

Abstract
Treatment resistance in T-cell acute lymphoblastic leukemia (T-ALL) is associated with PTEN deletions and resultant PI3K-AKT pathway activation, as well as MYC overexpression, and these pathways repress mitochondrial apoptosis in established T-lymphoblasts through poorly defined mechanisms. Normal T-cell progenitors are hypersensitive to mitochondrial apoptosis, a phenotype that is dependent on expression of proapoptotic BIM. In a conditional zebrafish model, MYC downregulation induced BIM expression in T-lymphoblasts, an effect that was blunted by expression of constitutively active AKT. In human T-ALL cell lines and treatment- resistant patient samples, treatment with MYC or PI3K-AKT pathway inhibitors each induced BIM upregulation and apoptosis, indicating that BIM is repressed downstream of MYC and PI3K-AKT in high-risk T-ALL. Restoring BIM function in human T-ALL cells using a stapled peptide mimetic of the BIM BH3 domain had therapeutic activity, indicating that BIM repression is required for T-ALL viability. In the zebrafish model, where MYC downregulation induces T- ALL regression via mitochondrial apoptosis, T-ALL persisted despite MYC downregulation in 10% of bim wild-type zebrafish, 18% of bim heterozygotes, and in 33% of bim homozygous mutants (P = 0.017). We conclude that downregulation of BIM represents a key survival signal downstream of oncogenic MYC and PI3K-AKT signaling in treatment-resistant T-ALL.
Keywords: T-cell acute lymphoblastic leukemia, BIM, AKT, MYC, apoptosis
INTRODUCTION
Human treatment-resistant T-ALL is associated with characteristic molecular lesions, including PTEN deletions and associated PI3K-AKT pathway activation,1–3 as well as MYC overexpression, which typically occurs downstream of activated NOTCH1 in all molecular subtypes of the disease.4–7 Also central to T-ALL pathobiology is inactivation of key tumor suppressors, which occurs in virtually all cases. For example, the majority of T-ALL cases harbor biallelic inactivation of CDKN2A (encoding INK4a and ARF),8–10 and numerous additional tumor suppressors are also commonly inactivated in this disease (reviewed in ref. 11). Nevertheless, despite tumor suppressor inactivation, the survival of T-ALL cells remains dependent on the persistent activity of key oncogenes, including MYC and AKT. Indeed, we have previously shown that ongoing activity of the MYC oncogene is required to repress mitochondrial apoptosis in established T-ALL cells, and that activated AKT can substitute for key survival signals downstream of MYC, thus preventing T-lymphoblast apoptosis despite MYC downregulation.12 Similarly, activated AKT can substitute for NOTCH1 signaling in human T-ALL,13 and T-ALL cells with AKT pathway activation (as a result of PTEN inactivation) are dependent on ongoing PI3K-AKT pathway activity.14 However, despite the dependence of established T-ALL cells on MYC and AKT, the mechanisms though which these repress mitochondrial apoptosis in established tumor cells remain poorly understood.
Normal T-cell progenitors are hypersensitive to mitochondrial apoptosis, a phenotype that is dependent on expression of BIM, a pro-apoptotic BCL-2 family member.15–17 Normal T-cell development involves an error-prone somatic mutagenesis process, known as V(D)J recombination, which introduces extensive variation in the antigen-recognition domain of T-cell receptor genes, thus allowing the generation of a diverse population of T-cells capable of responding to a countless range of foreign antigens.18,19 However, this process also generates a large number of T-cell progenitors that fail to express a functional T-cell receptor, or whose T-cell receptor reacts against self-antigens. Elimination of these cells is required for appropriate immune homeostasis and prevention of autoimmunity, and BIM-dependent mitochondrial apoptosis plays a key role in such T-cell selection, as evidenced by the development of fatal autoimmunity in Bim-deficient mice.15,16
In this study, we show that BIM is repressed downstream of MYC and AKT in a zebrafish model of T-ALL, and we confirm that this effect is conserved in human T-ALL cell lines and in primary T-ALL cells obtained from patients with treatment-resistant disease. Therapeutic restoration of BIM activity using a stapled peptide mimetic of its BH3 domain impairs the viability of human T-ALL cells. Finally, using a genetically engineered zebrafish model, we show that BIM is required for T-ALL regression following MYC oncogene downregulation. These findings demonstrate that repression of proapoptotic BIM mediates survival signaling downstream of MYC and AKT in the molecular pathogenesis of high-risk T-ALL.
MATERIALS AND METHODS
Transgenic and mutant zebrafish lines
The rag2:MYC-ER, rag2:EGFP and rag2:EGFP-bcl2 stable transgenic zebrafish lines have previously been described.12,20 To generate rag2:MYC-ER; rag2:EGFP double-transgenic zebrafish that also expressed either rag2:mCherry or rag2:myr-Akt2 transgenes, 1-cell stage embryos expressing rag2:MYC-ER and rag2:EGFP were injected with 30 pg of a pI-Sce-modified pBluescript vector harboring the transgene of interest, as previously described.12
The BIM mutant zebrafish line was generated by retroviral insertional mutagenesis as previously described,21 and identified from a sperm library maintained by Znomics (Portland, OR USA). Genotyping for the bim wild-type and mutant alleles was performed by genomic PCR using the following primers: bim wild-type forward, 5′-GAGCAAACGCTGGCCAATGGCCCGG, and reverse, 5′-GTCCGTCTTGCGCTTCGGAAATATT; and bim mutant forward, 5′-CGACAGCGATTCTGTGCCAGGTTC, and reverse, 5′-GACGCAGGCGCATAAAATCAGTC.
