

Abstract
An increase in intramuscular adipocyte tissue (IMAT) is associated with glucose dysregulation, decreased muscle strength, and increased risk of disability. Unfortunately, the mechanisms stimulating intramuscular adipogenesis remain unclear. We found that dexamethasone (Dex) administration to mice with injured muscles stimulates the accumulation of IMAT. To identify precursors of these adipocytes, we isolated satellite cells and fibro/adipogenic progenitors (FAPs) from muscle; satellite cells did not differentiate into adipocytes even following Dex treatment. In contrast, Dex stimulated FAP differentiation into adipocytes. In vivo, we transplanted purified FAPs from transgenic, EGFP mice into the injured muscles of C57/BL6 mice and found that Dex administration stimulated adipogenesis from FAP-EGFP. The increase in adipogenesis depended on Dex-induced inhibition of interleukin-4 (IL-4). In the injured muscle of IL-4-knockout mice, the levels of adipocytes were increased, while in the injured muscles of Dex-treated mice with IL-4 injections, adipogenesis was suppressed. In cultured FAPs, IL-4 inhibited Dex-induced conversion of FAPs into adipocytes; this did not occur in FAPs expressing knockdown of the IL-4 receptor. Thus, we concluded that glucocorticoids stimulate FAPs to differentiate into adipocytes in injured muscles. This process is blocked by IL-4, suggesting that interfering with IL-4 signaling could prevent adipogenesis in muscle.—Dong, Y., Silva, K. A. S., Dong, Y., Zhang, L. Glucocorticoids increase adipocytes in muscle by affecting IL-4 regulated FAP activity.
Keywords: progenitors, dexamethasone
In pathological conditions such as Duchenne muscular dystrophy (DMD), type II diabetes, obesity, or aging-related sarcopenia, skeletal muscles develop intramuscular adipose tissue (IMAT; refs. 1–3). These adipocytes are important because they lead to poor quality and dysfunctional performance of muscles (4, 5). The mechanisms causing the intramuscular adipocytes are not fully understood. A potential mechanism is that muscle cells differentiate into adipocytes. In fact, several types of skeletal muscle cells have been reported to possess the potential for adipogenic differentiation, including muscle progenitor cells (satellite cells), “side” population cells, and mesenchymal stem cells (6, 7). For example, satellite cell-derived primary myoblasts, expressing myogenic markers such as MyoD, Myf5, Pax7, and desmin, can reportedly differentiate into myotubes, osteocytes, or adipocytes following treatment with bone morphogenetic proteins or adipogenic inducers, respectively (8, 9). Side population cells isolated from skeletal muscles (10, 11) also exhibit stem cell activities, including self-renewal and the potential for dentinogenesis, chondrogenesis, adipogenesis, and neurogenesis (12). In addition, nonmyogenic, mesenchymal progenitor cells from skeletal muscle can develop into adipocytes (13). Recently, two groups have isolated mesenchymal progenitors from skeletal muscle and concluded that these cells have the potential to undergo fibrogenesis or adipogenesis, [i.e., fibro/adipogenic progenitors (FAPs); refs. 14, 15]. However, despite all this research, the triggers and the signaling pathways that stimulate FAPs to undergo adipogenic differentiation remain unclear.
Understanding the pathogenesis of adipocyte accumulation in muscle is important because both obese and aging patients have been shown to have increased IMAT associated with increased insulin resistance (16). These patients also showed increased glucocorticoid (GC) excretion (17–19). Injuries including burn injury and spinal cord injury also stimulates GCs production (20). GCs increase the differentiation of preadipocytes into mature adipocytes (21). A link between GCs and adipocyte accumulation in muscle is present in patients with Cushing's syndrome and adrenal adenomas (22, 23). In mice, experimental administration of dexamethasone (Dex) not only stimulates muscle protein catabolism but also increases adiposity and promotes insulin resistance (24–27). The mechanism by which GCs increase adiposity is not clear.
Muscle injury activates the innate immune system to recruit Th2 lymphocytes, macrophages, mast cells, eosinophils, etc. to the injury sites (28, 29). Eosinophils are reported to be the dominant cell type secreting interleukin 4 (IL-4) in injured muscle (30), and GCs are known to suppress eosinophils (31). IL-4 was shown to inhibit lipid accumulation in adipose tissue by a mechanism that includes activation of Stat6, which suppresses PPARα transcriptional activity (32, 33). We found that GCs suppress IL-4 expression in injured muscles. The decrease in IL-4 is associated with the accumulation of adipose tissue in injured muscles. We also found that IL-4 signaling serves as a key switch to control the fate and function of FAPs. These relationships among GCs, IL-4 expression, and the function of FAPs uncover a new mechanism that increases the accumulation of adipocytes in muscle.
MATERIALS AND METHODS
Animal experiments
All animal experiments and procedures were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. To study responses to GCs, male C57/BL6 mice (10–12 wk old; Jackson Laboratories, Bar Harbor, ME, USA) were injected intraperitoneally with 1 mg/kg Dex (Sigma-Aldrich, St. Louis, MO, USA) daily for 14 d (24, 34, 35). Dex-injected mice were pair-fed with control mice and body weights were assessed daily. Enhanced green fluorescent protein (EGFP) transgenic and IL-4-knockout (KO) mice (B57/BL6 background) were purchased from Jackson Laboratories. In some experiments, IL-4 (2 μg; Peprotech, Rocky Hill, NJ, USA) was injected intraperitoneally into mice 1 d before and then every day following muscle injury. Muscle injury was produced by injecting of 80 μl of 10 μM cardiotoxin (CTX; Sigma-Aldrich) into the tibialis anterior (TA) muscle; the contralateral TA muscles of mice were injected with same volume of phosphate-buffered saline (PBS) as a control (36–38).
