

Abstract
This study was undertaken to reveal the mechanisms by which RLIP76 regulates endothelial cell angiogenic responses. RLIP76 is an effector of the angiogenic modulator, R-Ras. RLIP76 is overexpressed in many tumors, required for tumor angiogenesis, and blockade of RLIP76 results in tumor regression in multiple models. We report here that RLIP76 was required for expression and secretion of vascular endothelial growth factor (VEGF) in carcinoma and melanoma cells. Conditioned medium derived from RLIP76-depleted tumor cells, but not control knockdown cells, could not stimulate proliferation, migration, or Matrigel cord formation in endothelial cell cultures, which indicates that RLIP76 regulates angiogenic components of the tumor cell secretome. Recombinant VEGF added to conditioned medium from RLIP76-knockdown tumor cells restored these endothelial cell functions. Transcriptional activity of hypoxia-inducible factor 1 (HIF-1), which drives VEGF expression, was blocked in RLIP76-depleted tumor cells. RLIP76 was required for PI3-kinase activation, known to regulate HIF-1, in these cells. However, HIF-1α expression and nuclear localization were unaffected by RLIP76 knockdown, which suggests that RLIP76 regulates HIF-1 at the functional level. Thus, RLIP76 regulates tumor cell transactivation of endothelial cells via control of VEGF expression and secretion, providing a new important link in the mechanism of tumor cell induction of angiogenesis.—Lee, S., Goldfinger, L. E. RLIP76 regulates HIF-1 activity, VEGF expression and secretion in tumor cells, and secretome transactivation of endothelial cells.
Keywords: angiogenesis, transcription factor, growth factors
Angiogenesis, the outgrowth of new blood vessels from existing ones, is required for progression of solid tumor growth and for metastatic dissemination. Nascent vessel growth is driven initially by vascular endothelial cells, which proliferate and migrate to form new vessels, providing nutrients essential for the growing tumor. Thus, a major goal in the therapeutic intervention in cancer is the identification and characterization of specific modulators of tumor angiogenesis (1). Stimulation and regulation of the endothelial cells and vessel development within tumors involves an intricate interplay between the endothelium and the tumor stromal cells. Endothelial cell proliferation, migration, and vessel formation are regulated by cytokines such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), fibronectin, and angiopoietins, which are synthesized and secreted by many cells including tumor cells (2, 3).
We have recently identified the oncoprotein Ral-interacting protein of 76 kDa (RLIP76) as an essential mediator of angiogenesis and tumor growth. RLIP76 is a multifunctional protein, originally identified as a Ral GTPase effector protein linking Ral to Rho pathways (4–6). RLIP76 also functions as an ATP-dependent glutathione-conjugate transporter for small molecules, including anticancer drugs and endogenous metabolites (7, 8), and mediates cell spreading and migration (9). RLIP76 is expressed in most human tissues including liver, heart, lung, muscle, and kidney as well as in most human tumor cell lines (10–12). Blockade of RLIP76 with targeting antibodies or antisense is associated with increased sensitivity to radiation and chemotherapy and leads to tumor regression in non-small-cell lung and colon carcinomas (13), prostate cancer (14), and B16 melanomas (15) in mice. RLIP76 is overexpressed in many tumor cells and types, including multiple carcinomas and melanomas (16, 17). Previously, we found that RLIP76 regulates tumor angiogenesis in primary solid tumors in vivo and isolated endothelial cells in vitro: Tumor growth and tumor angiogenesis were significantly diminished in RLIP76-knockout mice (18). Thus, RLIP76 expression in host vascular cells is required for efficient angiogenesis in primary tumors. In addition, we found that RLIP76 depletion in the tumor cells led to a further decrease in mass of the xenografted primary tumors (18), indicating that RLIP76 expression in tumor cells or other stromal cells also contributes importantly to tumor growth.
VEGF is a strong promoter of angiogenesis, inducing activation of endothelial cells following ligation of VEGF receptors on the endothelial cell surface (19, 20). VEGF is secreted by tumor cells, promoting recruitment of endothelial cells, tumor angiogenesis, and tumor growth (21–23). Hence, VEGF is also an important target in cancer therapies (3, 21, 23). VEGF transcription is controlled by hypoxia-inducible factor 1 (HIF-1; refs. 24, 25). HIF-1 also regulates tumor angiogenesis as well as endothelial cell apoptosis (26). Normally, HIF-1 becomes activated in hypoxic conditions within tumors, inducing tumor growth and angiogenesis, but HIF-1 can also be activated by nonhypoxic stimuli (27, 28). HIF-l in tumor cells stimulates the transcription of many genes involved in tumor growth as well as angiogenesis, including growth factors, cytokines, extracellular matrix (ECM) proteases, and signaling molecules (29). HIF-1 can be activated by several pathways, primarily the phosphatidylinositol-3 kinase (PI3K)/molecular target of rapamycin (mTOR) and Ras/Raf/MEK/ERK pathways, both of which converge on elF4E1 to regulate HIF-1 expression (27, 30, 31). On activation, HIF-1α translocates from the cytosol to the nucleus, where it binds cofactor CBP/p300 (27, 32, 33). The HIF-1/p300 complex then binds to HIF-1-responsive elements (HREs) in target genes to stimulate their expression (27, 32, 33). The VEGF gene promoter contains a HRE and is primarily transcribed by activated HIF-1 in tumor cells (27, 29, 34–36). Thus, understanding of the regulation of the VEGF and HIF-1 pathway is important to the overall understanding of angiogenesis, particularly in solid tumors. In this study, we show that RLIP76 is required for VEGF expression and secretion in two tumor cell types, for HIF-1 transcriptional activity in these cells, and for VEGF-dependent transactivation of endothelial cells by the tumor cell conditioned medium, pointing to a direct role of RLIP76 expression in tumor cell transactivation of angiogenic endothelium through control of VEGF.
MATERIALS AND METHODS
Antibodies and reagents
RLIP76, VEGF, and RhoGDI antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). HIF-1α antibody was from Abcam (Cambridge, MA, USA). p42/44 MAPK (Erk1/2), Akt, and phospho-Akt (Ser473) antibodies and recombinant VEGF165 were from Cell Signaling Technology (Danvers, MA, USA). Fluorophore-conjugated secondary antibodies were from LI-COR Biosciences (Lincoln, NE, USA). A RLIP76 murine-specific short hairpin RNA (shRNA)-targeting plasmid was generated in the pSUPER.retro puro vector (OligoEngine, Seattle, WA, USA) as described previously (18).
