Bennett Corp., Finland). Intravenous anesthetics (pentobarbital, thiopental and propofol) were made up fresh before each experiment. They were solubilized using 0.5% dimethyl sulfoxide and vortexed into stock solutions before being serially diluted into pre-bubbled (carbogen) ACSF solutions to achieve final concentrations, just before testing. Dimethyl sulfoxide at a concentration of 2.0% did not alter recorded responses (n = 5). GABA antagonists (bicuculline, gabazine, or CGP 35348) or the chloride channel blocker (picrotoxin) were added to pre-gassed (carbogen) ACSF solutions that had been pre-equilibrated with a given anesthetic. All drugs were added as at least a 1/500 final dilution from concentrated stock solutions in water. A single concentration of an anesthetic (and antagonist/blocker) was tested on each slice preparation to avoid tachyphylaxis or cross contamination of drug effects. The entire perfusion system was cleaned and replaced on a regular basis to avoid cross contamination between anesthetics, antagonists, or blockers.
Data Analysis
Time-matched experiments were averaged together using IgorPro software and graphed as mean ± SD of measured responses versus experimental time (in minutes; sampled three times per minute). Averaged experimental data are displayed as a percentage of control responses normalized to the size of baseline responses for ten minutes preceding the start of each experiment. Statistical analyses (ANOVA with post Tukey test) comparing non-normalized control, anesthetic, antagonist + anesthetic, or blocker + anesthetic responses were performed using Igor Pro software. In all cases “n” refers to individual experiments conducted on separate slices, usually from different rats. A two-tailed repeated measures analysis was used and data were considered significant at a level of p <0.01 and post hoc p > 0.15 was considered as not significant. Each experiment contributed a single “measure” and at least five measures were repeated for each statistical comparison. For the main effect of each anesthetic and anesthetic + antagonist, p values of < 0.0001 were evident, also seen as non-overlapping standard deviations in all graphs, so these p values were typically not stated. If analysis of the initial 5 experiments suggested that a significant difference was close to being achieved (i.e., p > 0.01 but < 0.05), then additional experiments were performed and added to the final analysis. Linear regression analysis and correlation coefficients were determined using a least squares approach in Igor Pro.
Results
Effective Concentrations for CA1 Circuit Depression
Initial studies determined the effective concentrations needed to produce a complete block of single pulse population spike responses for the five anesthetics investigated. Different anesthetics exhibit profoundly different diffusion characteristics for penetrating into brain slices.(28-30) In the present experiments, relatively aqueous soluble anesthetics like halothane and isoflurane produced rapid population spike depression, while highly lipid soluble agents like propofol and thiopental required several hours to achieve steady-state effects. For this reason, concentration-effect profiles were determined using a criterion of 100% population spike depression within 30 minutes to allow comparisons across anesthetic agents at an equi-effective concentration, within a reasonable time period. The volatile agents produced this level of depression with concentrations that were very close to the effective concentrations for producing anesthesia in rats (1.0 rat MAC – minimum alveolar concentration for blocking a tail clamp reflex). Halothane produced complete population spike depression at 1.21 ± 0.09 vol % (∼ 250 μM) close to MAC (1.26 ± 0.08 vol % for adult Sprague-Daley rats(31)). Isoflurane required higher concentrations to produce a comparable degree of population spike depression, 1.43 ± 0.14 vol % (∼350 μM), and these concentrations were also close to in vivo MAC concentrations for this agent (1.55 ± 0.08 vol %(31)).
The IV anesthetics also produced complete population spike depression at concentrations near their predicted in vivo clinically relevant levels. Propofol was the most potent agent tested and produced complete depression at an applied concentration of 30 μM. This would correspond to a brain slice effect-site concentration of 1.0 to 3.0 μM at the recording depth of 250 – 300 microns used in the present experiments.(28) Thiopental produced a complete population spike depression at 80 μM and pentobarbital was the least potent anesthetic studied, producing depression at a concentration of 400 μM. The brain slice effect-site concentrations for these barbiturates would be approximately 30 to 40 μM for thiopental and 350 to 375 μM for pentobarbital, using a simple ratio calculation based on the lipid solubility of these agents compared to that of propofol.(28)
Anesthetics Exhibit Differential Effects on GABAA-Mediated Inhibition at a Dendritic Level
To determine whether anesthetics altered glutamate-mediated excitatory synaptic transmission, field EPSPs were recorded in stratum radiatum, at the level of Schaffer-collateral fiber synapses onto CA1 neuron apical dendrites. Paired pulse stimuli were used to test for anesthetic effects on tonic as well as synaptic GABA inhibition, responses to the second pulse provide a measure of additional GABAA slow synaptic inhibition generated by the first stimulus pulse (Fig 1), as reported earlier in vivo and in vitro.(2,32) The ability of a GABAA receptor antagonist, bicuculline, to reverse anesthetic effects was studied at a concentration that has previously been shown to completely block GABA inhibition in the CA1 region (10 μM).(33) Each of the anesthetics studied produced a depression of EPSP responses; however, marked differences in degree of depression and bicuculline reversal of effects were apparent.
