

Summary
Controlled activation of the Aurora kinases regulates mitotic progression in normal cells. Overexpression and hyperactivation of the Aurora-A and -B kinases play a leading role in tumorigenesis, inducing aneuploidy and genomic instability. In squamous cell carcinomas of the head and neck (SCCHN), overexpression of Aurora-A is associated with decreased survival, and reduction of Aurora-A and -B expression inhibits SCCHN cell growth and increases apoptosis. In this article, we provide a basic overview of the biological functions of Aurora kinases in normal cells and in cancer, and review both small studies and high throughput datasets that implicate Aurora-A, particularly, in the pathogenesis of SCCHN. Early phase clinical trials are beginning to evaluate the activity of small molecule inhibitors of the Aurora kinases. We summarize the state of current trials evaluating Aurora inhibitors in SCCHN, and discuss rational directions for future drug combination trials and biomarkers for use with Aurora-inhibiting agents.
Introduction
Squamous cell carcinomas of the head and neck (SCCHN) encompass a heterogeneous group of malignancies involving the oral cavity, oropharynx, larynx and hypopharynx. Incidence of SCCHN worldwide accounts for more than 550,000 malignancies, and in the United States, SCCHN occurs in an estimated 52,000 patients annually.[1, 2] Use of tobacco and alcohol has traditionally been implicated as causative risk factors in the pathogenesis of SCCHN. However, it is now known that infection with oncogenic HPV is also a leading risk factor specifically for oropharyngeal disease[3], with the incidence of HPV-positive (HPV+) SCCHN increasing in epidemic proportions.[4] While HPV+ oropharynx cancer typically has a high chance of response to treatment and a good prognosis [5-7], HPV-negative (HPV-) disease, with an incidence rate of 1.0 per 100,000 in the United States [4], is notoriously refractory to the current standard of chemotherapy and radiation. In addition, the treatment options for recurrent/metastatic disease are limited, with a median survival of 10 months.[8] Thus, there is a need for the study of novel therapeutic approaches in SCCHN to improve these abysmal results.
Standard chemotherapies for SCCHN include platinum and taxanes, which exhibit direct cytotoxic effects. Targeted therapies are emerging as potent tools for many cancers, and have begun to impact therapeutic management of SCCHN. As one example, the epidermal growth factor receptor is overexpressed on over 90% of SCCHN cells and targeting this receptor is now a standard therapeutic approach.[8, 9] However, the single agent activity of the monoclonal antibody cetuximab, which targets EGFR, is seen in only about 10% of SCCHN.[10] A growing body of literature indicates that Aurora kinases are potentially valuable targets in multiple malignancies, and are actionable given the development of several Aurora kinase inhibitors. This review will focus on the role of Aurora kinases in cancer development, particularly emphasizing data regarding the role of Aurora in SCCHN.
Biological Rationale for Aurora kinase inhibition in SCCHN
Aurora kinase function in normal cells and tumors
