

Abstract
Amyloid precursor protein (APP) is widely expressed across many tissue and cell types. Proteolytic processing of the protein gives rise to a plethora of protein fragments with varied biological activities. Although a large amount of data has been generated describing the metabolism of the protein in neurons, its role in regulating the phenotype of other cells remains unclear. Based upon prior work demonstrating that APP regulates the activation phenotype of monocytic lineage cells, we hypothesized that APP can regulate macrophage activation phenotype in tissues other than brain. Ileums of the small intestines from C57BL6/J wild type and APP−/− mice were compared as a representative tissue normally associated with abundant macrophage infiltration. APP−/− intestines demonstrated diminished CD68 immunoreactivity compared to wild type mice. This correlated with significantly less cycloxygenase-2 (cox-2), CD68, CD40, CD11c, and βIII-tubulin protein levels. Peritoneal macrophage from APP−/− mice demonstrated decreased in vitro migratory ability compared to wild type cells and diminished basal KC cytokine secretion. Whereas, APP−/− intestinal macrophage had an increase in basal KC cytokine secretion compared to wild type cells. Conversely, there was a significant decrease in multiple cytokine levels in APP−/− compared to wild type ileums. Finally, APP−/− mice demonstrated impaired absorption and increased motility compared to wild type mice. These data demonstrate the APP expression regulates immune cell secretions and phenotype and intestinal function. This data set describes a novel function for this protein or its metabolites that may be relevant not only for Alzheimer’s disease but a range of immune-related disorders.
Keywords: amyloid, inflammation, Alzheimer, macrophage, cytokine, cyclooxygenase
Introduction
Amyloid precursor protein (APP) is a single pass transmembrane protein widely expressed in a variety of cells. A large amount of interest in this protein focuses on its expression in neurons and its ability to be proteolyzed to produce the amyloidogenic peptide, Aβ, characteristic of amyloid plaques in the brains of Alzheimer’s disease (AD) patients (Perry et al., 1989; Joachim et al., 1991). In addition, a variety of mutations in the gene coding for APP are responsible for a rare familial form of AD (Kamino et al., 1992; Tanzi et al., 1992). Therefore, many studies have examined expression and proteolytic processing of APP in central neurons in an effort to understand possible mechanisms of disease.
However, our prior studies (Sondag and Combs, 2004, 2006, 2010) as well as others (Bauer et al., 1991; Bullido et al., 1996; Vehmas et al., 2004; Spitzer et al., 2010) have demonstrated that APP is also expressed on immune cells where it appears to play a role in regulating cellular phenotype. For example, expression on monocytic lineage cells appears to regulate the ability of these cells to interact with extracellular matrix and mediate various cell-cell interactions (Austin and Combs, 2010; Sondag and Combs, 2010). In addition, APP expression increases in both macrophage and microglia in the presence of a reactive, stimulatory environment (Haass et al., 1991; Banati et al., 1994; Monning et al., 1994; Banati et al., 1995b; Banati et al., 1995a; Gehrmann et al., 1995a; Gehrmann et al., 1995b; Monning et al., 1995; Itoh et al., 2009). We have also observed that agonist antibody stimulation of APP leads to a diverse change in proinflammatory protein expression and cytokine secretion as well as release of Aβ peptides from monocytic cells (Sondag and Combs, 2004, 2006). Recently, others have suggested that secreted Aβ peptides are anti-microbial (Soscia et al., 2010) fitting well with the ability of immune cells to make the peptides upon activating ligand stimulations (Sondag and Combs, 2006; Spitzer et al., 2010). Finally, a variety of studies have demonstrated that APP metabolites and Aβ peptides in both their fibrillar and oligomeric forms are potent activating stimuli for macrophage, monocytes, and microglia (Klegeris et al., 1994; Yan et al., 1998; Combs et al., 1999; Yates et al., 2000; Smits et al., 2001; Yazawa et al., 2001; Ikezu et al., 2003; Uryu et al., 2003; Xiong et al., 2004; Floden and Combs, 2006; Sondag et al., 2009; Maezawa et al., 2010). Collectively, these data suggest that APP and its proteolytic products may serve as regulators of proinflammatory phenotype in particular immune cells.
The mammalian intestines are not only characterized by a unique enteric nervous system (Harrington et al., 2010; Tomita et al., 2010) and APP isoform expression (Yamada et al., 1989; Arai et al., 1991) but also an abundance of resident immune cell types, including macrophage necessary for monitoring resident and foreign microbial exposure (Santaolalla et al., 2010). In order to compare an environment in which neurons and macrophage are expected to abundantly express APP and a range of activating stimuli are continually being presented, we chose in this study to compare small intestine of wild type C57BL6/J and APP−/− mice. Based on our previously demonstrated role for APP in regulating monocytic lineage cell function, a lack of this protein in the dynamically activated immune environment of the intestine should result in significant alteration of immune parameters and perhaps neuronal and gut function in animals that lack the protein.
Materials and methods
Materials
Anti-β-Amyloid Precursor Protein (APP) and anti-occludin antibodies was purchased from Zymed Laboratories (San Francisco, CA). Anti-mouse IgM (goat), anti-rabbit (goat), anti-goat (bovine), anti-rat (goat), and anti-mouse (bovine) horseradish peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Cox-2, CD40, TLR4, CD36 and actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Elite Vectastain ABC Avidin and Biotin, Vector Vip, biotinylated anti-rabbit, anti-mouse, and anti-rat antibodies were purchased from Vector Laboratories Inc (Burlingame, CA, USA). Synaptophysin and βIII tubulin antibodies were purchased from Chemicon international, Inc (Temecula, CA, USA). CD68 antibody was purchased from AbD Serotec (Oxford, UK). PSD95 and GFAP antibodies were purchased from Cell Signaling Technology Inc (Danvers, MA, USA). MAP2 antibody and MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) were purchased from Sigma-Aldrich (St. Louis, MO, USA). CD3, CD11c and FATP4 antibodies were purchased from Abcam Inc (Cambridge, MA, USA). TLR2 antibody was purchased from Imgenex (San Diego, CA, USA). pTyr antibody was purchased from Millipore (Billerica, MA, USA).
Mice
APPtm1Dbo/J homozygous (APP−/−) mice and wild type (C57BL6/J) mice were purchased from Jackson Laboratory. Mice were provided food and water ad libitum and housed in a 12 hour light/dark cycle. Mice were maintained until 2 and 7 months of age then euthanized via CO2 asphyxiation followed by cervical dislocation or cardiac perfusion. Animals were weighed prior to collection at 7 months of age. The investigation conforms to the Guide for the Care and Use of Laboratory Animals (eighth edition, 2010). Animal use was approved by the University of North Dakota IACUC.
Blood collection and LAL assay
At the time of perfusion, blood was collected in heparinized tubes via heart puncture and spun at 190g~7400rpm for 10min at 4°C. Plasma was then transferred to sterile centrifuge tubes which were flash frozen to be analyzed the following day for lipopolysaccharide (LPS) quantitation according to the Genscript ToxinSensor™ Chromogenic LAL Endotoxin Assay Kit protocol (GenScript, Piscataway, NJ).
Western Blotting
At seven months, the animals were perfused with Ca2+/Mg2+ free PBS and ileum of intestine was collected, rinsed to remove lumen contents, flash frozen, pulverized and lysed using ice cold RIPA buffer (20mM Tris, pH 7.4, 150mM NaCl, 1mM Na3VO4, 10mM NaF, 1mM EDTA, 1mM EGTA, 0.2mM phenylmethylsulfonyl fluoride, 1% Triton, 0.1% SDS, and 0.5% deoxycholate) with protease inhibitors (AEBSF 104mM, Aprotinin 0.08mM, Leupeptin 2.1mM, Bestatin 3.6mM, Pepstatin A 1.5mM, E-64mM) and 50U/mL DNAse1 (Amresco Inc, Solon, OH, USA). To remove insoluble material cell lysates were sonicated and centrifuged (14,000 rpm, 4°C, 10 min). The Bradford method (Bradford, 1976) was used to quantitify protein concentrations. Proteins were resolved by 7 and 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (PVDF) for Western blotting using anti-APP, occludin, cox-2, synaptophysin, PSD95 (1:10,000), CD68 (1:2000), CD40, CD3, CD11c, GFAP (1:5000), TLR2, TLR4, CD36, FATP4, pTyr, βIII tubulin (loading control), and actin (loading control) antibodies at 1:1000 unless otherwise indicated. Antibody binding was detected with enhanced chemiluminescence (GE Healthcare, Piscataway, NJ). In some instances, blots were stripped in 0.2 NaOH, 10 min, 25°C, for reprobing. Western blots were quantified using Adobe Photoshop software. Optical densities of bands were normalized against their respective loading controls and averaged (+/−SD).
Histological Stain
At seven months, the animals were perfused with Ca2+/Mg2+ free PBS and ileum of the small intestine was collected and immersion fixed for 24hrs in 4% paraformaldehyde and cryoprotected through two successive 30% sucrose changes. A 1cm piece of ileum of small intestine was cut, then cut lengthwise and flattened for sectioning. Ileum samples were serially cryosectioned (10μm) for routine histology H & E (hematoxylin and eosin) and Alcian blue stains. For H and E stain, slides were rinsed in xylene three times for 1 min each, followed by rehydration in 2 changes of absolute alcohol for 2 min, then 95%, 80%, 70% alcohol for 1 min each, followed by a rinse in DH2O for 1 min. Slides were then stained with Hematoxylin Regulars for 40 seconds followed by a rinse in running tap water for 10 dips, then differentiated with 3% acid alcohol for 2 dips, followed by rinse in running tap water for 10 dips. Slides were then blued in lithium carbonate (1% sodium bicarbonate) solution for 3–5 dips followed by rinsing again in running tap water before a 1 min rinse in 70% alcohol and then 5–10 seconds in Eosin. Slides were dehydrated two times each for 1 min in 95% alcohol, 100% alcohol and xylene followed by covering with permount. For Alcian blue stain, slides were placed in 100% alcohol, 95% alcohol, 70% alcohol, and DH2O for 1 min each then mordant in 3% aqueous acetic acid for 1 min, 1% Alcian blue in 3% acetic acid pH 2.5 for 10 min followed by rinse in running water for 5–6min and rinse in DH2O. Slides were counterstained in nuclear fast Red 0.1% (kerechtrot) for 1.5 min then rinsed in running water for 10 min before dehydrating in 95% alcohol, 100% alcohol and xylene 2 times each for 1 minute before being covered with permount.