Small molecules
4-hydroxytamoxifen, doxycycline hyclate and dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich (St. Louis, MO, USA). BEZ235 was obtained from Haoyuan Chemexpress (Shanghai, China). JQ1 was synthesized as previously described.22
4-hydroxytamoxifen treatment and T-ALL monitoring of transgenic zebrafish
4-hydroxytamoxifen treatment of zebrafish, monitoring for T-ALL onset, and zebrafish image capture and analysis was performed as described.12 All images shown represent merged fluorescence (shown in green) and brightfield (shown in grayscale) images; image merging was performed using Photoshop version 7.0 (Adobe, San Jose, CA, USA). Following development of disseminated T-ALL, zebrafish with T-ALL were removed from 4-hydroxytamoxifen and placed into individual tanks, and tumors were imaged weekly for a total of 8 weeks. Tumor phenotypes after 4-hydroxytamoxifen removal were classified as tumor regression (defined as a >50% reduction in the diameter of the largest contiguous tumor mass by the end of the 8-week monitoring period), or tumor persistence (all tumors failing to meet the definition of regression). Fish that became moribund with leukemia less than 8 weeks after tamoxifen were euthanized and classified into the tumor persistence category.
T-ALL cell lines and patient samples
T-ALL cell lines were obtained from ATCC (Manassas, VA, USA), DSMZ (Braunschweig, Germany) or the A. Thomas Look laboratory (Boston, MA, USA) and cultured in RPMI 1640 (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (Sigma-Aldrich) and 1% penicillin/streptomycin (Invitrogen). The murine T-ALL cell line 4188, which is induced by a doxycycline-repressible MYC transgene, was obtained from Dean Felsher,23 and grown in RPMI 1640 (Invitrogen) with 10% fetal bovine serum (Sigma-Aldrich), 1% penicillin/streptomycin (Invitrogen), and 50 μM 2-mercaptoethanol (Invitrogen). Doxycycline (20 ng/ml) was added to the media to downregulate MYC transgene expression in 4188 cells.
Primary human T-ALL samples were obtained from children with T-ALL enrolled on clinical studies of the Dana-Farber Cancer Institute, with informed consent and DFCI Institutional Review Board approval. Induction failure samples were collected at the time of leukemia diagnosis from patients in whom initial induction chemotherapy failed to achieve a clinical remission. Relapse samples were obtained at the time of disease recurrence following the failure of front-line T-ALL therapy. Leukemic blasts were isolated from peripheral blood or bone marrow samples by Ficoll-Hypaque centrifugation and cryopreserved in fetal bovine serum (FBS) containing 10% DMSO and stored in liquid nitrogen. Fresh or frozen leukemic blasts were expanded in NOD scid IL2rγ−/− (NSG) by transplanting 0.5–5 million cells via intravenous injection. Mice were sacrificed following development of signs of leukemia, and leukemic blasts were isolated from the spleen and bone marrow. Percent human engraftment and immunophenotype was determined by flow cytometry staining for human CD45 (APC), CD4 (PE), CD8 (FITC) and CD34 (PE-CY7) and acquired on a LSRII (BD Bioscience, San Jose, CA, USA), and was greater than 80% in all samples. Primary human T-ALL samples were cultured in reconstituted alpha-minimum essential media supplemented with 10% fetal bovine serum, 10% human AB serum (Invitrogen), 1% L-glutamine, 1% penicillin/streptomycin in the presence of recombinant human cytokines stem cell factor (50 ng/mL), Flt3-L (20 ng/mL) and IL-7 (10 ng/mL) (R&D Systems, Minneapolis, MN, USA) at 37° under 5% CO2. Primary T-ALL samples were cultured at a density of 1–3 × 106 per mL in a 12 well plate for the indicated time in the presence of DMSO (Sigma-Aldrich), 500 nM BEZ235, or 1 μM JQ1.
RNA extraction and quantitative RT-PCR analysis
For quantitative reverse-transcriptase polymerase chain reaction (Q-RT-PCR) analysis of mRNA transcript levels, total RNA was extracted using Trizol (Invitrogen) for zebrafish T-ALL cells, or the RNeasy Mini Kit (Qiagen, Venlo, Netherlands) for human T-ALL cells, according to the manufacturer’s instructions. cDNA was synthesized using the SuperScript III First-Strand Synthesis system according to the manufacturer’s instructions (Invitrogen). Q-RT-PCR analysis was performed with the SYBR Green PCR Core Reagents kit (Applied Biosystems, Life Technologies, Grand Island, NY, USA) and a 7500 Real Time PCR System instrument (Applied Biosystems) according to the manufacturer’s instructions. All Q-RT-PCR reactions were performed in triplicate using primers listed in Supplementary Table 1. Cycle threshold (CT) for each condition, in triplicate, was compared to β-actin control according to the 2ΔCT comparative method.
For Q-RT-PCR analysis of microRNA expression, RNA was extracted from human T- ALL cells using the RNeasy Mini Kit (Qiagen), and cDNA synthesis was performed using the miScript Reverse Transcription Kit (Qiagen), according to the manufacturer’s instructions. Q- RT-PCR analysis for miR-19a and miR-19b was performed using the hsa-mir-19a and hsa-mir- 19b primers (Qiagen). The small nucleolar RNA SNORD61 was used as control, whose expression was assessed using the Hs_SNORD61_11 miScript Primer Assay (Qiagen). Cycle threshold (CT) for each condition was calculated according to the 2ΔCT comparative method.