Isoflurane depressed first pulse EPSP responses by 47.8 ± 14.1% and second pulse EPSP responses by 42.4 ± 8.3% (Figure 2A; p < 0.001, ANOVA, n = 6 for both 1st and 2nd responses compared to control). Neither of these paired pulse response depressions were reversed by bicuculline (p > 0.15), indicating that the isoflurane-induced depression was not mediated by enhanced GABAA inhibition. Pentobarbital also depressed both first and second pulse EPSP responses by 56.2 ± 16.2% and 60.1 ± 8.5% respectively (p < 0.001 for both, compared to control values, ANOVA, n = 7). Blocking GABAA receptors was able to partially reverse the pentobarbital-induced depression, by 9.3 ± 4.6% for first pulse responses and 19.8 ± 6.1% for second pulse responses. Thus, pentobarbital appeared to increase both a tonic GABAA-mediated inhibition, to depress first pulse responses, and also enhanceGABAA-mediated synaptic inhibition, accounting for the additional bicuculline-sensitive depression evident for second pulse EPSP responses (Figure 2B). Halothane produced a 37.9 ± 8.8% depression of first pulse EPSP responses and a 29.5 ± 7.7% depression of second pulse responses (p < 0.005, ANOVA, n = 5). Neither of these halothane-induced depressions were reversed by bicuculline (depression remained at 41.9 ± 10.4% and 32.3 ± 11.9% for first and second pulse responses respectively, in the presence of the GABA antagonist, p > 0.15). Propofol was less effective at depressing EPSP responses, producing a 27.7 ± 21.8% and 41.7 ± 12.6% depression of first and second pulse responses, respectively (p < 0.05 and p < 0.001, ANOVA, n = 8). Nearly all of the propofol-induced EPSP depression was reversed by bicuculline and responses returned to 98.7 ± 17.0% and 93.5 ± 14.3% of control for first and second pulse responses, indicating that propofol enhanced tonic as well as synaptic GABA-mediated inhibition in the CA1 neuron dendrites (p < 0.01 for both pulses comparing pre- and post-bicuculline values). Similar effects were evident for thiopental, first pulse EPSP responses were depressed by 21.3 ± 8.9% and second pulse responses by 32.4 ± 7.9% (p < 0.005 and p < 0.001, ANOVA, n = 5) and both were completely reversed by bicuculline (to 102.3 ± 10.4% and 97.5 ± 11.4% of control, p < 0.01). Thus, the volatile anesthetics were more effective at depressing glutamate-mediated EPSP responses and did not enhance either tonic or synaptic GABA-mediated inhibition at the level of CA1 apical dendrites. The IV anesthetics, in contrast, produced EPSP depression by enhancing both tonic and synaptic GABA-mediated inhibition, either entirely like propofol and thiopental, or partially, like pentobarbital.
Figure 2.
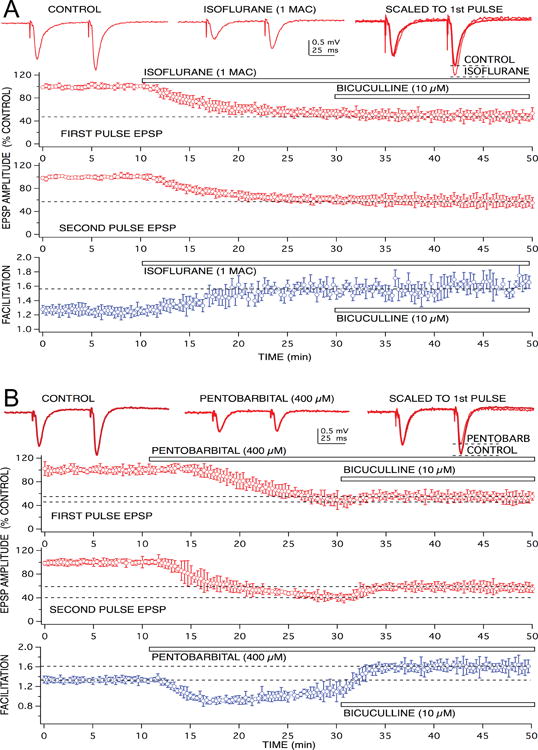
Anesthetics produced different effects on GABAA receptor-mediated feedforward inhibition at the level of CA1 neuron apical dendrites. Recordings of glutamate-mediated excitatory postsynaptic potentials (EPSPs) are shown at the top, in response to paired pulse stimulation of Schaffer-collateral fiber inputs. The volatile anesthetic, isoflurane (1.0 rat MAC = 1.5 vol % ∼ 350 μM) did not appear to enhance feedforward inhibition – evident as a lack of effect on paired pulse facilitation of EPSPs (A). Although EPSPs were depressed by isoflurane, responses still exhibited facilitation. In addition, the EPSP depression produced by isoflurane was not reversed using a GABAA receptor antagonist, bicuculline. This was evident in the time vs EPSP response amplitude graphs for both first and second paired pulse responses. In these and subsequent graphs, symbols represent the mean ± SD from at least five, time matched, experiments from separate slices. Isoflurane produced a 50% depression of EPSP amplitudes (when applied at the time indicated by the bar), when bicuculline was added to the preparation, in the continued presence of isoflurane, there was no additional change in response amplitude. Pentobarbital, in contrast, did appear to enhance GABAA receptor-mediated feedforward inhibition, because a clear depression of EPSP facilitation was produced by the barbiturate (B). In addition, pentobarbital-induced EPSP depression was partially reversed when bicuculline was applied in the continuing presence of the anesthetic.
Most of the volatile anesthetic-induced EPSP depression appeared to involve a presynaptic action as previously described,(34,35) because paired pulse facilitation was increased by these agents. Neither propofol nor thiopental increased paired pulse facilitation, however, pentobarbital also produced an increase in facilitation, but this was only revealed after block of GABA inhibitory effects with bicuculline (Fig 2B). Thus, pentobarbital depressed EPSP responses by both enhancing dendritic inhibition, as well as by a presynaptic action to reduce glutamate release as previously reported.(36)
Concentration-Dependent Effects on Paired Pulse Population Spike Responses
The anesthetics studied produced differential, concentration-dependent, effects on paired pulse population spike responses. For the IV agents, propofol, thiopental and pentobarbital, second pulse responses were considerably more sensitive than first pulse responses to anesthetic-induced depression. Pentobarbital, for example, produced a 77.9 ± 18.3% depression of second pulse responses at a concentration (50 μM; Figure 3; p < 0.001, ANOVA, n = 5) that had no appreciable effect on first pulse responses (99.5 ± 3.7% of control). Bicuculline fully reversed the depression of second pulse responses produced by 50 μM, and responses returned to about 10% above the control baseline. This indicates that all of the population spike depression produced by the low 50 μM concentration of pentobarbital could be attributed to enhanced synaptically driven GABA-mediated slow inhibition, since any effects on GABA fast or tonic receptors would have been seen on first pulse responses. Higher concentrations of pentobarbital produced depression of first pulse responses and also completely blocked second pulse responses (Figure 3B, C, D, E). Comparing concentration-effect relationships for first and second pulse responses indicated that pentobarbital was approximately 10-fold more potent at depressing second pulse responses (Fig 3F). Thus, the increased slow synaptic inhibition seen for second pulse responses dramatically increases the effectiveness of pentobarbital for depressing CA1 neuron discharge.