Cell culture and transfection
Bovine aortic endothelial cells (BAECs), B16F10 mouse melanoma (B16F10) cells, and Lewis lung carcinoma (LLC; LL/2) cells were obtained from American Type Culture Collection (Manassas, VA, USA). BAECs were maintained in endothelial cell growth medium [Dulbecco's modified Eagle's medium/F12 (DMEM/F) supplemented with 20% FBS, 5% microvascular growth supplement, and 0.1% gentamicin/amphotericin). B16F10 and LLC cells were cultured in DMEM supplemented with 10% FBS in a humidified, 5% CO2 atmosphere at 37°C. Cells were transfected with 5–10 μg plasmids/100 mm dish using Lipofectamine 2000 in Opti-MEM (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Cells were analyzed 24–48 h after transfection except where indicated otherwise.
Conditioned medium production and cell growth
B16F10 cells, LLC cells, and BAECs were cultured in minimal endothelial cell growth medium consisting of serum-free medium (SFM; 11111-044; Gibco, Carlsbad, CA, USA) supplemented with 20 ng/ml human recombinant basic fibroblast growth factor (rbFGF), 10 ng/ml recombinant epidermal growth factor (rEGF), and 10 μg/ml human plasma fibronectin, but without serum or VEGF, for 48 h before culture medium was collected and processed. In some cases, rVEGF was added at the start of the experiment. All media were filtered through a 0.2 μm borosilicate filter and stored at −80°C prior to being thawed and used.
Cell fractionation
Cultured cells were rinsed 3× with PBS and scraped into lysis buffer [10 mM Tris, pH 7.5; 100 mM NaCl; 2 mM MgOAc; and 5 mM KCl, plus 10 μM GTP and a protease inhibitor cocktail (GE Healthcare, Waukesha, WI, USA), added fresh] and lysed by dounce homogenization through a 27-gauge needle. Nuclear pellets were obtained from the whole-cell lysate by centrifugation for 10 min at 2000 rpm. The supernatants were further clarified by centrifugation for 10 min at 14,000 rpm to remove insoluble material, yielding cytosolic fractions in the remaining supernatants. For nuclear samples, pellets were washed with 500 μl lysis buffer, centrifuged for 10 min, then resuspended in 18 μl lysis buffer with 1 M sucrose, plus 300 μl lysis buffer with 1% Nonidet P-40, and incubated on ice for 1 h prior to processing for SDS-PAGE.
Protein precipitation with perchloric acid
Cells were cultured in 100 mm culture dishes. Culture medium (10 ml) was collected after 48 h and filtered, and proteins were precipitated from the filtrates with 1 ml of 6.6 M perchloric acid added to each sample for a final concentration of 0.66 M. Samples were incubated for 20 min at −80°C and centrifuged at 4000 rpm for 5 min. Pellets were rinsed with water and centrifuged again, and SDS buffer was added to the samples for SDS-PAGE.
Cell lysates and Western blotting
Cells were transiently transfected and, in some cases, serum-starved by culturing in DMEM/0.5% serum beginning 24 h after transfection. Cells were rinsed 3× in PBS, and cell lysates were harvested by scraping in lysis buffer (10 mM Tris-Cl, pH 7.5; 100 mM NaCl; 2 mM MgOAc; 10 μM GTP; and 0.5% Nonidet P-40; plus a cocktail of protease inhibitors) and maintained at 4°C. Insoluble material was removed by centrifugation. Fractions of the lysates were separated by SDS-PAGE, followed by Western blotting with appropriate antibodies.
Luciferase reporter assay
HIF-1 activity was measured using the Cignal reporter assay kit (Qiagen, Valencia, CA, USA) and processed according to the manufacturer's instructions. Briefly, the Cignal HIF reporter, Cignal negative control, or positive control plasmids were transfected into 4 × 105 cells/well in a 96-well plate, cotransfected with empty vector or with RLIP76 shRNA plasmids as indicated. After 40–48 h, the cell medium was removed, and the cells were rinsed 3× with PBS and resuspended in PBS without calcium and magnesium. Luciferase luminescence was monitored in a microplate reader (480 nm for Renilla luciferase, 560 nm for firefly luciferase).
BAEC proliferation
BAEC proliferation was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay using the Vybrant MTT cell proliferation assay kit (Life Technologies) according to the manufacturer's instructions (18). Briefly, 1 × 104 BAECs were seeded in each well in the presence of growth medium or tumor cell conditioned medium for up to 96 h. Cells at each time point were rinsed and incubated with 12 mM MTT for 3 h at 37°C. The amount of MTT formazan product was determined by measuring absorbance at 570 nm using a microplate reader.
BAEC transwell migration
BAEC migration was assessed in modified Boyden chambers. Cells (1×104/well) were suspended in 250 μl complete BAEC medium. The cells were placed in the top compartment of a standard Boyden chamber with 8 μm membrane pores and coated on the top of the filter with 1 μg/ml fibronectin, and 500 μl of conditioned medium was added to the bottom compartment. Chambers were returned to the incubator, and nonmigrating BAECs were removed from the top compartment with 0.25% trypsin at 3, 6, and 24 h after adding the cells. BAECs that had migrated to the bottom compartment were fixed and stained using 0.05% crystal violet. The stained BAECs in each well were photographed with the aid of a phase-contrast microscope, and staining intensities were determined with ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA).
In vitro cord formation
A total of 80 μl of growth factor-reduced Matrigel was added to each well of a 24-well tissue culture plate, and the plates were incubated at 37°C for 30 min to solidify the gel. BAECs (1×104/well) were seeded in each well in 100 μl of medium. After 3, 6, and 24 h, the center of each well was photographed under a microscope. Branch numbers were counted as branches in each field at 24 h.
Statistical analysis
One-way ANOVA followed by Fisher protected least significant difference analysis was used for all statistical data analysis, using StatView (SAS Institute, Cary, NC, USA). A 5% probability was considered significant. Results are representative of 3 independent experiments unless indicated otherwise.