Microarray-based gene expression analysis, data extraction and normalization of our previously published cohort of primary T-ALL patient samples was previously described.24
Western Blotting
T-ALL cells were lysed in RIPA buffer (EMD Millipore Corporation, Billerica, MA, USA) supplemented with cOmplete protease inhibitor (1 tablet/10 mL; Roche, Indianapolis, IN, USA) and PhosSTOP phosphatase inhibitor (1 tablet/10 mL; Roche). 10 μg protein lysate was mixed with Laemmli sample buffer (Bio-Rad, Hercules, CA, USA) and β-mercaptoethanol (Sigma-Aldrich) in appropriate proportions before being run on a 4–12% Novex bis-tris polyacrylamide gel (Invitrogen) at 200V for 45 minutes. Blots were transferred to a nitrocellulose membrane (Invitrogen) at 30V for 1 hour, blocked with 5% milk in phosphate-buffered saline with 0.1% tween (Boston Bioproducts, Ashland, MA, USA) and probed with the following antibodies: anti-BIM (1:1000; Cell Signaling Technology #2933, Danvers, MA, USA), anti-β-tubulin (1:1000; Cell Signaling #2128), anti-cleaved-PARP (1:1000; Cell Signaling #5625), anti-MCL1 (1:1000; Cell Signaling #5453), or anti-GAPDH (1:2000; Abcam, Cambridge, MA, USA). Secondary detection was performed with HRP-linked antibodies (Cell Signaling), HRP substrate was SuperSignal West Pico (Thermo Fisher Scientific, Rockford, IL, USA), and films were developed using a KODAK X-OMAT 2000A Processor.
Analysis of NOTCH1, PTEN, PI3K and AKT status in human T-ALL samples
The NOTCH1, PTEN and AKT status of all human T-ALL cell lines and primary patient samples is indicated in Supplementary Table 2. In the cell lines, NOTCH1 status was analyzed by Sanger sequencing as previously described,25 and Western blot analysis for PTEN and phospho-AKT (Ser473) was used to analyze PTEN-AKT status. In the primary T-ALL patient samples, NOTCH1, PTEN, PI3K and AKT status was analyzed using Sanger sequencing of these genes, as previously described.1
BIM SAHB treatment
Stabilized alpha-helix of BCL-2 domain (SAHB) modeled after amino acids 146-166 of the BIM BH3 helix (BIM SAHBA), as well as an analogous negative control peptide harboring an R153D reverse polarity substitution, were synthesized, purified and characterized by circular dichroism, as previously described.26 The effect of these SAHB stapled peptides on T-ALL cell viability was assessed as previously described.26 Briefly, T-ALL cells (10,000 cells in 50 μL) were placed in 96-well opaque plates and treated with serial dilutions of vehicle (0.8% DMSO, BIM SAHBA (amino acids 146-166), or BIM SAHBA (R153D) in serum-free RPMI media for 2 hours, followed by serum replacement with 20% FBS-containing RPMI media (50 μL) for a final volume of 100 μL containing 10% FBS. Cell viability was assayed at 24 hours by addition of CellTiter-Glo chemiluminescence reagent (Promega, Fitchburg, WI, USA), and luminescence measured by a DTX 880 Multimode Detector plate reader (Beckman Coulter, Brea, CA, USA).
Annexin V and 7AAD analysis for apoptosis induction
Primary human T-ALL samples were engrafted in immunodeficient NSG mice, harvested, cultured and treated with DMSO, JQ1, or BEZ235 as indicated. Cells were washed and resuspended in Annexin binding buffer and stained with Annexin V and 7AAD (7-amino-actinomycin D) following the manufacturer’s protocol (BD Biosciences). Samples were acquired on a LSRII (BD Biosciences) and analyzed using Flowjo software (Treestar, Ashland, OR, USA). Early apoptotic cells were defined as annexin V-positive, 7AAD-negative cells. Late apoptosis was defined as annexin V-positive, 7AAD-positive cells.
Retrovirus production and infection of cell lines
Retroviruses were generated by co-transfection of 293T cells (ATCC) with either MSCV-puro or MSCV-19a-20-19b (encoding miR-19a, miR-20 and miR-19b, generated by Lin He and obtained from Addgene; http://www.addgene.org/24827/), together with the packaging plasmids pMD (1 μg), and pCMV-VSV-G (1 μg), using Fugene6 (Promega). Retroviral transduction of CCRF-CEM cells was performed by incubating cells for 24 hours in virus-containing media at 37°C in the presence of 8 μg/mL polybrene (EMD Millipore). Infected cells were allowed to expand in normal growth media for 48 hours before selection in 2 μg/mL puromycin (Invitrogen) for at least 7 days before treatment with JQ1.
Transfections and Flow Cytometry
For siRNA experiments, CCRF-CEM cells were transfected with Lipofectamine RNAiMAX Reagent (Invitrogen) according to manufacturer’s instructions, using 10 nM SignalSilence control or Akt siRNA (Cell Signaling). 72 hours after transfection, cells were harvested, fixed in 16% paraformaldehyde (Alfa Aesar, Ward Hill, MA, USA) for 10 minutes at 37°C, and permeabilized in 90% methanol (Fisher Chemical, USA) for 30 minutes at 4°C prior to staining. Cells were blocked in 0.5% bovine serum albumin (Fisher Chemical, USA) in phosphate buffered saline and stained with the following primary antibodies: anti-AKT (1:100; Cell Signaling #9272), detected with AlexaFluor 488 anti-rabbit (1:400; Cell Signaling) and anti-BIM (1:200; Cell Signaling #2933), detected with Cy5 anti-rabbit (1:200; Invitrogen). Cells were analyzed on a LSRFortessa (BD Biosciences), and gating and data analysis were performed using FlowJo software (TreeStar, Ashland).
Transfection with dominant-negative FOXO was performed using Lipofectamine LTX with Plus Reagent (Invitrogen) according to the manufacturer’s instructions. PF382 cells were transfected using a FOXO-D256Δ expression construct developed by Domenico Accili27 and obtained from Addgene (http://www.addgene.org/12145), or pCS2 which was used as the vector control. Twenty-four hours post-transfection, cells were treated with 500 nM BEZ235 for an additional 24 hours, before RNA harvesting and Q-RT-PCR analysis for BIM mRNA expression, as described above.