Cell isolation and transplantation
Muscles from both hind limbs of 4- to 6-wk-old C57/BL6 mice were isolated, and nonmuscle tissue was removed before muscles were digested with 2 ml collagenase type 2 (Sigma-Aldrich; 2.5 U/ml) in 10 mM CaCl2 at 37°C for 30 min. After centrifugation and washing with PBS, 1 ml of collagenase D (1.5 U/ml) and dispase II (2.4 U/ml; Roche Applied Science, Indianapolis, IN, USA) was added, and the mixture was incubated at 37°C for 1 h. The muscle slurry was passed through a 40 μm strainer (BD Biosciences, San Jose, CA, USA) and the filtered material was centrifuged at 1600 rpm for 5 min. The pellet was resuspended with PBS containing 2 mM EDTA and 2% fetal bovine serum (FBS) into ∼3 × 107 cells/ml. Cells were then incubated with fluorescent-conjugated primary antibodies for 30 min at 4°C. Cell viability was assessed by propidium iodide and Hoechst 33342 staining. After washing, cells were resuspended at ∼1 × 107 cells/ml with PBS containing 0.5% bovine serum albumin (BSA) for sorting at the Cell Sorting Core Facility of Baylor College of Medicine. Sorting gates were defined based on isotype controls.
To isolate cells from muscles of the EGFP-transgenic mice, Pacific blue-labeled CD45 and CD31 were used (antibodies are listed in Table 1). Cells (5×104) isolated from EGFP-transgenic mice were resuspended in 30 μl Matrigel (BD Biosciences) and transplanted into TA muscles of mice that had been treated with Dex or injury. At 7 and 10 d after transplantation, TA muscles were removed from euthanized mice and placed in optimum cutting temperature (OCT) compound (Tissue-Tek; Sakura Finetek, Torrance, CA, USA) and frozen in dry ice-chilled isopentane for histological analysis, or TA muscles were stored in liquid nitrogen until proteins or RNAs were evaluated.
Table 1.
Antibodies
| Antibody | Source | Cat. no. | Clone | Application |
|---|---|---|---|---|
| CD31-FITC | BD Pharmingen (San Diego, CA, USA) | 553372 | MEC 13.3 | FACS |
| CD45-FITC | BD Pharmingen | 553080 | 30-F11 | FACS |
| Sca1-PE | BD Pharmingen | 553336 | E13-161.7 | FACS |
| Integrin α-7- APC | R&D Systems (Minneapolis, MN, USA) | FAB3518A | 334908 | FACS |
| FITC isotype control (CD31) | BD Pharmingen | 553929 | R35-95 | FACS |
| PE isotype control | BD Pharmingen | 553930 | R35-95 | FACS |
| FITC isotype control (CD45) | BD Pharmingen | 553988 | A95-1 | FACS |
| APC isotype control | R&D Systems | IC013A | 141945 | FACS |
| CD45-Pacific Blue | Biolegend (San Diego, CA, USA) | 103126 | 30-F11 | FACS |
| CD31-Pacific Blue | Biolegend | 102422 | 390 | FACS |
| Pacific Blue isotype control (CD31) | Biolegend | 400527 | RTK2758 | FACS |
| Pacific Blue isotype control (CD45) | Biolegend | 400627 | RTK4530 | FACS |
| Laminin | Sigma-Aldrich (St. Louis, MO, USA) | L8271 | Immunostaining | |
| eMyHC | DSHB | F1.652 | F1.652 | Immunostaining |
| Perilipin | Cell Signaling (Danvers, MA, USA) | 9349 | D1D8 | Immunostaining Western blots |
| PPARγ | Cell Signaling | 2435 | C26H12 | Western blots |
| C/EBPα | Cell Signaling | 2295 | Western blots | |
| PDGFRα | R&D Systems | AF1062 | Immunostaining Western blots | |
| Ki67-Alexa Fluor 555 | BD Biosciences (San Jose, CA, USA) | 558617 | B56 | Immunostaining |
| Pref-1 | Abcam (Cambridge, MA, USA) | Ab21682 | Western blots | |
| FSP-1 | Abcam | ab27957 | Immunostaining Western blots | |
| GAPDH | Millipore (Billerica, MA, USA) | MAB374 | 6C5 | Western blots |
Cell culture
Isolated satellite cells or FAPs were plated in Matrigel-coated tissue culture plates and cultured with growth medium (GM) containing high-glucose Dulbecco's modified Eagle medium (DMEM) supplemented with 2.5 ng/ml basic fibroblast growth factor (bFGF, Invitrogen, Grand Island, NY, USA), 20% FBS, and 10% heat-inactivated horse serum. After 3 d, medium was changed with fresh medium; this was repeated every 2 d. To induce adipogenic differentiation, we incubated cells in differentiation medium (DM) of DMEM with 20% FBS supplemented with 11.5 μg/ml isobutylmethylxanthine and 1 μg/ml insulin, or DM plus 1 μM Dex (DDM). For myoblast differentiation into myotubes, 95% confluent satellite cells were incubated in DMEM supplemented with 2% horse serum for 96 h.
For knockdown of IL-4 receptor (IL-4R) in FAPs, cells were cultured in GM for 3 d; at ∼50% confluence, they were treated to knock down IL-4R. Subsequently, FAPs were cultured in new medium plus 8 μg/ml polybrene and 1 × 105 infectious units of shRNA IL-4R lentivirus (Santa Cruz Biotechnology, Dallas, TX, USA). After 24 h, the medium was changed to GM, and cells were incubated for another 24 h. Adipogenic differentiation was induced over 3 d while being cultured in DM with Dex or DM with Dex plus IL-4.
Histology and imaging
Anesthetized mice were perfused through the left ventricle with PBS. Muscles were removed and processed for cryosectioning, followed by staining with hematoxylin and eosin (H&E) or Oil-Red-O. Immunostaining was performed using antibodies against perilipin, platelet-derived growth factor receptor α (PDGFRα) or Ki-67 (Table 1). Briefly, the cross-section of TA muscles were fixed in 4% paraformaldehyde (PFA) and permeabilized by incubating in 0.3% Triton X-100 in PBS followed by blocking with protein block (Dako, Carpinteria, CA, USA) for 1 h at room temperature. Sections were then incubated with a primary antibody mixed in antibody dilute (Dako) overnight at 4°C, followed by detection of primary antibodies using secondary antibodies that were conjugated to Alexa 568 or 488 (Invitrogen). Tissues were visualized using a Nikon 80i microscope (Nikon, Melville, NY, USA), and images were acquired using a DS-cooled camera and NIS-Elements Br 3.0 software (Nikon).