RESULTS
RLIP76 regulates VEGF expression and secretion in tumor cells
To investigate a potential role for RLIP76 in tumor cell function, we considered whether RLIP76 may participate in regulation of the tumor cell secretome, which could affect vascular cells and angiogenic responses. As VEGF is synthesized and secreted by many cells and is a potent angiogenic factor, we assessed the protein expression levels of VEGF in two murine tumor cell lines, B16F10 melanoma cells and LLC cells, depleted of RLIP76 expression by transfection with an shRNA targeting RLIP76 (18). VEGF expression was monitored for 24, 48, and 72 h after transfection of RLIP76 shRNA. VEGF levels were substantially diminished by RLIP76 knockdown in both melanoma and carcinoma cells. The degree of VEGF suppression mirrored the levels of RLIP76 knockdown, as by 72 h after transient transfection with the shRNA plasmid, RLIP76 expression, which had been knocked down, returned to ∼65% of baseline levels, and VEGF expression was also partially restored (Fig. 1A). Thus, RLIP76 expression in tumor cells is required for robust VEGF expression.
Figure 1.

RLIP76 depletion in tumor cells inhibits VEGF expression and secretion. RLIP76 shRNA was transfected in B16F10 and LLC tumor cells, and RLIP76, VEGF, and Erk expression were assessed in cell lysates at 24, 48, and 72 h post-transfection. A) Relative expression levels of VEGF and RLIP76 in each sample compared to untransfected control are shown below the respective panels, and VEGF expression levels from 3 independent experiments are shown at bottom; bars represent means + sem. B) VEGF secretion in precipitates of conditioned medium from both tumor cell lines either untransfected (WT) or transfected with RLIP76 shRNA (KD). VEGF concentration in the conditioned medium was extrapolated from a standard curve derived from a blot of the same membrane (bottom panel) with recombinant VEGF165 (rVEGF).
We next assessed the levels of VEGF secreted by the tumor cells into the culture medium. Cells were grown in medium for 48 h after transfection, the conditioned medium was collected and filtered, and the proteins in the medium were concentrated by precipitation with perchloric acid. Western blotting of the precipitates showed that the B16F10 and LLC conditioned media contained VEGF at ∼10–15 ng/ml, using purified recombinant VEGF as a standard (Fig. 1B). However, VEGF was not detected in blots of conditioned medium precipitates from each of these cell lines transfected with RLIP76 shRNA (Fig. 1B). Thus, RLIP76 depletion suppressed VEGF expression and completely blocked VEGF secretion from these tumor cell lines.
RLIP76 in tumor cells regulates endothelial cell functions by the tumor cell secretome in trans
Based on the above results, we considered whether RLIP76 expression in tumor cells affects the transactivation of vascular endothelial cells by the tumor cell secretome. To investigate this possibility, we tested the effects of tumor cell conditioned medium on the stimulation of proliferation, migration, and cord formation of endothelial cells in vitro. First, we optimized culture medium conditions for growing tumor cells with endothelial cell medium and for growing endothelial cells with the conditioned medium from the tumor cells. For this purpose, we used serum-free endothelial cell growth medium supplemented with defined growth factors but not including VEGF. The tumor cell lines, as well as BAECs, grew at normal doubling rates in endothelial cell culture medium (SFM plus bFGF, EGF, and fibronectin but without VEGF; results not shown). Therefore, we used conditioned culture medium from the tumor cells grown under these conditions applied to BAECs, to test the effects of RLIP76 knockdown in the tumor cells on endothelial cell transactivation by the tumor cell secretome. RLIP76 shRNA was transfected in tumor cells, and the conditioned medium was collected after 48 h. Using this approach, we examined proliferation of BAECs cultured under 4 different medium conditions: normal endothelium growth medium (containing serum), conditioned medium from untransfected [wild-type (WT)] tumor cells, conditioned medium from RLIP76-knockdown tumor cells, and SFM. Proliferation of BAECs under these conditions was monitored for 4 d. Conditioned medium collected from WT tumor cells promoted endothelial cell growth in vitro similar to the normal growth medium. However, conditioned medium from either B16F10 or LLC cells transfected with RLIP76 shRNA cells could not stimulate BAEC proliferation, and the cells began dying after 2 d in this medium, similar to serum-free growth conditions (Fig. 2). Thus, RLIP76 expression in tumor cells is required for the tumor cell conditioned medium to stimulate endothelial cell proliferation in trans, in vitro.
Figure 2.
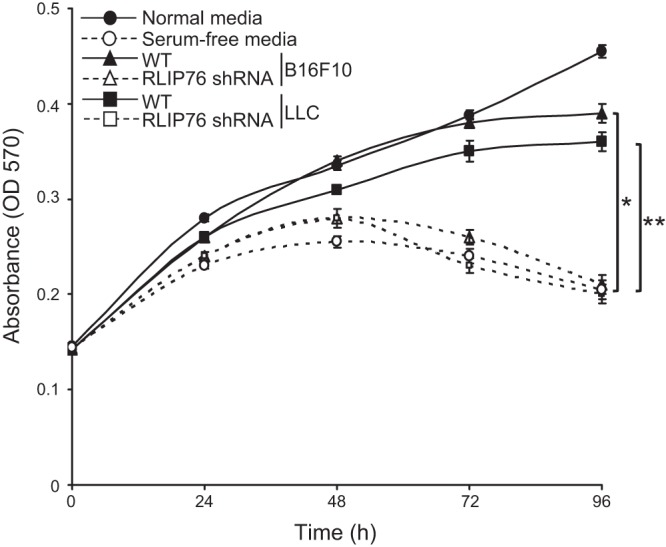
Endothelial cell proliferation in tumor cell conditioned medium requires RLIP76 expression in the tumor cells. Proliferation rates of endothelial cells cultured in endothelial growth medium (normal), conditioned medium from untransfected tumor cells (WT), conditioned medium from tumor cells transfected with RLIP76 shRNA, or SFM, measured by MTT assay are shown. Error bars = sem. *P < 0.0001; **P < 0.0002 (n=3, duplicate wells per sample).