Figure 3.
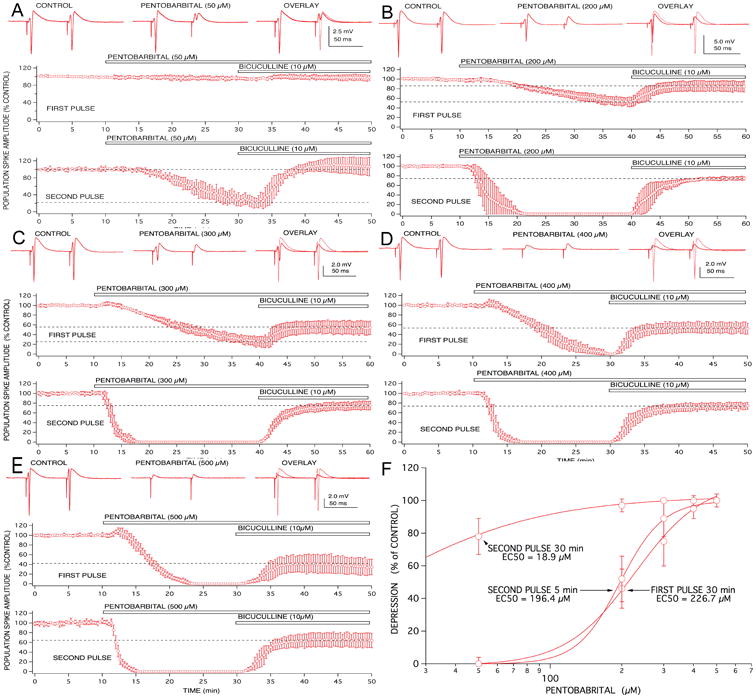
Anesthetic effects on paired pulse population spike responses recorded from CA1 neurons were concentration-dependent and exhibited differential involvement of GABAA receptor-mediated inhibition. Low concentrations of pentobarbital (50 μM; A) produced little effect on first pulse responses, but reduced second pulse responses to approximately 20% of control. This depression of second pulse responses appeared to be mediated entirely by enhanced GABAA-mediated inhibition, because it was completely reversed by bicuculline. Higher concentrations of pentobarbital (200 μM; B and 300 μM; C) produced increasing degrees of first pulse response depression that was only partially reversed by bicuculline. Similarly, second pulse depression produced by these concentrations was only partially reversed by the GABAA receptor antagonist. A transient excitatory effect on first pulse responses became evident at the highest concentrations of pentobarbital studied (300 and 500 μM; C and D). Symbols represent the mean ± SD for at least five time matched experiments from separate brain slices for each concentration studied.
The ability of bicuculline to reverse anesthetic-induced depression was also dependent on the anesthetic concentration. The GABA antagonist was much more effective at reversing effects produced by low concentrations, compared to higher concentrations, and this was true for both first and second pulse responses (Fig 3). Similar results were evident for thiopental and propofol, however, the volatile agents produced a comparable depression of both first and second pulse responses, at all effective concentrations. The threshold concentration for population spike depression with halothane and isoflurane was approximately 0.2 MAC and the small depressions produced (∼ 10%) were not reversed by bicuculline. The lowest effective applied concentrations for propofol and thiopental were 5.0 and 15 μM, respectively, and the small depressions of second pulse responses were completely reversed by bicuculline.
All of the Enhanced Synaptic Inhibition Produced by Anesthetics was GABAA-Mediated
To better characterize the anesthetic-induced effects on paired pulse population spike responses, the actions of GABAA and GABAB receptor antagonists were compared. The GABAB receptor antagonist CGP 35348 did not reverse any of the depression produced by any anesthetic studied, indicating that enhanced inhibition via GABAB receptor-mediated synapses does not contribute to anesthetic-induced depression of population spike responses over the time course measured here (Fig 4A: 60 ms). The GABAA synaptic receptor antagonist, gabazine (10 μM) was just as effective as bicuculline for reversing the effects of isoflurane and pentobarbital, indicating little additional contribution from tonic receptors (gabazine does not block CA1 tonic GABA receptors(15,16,23)). The chloride channel blocker, picrotoxin, was slightly more effective at reversing the first pulse depression produced by pentobarbital and equi-effective at reversing second pulse responses (Figure 4B). Thus, the anesthetic-induced enhanced inhibition seen in these experiments appears to involve GABAA-gated chloride channels. The difference between bicuculline and picrotoxin reversal of first pulse responses was significant (p < 0.05; ANOVA with post Tukey test) suggesting that the GABAA receptors involved in first versus second pulse responses probably include a somewhat different mix, exhibiting different sensitivities to the two GABA blockers. A comparison of 50 μM versus 200 μM picrotoxin effects on the paired pulse responses also supports this, as does the apparent difference in kinetics for first and second pulse response reversal by picrotoxin (Figure 4B). In any case, most of the depression produced by pentobarbital appeared to involve synaptically driven neuronal activity in CA1 neurons, because when CA1 neuron excitability was directly tested using antidromic stimulation of axons, there was only a slight depression of population spikes at concentrations that completely blocked orthodromic population spikes (Figure 4C). Second pulse antidromic responses where more sensitive to pentobarbital than were first pulse responses, consistent with a stronger role for feedback inhibition in depressing CA1 neuron excitability. All of the depression of antidromic responses was reversed by picrotoxin (Fig 4C).