RESULTS
BIM is downregulated by AKT and MYC in high-risk T-ALL
We have previously shown that activated AKT and MYC each repress mitochondrial apoptosis in established T-ALL cells.12 To confirm our previous findings and define the mechanisms involved, we generated rag2:MYC-ER; rag2:EGFP double-transgenic zebrafish that also expressed either rag2:mCherry or rag2:myr-Akt2 (encoding myristoylated, constitutively active Akt). Zebrafish were grown in the presence of 4-hydroxytamoxifen until T-ALL development, at which point 4-hydroxytamoxifen was removed, and T-ALL regression was monitored by weekly fluorescence imaging. We found that T-ALL persisted after MYC oncogene downregulation in 72% (n = 13 of 18) of zebrafish expressing a constitutively active myr-Akt2 transgene, whereas T-ALL persisted in only 22% (n = 2 of 9) of zebrafish expressing mCherry control (P = 0.036; Figure 1a–e). We have previously shown that T-ALL regression in this model is entirely dependent on mitochondrial apoptosis,12 thus these data indicate that AKT and MYC each repress mitochondrial apoptosis in established zebrafish T-ALL cells.
Figure 1.

BIM is downregulated downstream of AKT and MYC in a conditional zebrafish model of MYC-induced T-ALL. (a and c) One representative rag2:MYC-ER; rag2:EGFP; rag2:mCherry triple-transgenic zebrafish shown at the time of T-ALL onset (a) and 4 weeks after removal from 4-hydroxytamoxifen (4HT). (b and d) Representative rag2:MYC-ER; rag2:EGFP; rag2:myr-Akt2 zebrafish, shown at the time of T-ALL onset and 4 weeks after 4-hydroxytamoxifen removal (-4HT). (e) Quantitation of T-ALL phenotypes after MYC-ER downregulation (by removal from 4-hydroxytamoxifen), comparing animals expressing a rag2:myr-Akt2 transgene (encoding constitutively active Akt) to rag2:mCherry controls. P value calculated using Fisher’s exact test. (f) Q-RT-PCR analysis of bim mRNA expression, performed using RNA from T-ALL cells isolated from rag2:MYC-ER; rag2-EGFP-bcl2 zebrafish that also expressed either rag2:myr-Akt2 or rag2:mCherry control. T-ALL cells were sorted from animals in 4-hydroxytamoxifen (“MYC On”), or 4 days after tamoxifen removal (“MYC Off”). Bcl2-transgenic T-ALL cells were used in all conditions to avoid comparing live versus dying cells. bactin2 was the control for Q-RT-PCR analysis. Error bars represent mean +/− standard error of the mean for experiments performed in triplicate. Significance was assessed using the Kruskal-Wallis test, a statistical method designed to assess whether at least one of the conditions is significantly different from the others.
Mitochondrial apoptosis is regulated by the relative activity of pro-apoptotic and anti-apoptotic BCL2 family members,28 thus suggesting that AKT and MYC might repress apoptosis by altering the expression of BCL2 family members. To test this possibility, we induced leukemia in rag2:MYC-ER; rag2:EGFP-bcl2 double-transgenic zebrafish that also expressed either rag2:myr-Akt2 or rag2:mCherry transgenes, by raising these animals in 4-hydroxytamoxifen to activate the MYC-ER oncoprotein. Following T-ALL development, zebrafish were either kept in 4-hydroxytamoxifen, or removed from tamoxifen to downregulate MYC activity. Leukemic cells were EGFP-sorted four days later, and we used Q-RT-PCR to analyze the expression of the zebrafish BCL2-family apoptotic regulators that are known to be functionally conserved with their human counterparts.29 This analysis revealed bim as the only BCL2 family member whose expression was significantly altered in the presence of activated MYC and/or AKT transgenes in our zebrafish model (Figure 1f; P = 0.026). Although we were unable to accurately assess expression of endogenous bcl2 due to the overexpressed EGFP-bcl2 transgene (which we included to avoid comparing live versus dying cells), we found no significant change in expression of any other zebrafish BCL2 family members in the setting of activated MYC and/or AKT transgenes (Supplementary Figure 1).
To test the relevance of these findings to human T-ALL, we treated human T-ALL cell lines with BEZ235, a small molecule inhibitor of PI3K and mTOR,30 or with JQ1, a bromodomain inhibitor that effectively downregulates MYC activity.22,31 Treatment of the human T-ALL cell lines PF382, CCRF-CEM, ALL-SIL and MOLT4 with the PI3K-AKT inhibitor BEZ235 led to significant BIM mRNA upregulation in three of these four cell lines, whereas treatment with the MYC inhibitor JQ1 led to BIM upregulation at the mRNA level in all cell lines tested (Figure 2a and b). Further, Western blot analysis of these T-ALL cell lines following treatment with JQ1, BEZ235 or both drugs in combination revealed cooperative upregulation of BIM protein expression coupled to induction of apoptosis, as assessed by PARP cleavage (Figure 2, c–f).
Figure 2.

BIM is repressed downstream of AKT and MYC in human T-ALL cell lines. (a and b) Q-RT-PCR analysis of BIM mRNA expression in four human T-ALL cell lines treated with 500 nM BEZ235, a dual ATP-competitive PI3K and mTOR inhibitor versus DMSO for 24 hours (a), or 1 μM JQ1 (a bromodomain inhibitor that downregulates MYC activity) versus DMSO for 24 hours (b). P values calculated using the Welch t test. (c to f) Western blot analysis of human T-ALL cell lines treated for 24 hours using DMSO (vehicle control), 1 μM JQ1, 500 nM BEZ235, or both drugs in combination, using the indicated antibodies.