Next, we examined the effects of the conditioned medium on endothelial cell migration using modified Boyden chambers with the conditioned medium in the lower compartment as a chemoattractant. Similar to the proliferation effects, tumor medium from RLIP76-depleted cells was a weak chemoattractant compared to control, as cell migration toward RLIP76-knockdown conditioned medium was inhibited compared to that for conditioned medium from control cells (Fig. 3). Thus, RLIP76 regulates tumor cell secretion of promigratory factors for endothelial cells. Finally, as an in vitro approximation of angiogenic function, we tested cord formation by endothelial cells using growth factor-reduced Matrigel as a substrate, in the presence of different conditioned media (18). Whereas the BAECs formed cords in the presence of normal or WT conditioned medium, cord formation was significantly blocked with conditioned medium from the RLIP76-depleted tumor cells (Fig. 4). Together, these data demonstrate that RLIP76 regulates tumor cell expression and secretion of angiogenic genes including VEGF, leading to transactivation of endothelial cells by the tumor cell secretome.
Figure 3.

Migration of endothelial cells toward tumor cell conditioned medium. BAEC migration through fibronectin-coated filters in modified Boyden chambers, with the indicated medium in the lower chambers. A) Migration of transfected cells in Boyden chambers was imaged at the indicated times after seeding cells on the tops of the filters (3, 6, and 24 h). The undersides of the filters were fixed and stained with crystal violet solution. B) Staining intensities, indicating densities of migrated cells in duplicate wells from 3 independent experiments. *P < 0.001; **P < 0.003.
Figure 4.

Matrigel cord formation by BAECs in the presence of tumor cell conditioned medium requires RLIP76 expression in the tumor cells. Growth factor-reduced phenol-free Matrigel was allowed to polymerize. A) Endothelial cells were seeded on top of the matrix surface in the presence of each indicated medium and returned to the incubator for 3, 6, and 24 h before being photographed (24 h are shown). B) Average branch points of BAECs at 24 h in Matrigel; bars represent means ± sem. *P < 0.003 (n=3).
Reconstitution of endothelial cell function in RLIP76-knockdown conditioned medium with VEGF
The above results indicate that the secretome from tumor cells depleted of RLIP76 cannot support activation of endothelial cell functions. Since VEGF expression was decreased, and VEGF secretion was completely ablated in these tumor cells, we investigated whether addition of recombinant VEGF to the tumor cell conditioned medium could restore its ability to stimulate endothelial cells. BAEC proliferation, migration, and Matrigel cord formation were assessed in the presence of conditioned medium from B16F10 or LLC cells, with or without RLIP76 knockdown, and with rVEGF added to the conditioned medium at 5 ng/ml. As shown in Fig. 5A, whereas BAEC proliferation was suppressed in RLIP76-knockdown conditioned medium compared to WT medium, addition of rVEGF to the RLIP76-knockdown medium resulted in substantial reconstitution of BAEC proliferation close to WT levels. Similarly, suppression of BAEC chemotactic migration toward the RLIP76-knockdown conditioned medium was restored in the presence of rVEGF (Fig. 5B). Finally, cord formation and branching of BAECs on growth factor-reduced Matrigel, suppressed in the presence of RLIP76-knockdown medium, were restored close to WT levels when rVEGF was added to the RLIP76-knockdown conditioned medium (Fig. 5C). These results demonstrate that VEGF is sufficient to restore endothelial cell transactivation by the secretome of RLIP76-depleted tumor cells. Taken together, these data indicate that a major function of RLIP76 in tumor cell/endothelial interaction is the regulation of VEGF production and secretion by the tumor cells.
Figure 5.

VEGF is sufficient to restore endothelial function in conditioned medium from RLIP76-depleted tumor cells. A) Proliferation of BAECs in conditioned medium from untransfected tumor cells (WT), or tumor cells transfected with RLIP76 shRNA, with or without addition of 5 ng/ml rVEGF assessed at 96 h. rVEGF was added to RLIP76-knockdown cells only. *P < 0.006, **P < 0.01 vs. shRNA without rVEGF. B) BAEC migration (24 h) in modified Boyden chambers. *P < 0.01. C) BAEC cord formation in Matrigel (24 h). *P < 0.01 vs. shRNA without rVEGF. All results shown as means ± sem (n=3).
RLIP76 is required for HIF-1 transcriptional activity and PI3K activation in tumor cells
Since VEGF transcription is induced by HIF-1 in tumor cells (23), we sought to determine whether RLIP76 regulates HIF-1 transcriptional activation in these cells under normal growth conditions. We used a luciferase reporter assay comprising a firefly luciferase cassette under the control of an HRE, and Renilla luciferase as a transfection marker (37). Interestingly, RLIP76 depletion led to a significant blockade of HIF-1 transcriptional activity in both B16F10 and LLC tumor cells under normoxic conditions (Fig. 6A). Thus, RLIP76 is required for efficient HIF-1 activation in several tumor cell lines. To investigate potential mechanisms for this function of RLIP76, we evaluated whether RLIP76 regulates HIF-1α protein expression, as well as nuclear localization of HIF-1α required for its ability to stimulate transcription (30). Cells transfected with RLIP76 shRNA were maintained under normal growth conditions and lysed by shear homogenization in hyposmotic buffer, and cytosolic and nuclear fractions were separated by centrifugation. As shown in Fig. 6B, total cellular expression of HIF-1α (whole-cell lysate samples) was unaffected by RLIP76 knockdown, indicating that RLIP76 does not regulate HIF-1α at the level of protein expression. Steady-state levels of HIF-1α in the cell nuclei were also similar between control and RLIP76-knockdown cells, demonstrating that HIF-1α localizes to the cell nucleus in the absence of RLIP76 expression (Fig. 6B–D). These results show that RLIP76 expression is not required for HIF-1α expression or nuclear translocation, which suggests that RLIP76 regulates HIF-1α transcriptional activation at the functional level in these tumor cell lines. Since HIF-1α can be regulated through PI3K pathways, we next investigated whether RLIP76 is important for PI3K activation in these tumor cells. As shown in Fig. 6E, suppression of RLIP76 expression by shRNA resulted in an inhibition of phosphorylation of the downstream PI3K target, Akt. Together, these results indicate that RLIP76 regulates HIF-1 transcriptional activity but not expression or nuclear translocation, and RLIP76 is necessary for PI3K activity in these tumor cells.
Figure 6.