Immunohistochemistry
At seven months, the animals were perfused with Ca2+/Mg2+ free PBS and ileum of the small intestine was collected and immersion fixed for 24hrs in 4% paraformaldehyde and cryoprotected through two successive 30% sucrose changes. A 1cm piece of ileum of small intestine was cut, then cut lengthwise and flattened for sectioning. Ileum samples were serially cryosectioned (10μm) for immunostaining with anti-APP (1:1000), CD68 (1:500), cox-2 (1:500), occludin (1:100), MAP2 (1:500), pTyr (1:1000), and GFAP (1:1000) antibodies or respective secondary only antibodies (1:2000). Antibody binding in the intestine was visualized using the Vector VIP chromagen (Vector Laboratories, Burlingame, CA). Images were taken using an upright Leica DM1000 microscope and Leica DF320 digital camera system. Figures were made using Adobe Photoshop 7.0 software.
Cytokine Array
At seven months, the animals were perfused with Ca2+/Mg2+ free PBS and ileum of intestine was collected, flash frozen, pulverized and lysed using lysis buffer according to the manufacturer’s protocol (Ray Biotech, Norcross, GA, USA). Insoluble material was removed from cell lysates via centrifugation (14,000 rpm, 4°C, 10 min). The Bradford method (Bradford, 1976) was used to quantitify protein concentrations. 1mg per sample was used to probe a 40 cytokine antibody-based membrane array according to the manufacturer’s protocol (Ray Biotech, Norcross, GA, USA). Optical density of dots were normalized against their respective positive controls and averaged (+/−SD).
Stool Weight
Total stool weight was measured by placing the animals in a clean empty cage and collecting all stool produced over a 1hr time period. Stool was then desiccated at 80°C overnight to determine dry weight. Stool water was calculated as the difference between these two measurements.
Macrophage Isolation and Stimulation
At 2 months, non-elicited peritoneal macrophage were rinsed from the peritoneal cavity of the collected mice with sterile PBS, spun, resuspended, and allowed to adhere to tissue culture wells for 2–3 hours in DMEM/F12 plus PSN antibiotics (0.05mg/mL Penicillin, 0.05mg/mL Streptomycin, 0.1mg/mL Neomycin (Gibco, Carlsbad, CA, USA)). Ileum of the intestines were removed from the abdominal cavity and rinsed with sterile PBS inside and outside to remove residual contents. Intestines were then rinse again with HBSS (8g NaCl, 0.4g KCl, 3.57g Hepes, 0.06g Na2HPO4 × 2 H2O, 0.06 g KH2PO4 in 1 liter DH2O) and then placed in sterile enzyme solution (5mM Glucose, 1.5% BSA, 5mg/mL Collagenase, 20% FBS in HBSS), minced and placed in 37°C incubator for 30 min to allow digestion to occur. Tissue was then triturated, filtered through a 70μm cell strainer, quenched with HBSS and spun for 4 min at full speed. The pellet was resuspended in HBSS, triturated and spun for another 4 min at full speed. The pellet was resuspended in DMEM/F12 plus PSN antibiotics and 400uL of suspended cells was plated in another 1mL of DMEM/F12 plus PSN antibiotics for 2–3 hours to allow adherence of macrophage. Non-adherent and non-macrophage cells were rinsed from the wells using ice-cold DMEM/F12.
Enzyme-linked Immunosorbent Assay (ELISA)
Following rinses, DMEM/F12 plus PSN antibiotics with or without 25ng/mL LPS (Sigma) was added for overnight stimulation, then media was collected for quantifying secreted keratinocyte chemokine (KC), Regulated upon Activation, Normal T-cell Expressed, and Secreted (RANTES), Interleukin-6 (IL-6), and Interleukin-1 beta (IL-1β) according to the manufacturer’s protocol (R&D Systems, Minneapolis, MN, USA). Secretion was normalized to cell protein amount as determined by the Bradford method (Bradford, 1976) after cells were lysed in ice cold RIPA buffer with protease inhibitors, sonicated, and spun to remove insoluble material.
Migration and Proliferation Assay
After rinses, cells were removed with ice cold dissection media and gentle trituration. One set of macrophage (5,000 cells/well/96 well) had DMEM/F12 containing PSN antibiotics, 10% FBS and 5% horse serum added for 72hrs followed by an MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reduction assay to indirectly measure cellular proliferation. The media was removed and replaced with 0.1mg/mL MTT for 4 hr. The precipitated formazan was dissolved in isopropanol and absorbance read at 560nm. Another set of macrophage (5,000 cells/well) were resuspended in DMEM/F12 and transferred to transwell permeable support inserts (3μm, Corning Inc. Corning, NY, USA) in collagen coated 24 well dishes containing 25ng/mL LPS to measure transmigration by counting the number of migrated cells after 24 hours.
Statistical Analysis
The data were analyzed by unpaired two-tailed t-test with or without Welch correction for unequal variance as required or by a two-way ANOVA with Holm-Sidak post hoc test.
Results
Immunoreactivity differences between the ileum of APP−/− and C57BL6/J control mice
In order to determine histological differences between APP−/− and wild type C57BL6/J mice, the ileum of small intestine was collected from adult (7 months of age) mice and was fixed, sectioned and stained with hematoxylin and eosin and Alcian blue. Mice were collected at this age based upon prior observations that transgenic animals over-expressing mutant forms of APP demonstrate pathology in the gut at 6 months of age. This suggested that expression or function of endogenous APP, as was our study intent, may also be involved in regulating intestinal physiology at this age (Van Ginneken et al., 2011). There were no histological structural differences between the wild type C57BL6/J and APP−/− mice (Fig. 1). In order to characterize APP immunoreactivity in the intestine, wild type C57BL6/J and APP−/− mice ileum of the small intestine was immunostained. Wild type mice demonstrated robust APP immunoreactivity within enterocytes and neurons and diffuse immunoreactivity within the smooth muscle of the muscularis externa (Fig. 2). The tissue was then immunostained with a common intestine regulatory protein, cyclooxygenase-2 (cox-2) as well as the tight junction protein, occludin, to gain an impression of intestinal phenotype. Both cox-2 and occludin immunoreactivity again appeared primarily localized to the enterocyte layer with some areas of robust immunoreactivity for cox-2 in the wild type intestines compared to APP−/− mice (Fig. 2). This supported the idea that APP expression regulates some aspects of intestinal immune cell behavior as well as that of enterocytes. Based upon the fact that a portion of the APP immunoreactivity was clearly localized to neurons in wild type mice, the tissue was also stained with an anti-microtubule associated protein 2 (MAP2) antibody to visualize the integrity of the enteric plexi in the intestines. As expected, wild type mouse sections demonstrated robust immunoreactivity throughout the submucosa and lamina propria while the APP−/− sections had visibly less immunoreactivity (Fig. 2). This suggests that lack of APP expression also modulates neuronal integrity and likely enteric function in the small intestine. Since there was no obvious immunoreactivity for APP observed within macrophage or other cell types throughout the intestinal layers (Fig. 2) we then examined other cell types in the ileum of the small intestine. Wild type intestines demonstrated robust anti-CD68 immunoreactivity to identify macrophage while APP−/−mice presented with a markedly decreased staining (Fig. 3) suggesting either decreased activation or decreased numbers in the intestine. Because the phenotype of astrocyte-like enteric glia can contribute to maintenance of gut barrier integrity and inflammatory state (Savidge et al., 2007; Cirillo et al., 2011) we next assessed activation state of these cells. Based upon the fact that a population of enteric glia are astrocytic in nature (Jessen and Mirsky, 1985) we immunostained the ileum of the small intestine with anti-glial fibrillary acidic protein (GFAP) antibodies (Fig. 3). The GFAP immunoreactivity appeared to localize to the external portion of the muscularis externa. Unlike neuron and macrophage immunoreactivities there was no visible difference in astrocyte-like immunoreactivity between the wild type and APP−/− intestine (Fig. 3). To assess if APP expression resulted in tyrosine kinase activation in the ileum of the small intestine as another potential marker of immune cell macrophage activation, the ileum was stained with anti-pTyr antibodies. However, there was no obvious difference in pTyr immunoreactivity between wild type and APP−/− intestines (Fig. 3).
Fig. 1.
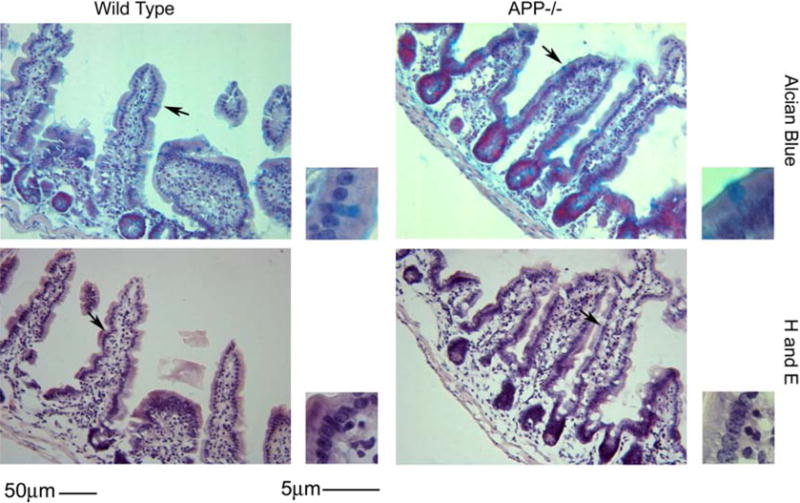
Histology of the small intestine was similar in both APP−/− and wild type controls. Ileum tissue samples were collected from C57BL6/J wild type and APP−/− mice, fixed in 4% paraformaldehyde, serially sectioned, and stained using routine histology H & E (hematoxylin and eosin) and Alcian blue. Arrows indicate the location of the region of interest shown as an enlarged inset to the right of each panel. Representative images from 5 animals per condition are shown.
Fig. 2.