RT-PCR analysis
RT-PCR was performed as described previously (39). The relative gene expression was calculated from cycle threshold (Ct) values using GAPDH as the internal control (relative expression=2(sample Ct − GAPDH Ct)). Primers and their sequences are listed in Table 2.
Table 2.
RT-PCR primers
| Gene | Forward, 5′–3′ | Reverse, 5′–3′ |
|---|---|---|
| C/EBPδ | CTCCAGGGTCTAAATACATAGC | CTCACAGCAGTCCACAAG |
| C/EBPα | AGAAGTCGGTGGACAAGAACAGCA | GCGTTGTTTGGCTTTATCTCGGCT |
| FABP4 | ACATGAAAGAAGTGGGAGTGGGCT | TCCTGTCGTCTGCGGTGATTTCAT |
| PPARγ | ATGAAGACATTCCATTCACAAGAGC | ATAGTGGAAGCCAGATGCTTTATCC |
| IL-4 | ATGCTTGAAGAAGAACTCTA | GTGGACTTGGACTCATTC |
| CD68 | CACCTGCTTCTCTCATTC | GTAGACAACCTTCTGCTG |
| CD206 | ATCCACTCTATCCACCTTCA | TGCTTGTTCATATCTGTCTTCA |
| CD163 | AAGCATTACTGTCATCATAG | CTCCACCTACAAGTCTAA |
| F4/80 | AAGCATCCGAGACACACACAGTCT | TGACTGTACCCACATGGCTGATGA |
| MyHC | CCTTGGCACCAATGTCCCGGCTC | GAAGCGCAATGCAGAGTGGGTG |
| GAPDH | ACCACCATGGAGAAGGCCGG | CTCAGTGTAGCCCAAGATGC |
Oil Red O staining
Two methods of Oil-Red-O staining were used on cells and muscle tissues. Briefly, cells were washed with PBS, fixed in 4% PFA for 30 min, and rinsed in dH2O and then 60% isopropanol before being stained with Oil-Red-O in 60% isopropanol. Extra dye was removed by dH2O washes. To obtain Oil-Red-O staining of TA muscle, sections were fixed in 4% PFA for 1 h, washed 3 times in dH2O (5 min each) and then stained with an Oil-Red-O solution in triethyl pentane for 30 min. Excess stain was removed by dH2O, and the slides were mounted using 10% glycerol solution.
Statistical analysis
Results are expressed as means ± se. Results across different treatment and time points were compared using 1-way or 2-way ANOVA. Student's t test was performed when results of 2 groups or measurements were compared, and statistical significance was established at P < 0.05. The number of replicates for each experimental condition is provided in the figure captions.
RESULTS
In mice, GCs stimulate adipogenesis in injured muscles
We recently reported that GCs cause loss of muscle mass by a mechanism involving inhibition of satellite cell activation and impaired muscle regeneration (35). Histologically, impairment in muscle regeneration leads to large spaces between regenerating myofibers. To identify cells present in the injured muscles, we evaluated perilipin, a marker of adipocytes. At 5 d after muscle injury, we found increased adipocyte accumulation in the regenerating muscles of Dex-treated mice vs. e non-Dex-treated mice. At 14 d after injury, few perilipin+ cells per field were found in muscles of control mice, but more and larger perilipin+ cells were present in the injured muscles of Dex-treated mice (P<0.05 vs. control; Fig. 1A, top panel; B). Oil-Red O staining confirmed these results (Fig. 1A, bottom panel). Dex treatment also significantly increased the mRNA expression of adipogenic genes (PPARγ and C/EBPα) in injured muscle vs. the results in injured muscles of mice not being treated by Dex (P<0.05 vs. control; Fig. 1C). We also immunostained the TA sections with anti-embryonic myosin heavy chain (eMyHC; marker for newly formed myofibers) and found fewer newly formed, smaller myofibers (eMyHC+ cells) in injured muscles of Dex-treated mice (Fig. 1A, middle panel, green). These results are consistent with our earlier report that GCs decrease myogenic gene expression in injured muscles (35).
Figure 1.

GCs stimulate adipogenesis in injured muscle. A) At 5 or 14 d after muscle injury of control or Dex-treated-mice, cryo-cross-sections of TA muscles were immunostained with anti-perilipin (red, top panels) plus either anti-laminin (green, uninjured muscle) or anti-eMyHC (green, injured muscle, middle panels), or stained with Oil-Red-O (bottom panels). Scale bars = 50 μm. B) The number of perilipin+ cells per view was counted and averaged (n=5 mice). *P < 0.05 vs. control (Ctrl). C) mRNAs of C/EBPα and PPARγ were evaluated by RT-PCR in injured and uninjured muscles of control or Dex-treated mice. Folds of mRNA expression in injured to uninjured muscles are indicated (n=5 mice). GAPDH was used as internal control. *P < 0.05 vs. control.
In vitro, GCs induce FAPs to differentiate into adipocytes
A potential source of the adipocytes found in injured muscles of Dex-treated mice is the satellite cells, because they exhibit potential for myogenic, osteogenic, and adipogenic differentiation (8). To examine whether Dex stimulates satellite cell differentiation into adipocytes, we separated satellite cells from muscles of C57/BL6 mice using FACS; satellite cells were positive for integrin α7 (a satellite cell marker) but negative for stem cell antigen 1 (Sca-1), CD31 (endothelial marker), and CD45 (a pan-hematopoietic marker) (Fig. 2A). Satellite cells were cultured in growth medium until 90% confluence; they were then placed in DM or DM plus Dex media for another week. Immunostaining with antiperilipin and eMyHC revealed that satellite cells do not contain perilipin under any culture conditions, but Dex did suppress myotube formation from the cultured satellite cells (Fig. 2B). The mRNA expression results were consistent with the immunostaining; no expression of adipocyte-specific mRNAs, such as C/EBPα or PPARγ, was detected in satellite cells. The satellite cells only expressed MyHC (Fig. 2F).
Figure 2.