The Aurora serine-threonine kinases are important for cell cycle regulation. The Aurora kinase family was initially discovered in 1995 and includes Aurora-A, -B and -C in mammals.[11] Each of these kinases play multiple roles in the regulation of cell division, including most importantly mitotic entry, assembly of the microtubule spindle, and completion of cytokinesis. Aurora-A predominantly localizes to the centrosome, and regulates centrosome maturation, entry into mitosis, formation and function of the bipolar spindle, and cytokinesis. During the S-phase, Aurora-A starts to accumulate at the centrosomes, with expression peaking in late G2. Aurora-A supports the centrosomal maturation process, necessary to allow nucleation of microtubules to form the mitotic spindle, and also for the centrosome to act as a signaling platform for mitotic regulators [12, 13]. During G2, Aurora-A physically associated with another critical mitotic kinase, PLK1, [14-16], which promotes the recruitment of Aurora-A to centrosome and activation of the CDK-activating phosphatase CDC25B (cell division cycle 25B) leading to mitotic entry [17]. Aurora-B, localized at the centromere kinetochore, is a chromosomal passenger protein that contributes to appropriate attachment and alignment of the mitotic spindle through interaction with a distinct set of partners.[18] Aurora-B and -C share similar substrates and functions and likely complement each other; however, as Aurora-C expression is predominantly restricted to the testis, it has attracted much less interest.[19, 20] The basic biology of Aurora-A and -B function has been extensively and recently reviewed.[21, 22]
Overexpression of Aurora kinases induces aneuploidy and genomic instability, which plays a leading role in the pathogenesis of malignancy for many types of tumor.[23] Aurora-A overexpression overrides the mitotic spindle checkpoint and leads to tetraploid cells with increased centrosome number that continue to divide in cells that lack a functioning G1 checkpoint and p53 pathway.[24, 25] The aneuploidy associated with overexpression of Aurora-A is by itself detrimental and triggers the mitotic checkpoints and apoptosis [25-27]. Aurora-A physically or functionally associate with many other key targets involved in tumorigenesis, with over 60 interacting partners have been defined (reviewed in [21, 28]). Key functional interactions include NMyc, IkBa, AKT, RalA, p53, and others (Figure 1). For example, Aurora-A phosphorylation of IκBα removes inhibition of NFκB, supporting its transcription of pro-survival genes [29]; Aurora-A-dependent phosphorylation of RalA stimulates cell migration. [30] Overexpression of Aurora-B correlates with poor outcomes in colon cancer, anaplastic thyroid cancer, and glioblastoma.[31-33] For Aurora-B, survivin is an important partner [34]. Finally, both Aurora kinases associate with Polo-like kinases and some additional partners critical for oncogenic activity. For example, one mechanism by which Aurora-A upregulation can contribute to cancer involves phosphorylation of p53 on S315, which increases MDM2-dependent degradation of p53 [35]; conversely, binding of p53 to Aurora-A can suppress Aurora-A activity, leading to elevated Aurora-A activation in p53 mutant tumors.[36] Aurora-B also phosphorylates p53, at S183, T211, and S215, accelerating its degradation through the polyubiquitination-proteasome pathway [37].
Figure 1. Localization and function of Aurora-A and Aurora-B.
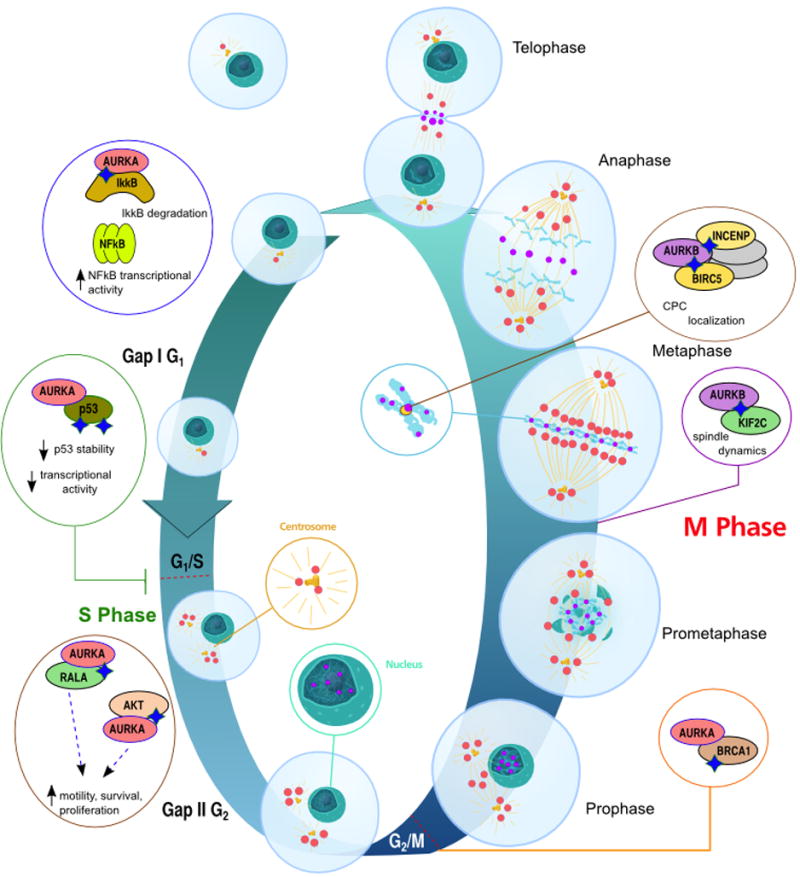
Representation of Aurora-A (red) and Aurora-B (purple) kinase association with intracellular structures during cell cycle. Association with partners of specific relevance for cancer are indicated in inset circles. DNA/chromatin is indicated in teal; the centrosome and mitotic spindle in gold. See text for details regarding functional interactions. CPC, Chromosomal passenger complex. Graphic representation adapted from http://en.wikipedia.org/wiki/File:Animal_cell_cycle.svg in Wikimedia Commons.