RLIP76 regulates HIF-1 transcriptional activity and PI3K in tumor cells. A) Cignal HIF firefly luciferase reporter and negative or positive control plasmids were cotransfected in 4 × 105 cells/well in tumor cell lines (B16F10 and LLC) with control or RLIP76 shRNA and Renilla luciferase plasmids. HIF activity was assessed in cell lysates 16 h post-transfection, shown as ratio of firefly to Renilla luciferase emission compared to negative control; bars represent means ± sem. *P < 0.005; **P < 0.03 (n=3). B) B16F10 and LLC cells were transfected as indicated and osmotically lysed, and cytosol (Cyt) and nuclear (Nuc) components were separated by centrifugation. HIF-1α total expression in the whole-cell lysate (WCL) and cellular distribution (Cyt and Nuc) were unaffected by RLIP76 knockdown. Band intensities in Cyt and Nuc fractions relative to WCL are shown. Erk1/2 is shown as a loading control. C) RLIP76 knockdown by shRNA, with Erk1/2 as a loading control. D) RhoGDI as a cytosol marker in the separated samples. E) Akt phosphorylation (p-Akt, Ser473), total Akt, and RLIP76 levels in B16F10 and LLC cells transfected as indicated. All results are representative of 3 independent experiments.)
DISCUSSION
The results from this study describe a new link in the regulation of proangiogenic factors by the RLIP76 oncogene in tumor cells. We previously showed that RLIP76 deletion in mice inhibits angiogenesis in xenografted tumors due to defects in the host angiogenic vascular cells, and RLIP76 depletion in the tumor cells further limits tumor growth in these mice (18). We now find that RLIP76 knockdown in two tumor cell lines by transient transfection with a targeting shRNA resulted in substantial loss of VEGF expression, which was later partially restored in these cells as the RLIP76 expression recovered from shRNA suppression over time. This result suggests that VEGF dosage is dependent on RLIP76 in tumor cells. This suppression of VEGF in the tumor cells by RLIP76 suppression could affect the tumor cell function, as VEGF plays important roles within tumor cells, such as regulating receptor surface expression (38). However, whereas VEGF expression was decreased but not eliminated in the knockdown cells, no VEGF could be detected in the conditioned medium from these cells, indicating that RLIP76 regulates both the expression and secretion of VEGF in these cells. Whereas RLIP76 appears to control VEGF expression at the level of transcription (see below), the mechanisms by which RLIP76 regulates VEGF secretion remain to be explored. One potential mechanism is through its interaction with the Ras family small GTPase R-Ras, and activation of PI3K. We have previously shown that activated R-Ras, which binds RLIP76, traffics to the plasma membrane in order to stimulate PI3K activation (9, 39), and we found here that RLIP76 is required for PI3K activity in the tumor cells. PI3K has been implicated in VEGF secretion in many cells, independent from its effects on VEGF expression, and PI3K may be a major regulator of VEGF secretion in angiogenic environments (40–43). It will be interesting to investigate how RLIP76 contributes to this process.
RLIP76-dependent VEGF production and secretion in melanoma and carcinoma cells were required for stimulation of endothelial cell angiogenic functions, including proliferation, migration, and cord formation in vitro, by the tumor cell secretome. Addition of recombinant VEGF to the conditioned medium from RLIP76-depleted tumor cells restored these cell functions in endothelial cells, indicating that regulation of VEGF expression and secretion is a major role of RLIP76 in tumor cell secretome stimulation of endothelium. VEGF is a key promoter of angiogenesis in physiological and pathological settings, stimulating angiogenic signaling in vascular endothelial cells via two receptor tyrosine kinases (VEGFR-1/Flt-1 and VEGFR-2 or Flk-1/KDR in mice; refs. 20, 21). The levels of VEGF must be precisely regulated to prevent vascular dysfunction. VEGF has long been recognized as the major factor in promoting angiogenesis in carcinomas and other tumors and is well known to be overexpressed at the mRNA and protein levels in many tumors and tumor cell lines, including lung carcinomas and melanomas, leading to promotion of neovascularization and tumor growth (3, 21, 22, 44–46). Although VEGF in the tumor environment is derived from many cells, including tumor stromal cells, vascular cells, and peripheral blood mononuclear cells (47–50), tumor cells are the major source of VEGF at least in murine tumor models (21, 38, 51, 52). However, VEGF derived from the tumor stroma also supports tumor angiogenesis and progression, as evidenced by findings that VEGF blockade in the tumor cells alone significantly reduces, but not does eliminate, tumor growth (3, 21). RLIP76 is also overexpressed in many tumor cells, and it is highly expressed in endothelial cells. Depletion or inhibition of RLIP76 causes regression of primary tumors and prolonged survival in a variety of murine tumor models (13–18, 53, 54). We previously found that angiogenesis and tumor growth were inhibited in B16F10 and LLC xenografts in RLIP76-knockout mice, due in part to growth and migration defects of the host endothelial cells, and further inhibition of tumor growth was observed when RLIP76 was depleted from the xenografted tumor cells by shRNA inhibition (18). Together with our results here, this finding suggests that an important angiogenic role of RLIP76 is the regulation of VEGF production and secretion by the tumor cells, but in addition RLIP76 also regulates functions in the tumor stromal cells and vascular cells to modulate angiogenesis and tumor growth.