In vitro, GC only induce differentiation of muscle-derived FAPs into adipocytes. A) Satellite cells (integrin-α7+/CD31−/CD45−/Scal-1−) and FAPs (integrin-α7−/CD31−/CD45−/Scal-1+) were isolated using FACS (left panel; see Materials and Methods). B) Isolated satellite cells were cultured in DM or DM plus Dex and coimmunostained with anti-eMyHC (red). The differentiation index was calculated as the percentage of nuclei within myotubes that stained positively for eMyHC plus the number of eMyHC+ mononuclear cells to the total number of nuclei in the area (right panel; n=3 repeat, medium without Dex). *P < 0.05 vs. control (Ctrl). C) Isolated FAPs were cultured in GM or DM plus Dex; cells were photographed under light microscopy (left panels). At 7 d after treatment, cells were stained with Oil-Red-O (middle panels), or were immunostained with antiperilipin at 14 d after treatment (right panels). D) mRNAs were evaluated by RT-PCR in FAPs (adipogenic differentiation in 2–4 d; n=3 repeat). *P < 0.05 vs. FAPs cultured in GM. E) FAPs were cultured in medium plus Dex for 4 d; protein levels of adipogenic proteins were evaluated by Western blots from the cell lysates. F) Satellite cells and FAPs were cultured in same conditions, and mRNAs from these cells were analyzed by RT-PCR (n=3 repeat).*P < 0.05 vs. FAPs in Dex.
Next, we tested whether Dex influences adipogenic differentiation from mesenchymal progenitors because these cells can contribute to adipocyte formation (14). We isolated FAPs from muscles of C57/BL6 mice by FACS (cells positive for Sca-1 and negative for CD31, CD45, and integrin α7; Fig. 2A). FAPs were cultured in GM for 5 d before adding DM plus Dex. We found that Dex vigorously converted FAPs into adipocytes vs. results in FAPs not exposed to Dex (Fig. 2C). Results of the expression of adipocyte mRNAs were consistent with the immunostaining: after 2–4 d exposure of FAPs to Dex, the mRNAs of C/EBPα, C/EBPδ, FABP4, and PPARγ were increased vs. cells not exposed to Dex (Fig. 2D). Protein levels of adipogenic genes (e.g., C/EBPα, PPARγ, and perilipin) in FAPs were increased, while the levels of the preadipocyte markers, PDGFRα or Pref-1, were decreased (Fig. 2E and ref. 40). Therefore, we conclude that Dex stimulates FAP differentiate into adipocytes in vitro.
In vivo, Dex stimulates FAPs to differentiate into adipocytes
To test whether FAPs, but not satellite cells, can differentiate into adipocytes in vivo, we transplanted 5 × 104 satellite cells (integrin-α7+/CD31−/CD45−/Scal-1−) isolated from muscles of EGFP-transgenic mice into TA muscles of control C57/BL6 mice. The control mice were not treated with Dex. We found that satellite cells from EGFP-transgenic mice engrafted into injured or uninjured TA muscles (Fig. 3A). Dex treatment decreased the incorporation of the transplanted satellite cells into muscle (Fig. 3B); it also inhibited the growth of new myofibers (Fig. 3C). Results from coimmunostaining of TA muscles with anti-GFP and anti-perilipin revealed no cells expressing both GFP and perilipin (data not shown). These results demonstrate that satellite cells do not differentiate into adipocytes in vivo even when Dex is added.
Figure 3.
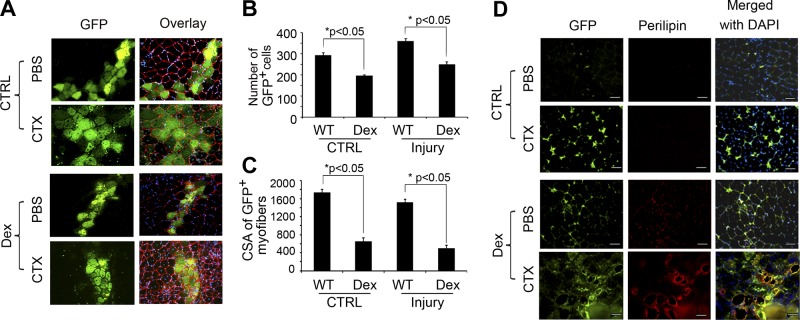
In vivo, Dex stimulates FAPs to differentiate into adipocytes. A) At 10 d after the transplantation of satellite cells isolated from EGFP mice into muscle, GFP-expressing myofibers were found along the needle track; laminin identifies the basement membranes of myofibers. B) Numbers of green myofibers were counted. C) Sizes of green myofibers were measured (n=100 myofibers). D) FAPs isolated from EGFP mice were injected into TA muscles of mice that had been treated with Dex or CTX to induce muscle injury (bottom panels). Cross-sections were immunostained with anti-perilipin (red) and GFP. Scale bars = 50 μm.
To determine whether FAPs differentiate into adipocytes in mouse muscle, we isolated FAPs from muscles of EGFP-transgenic mice (i.e., CD45−/CD31−/α7-integrin−/Sca-1+). The isolated FAPs were implanted into uninjured or injured TA muscles of mice that were being treated with or without Dex. At 10 d following transplantation of the FAPs, we observed that in uninjured muscles of control mice, the transplanted FAPs (GFP+ cells) were located in the interstitial spaces but not in myofibers (Fig. 3D, top panels). These FAPs also did not express perilipin, α-SMA or PDGFRα, but were terminal deoxynucleotidyl transferase dUTP nick-end labeling positive (TUNEL+; data not shown). In uninjured muscles of Dex-treated mice, the FAP cells from EGFP mice expressed PDGFRα and low levels of perilipin, and were TUNEL− (Fig. 3D, bottom middle panels). In injured muscles, the transplanted FAPs from EGFP mice were more plentiful vs. results from uninjured muscles (Fig. 3D, top middle vs. top panels). In injured muscles of Dex-treated mice, transplanted FAPs were positive for perilipin (Fig. 3D, bottom panels). These results indicate that Dex stimulates the differentiation of FAPs into adipocytes in injured muscle (Fig. 3D); however, FAPs that do not proliferate or differentiate will undergo apoptosis.