Figure 4.
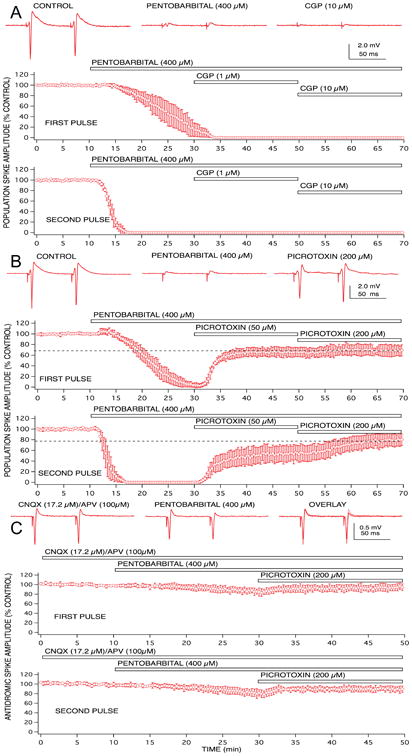
Population spike depression produced by pentobarbital was partially mediated by enhanced GABAA receptor gated chloride channels, but GABAB receptors and non-synaptic effects did not appear to play any role. A concentration of 400 μM pentobarbital was sufficient to produce complete depression of first and second pulse population spike responses. The GABAA receptor antagonist, bicuculline partially reversed the depression for both paired pulse responses (A). The GABAB receptor antagonist, CGP 35348 did not reverse any of the pentobarbital-induced depression, even in the presence of supra-maximally effective concentrations of the antagonist (10 μM; B). The chloride channel blocker, picrotoxin, produced a similar – but not identical – degree of reversal of the pentobarbital-induced depression of population spike responses (C). Changes in postsynaptic excitability of CA1 neurons did not appear to play a major role in the pentobarbital-induced depression, since only a small depression of antidromically-evoked population spike responses was evident at concentrations that produced complete depression of orthodromically-evoked responses (D), and all of this depression was reversed by picrotoxin. Symbols represent the mean ± SD for at least five time matched experiments from separate brain slices.
Anesthetics Exhibited Differential Effects on Paired Pulse Responses
Unlike pentobarbital, the isoflurane-induced depression of first pulse responses was hardly reversed at all by bicuculline (Fig 5), but second pulse response depression was fully reversed, reversed even more than the reversal of second pulse responses seen with pentobarbital. Thus, the contribution of increased GABA-mediated slow synaptic inhibition for isoflurane-induced depression of second pulse responses was considerably greater than that for first pulse responses. It should be noted that the increased paired pulse facilitation of EPSP responses produced by isoflurane would contribute, in part, to increase second pulse response amplitudes after application of bicuculline. However, a similar increase in facilitation was produced by pentobarbital (Fig 2) and by halothane, yet neither of these anesthetics exhibited the same degree of disproportional bicuculline reversal of first and second pulse responses. It should also be noted that there did not appear to be any concentration-dependency for GABA antagonist reversal of isoflurane effects (Fig 5B), nor did different GABA blockers exhibit different degrees of effect, unlike the case with pentobarbital. Thus, the GABA receptor subtype mix that these two anesthetics act on appear to be different. Alternatively, it is possible that isoflurane might have a stronger effect than pentobarbital on excitation of feedforward inhibitory interneurons, leading to a selective depression of these neurons and an apparent reduction of their inhibitory effectiveness. Little work has been done to compare anesthetic effects on different types of neurons in the CA1 circuit,6 or elsewhere in the brain, so this possibility will await further detailed single cell studies.
Figure 5.
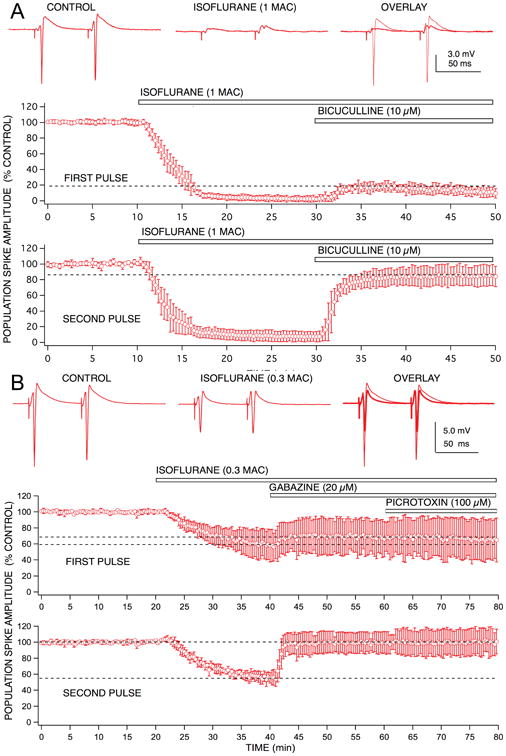
Population spike depression produced by isoflurane exhibited marked differences in GABAA receptor involvement for first vs second pulse responses. Isoflurane (1.0 rat MAC = 1.4 vol % ∼ 350 μM) produced a similar degree of depression for both first and second pulse responses, unlike pentobarbital, there did not appear to be any preferential effect on second pulse population spike depression. The GABAA receptor antagonist, bicuculline produced a small reversal of the depression observed for first pulse responses (< 20% recovery), and a marked reversal of the depression for second pulse responses (> 90% recovery). Thus, isoflurane was able to discriminate between effects on 1st pulse vs 2nd pulse GABAA receptor-mediated inhibition, and appear to markedly enhance only feedback inhibition. Symbols represent the mean ± SD for at least five time matched experiments from separate brain slices.