VEGF transcriptional activator HIF-1 was dependent on RLIP76 in both the carcinoma and melanoma cells, as HIF-1 activity was inhibited by RLIP76 knockdown in both tumor cell lines. This finding represents a new function for RLIP76 and a new level of regulation of HIF-1-mediated gene expression, and VEGF expression in particular. The VEGF gene promoter contains an HRE, and the VEGF gene is primarily transcribed by activated HIF-1 in tumor cells (27, 29, 34–36). Although HIF-1 activation is stimulated by hypoxic conditions in tumors, basal VEGF expression by some tumor cells may be more important for angiogenesis than VEGF induced by hypoxic conditions (55–57). Like RLIP76, HIF-1 is also overexpressed in many tumor cells, and targeting the HIF-1/VEGF pathway has been shown to inhibit angiogenesis and growth in tumors (29, 58). Notably, HIF-1 couples to p300/CBP to drive transcription, and recently RLIP76 expression was shown to be regulated by p300; furthermore, p300 and RLIP76 correlate in many tumor types, suggesting the possibility of a transcriptional regulatory loop (52). HIF-1 in tumor cells stimulates the transcription of many genes downstream of the PI3K/mTOR and Ras/Raf/MEK/ERK pathways, both of which converge on elF4E1 to regulate HIF-1 expression (27, 30, 31). We observed an inhibition of PI3K activation by RLIP76 suppression in carcinoma and melanoma cells, consistent with a blockade of PI3K pathway activation seen with RLIP76 antisense or antibodies in pancreatic tumors (50). However, we observed that despite inhibition of PI3K, HIF-1α expression was unchanged in the knockdown cells, indicating that RLIP76 is not required for HIF-1α expression, and that RLIP76 regulates HIF-1 at the functional level. HIF-1α translocates from the cytosol to the nucleus, where it binds cofactor CBP/p300 followed by binding to HREs in target genes to initiate transcription (27, 33). Nuclear accumulation of HIF-1α at steady state was also unaffected by RLIP76 knockdown in the tumor cells in our study. Thus, RLIP76 is not required for HIF-1α expression or nuclear translocation. We are currently exploring the molecular mechanisms by which RLIP76 regulates HIF-1α function and its effects on expression of VEGF and other genes.
Taken together, our results indicate that the angiogenic roles of RLIP76 expressed in tumor cells involve a combination of effects on VEGF expression through HIF-1 activation, and on VEGF secretion, possibly through RLIP76 regulation of PI3K. Targeting RLIP76 or VEGF results in antitumor effects in multiple settings, and in the case of VEGF, combinatorial therapies such as anti-VEGF antibodies along with chemotherapy or radiation enhance these effects (16, 17, 59–61). Further research into incorporating RLIP76 as an additional therapeutic target may lead to enhanced effectiveness in combating tumor angiogenesis and progression (51).
Acknowledgments
This work was supported by U.S. National Institutes of Health grant HL093416 to L.E.G. and American Heart Association Postdoctoral Fellowship 12POST12040257 to S.L.
The authors thank Jeremy G.T. Wurtzel for technical assistance. The authors declare no conflict of interest.
Footnotes
- BAEC
- bovine aortic endothelial cell
- bFGF
- basic fibroblast growth factor
- DMEM
- Dulbecco's modified Eagle's medium
- EGF
- epidermal growth factor
- FGF
- fibroblast growth factor
- HIF-1
- hypoxia-inducible factor 1
- HRE
- HIF-1-responsive element
- LLC
- Lewis lung carcinoma
- mTOR
- molecular target of rapamycin
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PI3K
- phosphatidylinositol-3 kinase
- RLIP76
- Ral-interacting protein of 76 kDa
- SFM
- serum-free medium
- shRNA
- short hairpin RNA
- VEGF
- vascular endothelial growth factor
REFERENCES
- 1. Folkman J. (1992) The role of angiogenesis in tumor growth. Semin. Cancer Biol. 3, 65–71 [PubMed] [Google Scholar]
- 2. Weis S. M., Cheresh D. A. (2011) Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med. 17, 1359–1370 [DOI] [PubMed] [Google Scholar]
- 3. Hicklin D. J., Ellis L. M. (2005) Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 23, 1011–1027 [DOI] [PubMed] [Google Scholar]
- 4. Jullien-Flores V., Dorseuil O., Romero F., Letourneur F., Saragosti S., Berger R., Tavitian A., Gacon G., Camonis J. H. (1995) Bridging Ral GTPase to Rho pathways. RLIP76, a Ral effector with CDC42/Rac GTPase-activating protein activity. J. Biol. Chem. 270, 22473–22477 [DOI] [PubMed] [Google Scholar]
- 5. Park S. H., Weinberg R. A. (1995) A putative effector of Ral has homology to Rho/Rac GTPase activating proteins. Oncogene 11, 2349–2355 [PubMed] [Google Scholar]
- 6. Awasthi S., Cheng J., Singhal S. S., Saini M. K., Pandya U., Pikula S., Bandorowicz-Pikula J., Singh S. V., Zimniak P., Awasthi Y. C. (2000) Novel function of human RLIP76: ATP-dependent transport of glutathione conjugates and doxorubicin. Biochemistry 39, 9327–9334 [DOI] [PubMed] [Google Scholar]
- 7. Awasthi S., Cheng J. Z., Singhal S. S., Pandya U., Sharma R., Singh S. V., Zimniak. P., Awasthi Y. (2001) Functional reassembly of ATP-dependent xenobiotic transport by the N- and C-terminal domains of RLIP76 and identification of ATP binding sequences. Biochemistry 40, 4159–4168 [DOI] [PubMed] [Google Scholar]
- 8. Awasthi S., Sharma R., Yang Y., Singhal S. S., Pikula S., Bandorowicz-Pikula J., Singh S. V., Zimniak P., Awasthi Y. C. (2002) Transport functions and physiological significance of 76 kDa Ral-binding GTPase activating protein (RLIP76). Acta Biochim. Pol. 49, 855–867 [PubMed] [Google Scholar]
- 9. Goldfinger L. E., Ptak C., Jeffery E. D., Shabanowitz J., Hunt D. F., Ginsberg M. H. (2006) RLIP76 (RalBP1) is an R-Ras effector that mediates adhesion-dependent Rac activation and cell migration. J. Cell Biol. 174, 877–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharma R., Gupta S., Singh S. V., Medh R. D., Ahmad H., LaBelle E. F., Awasthi Y. C. (1990) Purification and characterization of dinitrophenylglutathione ATPase of human erythrocytes and its expression in other tissues. Biochem. Biophys. Res. Commun. 171, 155–161 [DOI] [PubMed] [Google Scholar]