Dex stimulates FAP proliferation in vivo and in vitro
To examine the in vivo role of Dex in the functions of FAPs, we coimmunostained cross-sections of muscle with anti-PDGFRα (FAPs exclusively express PDGFRα) and anti-laminin (to show muscle myofiber structure) (14, 15). Dex treatment of mice with uninjured muscles did not change the number of PDGFRα+ cells (Fig. 4A, top panels). In contrast, at 3 d post-injury, there were more PDGFRα+ cells in TA muscles vs. results from uninjured muscles (Fig. 4A, bottom panela). To examine whether this increase in PDGFRα+ arose from proliferating FAPs at 3 d after injury of TA muscle of control or Dex-treated mice, we coimmunostained cross sections of TA muscles with anti-PDGFRα+ and Ki67 (a cell proliferation marker). The proliferation of FAPs was increased with Dex treatment (Fig. 4B).
Figure 4.

Dex stimulates FAP proliferation in vivo and in vitro. A) FAPs were evaluated by immunostaining muscle cross-sections with anti- PDGFRα and anti-laminin (uninjured muscles) or with anti-eMyHC (injured muscles). In sections from Dex-treated and control mice, means of PDGFRα cells per view were counted (right panel). *P < 0.05 vs. no injury/no Dex; #P < 0.05 vs. Dex no injury. B) FAP proliferation at 3 d after muscle injury was evaluated by coimmunostaining of cross-sections of muscles with anti-Ki67 and anti-PDGFRα. Right panel shows percentage of double-positive cells vs. PDGFRα+ cells. *P < 0.05 vs. control (Ctrl), no Dex. C) Cultured FAPs were stimulated with Dex (1 μM) for 24 h and then immunostained with anti-Ki67. Right panel shows proliferation rates as percentage of Ki67+ cells to total number of cells (n=3 repeats). *P < 0.05 vs. control, no Dex.
To document that Dex increases FAP proliferation, we isolated FAPs and cultured them with Dex for 24 h; we then immunostained them with anti-Ki67. The percentage of Ki67+ cells was significantly increased when the cells were treated with Dex (Fig. 4C). We thus concluded that Dex stimulates FAP proliferation in vitro and in vivo (Fig. 4).
Dex induces adipocyte formation in injured muscle via IL-4-dependent mechanism
Since injury stimulates infiltration of inflammatory cells into muscles, while Dex suppresses inflammation (35), we examined how muscle injury and Dex change the manner of inflammatory cell infiltration. Previously, we reported that Dex suppresses the mRNAs of cytokines and chemokines (IL-6, TNF-α, RANTES, and CXCL16) in injured muscles of mice (35). Dex treatment of mice inhibits the mRNA of IL-4 in injured muscles (Fig. 5A). To test whether the decrease in IL-4 was contributing to Dex-induced adipogenesis in injured muscle, we examined the injured muscles of IL-4-KO mice. At 5 or 7 d postinjury, H&E staining of cross-sections of muscle found fewer central nuclear myofibers in IL-4-KO mice vs. events in control mice (Fig. 5B). There were also fewer eMyHC+ cells in injured muscle of IL-4-KO mice (Fig. 5C). These results indicated a decrease in the maturation of myofibers. In contrast, perilipin+ adipocytes were more plentiful in injured muscles of IL-4-KO mice vs. results in control mice (Fig. 5C). Consistent with these immunostaining results, at 5 or 7 d postinjury we found increased mRNA expression of adipocyte genes PPARγ and C/EBPα in injured muscle of IL-4-KO mice vs. that in control mice (Fig. 5D). However, IL-4 KO did not affect IL-6 expression in injured muscle of mice (Fig. 5E).
Figure 5.

Dex induces adipocyte formation in injured muscle through suppression of IL-4 expression. A) IL-4 mRNA was evaluated from control or injured muscle of mice with or without Dex treatment by RT-PCR (n=3 mice/group). *P < 0.05 vs. control (Ctrl) no Dex. B) TA muscles of IL-4-KO and C57/BL6 mice were injured with CTX; 5 or 7 d later, the cross-sections of TA muscle were H&E stained. C) TA muscles of IL-4-KO and C57/BL6 mice were injured with CTX; 5 d later, the cross-sections of TA muscle were immunostained with anti-eMyHC (green) and perilipin (red). D) mRNAs of C/EBPα and PPARγ were measured from injured or control TA muscle of control or IL-4-KO mice (n=4)., *P < 0.05 vs. control mice. E) mRNA of IL-6 was measured from injured or control TA muscle of control or IL-4-KO mice (n=4),.*P < 0.05 vs. control mice.
IL-4 suppresses Dex-induced FAP differentiation into adipocytes
Next, we examined the possibility that IL-4 regulates the differentiation of FAPs into adipocytes (Fig. 6). FAP cells were cultured in growth medium for 7 d before switching to DM plus Dex and DM plus Dex and IL-4. The addition of IL-4 led to significantly fewer perilipin+ cells (Fig. 6A) as well as a suppressed level of PPARγ mRNA expression (Fig. 6B).
Figure 6.

IL-4 suppresses Dex-induced FAP adipogenic differentiation. A) FAPs were isolated by FACS and placed in GM for 7 d and then cultured in medium with Dex (1 μM) or Dex plus IL-4 for 4 d. Control (Ctrl) cells were continually cultured in GM. The fixed cells were immunostained with anti-perilipin (red) and DAPI (blue). B) In FAPs using similar treatment as in panel A, mRNA of PPARγ was evaluated by RT-PCR (n=3 repeat). *P < 0.05 vs. Ctrl. C) C57/BL6 mice were treated with Dex for 4 d and followed treatment with Dex plus IL-4 for 1 d before TA muscles were injured by CTX injection. At 8 d after muscle injury, cryo-cross-sections of TA muscles were H&E stained. D) Cross-sections of injured TA muscle from panel C were coimmunostained with anti-perilipin (red) plus anti-eMyHC (green). Scale bars = 50 μm. E) Top panel shows representative Western blots show the levels of C/EBPα and PPARγ in injured muscle of mice treated with Dex or Dex plus IL-4 at different days after injury. Bottom panel shows density (n=4). *P < 0.05 vs. Dex alone. F) FAPs were transfected with lentivirus of shRNA of IL-4R, or shRNA CTRL. After 4 d, cells were treated with Dex adipogenic differentiation medium with or without IL-4 for 7 d, and cells were photographed under light microscopy. G) Representative Western blots show levels of IL-4R in the cells from panel F.