The ability of GABA receptor antagonists and chloride channel blockers to reverse anesthetic-induced depression of first versus second pulse population spike responses varied widely across the agents studied (Fig 6A). For some anesthetics, like propofol, there was no significant difference between the bicuculline-induced reversal of first and second pulse responses. Isoflurane, in contrast exhibited a significant reversal of second pulse responses compared to first pulse responses (p < 0.001; ANOVA with post Tukey test for comparing bicuculline reversal of 1st and 2nd pulses). Across all the anesthetics studied, no consistent pattern of effects were evident, agents which exhibited strong effects on GABAA inhibition for second pulse responses had variable effects on first pulse responses (Fig 6). Propofol was the most effective agent for enhancing first pulse (probably tonic) inhibition,(16) but was less effective at enhancing second pulse inhibition (mostly synaptic) compared to thiopental and isoflurane. The rank order for enhancing first pulse inhibition was: propofol > thiopental > pentobarbital > halothane > isoflurane. The rank order for enhancing second pulse inhibition was: thiopental > isoflurane > pentobarbital > halothane > propofol. There was no apparent correlation (r < 0.22) between an anesthetic's ability to enhance inhibition associated with first and second pulse responses (Fig 6B).
Figure 6.
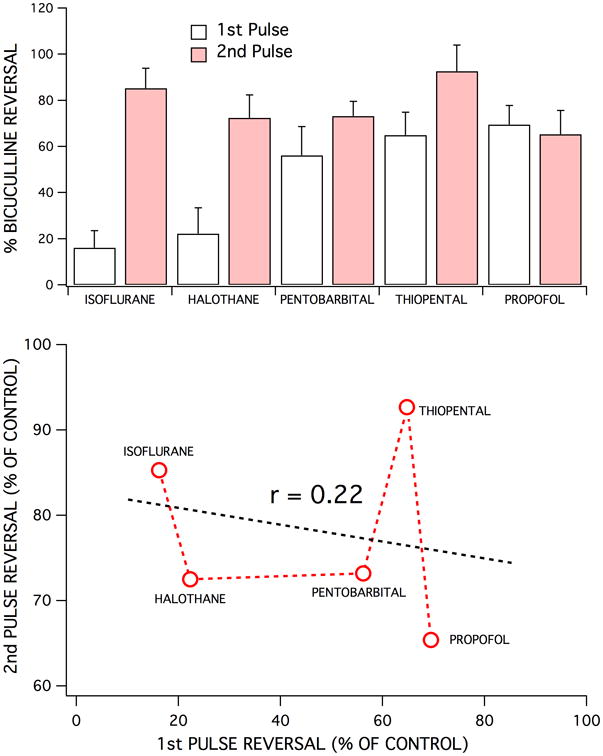
Each of the five anesthetics studied exhibited different effects on 1st pulse vs 2nd pulse GABAA receptor-mediated inhibition. This was evident in the degree of bicuculline-induced reversal of the depression of first and second pulse responses produced by each anesthetic – for comparable degrees of population spike depression (top bar graph). Isoflurane (350 μM) was least effective at enhancing feedforward inhibition (1st pulse responses), followed by halothane (250 μM), pentobarbital (400 μM), thiopental (80 μM) and propofol (30 μM). In contrast, isoflurane and thiopental were the most effective anesthetics at enhancing feedback inhibition (2nd pulse responses), followed by pentobarbital, halothane, and finally by propofol. Each bar represents the mean ± SD for the percent recovery produced by a maximally effective concentration of bicuculline (10 μM), for at least five experiments. Bicuculline reversal of 1st vs 2nd pulse responses were significantly different for isoflurane (p < 0.0001), halothane (p < 0.0001), pentobarbital (p < 0.01), thiopental (p < 0.001), but not for propofol (p > 0.05). There did not appear to be any correlation between the degrees of bicuculline-induced reversal of depression for first vs second pulse responses (bottom graph). There was also not an obvious correlation between lipid solubility vs anesthetic enhanced feedforward or feedback inhibition (not shown). However, lipid solubility did correlate with anesthetic effective concentrations for population spike depression with the following rank order of potency: propofol > thiopental > halothane > isoflurane > pentobarbital.
Discussion
Anesthetic Depression of CA1 Neurons Involves Different Mechanisms of Action
All anesthetics studied depressed CA1 neuron synaptically mediated excitation, as measured by population spike responses. In addition, population spike depression occurred at clinically relevant concentrations, indicating that this provides a surrogate cellular measure for a clinically relevant effect. Depressant effects on hippocampal synaptic responses would likely play a role for anesthetic-induced loss of recall during anesthesia,(37-39) and also appear to contribute to loss of righting reflex, and even to loss of tail pinch immobility, although that is mediated mainly, ∼80%, by spinal cord circuits.(40,41)
The depression of population spike responses came about via different mechanisms of action for the anesthetics studied. Volatile anesthetic effects resulted from a direct depression of glutamate-mediated synaptic excitation, since EPSP depression was not reversed by blocking GABA inhibition, consistent with previous findings.(33,42) This was partially true for pentobarbital as well, however, thiopental and propofol were less effective at depressing EPSP responses and most of the depression produced by these anesthetics was reversed by blocking GABA inhibition. These differential effects on excitatory synaptic responses are consistent with previous findings.(33-35)
The involvement of enhanced GABA-mediated inhibition for population spike depression also varied across the anesthetics studied. For propofol, thiopental and pentobarbital, most of the depression of first pulse responses could be reversed by the GABA antagonist, bicuculline. This indicates that the anesthetic-induced depression produced by these anesthetics comes about via enhanced GABA-mediated inhibition. Isoflurane and halothane, in contrast, produced a depression of first pulse responses that was only partially reversed by the GABA antagonist. Thus, depression of synapticallyevoked CA1 discharge produced by the volatile agents clearly involves non-GABA-dependent actions as well, including direct depression of glutamate-mediated excitation (Figure 2). The lack of involvement of GABA inhibition in the volatile anesthetic effect is in good agreement with in vivo findings showing that GABA antagonists fail to alter MAC determinations at the spinal cord level,(10) but do reverse the immobility produced by propofol.(43) These results add to well established in vivo(24) and in vitro(44) (25) evidence for selective, anesthetic-specific, mechanisms of action.