We then wanted to confirm that the effect of BEZ235 and JQ1 in mammalian T-ALL cells is mediated by inhibition of AKT and MYC pathways, respectively. To test whether direct AKT inhibition induces BIM upregulation, we transfected the human T-ALL cell line CCRF-CEM with control or anti-AKT siRNA. AKT siRNA transfection effectively downregulated AKT protein levels in a subset of cells, and the subpopulation in which effective AKT knock-down was achieved demonstrated BIM protein upregulation, as assessed by FACS analysis (P = 0.048; Supplementary Figure 2, a and b). To confirm that direct MYC inhibition leads to BIM upregulation in mammalian T-ALL cells, we took advantage of the murine MYC-induced T-ALL cell line 4188, which is induced by a human MYC transgene that can be specifically inactivated by doxycycline treatment.23 Inhibition of MYC transgene expression by doxycycline treatment for 48 hours induced BIM protein upregulation (Supplementary Figure 2c), indicating that the MYC-induced repression of BIM is conserved in mammalian T-ALL.
Inhibition of AKT signaling can decrease protein expression of the anti-apoptotic BCL2 family member MCL1, via both GSK3-dependent and mTOR-dependent pathways,32,33 leading us to investigate whether MCL1 protein downregulation might mediate apoptosis induction following treatment with MYC and AKT pathway inhibitors. We thus treated four human T-ALL cell lines using BEZ235 or JQ1, and assessed MCL1 protein levels and PARP cleavage (a marker of apoptosis induction) using Western blot analysis. Treatment with BEZ235 monotherapy led to MCL1 protein downregulation in all cell lines treated, but this was associated with little to no evidence of apoptosis induction, as assessed by PARP cleavage (Supplementary Figure 3). By contrast, monotherapy with JQ1 increased MCL1 protein levels, and combination treatment with both BEZ235 and JQ1 effectively induced apoptosis (as assessed by PARP cleavage) in all cell lines treated, despite MCL1 protein levels comparable to those in untreated cells (Supplementary Figure 3). Thus, although downregulation of MCL1 likely contributes to modest apoptosis induction by BEZ235, our data suggest that MCL1 downregulation is not the primary mechanism of apoptosis induction following treatment with the combination of BEZ235 and JQ1 in human T-ALL, and so we focused our attention on BIM.
To test whether regulation of BIM by these oncogenic pathways is conserved in primary T-ALL patient samples, we first analyzed the correlation between expression of BIM and MYC in our previously published cohort of 40 primary pediatric T-ALL patient samples analyzed by microarray gene expression analysis.24 This revealed that, although BIM expression appears to be relatively low in all T-ALL cases, its expression is inversely correlated with that of MYC (P = 0.019; Figure 3a). To test whether MYC and AKT repress BIM in primary treatment-resistant T-ALL, we took advantage of the panel of primary T-ALL samples that we have collected from children with relapsed or refractory T-ALL (samples collected at diagnosis from patients in whom initial induction chemotherapy failed to achieve a remission, or samples collected at the time of relapse) that have been engrafted and expanded in immunodeficient mice, without exposure to in vitro selection. Treatment of these T-ALL samples in short-term culture assays with BEZ235 induced BIM expression in all samples tested (Figure 3b), whereas treatment with JQ1 led to BIM upregulation in 3 of the 4 samples tested (Figure 3c).
Figure 3.
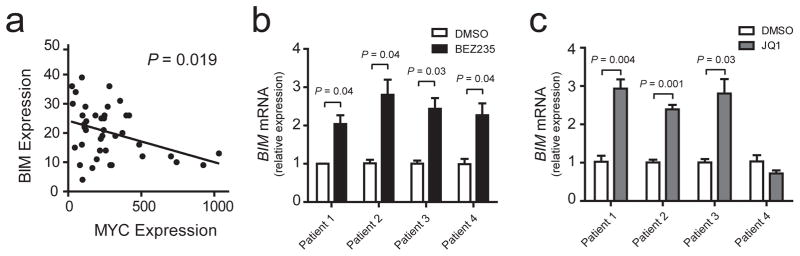
BIM is repressed by MYC and AKT in human treatment-resistant T-ALL. (a) Correlation of BIM and MYC expression levels, as assessed using microarray gene expression profiling of primary childhood T-ALL patient samples. Data are from the following probe sets: BIM, 1561844_at. MYC, 202431_s_at. Line shows results of a linear regression analysis, and P value was calculated by Pearson correlation analysis. (b and c) Q-RT-PCR analysis of BIM mRNA expression in four treatment-refractory primary pediatric T-ALL samples, expanded in immunocompromised mice and treated in short-term culture assays (24 hrs) with 500 nM BEZ235 (c) or 1 μM JQ1 (d), versus DMSO vehicle control. Error bars represent mean +/− standard error of the mean for experiments performed in triplicate. P values calculated using a Welch t test.
Taken together, these findings indicate that transcriptional repression of BIM is a key survival signal downstream of MYC and AKT in high-risk T-ALL, and we wanted to investigate the mechanisms through which these pathways repress BIM. We first tested the hypothesis that BIM upregulation following AKT pathway inhibition is mediated by FOXO transcription factors in T-ALL. FOXO transcription factors are directly phosphorylated and inactivated by AKT (reviewed in 34), and have been shown to mediate BIM upregulation following AKT pathway inactivation in normal T-cells.35 Thus, we transfected the human T-ALL cell line PF382 using either vector control or FOXO-D256Δ, encoding a dominant-negative FOXO mutant that lacks its C-terminal transactivation domain,27 cells were treated with DMSO or BEZ235, and RNA was harvested after 24 hours. Quantitative RT-PCR analysis revealed that BIM upregulation following BEZ235 treatment was blunted by expression of dominant-negative FOXO (P = 0.001; Supplementary Figure 4), implicating a role for FOXO proteins as mediators of BIM upregulation following AKT inactivation. We then turned our attention to the link between MYC and transcriptional repression of BIM, which we hypothesized might be mediated by miR-19a and mir-19b. These closely related microRNAs are both components the polycistronic miR-17-92 cluster that is directly upregulated by MYC,36 and miR-19a and miR-19b have been shown to directly downregulate BIM transcripts in murine T-ALL.37 We first used quantitative RT-PCR analysis of miR-19a and miR-19b expression to confirm that these are regulated by the MYC inhibitor JQ1 in human T-ALL cells (Supplementary Figure 5a). However, retroviral-mediated overexpression of the mir-19a, mir-19b and mir-20 components of the polycistronic miR-17-92 cluster in the human T-ALL cell line CCRF-CEM did not detectably rescue BIM mRNA upregulation following treatment with JQ1 (Supplementary Figure 5, b and c). These data thus suggest that BIM mRNA levels are also repressed by additional pathways downstream of MYC, whose elucidation will require additional investigation.