To examine whether similar events occur in vivo, we injected C57/BL6 mice with Dex; 2 d later, we began daily injections of Dex plus IL-4 for 10 d. Muscles were then injured on d 3 after Dex plus IL-4 injection. On H&E-stained slides taken 8 d after TA muscle injury, injections of Dex plus IL-4 were shown to increase muscle regeneration (i.e., myofibers with central nuclei in H&E staining) compared to results in pair-fed control mice injected with Dex alone (Fig. 6C). Administration of Dex plus IL-4 significantly decreased perilipin+ cells in the injured muscles (Fig. 6D) and suppressed the PPARγ and C/EBPα proteins (Fig. 6E).
To further explore this connection, we then knocked down the expression of the IL-4 receptors in cultured FAPs using a ShRNA lentivirus expressing IL-4R. After 3 d, cells were treated with either Dex or Dex plus IL-4. In cells where the IL-4 receptor was knocked down, the administration of IL-4 did not block adipocyte formation, whereas IL-4 did inhibit Dex-induced adipogenesis in control cells (Fig. 6F). The level of IL-4R was evaluated by Western blots (Fig. 6G). These results indicate that IL-4 signaling blocks the differentiation of FAPs into adipocytes.
DISCUSSION
Earlier, we reported that Dex impairs the activation of satellite cells, leading to decreased muscle regeneration (35). In this study, we examined whether FAPs are the source of Dex-induced adipogenesis in injured muscles. This study question was put forward in order to examine the impaired muscle regeneration observed in patients with DMD or during normal aging in whom muscle regeneration is often accompanied by the infiltration of adipocytes and fibroblasts in muscles (1, 41, 42). We have found that Dex stimulates the proliferation and differentiation of FAPs into adipocytes in vitro and in vivo (Figs. 3 and 4). Besides the identification of the source of adipocyte production, we have shown that IL-4 mediates the transformation of FAPs to adipocytes. We have also shown that Dex treatment of satellite cells does not stimulate their differentiation into adipocytes (Figs. 2 and 3). We chose to study FAPs because they reportedly facilitate muscle regeneration following injury (15). In addition, it is reported that FAPs can contribute to adipogenesis in glycerol-induced injury to muscles (14, 43). Potentially, the adipogenesis in these responses could be attributed to high blood glucose or to the activation of innate immunity (28, 44). Since our study results of muscle injury indicate that IL-4 can block adipogenesis, stimulation of IL-4 signaling could be a target for inhibiting adipocyte formation in injured muscles (Fig. 6).
Recently, we reported that GCs can impair satellite cell function and suppress muscle regeneration (35). GCs have also been shown to inhibit osteogenesis of human bone marrow stromal (45) and can stimulate a pluripotent cell line, C3H10T1/2, to undergo adipogenesis (46). In certain inherited diseases, such as DMD, muscle inflammation occurs and muscle tissue is lost, frequently being replaced by fat and fibrotic tissue. Inflammation in patients with DMD is often treated with prednisone or other GCs. From our results, it is tempting to suggest an increase in muscle adipose tissue in patients with DMD is linked to GC treatment. This hypothesis could be tested in the future.
Since Dex stimulates adipogenic differentiation of FAPs in injured but not in uninjured muscles, there must be activation of a second signal in response to muscle injury. This could be the type 2 innate signal by which muscle injury rapidly recruits inflammatory cells to infiltrate the muscle. Previously, we found suppression of macrophages, macrophage polarization and/or neutrophils inhibits muscle regeneration (37, 47). On the other hand, infiltration of eosinophils could be the source of the increase in IL-4 in injured muscles. Administration of Dex would diminish the infiltration of eosinophils and, hence, the production of IL-4 (31, 48). In this case, diminished production of IL-4 would stimulate adipocyte formation and the accumulation in injured muscle (Fig. 6 and ref. 28).
In summary, our results combined with the results of other researchers confirm that in response to muscle injury, an increase in GCs will stimulate FAPs to differentiate into adipocytes that then accumulate in muscle (14, 15). This resulting adipogenesis in muscle leads to negative downstream health effects, such as insulin resistance and muscle weakness, both of which are common sequelae observed in aging patients or those with muscle dystrophy. Targeting IL-4-induced intracellular signaling in catabolic conditions could provide new therapeutic strategies to reduce the health burden associated with adipogenesis in muscles.
Acknowledgments
The authors thank Dr. William E. Mitch for his critical review.
This work was supported by the generous support of Dr. and Mrs. Harold Selzman, the Norman S. Coplon extramural research grants from Satellite Health and the American Diabetic Association (1–11-BS-194) to L.Z., U.S. National Institutes of Health (NIH) grant R37 DK37175, and the Cytometry and Cell Sorting Core at Baylor College of Medicine, with funding from the NIH (AI036211, CA125123, and RR024574), and the expert assistance of Joel M. Sederstrom. The work was supported in part by the Pilot/Feasibility Program of the Diabetes Research Center at Baylor College of Medicine, with funding from the NIH (P30 DK079638).