Bicuculline and Picrotoxin Discriminate Between Feedforward and Feedback Inhibition
GABAA receptors appear to contribute to several forms of inhibition, first noted as GABAA, gated fast versus slow IPSCs in hippocampal cortex.(14) More recently, a new class of GABAA-gated tonic, presumed extrasynaptic, chloride channels have been characterized in hippocampal CA1 neurons.(15) These tonic GABAA-gated currents are insensitive to gabazine at concentrations that totally block synaptic receptors, but are blocked by bicuculline and picrotoxin.(15,16,23) Tonic GABAA receptors appear to differ in subunit composition compared to synaptic receptors, by incorporating alpha5 subunits.(17) In the present study, bicuculline and picrotoxin appeared to exhibit subtle, but statistically significant, differences in effect on 1st pulse versus 2nd pulse GABAA-gated responses. This was evident as both a difference in kinetics and in magnitude of the reversal of anesthetic effects on first pulse versus second pulse population spike responses (Figure 4). Picrotoxin produced a faster reversal of first pulse responses compared to bicuculline, but a much slower reversal of second pulse responses. The magnitude of reversal of first pulse responses produced by picrotoxin was greater than that produced by bicuculline, suggesting a difference that might come about from receptors made-up of different subunits. Thus, GABAA-gated chloride channels appear to exist in several “flavors” that likely reflect different subunit compositions distributed at different synaptic (fast vs slow) and extrasynaptic/perisynaptic sites.(13,45) Depression of population spike responses by the anesthetics did not appear to involve only tonic GABAA receptors, because gabazine was as effective at reversing anesthetic effects as were bicuculline and picrotoxin, although for other anesthetic effects like CA1 neuron hyperpolarization and depression of DC-evoked spiking, gabazine-insensitive tonic receptors can play a major role.(16,17,23) The population spike measure provides a better overall measure of anesthetic effects, because it is sensitive to both synaptic and tonic actions of anesthetics.
Anesthetics Discriminate Between Fast and Slow GABAA Inhibition
First pulse responses provide a measure of both feedforward and tonic inhibition, while second pulse responses include an additional contribution from GABAA slow inhibition, because the second pulse was timed to occur after GABAA fast inhibition was over, but at the peak anesthetic-induced prolongation of the GABAA slow IPSC. The GABAA receptors associated with fast and slow synaptic inhibition of CA1 neurons exhibited different pharmacological sensitivities to the anesthetics studied. This was most evident for isoflurane, which appeared to strongly enhance slow GABAA-gated inhibition while having little effect on fast and tonic inhibition (Figure 5). A similar profile was apparent for halothane, and less so for the barbiturates (Figure 6). Propofol was the only anesthetic studied that did not appear to discriminate between first pulse and second pulse responses. GABAA fast and slow IPSCs were previously shown to be equally sensitive to halothane.(46) However when equi-effective amnesic concentrations for different anesthetics were compared, isoflurane and etomidate exhibited opposite sensitivities for fast versus slow inhibition.(19) By analogy to the tonic GABAA-gated channels,(18) it is likely that these receptors have a unique subunit composition.(12,13)
Differential Anesthetic Effects on GABAA Receptors
The different anesthetic sensitivities exhibited by GABAA tonic, fast and slow receptors could account for some of the differences in effect profiles evident for volatile, barbiturate and other anesthetics that have been observed in vivo.(24) Further characterization of these differential sensitivities could lead to the development of selectively targeted new anesthetics that exhibit fewer unwanted side effects. It may eventually be possible to attribute different components of anesthesia, like analgesia, unconsciousness, amnesia, immobility, etc., to selective actions at one or more types of GABAA-gated channels.
Acknowledgments
I thank Professors J R Trudell and E J Bertaccini for helpful discussions and comments on this manuscript. Allison Himmel and Sky Pittson helped with experiments and with collecting data.
Funding: NIH NIGMSR01 GM095653
Footnotes
The author declares no conflicts of interest.
Reprints will not be available from the author.
Disclosures: Name: M. Bruce MacIver, MSc, PhD
Contribution: MacIver designed and conducted experiments, analyzed data and prepared the manuscript.
Attestation: MacIver approved the final manuscript, MacIver attests to the integrity of the original data and the analysis reported in this manuscript. MacIver is the archival author.