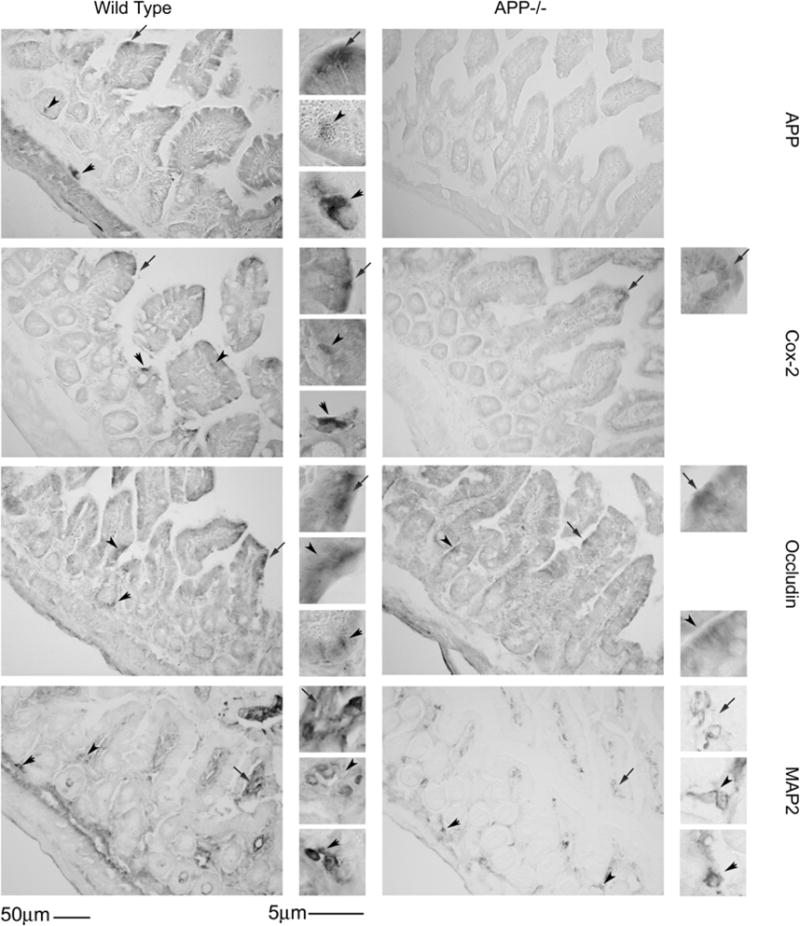
Small intestine immunoreactivity for cox-2 and MAP2 decreased in APP−/− mice compared to wild type controls with no change in occludin. Ileum tissue samples were collected from C57BL6/J wild type and APP−/− mice, fixed in 4% paraformaldehyde, serially sectioned, and immunostained. Tissue sections were immunostained using anti-APP, cox-2, occludin and MAP2 antibodies and antibody binding was visualized using Vector VIP as the chromagen. Arrows indicate the location of immunoreactivity shown as an enlarged inset to the right of each panel. Representative images from 5 animals per condition are shown.
Fig. 3.
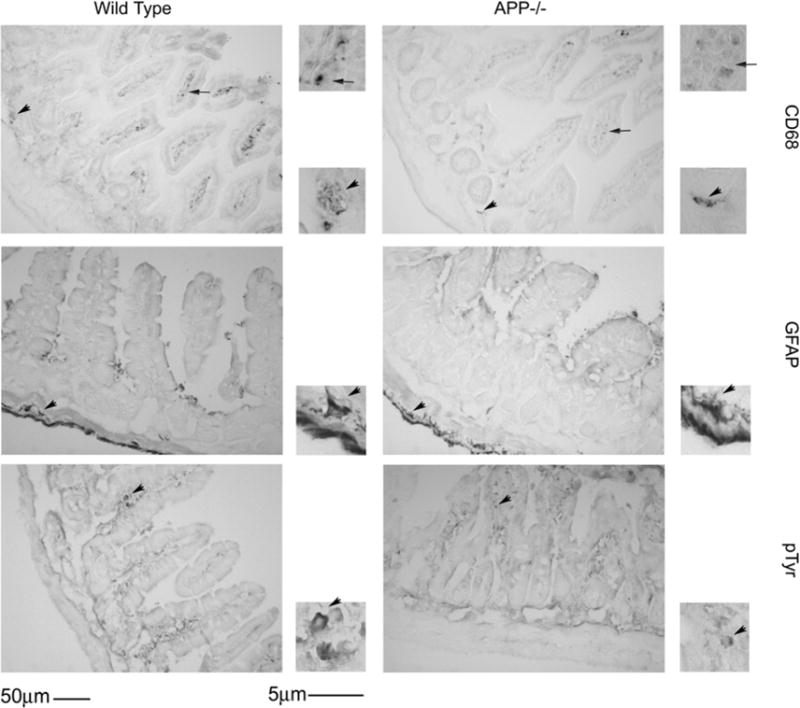
Small intestine immunoreactivity for CD68 decreased in APP−/− mice compared to wild type controls with no change in GFAP or pTyr. Ileum tissue samples were collected from C57BL6/J wild type and APP−/− mice, fixed in 4% paraformaldehyde, serially sectioned, and immunostained. Tissue sections were immunostained using anti-CD68, GFAP and pTyr antibodies and antibody binding was visualized using Vector VIP as the chromagen. Arrows indicate the location of immunoreactivity shown as an enlarged inset to the right of each panel. Representative images from 5 animals per condition are shown.
Quantitation of protein differences in the ileum of C57BL6/J wild type and APP−/− mice
Based upon the qualitative changes in immunoreactivity patterns observed, it was necessary to quantify whether there were indeed protein level changes in wild type versus APP−/− intestine. Ileum samples were Western blotted to determine whether the changes observed by immunohistochemistry were significant. As expected, Western blot analysis demonstrated that cox-2 and CD68 levels were significantly decreased in the APP−/− mice compared to wild type controls with no difference in occludin, GFAP, and pTyr (Fig. 4) consistent with the immunostaining observed (Fig. 2). To assess whether other immune cell types present in the intestine besides macrophage were altered in APP−/− mice, Western blots for markers of T-cells (CD3), B-cells (CD40), as well as dendritic cells (CD11c) were quantified. Although there was no change in CD3 levels there was a significant decrease in CD40 and CD11C protein levels in the APP−/− compared to the wild type mice (Fig. 4). As a measure of neuronal integrity, presynaptic and postsynaptic markers were also examined by Western blot. Interestingly, the total amount of neuronal βIII-tubulin protein levels was decreased between the wild type and APP−/− mice, with no difference in protein levels of the presynaptic (synaptophysin) or postsynaptic (PSD95) protein markers (Fig. 4). This decrease in βIII-tubulin levels was consistent with the observed decrease in dendritic marker, MAP2, immunoreactivity in APP−/− mice (Fig. 2). A change in macrophage, B cell, and dendritic cell protein levels could correlate with altered expression of immunomodulatory receptors in the intestine. Therefore, critical pattern recognition receptor, TLR2 and TLR4, protein levels were examined as well. However there was no change in expression of either TLR between wild type and APP−/− ileum of the small intestine (Fig. 4). Finally, protein levels of necessary fatty acid transport proteins was assessed as an additional means of examining changes in intestinal barrier integrity. CD36 and FATP4 protein levels were analyzed, with again, no difference in expression between the wild type and APP−/− ileum of the small intestine (Fig. 4) suggesting again that epithelial function may be preserved in the APP−/− intestines.
Fig. 4.

Western blot analysis of cox-2, CD68, CD40, CD11c and βIII-tubulin demonstrated decreased protein levels in the small intestine of APP−/− compared to wild type mice. Ileum of the small intestine was collected from APP−/− and C57BL6/J wild type mice.. The tissue was lysed, resolved by 10–15% SDS-PAGE and Western blotted using anti-CD68, CD3, CD40, CD11c, GFAP, TLR2, TLR4, CD36, FATP4, pTyr synaptophysin, PSD95, APP, cox-2, occludin, βIII tubulin (neuronal loading control), or actin (general loading control) antibodies. Antibody binding was visualized by chemiluminescence. Blots from 4 of the animals for each strain are shown. Optical densities of the Western blotted ileum proteins were normalized against their respective actin (cox-2, CD68, CD3, CD40, CD11c, GFAP, TLR2, TLR4, CD36, FATP4, pTyr and occludin) or βIII tubulin (synaptophysin and PSD95) loading controls and averaged (+/−SD) from 10 animals per each condition *p<0.05.
In vitro migration, proliferation, and differentiation comparison of macrophage from APP−/− and C57BL6/J wild type mice
Since Western blot analysis had validated that protein differences existed in neuronal proteins and enterocyte proteins in APP−/− compared to wild type mice, we next sought to further examine the changes in CD68 positive macrophage immunoreactivity observed in the APP−/− ileums. As intestinal macrophage have previously been shown to have very different responses to LPS (Grimm et al., 1995; Rogler et al., 1998), both peritoneal and intestinal macrophage have been included in our analysis as both contribute to the immunomodulatory behavior of the ileum of the intestine. One possibility for the decrease in CD68 immunoreactivity might simply be that APP−/− cells were less able to migrate into the ileum. As a means of testing this idea, we isolated non-elicited peritoneal macrophage and intestinal macrophage from wild type and APP−/− mice at 2 months of age and compared the ability of these cells to migrate through a transwell culture insert in vitro. As expected, APP−/− peritoneal macrophage demonstrated a significantly decreased ability to migrate through the insert into the LPS containing media below compared to wild type peritoneal cells. However, there was no significant difference in the migratory behavior of wild type and APP−/− intestinal macrophage (Fig. 5A and B). Another possibility for decreased CD68 immunoreactivity in the APP−/− ileums could be due to an impaired differentiation or proliferation ability of APP−/− cells. As a means of assessing proliferative ability of the cells, peritoneal and intestinal macrophage from wild type and APP−/− animals were grown in the presence of serum for 72 hours. However, there was no significant difference in proliferative ability in vitro of either type of macrophage (Fig. 5 C and D). To examine differentiative or activation phenotype differences between the two genotypes, collected macrophage were stimulated 24 hr in the absence or presence of the potent bacterial endotoxin LPS and media were collected to assay secretion of select cytokines. Based upon prior analysis of abundant peritoneal macrophage in vitro secretion, keratinocyte chemokine (KC), and interleukin-6 (IL-6) levels were quantified from the media as representative secretory factors from peritoneal and intestinal macrophage. Although there were no differences in LPS-stimulated levels between genotypes for KC cytokine secretion in the peritoneal macrophage, the APP−/− peritoneal macrophage secreted significantly less KC compared to wild type cells during basal, unstimulated conditions (Fig. 6A). LPS-stimulated wild type peritoneal macrophage had increased secretion of IL-6 compared to LPS-stimulated APP−/− and untreated wild type peritoneal macrophage (Fig. 6C). The LPS-stimulated intestinal macrophage had no change in secretion of KC or IL-6, suggesting that intestinal macrophage are unresponsive to LPS (Fig. 6B and D). However, basally APP−/− intestinal macrophage secreted more KC than wild type intestinal macrophage (Fig. 6B). As a follow up to the decreased migratory behavior of the APP−/− peritoneal macrophage, Regulated upon Activation, Normal T-cell Expressed, and Secreted (RANTES) was measured from peritoneal macrophage stimulated with and without LPS (Fig. 6E). As with the secretion of KC, LPS increased RANTES secretion with no significant difference between wild type and APP−/− peritoneal macrophage (Fig. 6E). To further compare phenotype differences particularly in intestinal macrophage, interleukin-1 beta (IL-1β) was next measured. Again as with KC and IL-6, LPS stimulation had no effect on wild type intestinal macrophage. However, LPS significantly increased secretion of IL-1β from the APP−/− intestinal macrophage as compared to the unstimulated APP−/− intestinal macrophage as well as the LPS stimulated wild type intestinal macrophage (Fig. 6F). Collectively, these findings suggest that peritoneal and intestinal macrophage of APP−/− mice are phenotypically different with impaired migratory ability in peritoneal macrophage compared to their intestinal counterparts. More importantly, in spite of an apparent decrease in numbers of CD68 positive macrophage in the ileum of APP−/− mice, these cells appear, to some degree, hyperreactive basally and in response to LPS stimulation when compared to wild type intestinal macrophage.