Aurora-A mRNA and protein expression
Elevated expression and enhanced activity of Aurora-A has been identified in solid tumors since the 1990s, and is often associated with spread of Aurora-A beyond its normal localization at the centrosomes throughout the cytoplasm and nucleus. In SCCHN cells, it has been shown that accumulation of Aurora-A in the nuclear compartment is essential for its role in oncogenesis.[38] In initial studies of the mechanism of overexpression, Reiter and colleagues used RT-PCR and immunohistochemistry to quantify Aurora-A mRNA and protein levels from 66 head and neck cancer tumors and normal squamous epithelium. This revealed a correlated increase between Aurora-A mRNA and protein expression (p=0.003) in tumors compared to normal tissue. Higher message levels of Aurora-A were associated with a decreased overall survival (p<0.001) and disease free survival (p=0.03).[39] In a retrospective analysis of laryngeal cancers, analysis of 37 paired samples by RT-PCR also revealed increased expression of Aurora-A mRNA was present in tumor versus normal tissue (p=0.001), correlated with lymph node metastasis and clinical stage. [40] An additional study used IHC and Western analysis to evaluate Aurora-A protein expression in 63 paired specimens of normal tissue and SCCHN tumors. 65% of the SCCHN tumors were strongly positive for Aurora-A, 19% moderately positive, and 15% negative, while the paired normal tissue had low overall staining and minimal nuclear staining for Aurora-A. Besides changes in expression, most tumors also showed elevated Aurora-A activity levels, based on in vitro kinase assays of 8 normal and SCCHN tumor paired specimens. [41] In explaining mechanism of overexpression, one small study reported amplification of Aurora-A in 4/11 oral squamous cell carcinomas.[42] Aurora-A expression is well known to be regulated by a balance of protein interactions that target it for, or protect it from, proteolytic degradation (reviewed in [21]). One study in SCCHN cell lines found constitutive phosphorylation of Ser-51 on Aurora-A resulted in decreased ubiquitylation and degradation.[43]
At Fox Chase Cancer Center, a retrospective analysis of Aurora-A and phospho-Aurora-A expression was performed on archival tissue from 89 surgical specimens of SCCHN tumors.[44] Utilizing the AQUA quantitative immunohistochemistry platform,[45] the median value was used as the cut point between high and low expression. p16 expression, a well characterized surrogate biomarker for the HPV+ disease [46-48], was used to sort tumors to 20 HPV+ (predominantly oropharyngeal) versus 69 subsets not associated with HPV (predominantly oral cavity and laryngeal sites). Among the tumors not associated with HPV, overall survival (OS) was greater among the tumors with low Aurora-A levels, with an OS of 93.6 months, compared to 35.9 months. Expression did not predict OS among the HPV+ tumors, but the lack of statistical power prohibited firm conclusions.
Aurora-B mRNA and protein expression
Few studies have characterized Aurora-B expression in SCCHN. In one analysis, forty SCCHN tumors were assayed for Aurora-B expression by IHC, compared to normal tissue controls. Higher Aurora-B was associated with a more poorly differentiated phenotype, increased multinuclear cells, increased lymph node metastases, and cellular proliferations.[49] Aurora-B function is closely linked to interaction with a partner, survivin, in the cell nucleus.[50] In SCCHN tissue, increased nuclear survivin expression correlated with increased Aurora-B (r=0.368, p=0.004), and was associated with a poorly differentiated phenotype (p=0.024) and decreased survival (p<0.0000).[34] These associations were not observed with cytoplasmic survivin expression.
Large scale, genomic and transcriptome profiles of SCCHN
As of 2013, large scale data sets describing DNA and mRNA changes that characterize SCCHN tumors removed by surgical resection, and HPV- SCCHN cell lines, are becoming available from resources maintained at the TCGA, the Broad Institute, and the Sanger Institute. A vast number of microarray data from smaller scale experiments, deposited in such public databases as Gene Expression Omnibus (GEO) and ArrayExpress, reached a “critical mass” for meaningful meta-analysis by such platforms as Oncomine and Genevestigator. In some databases such as the TCGA, changes in gene expression can be readily probed for predictive value in regard to overall survival. In general, this data confirms and expands observations made in the smaller studies, described above. (Figures 2, 3).