This manuscript was handled by: Marcel E. Durieux, MD, PhD
References
- 1.Nicoll RA, Eccles JC, Oshima T, Rubia F. Prolongation of hippocampal inhibitory postsynaptic potentials by barbiturates. Nature. 1975;258:625–7. doi: 10.1038/258625a0. [DOI] [PubMed] [Google Scholar]
- 2.Pearce RA, Stringer JL, Lothman EW. Effect of volatile anesthetics on synaptic transmission in the rat hippocampus. Anesthesiology. 1989;71:591–8. doi: 10.1097/00000542-198910000-00019. [DOI] [PubMed] [Google Scholar]
- 3.Tanelian DL, Kosek P, Mody I, MacIver MB. The role of the GABAA receptor/chloride channel complex in anesthesia. Anesthesiology. 1993;78:757–76. doi: 10.1097/00000542-199304000-00020. [DOI] [PubMed] [Google Scholar]
- 4.Trudell JR, Bertaccini E, Maciver MB. Teaching an old GABA receptor new tricks. Anesth Analg. 2012;115:270–3. doi: 10.1213/ANE.0b013e31824a0b3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maciver MB, Bronte-Stewart HM, Henderson JM, Jaffe RA, Brock-Utne JG. Human subthalamic neuron spiking exhibits subtle responses to sedatives. Anesthesiology. 2011;115:254–64. doi: 10.1097/ALN.0b013e3182217126. [DOI] [PubMed] [Google Scholar]
- 6.Nishikawa K, MacIver MB. Membrane and synaptic actions of halothane on rat hippocampal pyramidal neurons and inhibitory interneurons. J Neurosci. 2000;20:5915–23. doi: 10.1523/JNEUROSCI.20-16-05915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galarreta M, Hestrin S. Electrical and chemical synapses among parvalbumin fast-spiking GABAergic interneurons in adult mouse neocortex. Proc Natl Acad Sci U S A. 2002;99:12438–43. doi: 10.1073/pnas.192159599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banke TG, McBain CJ. GABAergic input onto CA3 hippocampal interneurons remains shunting throughout development. J Neurosci. 2006;26:11720–5. doi: 10.1523/JNEUROSCI.2887-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. Faseb J. 2003;17:250–2. doi: 10.1096/fj.02-0611fje. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Sonner JM, Eger EI, 2nd, Stabernack CR, Laster MJ, Raines DE, Harris RA. Gamma-aminobutyric acidA receptors do not mediate the immobility produced by isoflurane. Anesth Analg. 2004;99:85–90. doi: 10.1213/01.ANE.0000118108.64886.42. [DOI] [PubMed] [Google Scholar]
- 11.Werner DF, Swihart A, Rau V, Jia F, Borghese CM, McCracken ML, Iyer S, Fanselow MS, Oh I, Sonner JM, Eger EI, 2nd, Harrison NL, Harris RA, Homanics GE. Inhaled anesthetic responses of recombinant receptors and knockin mice harboring alpha2(S270H/L277A) GABA(A) receptor subunits that are resistant to isoflurane. J Pharmacol Exp Ther. 2011;336:134–44. doi: 10.1124/jpet.110.170431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capogna M, Pearce RA. GABA A, slow: causes and consequences. Trends in neurosciences. 2011;34:101–12. doi: 10.1016/j.tins.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Mody I, Pearce RA. Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends in neurosciences. 2004;27:569–75. doi: 10.1016/j.tins.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Pearce RA. Physiological evidence for two distinct GABAA responses in rat hippocampus. Neuron. 1993;10:189–200. doi: 10.1016/0896-6273(93)90310-n. [DOI] [PubMed] [Google Scholar]
- 15.Bai D, Zhu G, Pennefather P, Jackson MF, MacDonald JF, Orser BA. Distinct functional and pharmacological properties of tonic and quantal inhibitory postsynaptic currents mediated by gamma-aminobutyric acid(A) receptors in hippocampal neurons. Mol Pharmacol. 2001;59:814–24. doi: 10.1124/mol.59.4.814. [DOI] [PubMed] [Google Scholar]
- 16.Bieda MC, MacIver MB. Major role for tonic GABAA conductances in anesthetic suppression of intrinsic neuronal excitability. J Neurophysiol. 2004;92:1658–67. doi: 10.1152/jn.00223.2004. [DOI] [PubMed] [Google Scholar]
- 17.Caraiscos VB, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Rosahl TW, Wafford KA, MacDonald JF, Orser BA. Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 2004;101:3662–7. doi: 10.1073/pnas.0307231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orser BA. Lifting the fog around anesthesia. Sci Am. 2007;296:54–61. doi: 10.1038/scientificamerican0607-54. [DOI] [PubMed] [Google Scholar]
- 19.Dai S, Perouansky M, Pearce RA. Isoflurane enhances both fast and slow synaptic inhibition in the hippocampus at amnestic concentrations. Anesthesiology. 2012;116:816–23. doi: 10.1097/ALN.0b013e31824be0e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hentschke H, Benkwitz C, Banks MI, Perkins MG, Homanics GE, Pearce RA. Altered GABAA, slow inhibition and network oscillations in mice lacking the GABAA receptor beta3 subunit. J Neurophysiol. 2009;102:3643–55. doi: 10.1152/jn.00651.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallner M, Hanchar HJ, Olsen RW. Ethanol enhances alpha 4 beta 3 delta and alpha 6 beta 3 delta gamma-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc Natl Acad Sci U S A. 2003;100:15218–23. doi: 10.1073/pnas.2435171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei W, Faria LC, Mody I. Low ethanol concentrations selectively augment the tonic inhibition mediated by delta subunit-containing GABAA receptors in hippocampal neurons. J Neurosci. 2004;24:8379–82. doi: 10.1523/JNEUROSCI.2040-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bieda MC, Su H, Maciver MB. Anesthetics discriminate between tonic and phasic gamma-aminobutyric acid receptors on hippocampal CA1 neurons. Anesth Analg. 2009;108:484–90. doi: 10.1213/ane.0b013e3181904571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winters WD. Effects of drugs on the electrical activity of the brain: anesthetics. Annu Rev Pharmacol Toxicol. 1976;16:413–26. doi: 10.1146/annurev.pa.16.040176.002213. [DOI] [PubMed] [Google Scholar]