- 11. Awasthi S., Singhal S. S., Srivastava S. K., Zimniak P., Bajpai K. K., Saxena M., Sharma R., Ziller S. A., 3rd, Frenkel E. P., Singh S. V. (1994) Adenosine triphosphate-dependent transport of doxorubicin, daunomycin, and vinblastine in human tissues by a mechanism distinct from the P-glycoprotein. J. Clin. Invest. 93, 958–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Awasthi Y. C., Singhal S. S., Gupta S., Ahmad H., Zimniak P., Radominska A., Lester R., Sharma R. (1991) Purification and characterization of an ATPase from human liver which catalyzes ATP hydrolysis in the presence of the conjugates of bilirubin bile acids and glutathione. Biochem. Biophys. Res. Commun. 175, 1090–1096 [DOI] [PubMed] [Google Scholar]
- 13. Singhal S. S., Singhal J., Yadav S., Dwivedi S., Boor P. J., Awasthi Y. C., Awasthi S. (2007) Regression of lung and colon cancer xenografts by depleting or inhibiting RLIP76 (Ral-binding protein 1). Cancer Res. 67, 4382–4389 [DOI] [PubMed] [Google Scholar]
- 14. Singhal S. S., Roth C., Leake K., Singhal J., Yadav S., Awasthi S. (2009) Regression of prostate cancer xenografts by RLIP76 depletion. Biochem. Pharmacol. 77, 1074–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Singhal S. S., Awasthi Y. C., Awasthi S. (2006) Regression of melanoma in a murine model by RLIP76 depletion. Cancer Res. 66, 2354–2360 [DOI] [PubMed] [Google Scholar]
- 16. Awasthi S., Singhal S. S., Awasthi Y. C., Martin B., Woo J. H., Cunningham C. C., Frankel A. E. (2008) RLIP76 and cancer Clin. Cancer Res. 14, 4372–4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goldfinger L. E., Lee S. (2013) Emerging treatments in lung cancer – targeting the RLIP76 molecular transporter. Lung Cancer Targets Ther. 4, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee S., Wurtzel J. G., Singhal S. S., Awasthi S., Goldfinger L. E. (2012) RALBP1/RLIP76 depletion in mice suppresses tumor growth by inhibiting tumor neovascularization. Cancer Res. 72, 5165–5173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McMahon G. (2000) VEGF receptor signaling in tumor angiogenesis. Oncologist 5(Suppl. 1), 3–10 [DOI] [PubMed] [Google Scholar]
- 20. Neufeld G., Cohen T., Gengrinovitch S., Poltorak Z. (1999) Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 13, 9–22 [PubMed] [Google Scholar]
- 21. Ferrara N. (2004) Vascular endothelial growth factor: basic science and clinical progress. Endocrine Rev. 25, 581–611 [DOI] [PubMed] [Google Scholar]
- 22. Aonuma M., Saeki Y., Akimoto T., Nakayama Y., Hattori C., Yoshitake Y., Nishikawa K., Shibuya M., Tanaka N. G. (1999) Vascular endothelial growth factor overproduced by tumour cells acts predominantly as a potent angiogenic factor contributing to malignant progression. Int. J. Exper. Pathol. 80, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Terman B., Stoletov K. (2001) VEGF and tumor angiogenesis. Einstein Quar. J. Biol. Med. 18, 59–66 [Google Scholar]
- 24. Choi K. S., Bae M. K., Jeong J. W., Moon H. E., Kim K. W. (2003) Hypoxia-induced angiogenesis during carcinogenesis. J. Biochem. Mol. Biol. 36, 120–127 [DOI] [PubMed] [Google Scholar]
- 25. Forsythe J. A., Jiang B. H., Iyer N. V., Agani F., Leung S. W., Koos R. D., Semenza G. L. (1996) Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yan J., Liu Q., Dou Y., Hsieh Y., Liu Y., Tao R., Zhu D., Lou Y. (2013) Activating glucocorticoid receptor-ERK signaling pathway contributes to ginsenoside Rg1 protection against beta-amyloid peptide-induced human endothelial cells apoptosis. J. Ethnopharmacol. 147, 456–466 [DOI] [PubMed] [Google Scholar]
- 27. Nagy M. A. (2011) HIF-1 is the commander of gateways to cancer. Cancer Sci. Ther. 3, 35–40 [Google Scholar]
- 28. Dery M. A., Michaud M. D., Richard D. E. (2005) Hypoxia-inducible factor 1: regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell. Biol. 37, 535–540 [DOI] [PubMed] [Google Scholar]
- 29. Semenza G. L. (2009) HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20, 51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yi T., Papadopoulos E., Hagner P. R., Wagner G. (2013) Hypoxia-inducible factor-1alpha (HIF-1alpha) promotes cap-dependent translation of selective mRNAs through up-regulating initiation factor eIF4E1 in breast cancer cells under hypoxia conditions. J. Biol. Chem. 288, 18732–18742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fukuda R., Hirota K., Fan F., Jung Y. D., Ellis L. M., Semenza G. L. (2002) Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J. Biol. Chem. 277, 38205–38211 [DOI] [PubMed] [Google Scholar]
- 32. Lando D., Peet D. J., Whelan D. A., Gorman J. J., Whitelaw M. L. (2002) Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 295, 858–861 [DOI] [PubMed] [Google Scholar]
- 33. Ziello J. E., Jovin I. S., Huang Y. (2007) Hypoxia-inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 80, 51–60 [PMC free article] [PubMed] [Google Scholar]
- 34. Semenza G. L. (2009) HIF-1 inhibitors for cancer therapy: from gene expression to drug discovery. Curr. Pharm. Des. 15, 3839–3843 [DOI] [PubMed] [Google Scholar]
- 35. Semenza G. L. (2012) Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 33, 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muz B., Khan M. N., Kiriakidis S., Paleolog E. M. (2009) Hypoxia. The role of hypoxia and HIF-dependent signalling events in rheumatoid arthritis. Arthrit. Res. Ther. 11, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Malo M. S., Mozumder M., Chen A., Mostafa G., Zhang X. B., Hodin R. A. (2006) pFRL7: an ideal vector for eukaryotic promoter analysis. Anal. Biochem. 350, 307–309 [DOI] [PubMed] [Google Scholar]