Fig. 5.
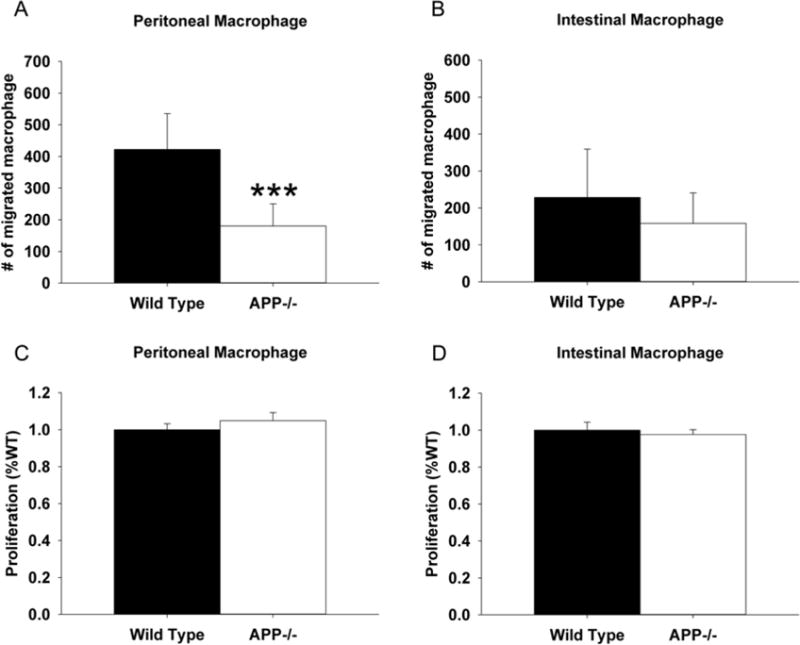
Peritoneal macrophage from APP−/− mice demonstrated significantly decreased migratory ability compared to wild type controls. Non-elicited peritoneal macrophage and intestinal macrophage were isolated from APP−/− and C57BL6/J control mice. (A) For migration assays, macrophage were grown in serum-free DMEM/F12. Cells were added to the top of transwell permeable support inserts placed into collagen coated dishes containing 25ng/mL LPS for 24hr in order to measure migration into the bottom well as assessed by the number of migrated cells. (B) For proliferation assays, macrophage were grown in DMEM/F12 with 10% FBS and 5% horse serum for 72 hours. MTT reduction assays were used as an indirect measure of cellular number to quantify proliferation. Results are mean (+/−SD) from six animals per condition ***p<0.05.
Fig. 6.
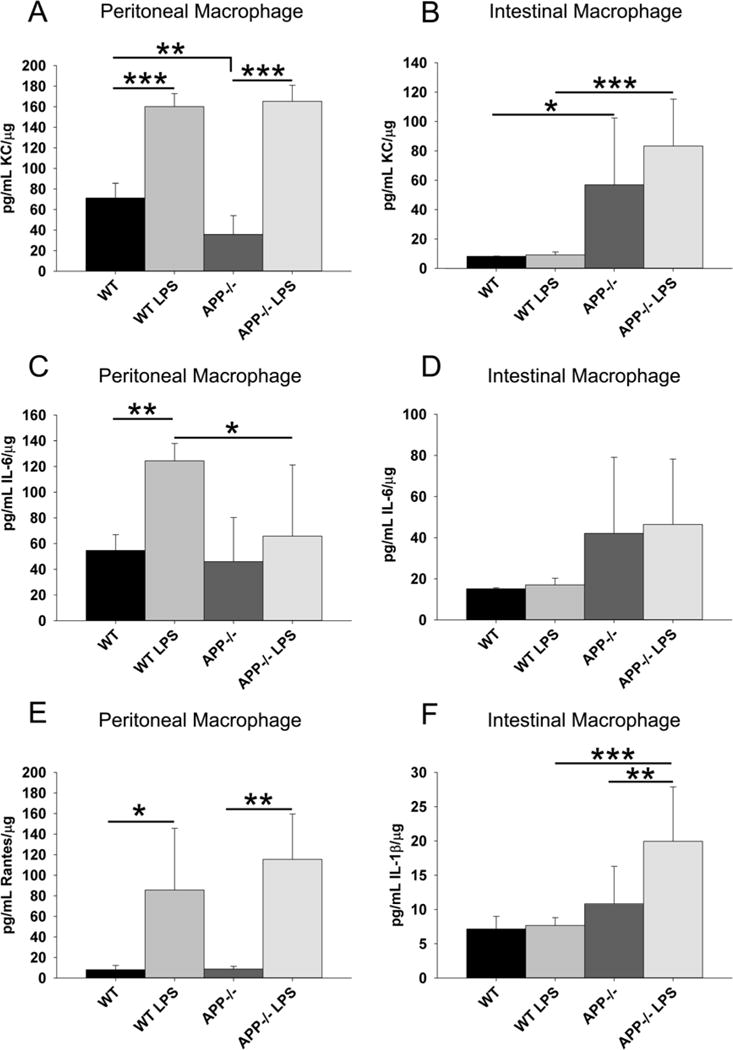
APP−/− and wild type peritoneal macrophage demonstrated different cytokine secretory profiles compared to intestinal macrophage. Non-elicited peritoneal macrophage and intestinal macrophage were isolated from C57BL6/J wild type and APP−/− mice. Macrophage were stimulated with or without 25ng/mL LPS, 24hr in serum-free DMEM/F12. Media was collected to quantify basal and stimulated secretion of KC, IL-6, RANTES and IL-1β. The results are displayed as mean (+/−SD) from 6 animals in each group *p<0.05, **p<0.01, ***p<0.001.
Profile of cytokine levels in ileum of APP−/− and C57BL6/J wild type mice
Based upon the in vitro macrophage secretory differences observed, the reduced CD68 ileum macrophage immunoreactivity, and the reduced protein levels of CD68, CD40, and CD11c it was reasonable to expect that levels of cytokines in the APP−/− versus wild type ileums as a whole would also differ. To examine this possibility, ileums were collected from 7 month old animals and used to perform antibody-based cytokine arrays profiling 40 different cytokines. As expected, the APP−/− ileums demonstrated significantly decreased levels of 12 different factors, CD30L, eotaxin, fas ligand, fractalkine, GCSF, IL-10, IL-12p40p70, IL-3, IL-9, MIG, MIP-1γ, and sTNFRI, compared to wild type mice (Fig. 7). Therefore, these findings were consistent with the immunohistochemistry and further supported the notion that APP expression regulates immune cell phenotype in the intestine.
Fig. 7.
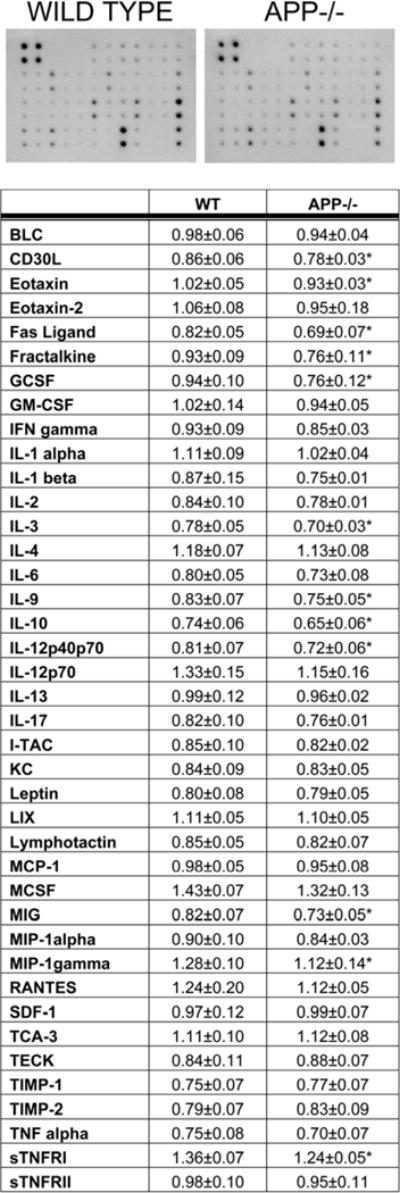
Small intestine of APP−/− mice demonstrated altered levels of multiple cytokines compared wild type controls. Ileum from C57BL6/J wild type and APP−/− mice were collected, lysed, and used in an antibody cytokine array. APP−/− mice demonstrated decreased CD30L, eotaxin, fas ligand, fractalkine, GCSF, IL-10, IL-12p40p70, IL-3, IL-9, MIG, MIP-1γ, and sTNFRI compared to wild type mice. A representative array for one animal from each genotype is shown. Optical densities of normalized dot intensities from 6 animals in each group are displayed as mean (+/−SD). *p<0.05
Differences in intestinal motility and absorption in APP−/− and C57BL6/J mice
A combined change in levels of multiple cytokines, proinflammatory and regulatory proteins, numbers of macrophage, and finally neuronal synaptic markers all suggest that the function of APP−/− intestines would differ significantly compared to wild type control animals. In order to address intestinal function, mean stool production and water content were compared between the genotypes. Although this was not exclusively an assessment of small intestine behavior, APP−/− animals generated significantly more stool per time period with increased water content (Fig. 8A). This suggested increased motility and decreased absorption in the intestine of APP−/− mice, which correlated with reduced body mass in these mice (8B). This suggests that APP−/− may have reduced weight gain ability due to increased motility and reduced absorption. Moreover, the adverse consequence of decreased absorption in the smaller APP−/− mice likely has a significant effect on overall physiology. As an additional means of assessing absorption, plasma levels of LPS were quantified from the blood of C57BL6/J and wild type mice. Trace amounts of LPS are known to be taken up into the blood from the intestines during normal absorption of fatty acids (Amar et al., 2008; Ghoshal et al., 2009). As now predicted, APP−/− mice demonstrated significantly less LPS in their plasma compared to wild type mice again suggesting impaired intestinal absorptive function (Fig. 8C).