Footnotes
- bFGF
- basic fibroblast growth factor
- CTX
- cardiotoxin
- Dex
- dexamethasone
- DM
- differentiation medium
- DMD
- Duchenne muscular dystrophy
- DMEM
- Dulbecco's modified Eagle medium
- EGFP
- enhanced green fluorescent protein
- eMyHC
- embryonic myosin heavy chain
- FAP
- fibro/adipogenic progenitor
- FBS
- fetal bovine serum
- GC
- glucocorticoid
- GM
- growth medium
- H&E
- hematoxylin and eosin
- IL-4
- interleukin 4
- IL-4R
- interleukin 4 receptor
- IMAT
- intramuscular adipose tissue
- KO
- knockout
- PBS
- phosphate-buffered saline
- PDGFRα
- platelet-derived growth factor receptor α
- PFA
- paraformaldehyde
- TA
- tibialis anterior
- Sca-1
- stem cell antigen 1
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick-end labeling
REFERENCES
- 1. Goodpaster B. H., Wolf D. (2004) Skeletal muscle lipid accumulation in obesity, insulin resistance, and type 2 diabetes. Pediatr. Diabetes. 5, 219–226 [DOI] [PubMed] [Google Scholar]
- 2. Goldspink G., Fernandes K., Williams P. E., Wells D. J. (1994) Age-related changes in collagen gene expression in the muscles of mdx dystrophic and normal mice. Neuromuscul. Disord. 4, 183–191 [DOI] [PubMed] [Google Scholar]
- 3. Pastoret C., Sebille A. (1995) mdx mice show progressive weakness and muscle deterioration with age. J. Neurol. Sci. 129, 97–105 [DOI] [PubMed] [Google Scholar]
- 4. Tuttle L. J., Sinacore D. R., Mueller M. J. (2012) Intermuscular adipose tissue is muscle specific and associated with poor functional performance. J. Aging Res. 2012, 172957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Delmonico M. J., Harris T. B., Visser M., Park S. W., Conroy M. B., Velasquez-Mieyer P., Boudreau R., Manini T. M., Nevitt M., Newman A. B., Goodpaster B. H. (2009) Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am. J. Clin. Nutr. 90, 1579–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Young H. E., Steele T. A., Bray R. A., Hudson J., Floyd J. A., Hawkins K., Thomas K., Austin T., Edwards C., Cuzzourt J., Duenzl M., Lucas P. A., Black A. C., Jr. (2001) Human reserve pluripotent mesenchymal stem cells are present in the connective tissues of skeletal muscle and dermis derived from fetal, adult, and geriatric donors. Anat. Rec. 264, 51–62 [DOI] [PubMed] [Google Scholar]
- 7. Da Silva M. L., Chagastelles P. C., Nardi N. B. (2006) Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 119, 2204–2213 [DOI] [PubMed] [Google Scholar]
- 8. Asakura A., Komaki M., Rudnicki M. (2001) Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation 68, 245–253 [DOI] [PubMed] [Google Scholar]
- 9. Seale P., Asakura A., Rudnicki M. A. (2001) The potential of muscle stem cells. Dev. Cell 1, 333–342 [DOI] [PubMed] [Google Scholar]
- 10. Asakura A., Seale P., Girgis-Gabardo A., Rudnicki M. A. (2002) Myogenic specification of side population cells in skeletal muscle. J. Cell Biol. 159, 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jackson K. A., Mi T., Goodell M. A. (1999) Hematopoietic potential of stem cells isolated from murine skeletal muscle. Proc. Natl. Acad. Sci. U. S. A. 96, 14482–14486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matsuzaki Y., Kinjo K., Mulligan R. C., Okano H. (2004) Unexpectedly efficient homing capacity of purified murine hematopoietic stem cells. Immunity 20, 87–93 [DOI] [PubMed] [Google Scholar]
- 13. Starkey J. D., Yamamoto M., Yamamoto S., Goldhamer D. J. (2011) Skeletal muscle satellite cells are committed to myogenesis and do not spontaneously adopt nonmyogenic fates. J. Histochem. Cytochem. 59, 33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uezumi A., Fukada S., Yamamoto N., Takeda S., Tsuchida K. (2010) Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 12, 143–152 [DOI] [PubMed] [Google Scholar]
- 15. Joe A. W., Yi L., Natarajan A., Le G. F., So L., Wang J., Rudnicki M. A., Rossi F. M. (2010) Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 12, 153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hilton T. N., Tuttle L. J., Bohnert K. L., Mueller M. J., Sinacore D. R. (2008) Excessive adipose tissue infiltration in skeletal muscle in individuals with obesity, diabetes mellitus, and peripheral neuropathy: association with performance and function. Phys. Ther. 88, 1336–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rask E., Olsson T., Soderberg S., Andrew R., Livingstone D. E., Johnson O., Walker B. R. (2001) Tissue-specific dysregulation of cortisol metabolism in human obesity. J. Clin. Endocrinol. Metab. 86, 1418–1421 [DOI] [PubMed] [Google Scholar]
- 18. Duclos M., Gatta B., Corcuff J. B., Rashedi M., Pehourcq F., Roger P. (2001) Fat distribution in obese women is associated with subtle alterations of the hypothalamic-pituitary-adrenal axis activity and sensitivity to glucocorticoids. Clin. Endocrinol. (Oxf.) 55, 447–454 [DOI] [PubMed] [Google Scholar]
- 19. Andrew R., Gale C. R., Walker B. R., Seckl J. R., Martyn C. N. (2002) Glucocorticoid metabolism and the metabolic syndrome: associations in an elderly cohort. Exp. Clin. Endocrinol. Diabetes 110, 284–290 [DOI] [PubMed] [Google Scholar]
- 20. Fang C. H., James H. J., Ogle C., Fischer J. E., Hasselgren P. O. (1995) Influence of burn injury on protein metabolism in different types of skeletal muscle and the role of glucocorticoids J. Am. Coll. Surg. 180, 33–42 [PubMed] [Google Scholar]
- 21. Cristancho A. G., Lazar M. A. (2011) Forming functional fat: a growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 12, 722–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tauchmanova L., Rossi R., Biondi B., Pulcrano M., Nuzzo V., Palmieri E. A., Fazio S., Lombardi G. (2002) Patients with subclinical Cushing's syndrome due to adrenal adenoma have increased cardiovascular risk. J. Clin. Endocrinol. Metab. 87, 4872–4878 [DOI] [PubMed] [Google Scholar]
- 23. Ross E. J., Linch D. C. (1982) Cushing's syndrome–killing disease: discriminatory value of signs and symptoms aiding early diagnosis. Lancet 2, 646–649 [DOI] [PubMed] [Google Scholar]