Restoration of BIM function is sufficient to impair T-ALL viability
To determine whether repression of BIM plays a role in survival signaling downstream of PI3K-AKT and MYC in T-ALL cells, we took advantage of a stabilized alpha-helix of BCL-2 domain (SAHB) modeled after amino acids 146-166 of the BIM BH3 helix to restore BIM function in human T-ALL cells. The stapled peptide designated BIM SAHBA contains non-natural amino acids inserted at positions 154 and 158, which are then cross-linked by ruthenium-catalyzed olefin metathesis to yield the bioactive peptide helix. Such stapling allows these relatively small peptides to maintain the alpha-helical conformation required for the BIM BH3 domain to engage its native targets while also enabling cellular uptake, resulting in dose-dependent and sequence-specific apoptosis induction.26,38 As a negative control, we used an analogous stapled peptide harboring a single R153D reverse polarity substitution that prevents target binding and apoptosis induction.38 Treatment of a panel of human T-ALL cell lines with BIM SAHBA impaired viability in all cell lines tested, whereas the R135D negative control had no effect (Figure 4). We then tested whether targeting BIM repression using small molecule inhibitors of MYC and PI3K-AKT pathways can induce therapeutic apoptosis in primary treatment-resistant T-ALL patient samples. BEZ235 variably induced apoptosis whereas JQ1 significantly induced apoptosis in all samples tested, and both drugs in combination had additive effects in 3 of the 4 samples treated (Figure 5).
Figure 4.

Restoration of BIM function has therapeutic activity in human T-ALL. Human T-ALL cell lines (a) ALL-SIL, (b) CCRF-CEM, (c) MOLT4 or (d) PF382 were treated with a stapled peptide mimetic of the BIM BH3 domain, BIM SAHBA (amino acids 146-166), or with the analogous peptide harboring an inactivating R153D reverse polarity mutation, for 24 hours at the indicated doses. Cell viability was assessed by CellTiter Glo, and is shown relative to DMSO control. Error bars represent mean +/− standard error of the mean for experiments performed in triplicate.
Figure 5.

Therapeutic apoptosis induction by inhibition of MYC and PI3K-AKT pathways in treatment-resistant human T-ALL. (a to d) Effect of BEZ235 (500 nM), JQ1 (1 μM), or both drugs in combination on apoptosis induction in primary human treatment-resistant T-ALL samples treated in short-term culture assays (48 hrs), as assessed using FACS analysis for Annexin V and 7AAD staining. Data for early apoptotic (annexin V-positive, 7AAD-negative) and late apoptotic (annexin V-positive, 7AAD-positive) cells are shown separately, and error bars indicate mean +/− standard error of the mean for each apoptotic subset. P values were calculated using 2-way ANOVA analysis on the combined data for all apoptotic cells (early + late). In all four patient samples, the effects of the drugs were at best additive; no interaction terms were statistically significant at the 0.10 level.
BIM is necessary for T-ALL apoptosis induction in vivo
To test whether BIM is required for T-ALL apoptosis in vivo, we took advantage of the conservation of bim function between zebrafish and mammals,29 leveraging a bim-mutant zebrafish line that was generated by retroviral insertional mutagenesis, as previously described.21 This line harbors a retroviral insertion into the coding sequence of bim exon 2, which is present in all known human and zebrafish BIM transcriptional variants and lies upstream of the BH3 domain required for apoptosis induction.39–42 This bim mutation was crossed into our conditional zebrafish model of MYC-induced T-ALL, in which MYC downregulation leads to regression of established leukemia, a phenotype that is completely dependent on mitochondrial apoptosis induction.12 We thus generated rag2:MYC-ER; rag2:EGFP double-transgenic siblings that were either bim wild-type, heterozygous, or homozygous mutant, animals were raised in 50 μg/L (129 nM) 4-hydroxytamoxifen to activate the MYC oncogene beginning at 5 days post-fertilization, and TALL onset was monitored by weekly fluorescence microscopy. After development of disseminated T-ALL, zebrafish were removed from 4-hydroxytamoxifen to downregulate activity of the MYC oncogene, and T-ALL phenotypes were assessed based on the change in tumor size by the end of the 8-week monitoring period (Figure 6a).
Figure 6.

Mutation of bim does not accelerate onset of MYC-induced T-ALL in the zebrafish. (a) Experimental design to test the effect of bim mutation on onset of MYC-induced T-ALL, and on tumor regression after MYC-ER downregulation following removal from 4-hydroxytamoxifen (4HT). (b) Analysis of T-ALL onset in zebrafish from the experiment described in a. Tumor-free survival was calculated using the method of Kaplan-Meier. and P value was calculated using the log rank test.