- 38. Goel H. L., Mercurio A. M. (2013) VEGF targets the tumour cell. Nat. Rev. Cancer 13, 871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wurtzel J. G., Kumar P., Goldfinger L. E. (2012) Palmitoylation regulates vesicular trafficking of R-Ras to membrane ruffles and effects on ruffling and cell spreading. Small GTPases 3, 139–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karar J., Maity A. (2011) PI3K/AKT/mTOR Pathway in angiogenesis. Front. Mol. Neurosci. 4, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moriya R, Takahashi K, Kitahara A, Onuma H, Handa K, Sumitani Y, Tanaka T., Katsuta H., Nishida S., Itagaki E., Inukai K., Ishida H. (2014) Possible involvement of PI3K-dependent pathways in the increased VEGF release from osteoblastic cells preloaded with palmitate in vitro. Biochem. Biophys. Res. Commun. 445, 275–281 [DOI] [PubMed] [Google Scholar]
- 42. Zhao W., Guo W., Zhou Q., Ma S. N., Wang R., Qiu Y., Jin M., Duan H. Q., Kong D. (2013) In vitro antimetastatic effect of phosphatidylinositol 3-kinase inhibitor ZSTK474 on prostate cancer PC3 cells. Int. J. Mol. Sci. 14, 13577–13591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takahashi K., Miyokawa-Gorin K., Handa K., Kitahara A., Moriya R., Onuma H., Sumitani Y., Tanaka T., Katsuta H., Nishida S., Yoshimoto K., Ohno H., Ishida H. (2013) Endogenous oxidative stress, but not ER stress, induces hypoxia-independent VEGF120 release through PI3K-dependent pathways in 3T3-L1 adipocytes. Obesity 21, 1625–1634 [DOI] [PubMed] [Google Scholar]
- 44. Volm M., Koomagi R., Mattern J., Stammler G. (1997) Angiogenic growth factors and their receptors in non-small cell lung carcinomas and their relationships to drug response in vitro. Anticancer Res 17, 99–103 [PubMed] [Google Scholar]
- 45. Marneros A. G. (2009) Tumor angiogenesis in melanoma. Hematol. Oncol. Clin. North Am. 23, 431–446, vii–viii. [DOI] [PubMed] [Google Scholar]
- 46. Mahabeleshwar G. H., Byzova T. V. (2007) Angiogenesis in melanoma. Semin. Oncol. 34, 555–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fukumura D., Xavier R., Sugiura T., Chen Y., Park E. C., Lu N., Selig M., Nielsen G., Taksir T., Jain R. K., Seed B. (1998) Tumor induction of VEGF promoter activity in stromal cells. Cell 94, 715–725 [DOI] [PubMed] [Google Scholar]
- 48. Gerber H. P., Kowalski J., Sherman D., Eberhard D. A., Ferrara N. (2000) Complete inhibition of rhabdomyosarcoma xenograft growth and neovascularization requires blockade of both tumor and host vascular endothelial growth factor. Cancer Res. 60, 6253–6258 [PubMed] [Google Scholar]
- 49. Kishimoto J., Ehama R., Ge Y., Kobayashi T., Nishiyama T., Detmar M., Burgeson R. E. (2000) In vivo detection of human vascular endothelial growth factor promoter activity in transgenic mouse skin. Am. J. Pathol. 157, 103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Leake K., Singhal J., Nagaprashantha L. D., Awasthi S., Singhal S. S. (2012) RLIP76 regulates PI3K/Akt signaling and chemo-radiotherapy resistance in pancreatic cancer PLoS One 7, e34582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Limaverde-Sousa G., Sternberg C., Ferreira C. G. (2014) Antiangiogenesis beyond VEGF inhibition: a journey from antiangiogenic single-target to broad-spectrum agents. Cancer Treat. Rev. 40, 548–557 [DOI] [PubMed] [Google Scholar]
- 52. Sehrawat A., Yadav S., Awasthi Y. C., Basu A., Warden C., Awasthi S. (2013) P300 regulates the human RLIP76 promoter activity and gene expression. Biochem. Pharmacol. 85, 1203–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Singhal S. S., Yadav S., Singhal J., Zajac E., Awasthi Y. C., Awasthi S. (2005) Depletion of RLIP76 sensitizes lung cancer cells to doxorubicin. Biochem. Pharmacol. 70, 481–488 [DOI] [PubMed] [Google Scholar]
- 54. Margutti P., Matarrese P., Conti F., Colasanti T., Delunardo F., Capozzi A., Garofalo T., Profumo E., Riganò R., Siracusano A., Alessandri C., Salvati B., Valesini G., Malorni W., Sorice M., Ortona E. (2008) Autoantibodies to the C-terminal subunit of RLIP76 induce oxidative stress and endothelial cell apoptosis in immune-mediated vascular diseases and atherosclerosis. Blood 111, 4559–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gille J. (2006) Antiangiogenic cancer therapies get their act together: current developments and future prospects of growth factor- and growth factor receptor-targeted approaches. Exp. Dermatol. 15, 175–186 [DOI] [PubMed] [Google Scholar]
- 56. Motl S. (2005) Bevacizumab in combination chemotherapy for colorectal and other cancers. Am. J. Health Syst. Pharm. 62, 1021–1032 [DOI] [PubMed] [Google Scholar]
- 57. Sparano J. A., Gray R., Giantonio B., O'Dwyer P., Comis R. L. (2004) Evaluating antiangiogenesis agents in the clinic: the Eastern Cooperative Oncology Group Portfolio of Clinical Trials. Clin. Cancer Res. 10, 1206–1211 [DOI] [PubMed] [Google Scholar]
- 58. Tsuzuki Y., Fukumura D., Oosthuyse B., Koike C., Carmeliet P., Jain R. K. (2000) Vascular endothelial growth factor (VEGF) modulation by targeting hypoxia-inducible factor-1alpha–> hypoxia response element–> VEGF cascade differentially regulates vascular response and growth rate in tumors. Cancer Res. 60, 6248–6252 [PubMed] [Google Scholar]
- 59. Klement G., Baruchel S., Rak J., Man S., Clark K., Hicklin D. J., Bohlen P., Kerbel R. S. (2000) Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J. Clin. Invest. 105, R15–R24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kozin S. V., Boucher Y., Hicklin D. J., Bohlen P., Jain R. K., Suit H. D. (2001) Vascular endothelial growth factor receptor-2-blocking antibody potentiates radiation-induced long-term control of human tumor xenografts. Cancer Res. 61, 39–44 [PubMed] [Google Scholar]
- 61. Lee C. G., Heijn M., di Tomaso E., Griffon-Etienne G., Ancukiewicz M., Koike C., Park K. R., Ferrara N., Jain R. K., Suit H. D., Boucher Y. (2000) Anti-vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. 60, 5565–5570 [PubMed] [Google Scholar]