Fig. 8.
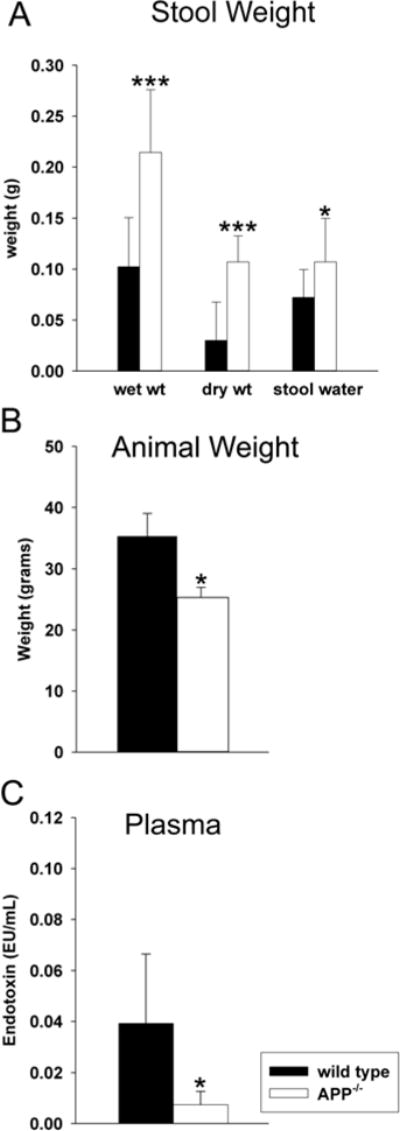
APP−/− mice demonstrated intestinal motility and absorption differences compared to wild type mice. To compare overall intestinal absorption and motility, (A) water and stool production, (B) animal weight and (C) uptake of LPS were quantified from C57BL6/J wild type and APP−/− mice. (A) To assess stool production and water absorption, wet and dry stool weights and the differences (water content) were collected over 1hr. Data from 13 animals in each group are displayed as mean (+/−SD). (B) To assess animal weight difference between strains, mice were weighed prior to collection. (C) To quantify uptake/transport of intestinal LPS into blood, whole blood was collected, plasma was separated and the LAL Endotoxin assay was performed according to manufacturer protocol. Data from 5 animals in each group are displayed as mean (+/−SD).*p<0.05, ***p<0.001.
Discussion
We have demonstrated that APP expression has profound regulatory effects on protein expression in a range of small intestine cells including neurons, CD68 positive macrophage, and enterocyte epithelial cells. This represents, to the best of our knowledge, the first demonstration of a function for this protein in this manner. Indeed, a scarcity of data exists regarding the function of APP in general. Based upon the quantified differences in multiple cytokine expression levels as well as changes in intestinal motility and absorption it appears that APP has a role in diverse tissue types. More importantly, mutations in the gene coding for APP or altered processing of APP could have effects in other tissues, such as the intestine, in addition or uniquely from any observed in the brain.
It is important to point out that at this point it is not clear whether lack of APP on one or multiple cell types is responsible for the changes in cytokine profile, enterocyte protein expression or motility and absorption observed. Also, in spite of the robust changes in macrophage behavior and cytokine levels in APP−/− versus wild type mice, little detectable APP immunoreactivity was observed in macrophage-like cells in ileums of wild type mice. Based upon prior evidence of clear APP expression in monocytic lineage cells (Bauer et al., 1991; Bullido et al., 1996; Sondag and Combs, 2004; Vehmas et al., 2004; Sondag and Combs, 2006, 2010; Spitzer et al., 2010) it is likely that the level of expression of APP in macrophage was simply not high enough to be detectable by the immunostaining procedure employed. Indeed, prior work from others has demonstrated in the related brain macrophage cell type, microglia, that APP expression is upregulated when the cells are provided the appropriate stimulatory environment (Haass et al., 1991; Banati et al., 1994; Monning et al., 1994; Banati et al., 1995b; Banati et al., 1995a; Gehrmann et al., 1995a; Gehrmann et al., 1995b; Monning et al., 1995; Bullido et al., 1996; Vehmas et al., 2004; Itoh et al., 2009). In this fashion, it is likely that APP expression still directly regulates macrophage phenotype in the intestine in spite of the limitations in detection using the visible light chromagen method. One intriguing hypothesis is simply that macrophage numbers are limited due to impaired ability of APP−/− cells to migrate into the submucosa and lamina propria. For instance, our prior work demonstrated that APP expression is required on both monocytes and endothelial cells for cell-cell interaction to occur necessary for monocytic adhesion required for activities such as diapedesis through the vasculature (Austin et al., 2009; Austin and Combs, 2010). It is possible that APP−/− macrophage are limited in the intestine due to decreased influx through the vasculature. The impaired migratory ability of APP−/− macrophage supports this notion.
On the other hand, it is also quite possible that APP and its proteolytic metabolites expressed on other cells, such as neurons or enterocytes are normally responsible for some level of CD68 positive macrophage numbers in the intestine. The robust detection of APP immunoreactivity that localized to enterocytes and neurons is entirely consistent with the fact that neurons particularly in the brain express large amounts of APP compared to other tissue types (Schmechel et al., 1988; Yamada et al., 1989; LeBlanc et al., 1991) and prior expression of at least the Aβ peptide, has been detected in rodent enterocytes (Galloway et al., 2007; Galloway et al., 2008; Galloway et al., 2009; Pallebage-Gamarallage et al., 2009). This suggests that lack of APP metabolites from neurons or enterocytes may lead to diminished stimulation of intestinal macrophage. Indeed, we as well as others have demonstrated in a plethora of studies that different multimeric forms of Aβ peptide or other APP fragments can directly stimulate immune cells such as monocytes, macrophage, and microglia to alter their phenotype (Klegeris et al., 1994; Yan et al., 1998; Combs et al., 1999; Yates et al., 2000; Smits et al., 2001; Yazawa et al., 2001; Ikezu et al., 2003; Uryu et al., 2003; Xiong et al., 2004; Floden and Combs, 2006; Sondag et al., 2009; Maezawa et al., 2010). In this scenario, limited CD68 immunoreactivity in the APP−/− intestine could be a consequence of not only loss of macrophage APP function but also loss of APP metabolite-stimulated signaling of macrophage by neurons or enterocytes. Indeed, a lack of APP-dependent activation of immune cells or a lack of Aβ-dependent stimulation of immune cells in the ileum could possibly explain not only the decrease in CD68 protein levels but also CD40 and CD11c as well as the dramatic decrease in multiple cytokines in APP−/− intestines. This scenario suggests that perhaps APP or its metabolites offer some sort of basal or inducible stimulatory signal to help regulate gut function. However, as the premise of the current study was to first document a novel change in intestinal immune phenotype in APP−/− animals, further mechanistic dissection of the precise reason for changes in immune cell numbers or activation states remains a future goal. In fact, assessing changes in intestinal immune cell behavior as a consequence of APP expression and metabolism might be best addressed in the context of including animals that over-express human mutant APP characteristic of AD. Provided that expression of the transgene occurs in the relevant cell types in the intestine, models such as these might offer insight into consequences of over-expression of APP or elevated Aβ levels on gut function and immune phenotype. More importantly, this might offer insight into consequences of mutant APP expression in the gut as it relates to AD. It has already been demonstrated that Aβ immunoreactive plaques accumulate in the intestines of AD patients (Joachim et al., 1989) although evidence of enteric neuron loss is not established (Shankle et al., 1993; Van Ginneken et al., 2010). A role for APP or its proteolytic products in intestine function would provide not only insight into the immunoregulatory role of APP and its metabolites but also begin characterization of disease-relevant changes in a tissue other than the brain.
Although the most straightforward clinical application of our data set is to extrapolate to conditions of AD in which mutant APP has a known role in the biology of disease, it is interesting to speculate that APP may ultimately have a role in modulating immune cell behavior in conditions other than AD. For instance, the changes in CD68 immunoreactivity observed in APP−/− mice combined with the cytokine profile changes suggests that systemic immune changes exist in these animals. We have only examined peritoneal macrophage and intestinal macrophage from the ileum in this study since our prior work has demonstrated a role for APP in monocytic lineage cell behavior (Sondag and Combs, 2004, 2006, 2010). However, unlike the brain, the gut is home to a diverse population of dynamically changing and regulated immune cells that will likely be affected by any alteration of macrophage behavior. Indeed the decreased protein levels of CD40 and CD11c in the ileum suggest that levels or activation state of other immune cells such as B cells and dendritic cells are also altered in the in APP−/− mice.
Furthermore, for simplicity and proof-of-concept we have limited this assessment to the ileum of the small intestine base upon the fact that it contains immune cell infiltrates and a robust enteric nervous system. However, future efforts to generate a comprehensive profile of all immune cell changes not only in the ileum but throughout the digestive tract particularly the large intestine would offer cell and region specific information that could be correlated with plasma and other organ changes in cytokine and immune cell differences in APP−/− versus wild type mice.