Deficiency of bim did not measurably accelerate the onset of MYC-induced T-ALL in the zebrafish model system (Figure 6b). By contrast, following development of established MYC-induced T-ALL, downregulation of MYC oncogene activity (by 4-hydroxytamoxifen withdrawal) was followed by T-ALL persistence in 10% of bim wild-type zebrafish, 18% of bim heterozygotes, and in 33% of bim homozygous mutants (Figure 7; P = 0.017), reflecting an inverse relationship between bim gene dosage and tumor maintenance following MYC oncogene inactivation.
Figure 7.

BIM mediates T-ALL regression following MYC inactivation in vivo. (a and c) One representative rag2:MYC-ER;rag2:EGFP double-transgenic, bim wild-type zebrafish, shown at the time of T-ALL onset (a) and 8 weeks after removal from 4HT (c). (b and d) One rag2:MYC-ER; rag2:EGFP double-transgenic zebrafish that harbored homozygous mutations of bim, shown at time of T-ALL onset (b) and 8 weeks after removal from 4-hydroxytamoxifen (d). (e) Quantitation of T-ALL persistence after downregulation of MYC by removal from 4-hydroxytamoxifen. P value was calculated using a Kruskal-Wallis test. (f) Proposed model to explain our findings.
DISCUSSION
Normal T-cell progenitors are characterized by a hypersensitivity to mitochondrial apoptosis in response to diverse stimuli, and this apoptotic phenotype is dependent on expression of BIM, a proapoptotic BCL2 family member.15–17 We have shown that BIM repression downstream of MYC and AKT is a key survival signal in the molecular pathogenesis of high-risk T-cell acute lymphoblastic leukemia. We demonstrate that MYC and AKT repress BIM mRNA expression in vivo in the zebrafish model system, as well as in human T-ALL cells, including those from patients with treatment-resistant disease. We further show that restoring BIM function using a stapled peptide mimetic of its BH3 domain is sufficient to impair the viability of human T-ALL cells, and targeting BIM repression using small molecule inhibitors of MYC and PI3K-AKT pathways can effectively induce apoptosis in primary treatment-resistant T-ALL. These small molecule inhibitors had activity in a broader range of treatment-resistant patient samples than predicted by mutational analysis of the PTEN-PI3K-AKT axis and of NOTCH1 (the most common mechanism of MYC activation in T-ALL)4,6,7,43 (Supplemental Table 2), suggesting the need for improved biomarkers to identify all patients who might benefit from these agents. Finally, we demonstrate that BIM is necessary for robust in vivo apoptosis induction upon MYC oncogene downregulation in a conditional zebrafish model of the disease. Taken together, these findings thus provide a key survival mechanism that explains the dependence of established T-ALL cells on ongoing signaling activity of the MYC and PI3K-AKT pathways (Figure 7f).
We show here that bim mutations do not accelerate the onset of MYC-induced T-ALL in the zebrafish model. Although these findings are discordant with studies in mice with an intact ARF-p53 pathway,44 our data are fully consistent with studies in mice harboring ARF-p53 pathway inactivation (as a result of p53 deletion), in which BIM-targeting mutations fail to accelerate the onset of MYC-induced lymphomagenesis.44 ARF is a mammalian tumor suppressor that links MYC-induced oncogenic stress to p53-dependent tumor suppression,45 and we have now shown that zebrafish lack a functional ortholog of ARF (manuscript under review). Biallelic inactivation of ARF (encoded by the CDKN2A locus) is the most common lesion in human T-ALL.8–10 Thus, we conclude that the role of BIM in the pathobiology of established human T-ALL is best defined in ARF-deficient models, such as the zebrafish.
Blockade of mitochondrial apoptosis is being increasingly recognized as a mechanism for chemotherapy resistance in diverse human cancers,46–48 and BIM repression has been shown to drive chemotherapy resistance in hematologic malignancies.49–51 Thus, cooperative repression of BIM by PI3K-AKT and MYC provides a molecular mechanism for the association between activation of both of these oncogenic pathways and treatment resistance in human T-ALL. Our findings thus highlight the potential therapeutic utility of targeting this survival pathway, using either combined inhibition of AKT and MYC or restoration of BIM activity using a stapled peptide such as BIM SAHBA, to reverse chemotherapy resistance in human T-ALL.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health/National Cancer Institute grant CA167124, and by a grant from the William Lawrence Blanche Hughes Foundation. A.G. is a Research Fellow of the Gabrielle’s Angel Foundation for Cancer Research. We thank Domenico Accili for the FOXO-D256Δ construct, which was obtained from Addgene.
Footnotes
CONFLICTS OF INTEREST
L.D.W. is a scientific advisory board member and consultant for Aileron Therapeutics. The Dana-Farber Cancer Institute has filed patents on and licensed drug-like derivatives of JQ1 to Tensha Therapeutics for clinical translation as cancer therapeutics. J.E.B. and the Dana-Farber Cancer Institute have been granted equity (minority) in Tensha, and J.E.B. serves on the Board of Directors. The authors have no other relevant conflicts of interest to disclose.