- 25.MacIver MB, Roth SH. Inhalation anaesthetics exhibit pathway-specific and differential actions on hippocampal synaptic responses in vitro. Br J Anaesth. 1988;60:680–91. doi: 10.1093/bja/60.6.680. [DOI] [PubMed] [Google Scholar]
- 26.Mikulec AA, Pittson S, Amagasu SM, Monroe FA, MacIver MB. Halothane depresses action potential conduction in hippocampal axons. Brain Res. 1998;796:231–8. doi: 10.1016/s0006-8993(98)00348-5. [DOI] [PubMed] [Google Scholar]
- 27.Hagan CE, Pearce RA, Trudell JR, MacIver MB. Concentration measures of volatile anesthetics in the aqueous phase using calcium sensitive electrodes. J Neurosci Methods. 1998;81:177–84. doi: 10.1016/s0165-0270(98)00029-6. [DOI] [PubMed] [Google Scholar]
- 28.Gredell JA, Turnquist PA, Maciver MB, Pearce RA. Determination of diffusion and partition coefficients of propofol in rat brain tissue: implications for studies of drug action in vitro. Br J Anaesth. 2004;93:810–7. doi: 10.1093/bja/aeh272. [DOI] [PubMed] [Google Scholar]
- 29.Chesney MA, Perouansky M, Pearce RA. Differential uptake of volatile agents into brain tissue in vitro. Measurement and application of a diffusion model to determine concentration profiles in brain slices. Anesthesiology. 2003;99:122–30. doi: 10.1097/00000542-200307000-00021. [DOI] [PubMed] [Google Scholar]
- 30.Benkwitz C, Liao M, Laster MJ, Sonner JM, Eger EI, 2nd, Pearce RA. Determination of the EC50 amnesic concentration of etomidate and its diffusion profile in brain tissue: implications for in vitro studies. Anesthesiology. 2007;106:114–23. doi: 10.1097/00000542-200701000-00020. [DOI] [PubMed] [Google Scholar]
- 31.Eger EI, 2nd, Xing Y, Laster M, Sonner J, Antognini JF, Carstens E. Halothane and isoflurane have additive minimum alveolar concentration (MAC) effects in rats. Anesth Analg. 2003;96:1350–1353. doi: 10.1213/01.ANE.0000055802.27976.8A. table of contents. [DOI] [PubMed] [Google Scholar]
- 32.Pearce RA. Volatile anaesthetic enhancement of paired-pulse depression investigated in the rat hippocampus in vitro. J Physiol. 1996;492(Pt 3):823–40. doi: 10.1113/jphysiol.1996.sp021349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pittson S, Himmel AM, MacIver MB. Multiple synaptic and membrane sites of anesthetic action in the CA1 region of rat hippocampal slices. BMC Neurosci. 2004;5:52. doi: 10.1186/1471-2202-5-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perouansky M, Baranov D, Salman M, Yaari Y. Effects of halothane on glutamate receptor-mediated excitatory postsynaptic currents. A patch-clamp study in adult mouse hippocampal slices. Anesthesiology. 1995;83:109–19. doi: 10.1097/00000542-199507000-00014. [DOI] [PubMed] [Google Scholar]
- 35.MacIver MB, Mikulec AA, Amagasu SM, Monroe FA. Volatile anesthetics depress glutamate transmission via presynaptic actions. Anesthesiology. 1996;85:823–34. doi: 10.1097/00000542-199610000-00018. [DOI] [PubMed] [Google Scholar]
- 36.Baudoux S, Empson RM, Richards CD. Pentobarbitone modulates calcium transients in axons and synaptic boutons of hippocampal CA1 neurons. Br J Pharmacol. 2003;140:971–9. doi: 10.1038/sj.bjp.0705519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacIver MB, Tauck DL, Kendig JJ. General anaesthetic modification of synaptic facilitation and long-term potentiation in hippocampus. Br J Anaesth. 1989;62:301–10. doi: 10.1093/bja/62.3.301. [DOI] [PubMed] [Google Scholar]
- 38.Simon W, Hapfelmeier G, Kochs E, Zieglgansberger W, Rammes G. Isoflurane blocks synaptic plasticity in the mouse hippocampus. Anesthesiology. 2001;94:1058–65. doi: 10.1097/00000542-200106000-00021. [DOI] [PubMed] [Google Scholar]
- 39.Perouansky M, Pearce RA. How we recall (or don't): the hippocampal memory machine and anesthetic amnesia. Can J Anaesth. 2011;58:157–66. doi: 10.1007/s12630-010-9417-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma J, Shen B, Stewart LS, Herrick IA, Leung LS. The septohippocampal system participates in general anesthesia. J Neurosci. 2002;22:RC200. doi: 10.1523/JNEUROSCI.22-02-j0004.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo T, Leung LS. Involvement of tuberomamillary histaminergic neurons in isoflurane anesthesia. Anesthesiology. 2011;115:36–43. doi: 10.1097/ALN.0b013e3182207655. [DOI] [PubMed] [Google Scholar]
- 42.Schlame M, Hemmings HC., Jr Inhibition by volatile anesthetics of endogenous glutamate release from synaptosomes by a presynaptic mechanism. Anesthesiology. 1995;82:1406–16. doi: 10.1097/00000542-199506000-00012. [DOI] [PubMed] [Google Scholar]
- 43.Sonner JM, Zhang Y, Stabernack C, Abaigar W, Xing Y, Laster MJ. GABA(A) receptor blockade antagonizes the immobilizing action of propofol but not ketamine or isoflurane in a dose-related manner. Anesth Analg. 2003;96:706–12. doi: 10.1213/01.ANE.0000048821.23225.3A. table of contents. [DOI] [PubMed] [Google Scholar]
- 44.Richards CD. The action of anaesthetics on synaptic transmission. General pharmacology. 1978;9:287–93. doi: 10.1016/0306-3623(78)90063-0. [DOI] [PubMed] [Google Scholar]
- 45.Banks MI, Pearce RA. Kinetic differences between synaptic and extrasynaptic GABA(A) receptors in CA1 pyramidal cells. J Neurosci. 2000;20:937–48. doi: 10.1523/JNEUROSCI.20-03-00937.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lukatch HS, MacIver MB. Voltage-clamp analysis of halothane effects on GABA(A fast) and GABA(A slow) inhibitory currents. Brain Res. 1997;765:108–12. doi: 10.1016/s0006-8993(97)00516-7. [DOI] [PubMed] [Google Scholar]