The change in cox-2 protein levels in APP−/− mice certainly supports the idea that APP expression regulates gut function. A host of studies have documented a role for cox-2 and its prostaglandin products in negatively regulating intestinal motility (Cong et al., 2007; Takechi et al., 2009; Fairbrother et al., 2011; Nylander, 2011; Shi et al., 2011). In addition, cox-2 activity positively regulates intestine macrophage infiltration in a rodent model of sepsis (Osterberg et al., 2006) perhaps offering an additional mechanism explaining why APP−/− mouse CD68 positive macrophage immunoreactivity was attenuated compared to wild type mice. Therefore the decrease in cox-2 protein levels observed in the APP−/− mice is entirely consistent with the increased motility and decreased water absorption observed in the collected stool samples. A change in absorptive capacity was further demonstrated by the significantly decreased levels of absorbed LPS in the APP−/− mice. The uptake of bacterial flora-derived LPS into plasma of rodents and humans as a consequence of lipid absorption is a well-characterized phenomenon (Amar et al., 2008; Ghoshal et al., 2009). Although this is not necessarily the only source for plasma LPS uptake, it correlates well with prior work and suggests that APP−/− intestines may also have impaired lipid absorption. In addition, although we did not detect significant differences in occludin protein levels between genotypes it is quite feasible that the integrity of the gut epithelial barrier is impaired in the APP−/− mice. Further work to provide additional measures of gut motility, barrier integrity, and absorption will better resolve the differences in gut function dependent upon expression of APP.
Another interesting difference between APP−/− and wild type ileums was the decreased staining for MAP2 and the decrease in βIII-tubulin protein levels in the APP−/− mice. Based upon the role of the enteric nervous system in regulating absorption, secretion, and motility in the intestine it is feasible that the decrease in βIII-tubulin levels reflects a dysfunction of the enteric nervous control of the intestine. Future work examining more refined assessment of intestinal function will offer insight into this question. This may have nothing to do with enterocyte or immune cell function but instead be the consequence of loss of neuronal APP expression. Certainly, prior work examining the brains of APP−/− mice has demonstrated a similar decrease in MAP2 immunoreactivity (Seabrook et al., 1999). Moreover, a number of studies have implicated a role for APP expression in regulating both presynaptic and postsynaptic integrity in the central and peripheral nervous system (Seabrook et al., 1999; Yang et al., 2005; Priller et al., 2006; Hoe et al., 2009; Wang et al., 2009; Lee et al., 2010; Weyer et al., 2011).
It is important to point out that we compared intestinal and peritoneal macrophage in isolation when examining cytokine secretory phenotypes. Although this analysis clearly demonstrated differences not only between peritoneal and intestinal macrophage, it also verified that intestinal macrophage from APP−/− mice are unique from their wild-type counterparts. Perhaps the more physiologically relevant comparison was derived from the cytokine array comparison of wild type and APP−/− ileums. This offered a broad screen of multiple cytokine changes that are a reflection of the more physiologically relevant multi-cell environment in vivo. Taking into account that the decreased levels of multiple cytokines in APP−/− ileums are still only a snapshot into the dynamic immune changes among multiple cell types that regulate the gut, the array nevertheless does offer an interesting speculative opportunity for hypothesizing how APP may have a role in diverse intestinal conditions. For instance, it has been previously shown that CD30L plays a critical role in controlling inflammatory bowel syndrome in mice by increasing production of T-cells (Sun et al., 2008). Eotaxin mediates colonic eosinophilic inflammation (Waddell et al., 2011) and fas ligand has been shown to sequester double negative T cells in the gut epithelium (Hamad, 2010). Fractalkine can regulate increased IL-6 and TNFα release in the intestine (Niess and Adler, 2010) and IL-12 production is increased by T-cells in irritable bowel disease (Uza et al., 2011). Therefore, since APP−/− mice have decreased levels of these multiple cytokines they may have decreased risk for inflammatory gut disease such as irritable bowel disease which could be investigated further. The decrease in GCSF could explain why APP−/− mice also have decreased dendritic cell marker protein, CD11c, as GCSF is the key factor in differentiating monocytes into dendritic cells (Metcalf, 1985). IL-10, an anti-inflammatory cytokine, may be decreased due to the decrease in T-cell and macrophage expression as IL-10 has been shown to be a potent suppressor of macrophage and T-cells functions in vitro (Kuhn et al., 1993). MIP-1γ may also be decreased due to the decrease in macrophage and dendritic cell numbers or activation as it is constitutively expressed in these cells (Mohamadzadeh et al., 1996; Haddad and Belosevic, 2009). IL-9 has been shown to promote IL-13 up-regulation of innate immune receptors (Steenwinckel et al., 2009). Therefore the decrease in both IL-9 and IL-13 observed is probably protective in these APP−/− mice as IL-13 has been shown to exert detrimental effects on epithelial barrier function by increasing epithelial cell apoptosis, derangement of tight junction integrity and acts as a key effector cytokine in ulcerative colitis (Heller et al., 2005). MIG has also been shown to be concomitantly expressed in ulcerative colitis (Egesten et al., 2007). TNFα has been implicated in the pathogenesis of Crohn’s disease, and by selectively blocking TNFR1 the severity of TNBS-induced colitis in rats is ameliorated (Yin et al., 2011). Again, the collective decrease in the multiple cytokines suggests that absence of APP attenuates gut inflammatory response perhaps influencing the susceptibility of these mice to gut inflammatory disease.
We ultimately expect that a number of conditions involving immune cell behavior in the gut, such as irritable bowel syndrome and Crohn’s disease may well be altered in the absence of APP expression. Moreover, a role for APP in regulating immune responses may extend well beyond the gut to include conditions of immune dysfunction as varied as asthma and respiratory allergies since immune cells within the gastrointestinal system are not confined to this organ system but are free to migrate throughout the body. Future work to identify the extent of various immune cell phenotype changes dependent upon specific functions of APP or its metabolites will validate the role of this protein not only during normal physiology but a variety of diseases.
Acknowledgments
We are grateful to Mr. Andrew Rebel for technical assistance, Dr. John Watt for helpful suggestions during tissue sectioning and Hongyan Wang for their technical expertise in histological staining. We thank Dr. Mark Musch for helpful suggestions regarding intestinal protein analyses. This work was supported by NIH/NCRR 1P20 RR17699 and NIH/NIA 1R01AG026330.
Sources of support: This work was supported by NIH/NCRR 1P20 RR17699 and NIH/NIA 1R01AG026330.
Footnotes
Conflict of Interest Disclaimer: The authors, Kendra L. Puig, Adam J. Swigost, Xudong Zhou, Mary Ann Sens and Colin K. Combs, claim that this data set is original and is not under consideration elsewhere. All authors have approved the final version of the manuscript and have no financial or other relationship that might lead to a conflict of interest.
Contributor Information
Kendra L. Puig, Email: kpuig@medicine.nodak.edu, Department of Pharmacology, Physiology and Therapeutics, University of North Dakota School of Medicine and Health Sciences, 504 Hamline Street, Neuroscience Building, Grand Forks, ND 58203
Adam J. Swigost, Email: Adam.J.Swigost@und.edu, Department of Pharmacology, Physiology and Therapeutics, University of North Dakota School of Medicine and Health Sciences, 504 Hamline Street, Neuroscience Building, Grand Forks, ND 58203
Xudong Zhou, Email: xudong.zhou@med.und.edu, Department of Pathology, University of North Dakota School of Medicine and Health Sciences, 501 North Colubmia Road, Grand Forks, ND 58203.
MaryAnn Sens, Email: mary.sens@med.und.edu, Department of Pathology, University of North Dakota School of Medicine and Health Sciences, 501 North Colubmia Road, Grand Forks, ND 58203.
Colin K. Combs, Department of Pharmacology, Physiology and Therapeutics, University of North Dakota School of Medicine and Health Sciences, 504 Hamline Street, Neuroscience Building, Grand Forks, ND 58203
References
- Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, Chamontin B, Ferrieres J. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr. 2008;87:1219–1223. doi: 10.1093/ajcn/87.5.1219. [DOI] [PubMed] [Google Scholar]
- Arai H, Lee VM, Messinger ML, Greenberg BD, Lowery DE, Trojanowski JQ. Expression patterns of beta-amyloid precursor protein (beta-APP) in neural and nonneural human tissues from Alzheimer’s disease and control subjects. Ann Neurol. 1991;30:686–693. doi: 10.1002/ana.410300509. [DOI] [PubMed] [Google Scholar]
- Austin SA, Combs CK. Amyloid precursor protein mediates monocyte adhesion in AD tissue and apoE(−)/(−) mice. Neurobiol Aging. 2010;31:1854–1866. doi: 10.1016/j.neurobiolaging.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin SA, Sens MA, Combs CK. Amyloid precursor protein mediates a tyrosine kinase-dependent activation response in endothelial cells. J Neurosci. 2009;29:14451–14462. doi: 10.1523/JNEUROSCI.3107-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banati RB, Gehrmann J, Kreutzberg GW. Glial beta-amyloid precursor protein: expression in the dentate gyrus after entorhinal cortex lesion. Neuroreport. 1994;5:1359–1361. doi: 10.1097/00001756-199406270-00016. [DOI] [PubMed] [Google Scholar]