References
- 1.Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114:647–650. doi: 10.1182/blood-2009-02-206722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gutierrez A, Dahlberg SE, Neuberg DS, Zhang J, Grebliunaite R, Sanda T, et al. Absence of biallelic TCRgamma deletion predicts early treatment failure in pediatric T-cell acute lymphoblastic leukemia. J Clin Oncol. 2010;28:3816–3823. doi: 10.1200/JCO.2010.28.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 5.Sharma VM, Calvo JA, Draheim KM, Cunningham LA, Hermance N, Beverly L, et al. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol Cell Biol. 2006;26:8022–8031. doi: 10.1128/MCB.01091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20:2096–2109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hebert J, Cayuela JM, Berkeley J, Sigaux F. Candidate tumor-suppressor genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous deletions in primary cells from T- but not from B-cell lineage acute lymphoblastic leukemias. Blood. 1994;84:4038–4044. [PubMed] [Google Scholar]
- 9.Haidar MA, Cao XB, Manshouri T, Chan LL, Glassman A, Kantarjian HM, et al. p16INK4A and p15INK4B gene deletions in primary leukemias. Blood. 1995;86:311–315. [PubMed] [Google Scholar]
- 10.Fizzotti M, Cimino G, Pisegna S, Alimena G, Quartarone C, Mandelli F, et al. Detection of homozygous deletions of the cyclin-dependent kinase 4 inhibitor (p16) gene in acute lymphoblastic leukemia and association with adverse prognostic features. Blood. 1995;85:2685–2690. [PubMed] [Google Scholar]
- 11.Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest. 2012;122:3398–3406. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutierrez A, Grebliunaite R, Feng H, Kozakewich E, Zhu S, Guo F, et al. Pten mediates Myc oncogene dependence in a conditional zebrafish model of T cell acute lymphoblastic leukemia. J Exp Med. 2011;208:1595–1603. doi: 10.1084/jem.20101691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203–1210. doi: 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Subramaniam PS, Whye DW, Efimenko E, Chen J, Tosello V, De Keersmaecker K, et al. Targeting Nonclassical Oncogenes for Therapy in T-ALL. Cancer Cell. 2012;21:459–472. doi: 10.1016/j.ccr.2012.02.029. [DOI] [PubMed] [Google Scholar]
- 15.Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, et al. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 2002;415:922–926. doi: 10.1038/415922a. [DOI] [PubMed] [Google Scholar]
- 16.Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 17.Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12895–12900. doi: 10.1073/pnas.0914878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 2002;109 (Suppl):S45–55. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- 19.Viret C, Janeway CA., Jr MHC and T cell development. Reviews in immunogenetics. 1999;1:91–104. [PubMed] [Google Scholar]
- 20.Langenau DM, Jette C, Berghmans S, Palomero T, Kanki JP, Kutok JL, et al. Suppression of apoptosis by bcl-2 overexpression in lymphoid cells of transgenic zebrafish. Blood. 2005;105:3278–3285. doi: 10.1182/blood-2004-08-3073. [DOI] [PubMed] [Google Scholar]
- 21.Golling G, Amsterdam A, Sun Z, Antonelli M, Maldonado E, Chen W, et al. Insertional mutagenesis in zebrafish rapidly identifies genes essential for early vertebrate development. Nat Genet. 2002;31:135–140. doi: 10.1038/ng896. [DOI] [PubMed] [Google Scholar]
- 22.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell. 2010;18:485–498. doi: 10.1016/j.ccr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutierrez A, Sanda T, Ma W, Zhang J, Grebliunaite R, Dahlberg S, et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood. 2010;115:2845–2851. doi: 10.1182/blood-2009-07-234377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012;122:2018–2031. doi: 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakae J, Barr V, Accili D. Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the forkhead transcription factor FKHR. EMBO J. 2000;19:989–996. doi: 10.1093/emboj/19.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davids MS, Letai A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J Clin Oncol. 2012;30:3127–3135. doi: 10.1200/JCO.2011.37.0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jette CA, Flanagan AM, Ryan J, Pyati UJ, Carbonneau S, Stewart RA, et al. BIM and other BCL-2 family proteins exhibit cross-species conservation of function between zebrafish and mammals. Cell Death Differ. 2008;15:1063–1072. doi: 10.1038/cdd.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 31.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei G, Twomey D, Lamb J, Schlis K, Agarwal J, Stam RW, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006;10:331–342. doi: 10.1016/j.ccr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, et al. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168:5024–5031. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- 36.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 37.Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol. 2010;12:372–379. doi: 10.1038/ncb2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, et al. Ensembl 2013. Nucleic Acids Res. 2013;41:D48–55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, et al. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu JW, Chandra D, Tang SH, Chopra D, Tang DG. Identification and characterization of Bimgamma, a novel proapoptotic BH3-only splice variant of Bim. Cancer Res. 2002;62:2976–2981. [PubMed] [Google Scholar]
- 42.Marani M, Tenev T, Hancock D, Downward J, Lemoine NR. Identification of novel isoforms of the BH3 domain protein Bim which directly activate Bax to trigger apoptosis. Mol Cell Biol. 2002;22:3577–3589. doi: 10.1128/MCB.22.11.3577-3589.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan SM, Weng AP, Tibshirani R, Aster JC, Utz PJ. Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood. 2007;110:278–286. doi: 10.1182/blood-2006-08-039883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–811. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–673. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 46.Chonghaile TN, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore VD, et al. Pretreatment Mitochondrial Priming Correlates with Clinical Response to Cytotoxic Chemotherapy. Science. 2011 doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 48.Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012;151:344–355. doi: 10.1016/j.cell.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bachmann PS, Piazza RG, Janes ME, Wong NC, Davies C, Mogavero A, et al. Epigenetic silencing of BIM in glucocorticoid poor-responsive pediatric acute lymphoblastic leukemia, and its reversal by histone deacetylase inhibition. Blood. 2010;116:3013–3022. doi: 10.1182/blood-2010-05-284968. [DOI] [PubMed] [Google Scholar]
- 50.Happo L, Cragg MS, Phipson B, Haga JM, Jansen ES, Herold MJ, et al. Maximal killing of lymphoma cells by DNA damage-inducing therapy requires not only the p53 targets Puma and Noxa, but also Bim. Blood. 2010;116:5256–5267. doi: 10.1182/blood-2010-04-280818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richter-Larrea JA, Robles EF, Fresquet V, Beltran E, Rullan AJ, Agirre X, et al. Reversion of epigenetically mediated BIM silencing overcomes chemoresistance in Burkitt lymphoma. Blood. 2010;116:2531–2542. doi: 10.1182/blood-2010-02-268003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.