- 24. Hu Z., Wang H., Lee I. H., Du J., Mitch W. E. (2009) Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J. Clin. Invest. 119, 7650–7659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Asensio C., Muzzin P., Rohner-Jeanrenaud F. (2004) Role of glucocorticoids in the physiopathology of excessive fat deposition and insulin resistance. Int. J. Obes. Relat. Metab. Disord. 28(Suppl 4), S45–S52 [DOI] [PubMed] [Google Scholar]
- 26. Gounarides J. S., Korach-Andre M., Killary K., Argentieri G., Turner O., Laurent D. (2008) Effect of dexamethasone on glucose tolerance and fat metabolism in a diet-induced obesity mouse model. Endocrinology 149, 758–766 [DOI] [PubMed] [Google Scholar]
- 27. Korach-Andre M., Gao J., Gounarides J. S., Deacon R., Islam A., Laurent D. (2005) Relationship between visceral adiposity and intramyocellular lipid content in two rat models of insulin resistance. Am. J. Physiol. Endocrinol. Metab. 288, E106–E116 [DOI] [PubMed] [Google Scholar]
- 28. Heredia J. E., Mukundan L., Chen F. M., Mueller A. A., Deo R. C., Locksley R. M., Rando T. A., Chawla A. (2013) Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153, 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tidball J. G., Villalta S. A. (2010) Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. 298, R1173–R1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mito N., Hosoda T., Kato C., Sato K. (2000) Change of cytokine balance in diet-induced obese mice. Metabolism 49, 1295–1300 [DOI] [PubMed] [Google Scholar]
- 31. Lamas A. M., Leon O. G., Schleimer R. P. (1991) Glucocorticoids inhibit eosinophil responses to granulocyte-macrophage colony-stimulating factor. J. Immunol. 147, 254–259 [PubMed] [Google Scholar]
- 32. Chang Y. H., Ho K. T., Lu S. H., Huang C. N., Shiau M. Y. (2012) Regulation of glucose/lipid metabolism and insulin sensitivity by interleukin-4. Int. J. Obes. (Lond.) 36, 993–998 [DOI] [PubMed] [Google Scholar]
- 33. Ricardo-Gonzalez R. R., Red E. A., Odegaard J. I., Jouihan H., Morel C. R., Heredia J. E., Mukundan L., Wu D., Locksley R. M., Chawla A. (2010) IL-4/STAT6 immune axis regulates peripheral nutrient metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. U. S. A. 107, 22617–22622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. May R. C., Kelly R. A., Mitch W. E. (1986) Metabolic acidosis stimulates protein degradation in rat muscle by a glucocorticoid-dependent mechanism. J. Clin. Invest. 77, 614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dong Y., Pan J. S., Zhang L. (2013) Myostatin suppression of Akirin1 mediates glucocorticoid-induced satellite cell dysfunction. PLoS One 8, e58554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dong Y., Lakhia R., Thomas S. S., Dong Y., Wang X. H., Silva K. A., Zhang L. (2013) Interactions between p-Akt and Smad3 in injured muscles initiate myogenesis or fibrogenesis. Am. J. Physiol. Endocrinol. Metab. 305, E367–E375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang L., Ran L., Garcia G. E., Wang X. H., Han S., Du J., Mitch W. E. (2009) Chemokine CXCL16 regulates neutrophil and macrophage infiltration into injured muscle, promoting muscle regeneration. Am. J. Pathol. 175, 2518–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang L., Wang X. H., Wang H., Du J., Mitch W. E. (2010) Satellite cell dysfunction and impaired IGF-1 signaling cause CKD-induced muscle atrophy. J. Am. Soc. Nephrol. 21, 419–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang L., Du J., Hu Z., Han G., Delafontaine P., Garcia G., Mitch W. E. (2009) IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J. Am. Soc. Nephrol. 20, 604–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smas C. M., Chen L., Zhao L., Latasa M. J., Sul H. S. (1999) Transcriptional repression of pref-1 by glucocorticoids promotes 3T3-L1 adipocyte differentiation. J. Biol. Chem. 274, 12632–12641 [DOI] [PubMed] [Google Scholar]
- 41. Mann C. J., Perdiguero E., Kharraz Y., Aguilar S., Pessina P., Serrano A. L., Munoz-Canoves P. (2011) Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 1, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakamura K., Nakano S., Miyoshi T., Yamanouchi K., Matsuwaki T., Nishihara M. (2012) Age-related resistance of skeletal muscle-derived progenitor cells to SPARC may explain a shift from myogenesis to adipogenesis. Aging (Albany, NY) 4, 40–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Uezumi A., Ito T., Morikawa D., Shimizu N., Yoneda T., Segawa M., Yamaguchi M., Ogawa R., Matev M. M., Miyagoe-Suzuki Y., Takeda S., Tsujikawa K., Tsuchida K., Yamamoto H., Fukada S. (2011) Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell Sci. 124, 3654–3664 [DOI] [PubMed] [Google Scholar]
- 44. Aguiari P., Leo S., Zavan B., Vindigni V., Rimessi A., Bianchi K., Franzin C., Cortivo R., Rossato M., Vettor R., Abatangelo G., Pozzan T., Pinton P., Rizzuto R. (2008) High glucose induces adipogenic differentiation of muscle-derived stem cell. Proc. Natl. Acad. Sci. U. S. A. 105, 1226–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carcamo-Orive I., Gaztelumendi A., Delgado J., Tejados N., Dorronsoro A., Fernandez-Rueda J., Pennington D. J., Trigueros C. (2010) Regulation of human bone marrow stromal cell proliferation and differentiation capacity by glucocorticoid receptor and AP-1 crosstalk. J. Bone Miner. Res. 25, 2115–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Feldman B. J., Streeper R. S., Farese R. V., Jr., Yamamoto K. R. (2006) Myostatin modulates adipogenesis to generate adipocytes with favorable metabolic effects. Proc. Natl. Acad. Sci. U. S. A. 103, 15675–15680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang L., Dong Y., Dong Y., Cheng J., Du J. (2012) Role of integrin-beta3 protein in macrophage polarization and regeneration of injured muscle. J. Biol. Chem. 287, 6177–6186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schleimer R. P., Bochner B. S. (1994) The effects of glucocorticoids on human eosinophils. J. Allergy Clin. Immunol. 94, 1202–1213 [DOI] [PubMed] [Google Scholar]