- Banati RB, Gehrmann J, Wiessner C, Hossmann KA, Kreutzberg GW. Glial expression of the beta-amyloid precursor protein (APP) in global ischemia. J Cereb Blood Flow Metab. 1995a;15:647–654. doi: 10.1038/jcbfm.1995.80. [DOI] [PubMed] [Google Scholar]
- Banati RB, Gehrmann J, Lannes-Vieira J, Wekerle H, Kreutzberg GW. Inflammatory reaction in experimental autoimmune encephalomyelitis (EAE) is accompanied by a microglial expression of the beta A4-amyloid precursor protein (APP) Glia. 1995b;14:209–215. doi: 10.1002/glia.440140306. [DOI] [PubMed] [Google Scholar]
- Bauer J, Konig G, Strauss S, Jonas U, Ganter U, Weidemann A, Monning U, Masters CL, Volk B, Berger M, et al. In-vitro matured human macrophages express Alzheimer’s beta A4-amyloid precursor protein indicating synthesis in microglial cells. FEBS Lett. 1991;282:335–340. doi: 10.1016/0014-5793(91)80508-z. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bullido MJ, Munoz-Fernandez MA, Recuero M, Fresno M, Valdivieso F. Alzheimer’s amyloid precursor protein is expressed on the surface of hematopoietic cells upon activation. Biochim Biophys Acta. 1996;1313:54–62. doi: 10.1016/0167-4889(96)00015-8. [DOI] [PubMed] [Google Scholar]
- Cirillo C, Sarnelli G, Esposito G, Turco F, Steardo L, Cuomo R. S100B protein in the gut: the evidence for enteroglial-sustained intestinal inflammation. World J Gastroenterol. 2011;17:1261–1266. doi: 10.3748/wjg.v17.i10.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Cannady SB, Lehman TM, Landreth GE. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J Neurosci. 1999;19:928–939. doi: 10.1523/JNEUROSCI.19-03-00928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong P, Pricolo V, Biancani P, Behar J. Abnormalities of prostaglandins and cyclooxygenase enzymes in female patients with slow-transit constipation. Gastroenterology. 2007;133:445–453. doi: 10.1053/j.gastro.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Egesten A, Eliasson M, Olin AI, Erjefalt JS, Bjartell A, Sangfelt P, Carlson M. The proinflammatory CXC-chemokines GRO-alpha/CXCL1 and MIG/CXCL9 are concomitantly expressed in ulcerative colitis and decrease during treatment with topical corticosteroids. Int J Colorectal Dis. 2007;22:1421–1427. doi: 10.1007/s00384-007-0370-3. [DOI] [PubMed] [Google Scholar]
- Fairbrother SE, Smith JE, Borman RA, Cox HM. Characterization of the EP receptor types that mediate longitudinal smooth muscle contraction of human colon, mouse colon and mouse ileum. Neurogastroenterol Motil. 2011 doi: 10.1111/j.1365-2982.2011.01727.x. [DOI] [PubMed] [Google Scholar]
- Floden AM, Combs CK. Beta-amyloid stimulates murine postnatal and adult microglia cultures in a unique manner. J Neurosci. 2006;26:4644–4648. doi: 10.1523/JNEUROSCI.4822-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway S, Jian L, Johnsen R, Chew S, Mamo JC. beta-amyloid or its precursor protein is found in epithelial cells of the small intestine and is stimulated by high-fat feeding. J Nutr Biochem. 2007;18:279–284. doi: 10.1016/j.jnutbio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Galloway S, Takechi R, Pallebage-Gamarallage MM, Dhaliwal SS, Mamo JC. Amyloid-beta colocalizes with apolipoprotein B in absorptive cells of the small intestine. Lipids Health Dis. 2009;8:46. doi: 10.1186/1476-511X-8-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway S, Pallebage-Gamarallage MM, Takechi R, Jian L, Johnsen RD, Dhaliwal SS, Mamo JC. Synergistic effects of high fat feeding and apolipoprotein E deletion on enterocytic amyloid-beta abundance. Lipids Health Dis. 2008;7:15. doi: 10.1186/1476-511X-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann J, Banati RB, Cuzner ML, Kreutzberg GW, Newcombe J. Amyloid precursor protein (APP) expression in multiple sclerosis lesions. Glia. 1995a;15:141–151. doi: 10.1002/glia.440150206. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Banati RB, Wiessner C, Hossmann KA, Kreutzberg GW. Reactive microglia in cerebral ischaemia: an early mediator of tissue damage? Neuropathol Appl Neurobiol. 1995b;21:277–289. doi: 10.1111/j.1365-2990.1995.tb01062.x. [DOI] [PubMed] [Google Scholar]
- Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. 2009;50:90–97. doi: 10.1194/jlr.M800156-JLR200. [DOI] [PubMed] [Google Scholar]
- Grimm MC, Pullman WE, Bennett GM, Sullivan PJ, Pavli P, Doe WF. Direct evidence of monocyte recruitment to inflammatory bowel disease mucosa. J Gastroenterol Hepatol. 1995;10:387–395. doi: 10.1111/j.1440-1746.1995.tb01589.x. [DOI] [PubMed] [Google Scholar]
- Haass C, Hung AY, Selkoe DJ. Processing of beta-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J Neurosci. 1991;11:3783–3793. doi: 10.1523/JNEUROSCI.11-12-03783.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad G, Belosevic M. Transferrin-derived synthetic peptide induces highly conserved pro-inflammatory responses of macrophages. Mol Immunol. 2009;46:576–586. doi: 10.1016/j.molimm.2008.07.030. [DOI] [PubMed] [Google Scholar]
- Hamad AR. Analysis of gene profile, steady state proliferation and apoptosis of double-negative T cells in the periphery and gut epithelium provides new insights into the biological functions of the Fas pathway. Immunol Res. 2010;47:134–142. doi: 10.1007/s12026-009-8144-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AM, Hutson JM, Southwell BR. Cholinergic neurotransmission and muscarinic receptors in the enteric nervous system. Prog Histochem Cytochem. 2010;44:173–202. doi: 10.1016/j.proghi.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Burgel N, Fromm M, Zeitz M, Fuss I, Strober W, Schulzke JD. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Fu Z, Makarova A, Lee JY, Lu C, Feng L, Pajoohesh-Ganji A, Matsuoka Y, Hyman BT, Ehlers MD, Vicini S, Pak DT, Rebeck GW. The effects of amyloid precursor protein on postsynaptic composition and activity. J Biol Chem. 2009;284:8495–8506. doi: 10.1074/jbc.M900141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikezu T, Luo X, Weber GA, Zhao J, McCabe L, Buescher JL, Ghorpade A, Zheng J, Xiong H. Amyloid precursor protein-processing products affect mononuclear phagocyte activation: pathways for sAPP- and Abeta-mediated neurotoxicity. J Neurochem. 2003;85:925–934. doi: 10.1046/j.1471-4159.2003.01739.x. [DOI] [PubMed] [Google Scholar]
- Itoh T, Satou T, Nishida S, Tsubaki M, Hashimoto S, Ito H. Expression of amyloid precursor protein after rat traumatic brain injury. Neurol Res. 2009;31:103–109. doi: 10.1179/016164108X323771. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. Glial fibrillary acidic polypeptides in peripheral glia. Molecular weight, heterogeneity and distribution. J Neuroimmunol. 1985;8:377–393. doi: 10.1016/s0165-5728(85)80074-6. [DOI] [PubMed] [Google Scholar]
- Joachim C, Games D, Morris J, Ward P, Frenkel D, Selkoe D. Antibodies to non-beta regions of the beta-amyloid precursor protein detect a subset of senile plaques. Am J Pathol. 1991;138:373–384. [PMC free article] [PubMed] [Google Scholar]
- Joachim CL, Mori H, Selkoe DJ. Amyloid beta-protein deposition in tissues other than brain in Alzheimer’s disease. Nature. 1989;341:226–230. doi: 10.1038/341226a0. [DOI] [PubMed] [Google Scholar]
- Kamino K, Orr HT, Payami H, Wijsman EM, Alonso ME, Pulst SM, Anderson L, O’Dahl S, Nemens E, White JA, et al. Linkage and mutational analysis of familial Alzheimer disease kindreds for the APP gene region. Am J Hum Genet. 1992;51:998–1014. [PMC free article] [PubMed] [Google Scholar]
- Klegeris A, Walker DG, McGeer PL. Activation of macrophages by Alzheimer beta amyloid peptide. Biochem Biophys Res Commun. 1994;199:984–991. doi: 10.1006/bbrc.1994.1326. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- LeBlanc AC, Chen HY, Autilio-Gambetti L, Gambetti P. Differential APP gene expression in rat cerebral cortex, meninges, and primary astroglial, microglial and neuronal cultures. FEBS Lett. 1991;292:171–178. doi: 10.1016/0014-5793(91)80861-v. [DOI] [PubMed] [Google Scholar]
- Lee KJ, Moussa CE, Lee Y, Sung Y, Howell BW, Turner RS, Pak DT, Hoe HS. Beta amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. Neuroscience. 2010;169:344–356. doi: 10.1016/j.neuroscience.2010.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Zimin PI, Wulff H, Jin LW. Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem. 2010;286:3693–3706. doi: 10.1074/jbc.M110.135244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf D. The granulocyte-macrophage colony-stimulating factors. Science. 1985;229:16–22. doi: 10.1126/science.2990035. [DOI] [PubMed] [Google Scholar]
- Mohamadzadeh M, Poltorak AN, Bergstressor PR, Beutler B, Takashima A. Dendritic cells produce macrophage inflammatory protein-1 gamma, a new member of the CC chemokine family. J Immunol. 1996;156:3102–3106. [PubMed] [Google Scholar]
- Monning U, Sandbrink R, Banati RB, Masters CL, Beyreuther K. Transforming growth factor beta mediates increase of mature transmembrane amyloid precursor protein in microglial cells. FEBS Lett. 1994;342:267–272. doi: 10.1016/0014-5793(94)80514-8. [DOI] [PubMed] [Google Scholar]
- Monning U, Sandbrink R, Weidemann A, Banati RB, Masters CL, Beyreuther K. Extracellular matrix influences the biogenesis of amyloid precursor protein in microglial cells. J Biol Chem. 1995;270:7104–7110. doi: 10.1074/jbc.270.13.7104. [DOI] [PubMed] [Google Scholar]
- Niess JH, Adler G. Enteric flora expands gut lamina propria CX3CR1+ dendritic cells supporting inflammatory immune responses under normal and inflammatory conditions. J Immunol. 2010;184:2026–2037. doi: 10.4049/jimmunol.0901936. [DOI] [PubMed] [Google Scholar]
- Nylander O. The impact of cyclooxygenase inhibition on duodenal motility and mucosal alkaline secretion in anaesthetized rats. Acta Physiol (Oxf) 2011;201:179–192. doi: 10.1111/j.1748-1716.2010.02196.x. [DOI] [PubMed] [Google Scholar]
- Osterberg J, Ljungdahl M, Haglund U. Influence of cyclooxygenase inhibitors on gut immune cell distribution and apoptosis rate in experimental sepsis. Shock. 2006;25:147–154. doi: 10.1097/01.shk.0000189843.78729.e2. [DOI] [PubMed] [Google Scholar]
- Pallebage-Gamarallage MM, Galloway S, Johnsen R, Jian L, Dhaliwal S, Mamo JC. The effect of exogenous cholesterol and lipid-modulating agents on enterocytic amyloid-beta abundance. Br J Nutr. 2009;101:340–347. doi: 10.1017/S0007114508012269. [DOI] [PubMed] [Google Scholar]
- Perry G, Siedlak S, Mulvihill P, Kancherla M, Mijares M, Kawai M, Gambetti P, Sharma S, Maggiora L, Cornette J, et al. Immunolocalization of the amyloid precursor protein within the senile plaque. Prog Clin Biol Res. 1989;317:1021–1025. [PubMed] [Google Scholar]
- Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26:7212–7221. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogler G, Hausmann M, Vogl D, Aschenbrenner E, Andus T, Falk W, Andreesen R, Scholmerich J, Gross V. Isolation and phenotypic characterization of colonic macrophages. Clin Exp Immunol. 1998;112:205–215. doi: 10.1046/j.1365-2249.1998.00557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaolalla R, Fukata M, Abreu MT. Innate immunity in the small intestine. Curr Opin Gastroenterol. 2010;27:125–131. doi: 10.1097/MOG.0b013e3283438dea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidge TC, Sofroniew MV, Neunlist M. Starring roles for astroglia in barrier pathologies of gut and brain. Lab Invest. 2007;87:731–736. doi: 10.1038/labinvest.3700600. [DOI] [PubMed] [Google Scholar]
- Schmechel DE, Goldgaber D, Burkhart DS, Gilbert JR, Gajdusek DC, Roses AD. Cellular localization of messenger RNA encoding amyloid-beta-protein in normal tissue and in Alzheimer disease. Alzheimer Dis Assoc Disord. 1988;2:96–111. doi: 10.1097/00002093-198802020-00002. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Smith DW, Bowery BJ, Easter A, Reynolds T, Fitzjohn SM, Morton RA, Zheng H, Dawson GR, Sirinathsinghji DJ, Davies CH, Collingridge GL, Hill RG. Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology. 1999;38:349–359. doi: 10.1016/s0028-3908(98)00204-4. [DOI] [PubMed] [Google Scholar]
- Shankle WR, Landing BH, Ang SM, Chui H, Villarreal-Engelhardt G, Zarow C. Studies of the enteric nervous system in Alzheimer disease and other dementias of the elderly: enteric neurons in Alzheimer disease. Mod Pathol. 1993;6:10–14. [PubMed] [Google Scholar]
- Shi XZ, Lin YM, Powell DW, Sarna SK. Pathophysiology of motility dysfunction in bowel obstruction: role of stretch-induced COX-2. Am J Physiol Gastrointest Liver Physiol. 2011;300:G99–G108. doi: 10.1152/ajpgi.00379.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits HA, van Beelen AJ, de Vos NM, Rijsmus A, van der Bruggen T, Verhoef J, van Muiswinkel FL, Nottet HS. Activation of human macrophages by amyloid-beta is attenuated by astrocytes. J Immunol. 2001;166:6869–6876. doi: 10.4049/jimmunol.166.11.6869. [DOI] [PubMed] [Google Scholar]
- Sondag CM, Combs CK. Amyloid precursor protein mediates proinflammatory activation of monocytic lineage cells. J Biol Chem. 2004;279:14456–14463. doi: 10.1074/jbc.M313747200. [DOI] [PubMed] [Google Scholar]
- Sondag CM, Combs CK. Amyloid precursor protein cross-linking stimulates beta amyloid production and pro-inflammatory cytokine release in monocytic lineage cells. J Neurochem. 2006;97:449–461. doi: 10.1111/j.1471-4159.2006.03759.x. [DOI] [PubMed] [Google Scholar]
- Sondag CM, Combs CK. Adhesion of monocytes to type I collagen stimulates an APP-dependent proinflammatory signaling response and release of Abeta1–40. J Neuroinflammation. 2010;7:22. doi: 10.1186/1742-2094-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondag CM, Dhawan G, Combs CK. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation. 2009;6:1. doi: 10.1186/1742-2094-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5:e9505. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer P, Herrmann M, Klafki HW, Smirnov A, Lewczuk P, Kornhuber J, Wiltfang J, Maler JM. Phagocytosis and LPS alter the maturation state of beta-amyloid precursor protein and induce different Abeta peptide release signatures in human mononuclear phagocytes. J Neuroinflammation. 2010;7:59. doi: 10.1186/1742-2094-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenwinckel V, Louahed J, Lemaire MM, Sommereyns C, Warnier G, McKenzie A, Brombacher F, Van Snick J, Renauld JC. IL-9 promotes IL-13-dependent paneth cell hyperplasia and up-regulation of innate immunity mediators in intestinal mucosa. J Immunol. 2009;182:4737–4743. doi: 10.4049/jimmunol.0801941. [DOI] [PubMed] [Google Scholar]
- Sun X, Somada S, Shibata K, Muta H, Yamada H, Yoshihara H, Honda K, Nakamura K, Takayanagi R, Tani K, Podack ER, Yoshikai Y. A critical role of CD30 ligand/CD30 in controlling inflammatory bowel diseases in mice. Gastroenterology. 2008;134:447–458. doi: 10.1053/j.gastro.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Takechi R, Galloway S, Pallebage-Gamarallage M, Wellington C, Johnsen R, Mamo JC. Three-dimensional colocalization analysis of plasma-derived apolipoprotein B with amyloid plaques in APP/PS1 transgenic mice. Histochem Cell Biol. 2009;131:661–666. doi: 10.1007/s00418-009-0567-3. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Vaula G, Romano DM, Mortilla M, Huang TL, Tupler RG, Wasco W, Hyman BT, Haines JL, Jenkins BJ, et al. Assessment of amyloid beta-protein precursor gene mutations in a large set of familial and sporadic Alzheimer disease cases. Am J Hum Genet. 1992;51:273–282. [PMC free article] [PubMed] [Google Scholar]
- Tomita R, Igarashi S, Fujisaki S, Koshinaga T, Kusafuka T. Are there any functional differences of the enteric nervous system between jejunum and ileum in normal humans? Hepatogastroenterology. 2010;57:777–780. [PubMed] [Google Scholar]
- Uryu S, Tokuhiro S, Oda T. beta-Amyloid-specific upregulation of stearoyl coenzyme A desaturase-1 in macrophages. Biochem Biophys Res Commun. 2003;303:302–305. doi: 10.1016/s0006-291x(03)00334-6. [DOI] [PubMed] [Google Scholar]
- Uza N, Nakase H, Yamamoto S, Yoshino T, Takeda Y, Ueno S, Inoue S, Mikami S, Matsuura M, Shimaoka T, Kume N, Minami M, Yonehara S, Ikeuchi H, Chiba T. SR-PSOX/CXCL16 plays a critical role in the progression of colonic inflammation. Gut. 2011;60:1494–1505. doi: 10.1136/gut.2010.221879. [DOI] [PubMed] [Google Scholar]
- Van Ginneken C, Schafer KH, Van Dam D, Huygelen V, De Deyn PP. Morphological changes in the enteric nervous system of aging and APP23 transgenic mice. Brain Res. 2010;1378:43–53. doi: 10.1016/j.brainres.2011.01.030. [DOI] [PubMed] [Google Scholar]
- Van Ginneken C, Schafer KH, Van Dam D, Huygelen V, De Deyn PP. Morphological changes in the enteric nervous system of aging and APP23 transgenic mice. Brain Res. 2011;1378:43–53. doi: 10.1016/j.brainres.2011.01.030. [DOI] [PubMed] [Google Scholar]
- Vehmas A, Lieu J, Pardo CA, McArthur JC, Gartner S. Amyloid precursor protein expression in circulating monocytes and brain macrophages from patients with HIV-associated cognitive impairment. J Neuroimmunol. 2004;157:99–110. doi: 10.1016/j.jneuroim.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Waddell A, Ahrens R, Steinbrecher K, Donovan B, Rothenberg ME, Munitz A, Hogan SP. Colonic eosinophilic inflammation in experimental colitis is mediated by Ly6C(high) CCR2(+) inflammatory monocyte/macrophage-derived CCL11. J Immunol. 2011;186:5993–6003. doi: 10.4049/jimmunol.1003844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wang B, Yang L, Guo Q, Aithmitti N, Songyang Z, Zheng H. Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J Neurosci. 2009;29:10788–10801. doi: 10.1523/JNEUROSCI.2132-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer SW, Klevanski M, Delekate A, Voikar V, Aydin D, Hick M, Filippov M, Drost N, Schaller KL, Saar M, Vogt MA, Gass P, Samanta A, Jaschke A, Korte M, Wolfer DP, Caldwell JH, Muller UC. APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. Embo J. 2011;30:2266–2280. doi: 10.1038/emboj.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, McCabe L, Costello J, Anderson E, Weber G, Ikezu T. Activation of NR1a/NR2B receptors by soluble factors from APP-stimulated monocyte-derived macrophages: implications for the pathogenesis of Alzheimer’s disease. Neurobiol Aging. 2004;25:905–911. doi: 10.1016/j.neurobiolaging.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Yamada T, Sasaki H, Dohura K, Goto I, Sakaki Y. Structure and expression of the alternatively-spliced forms of mRNA for the mouse homolog of Alzheimer’s disease amyloid beta protein precursor. Biochem Biophys Res Commun. 1989;158:906–912. doi: 10.1016/0006-291x(89)92808-8. [DOI] [PubMed] [Google Scholar]
- Yan SD, Stern D, Kane MD, Kuo YM, Lampert HC, Roher AE. RAGE-Abeta interactions in the pathophysiology of Alzheimer’s disease. Restor Neurol Neurosci. 1998;12:167–173. [PubMed] [Google Scholar]
- Yang G, Gong YD, Gong K, Jiang WL, Kwon E, Wang P, Zheng H, Zhang XF, Gan WB, Zhao NM. Reduced synaptic vesicle density and active zone size in mice lacking amyloid precursor protein (APP) and APP-like protein 2. Neurosci Lett. 2005;384:66–71. doi: 10.1016/j.neulet.2005.04.040. [DOI] [PubMed] [Google Scholar]
- Yates SL, Burgess LH, Kocsis-Angle J, Antal JM, Dority MD, Embury PB, Piotrkowski AM, Brunden KR. Amyloid beta and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microglia. J Neurochem. 2000;74:1017–1025. doi: 10.1046/j.1471-4159.2000.0741017.x. [DOI] [PubMed] [Google Scholar]
- Yazawa H, Yu ZX, Takeda, Le Y, Gong W, Ferrans VJ, Oppenheim JJ, Li CC, Wang JM. Beta amyloid peptide (Abeta42) is internalized via the G-protein-coupled receptor FPRL1 and forms fibrillar aggregates in macrophages. Faseb J. 2001;15:2454–2462. doi: 10.1096/fj.01-0251com. [DOI] [PubMed] [Google Scholar]
- Yin B, Hu X, Wang J, Liang H, Li X, Niu N, Li B, Jiang X, Li Z. Blocking TNF-alpha by combination of TNF-alpha- and TNFR-binding cyclic peptide ameliorates the severity of TNBS-induced colitis in rats. Eur J Pharmacol. 2011;656:119–124. doi: 10.1016/j.ejphar.2011.01.046. [DOI] [PubMed] [Google Scholar]