Figure 2. Expression and mutational profile of Aurora proteins in SCCHN tumors.

A. Publicly available microarray data from the Gene Expression Omnibus (GEO) [51] and ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) databases were used by Genevestigator portal https://genevestigator.com/[52] to generate expression meta-profiles, which make data highly comparable between different experiments. Expression levels of Aurora-A (AURKA) and Aurora-B (AURKB), and other genes noted in the text across a range of cancer types were plotted using Genevestigator software. X-axis, log2 scale. B. Analysis of the Head and Neck Squamous Cell Carcinoma subset of TCGA data[53] (from a provisional release of 310 samples) were analyzed using the cBio portal. [54] The displayed Oncoprint, generated by the cBio portal, shows individual samples as vertical columns, with the alterations on each gene as shown in the graphic legend embedded in the figure: not all specimens with only mutated p53, or with no mutations, are shown. High gene expression is defined as having a z score >2 based on reference (matched normal) sample population. IQR, interquartile range (defines expression of 25-75% of all genes) C. Kaplan-Meier plot comparing survival for high and low expression cohorts in regard to Aurora-A and Aurora-B, based on TCGA data.
Figure 3. A SCCHN network for Aurora-associated proteins.
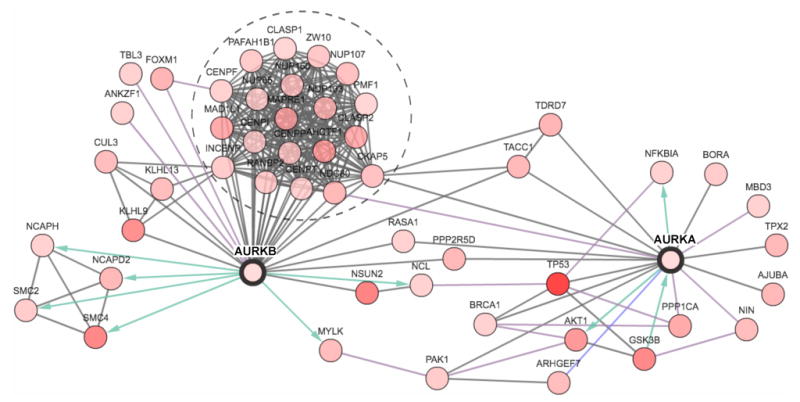
A network was generated around the Aurora-A (AURKA) and Aurora-B (AURKB) proteins was generated by the cBio portal [54] using data from Pathway Commons regarding protein-protein (HPRD), enzymatic (Reactome ) and pathway (NCI-Nature Pathway Interaction Database) interactions. Of all neighbors of the seed genes (AURKA & AURKB), the 50 most frequently altered in the SCCHN subset of TCGA data are shown. All proteins are identified by official gene symbol. Node coloring (white to red: low to high) reflects the combined frequency of all alterations for a given gene. Blue and green arrows represent enzyme-substrate interactions. The group of proteins circled by a dashed line represents a large group of coordinately regulated proteins found in a mitotic complex with Aurora-B at the centromere/kinetochore. Detailed information (i.e., copy number alterations, expression change, frequency of mutations) can be viewed for each gene in a network generated for AURKA and AURKB on http://www.cbioportal.org/.
Based on review of global mRNA expression data from 69 SCCHN tumors obtained using the Human Genome 47K array to benchmark expression between tumor types, the average expression of mRNA for Aurora-A is highest in SCCHN tumors compared to the other tumor types studied in a very large data set that included many different types of malignancies (Figure 2A). In contrast, Aurora-B expression did not differ significantly from the average across tumor types. Complementary analysis using the TCGA data set at the level of single SCCHN specimens indicates that among 292 SCCHN samples, only 7% overexpress Aurora-A (Figure 2B), but this overexpression is linked with inferior survival (Figure 2C). In contrast, while a similar fraction of tumors overexpress Aurora-B at some level, in this case there is no linkage to survival (Figure 2D).
These data confirm observations in earlier smaller studies indicating that Aurora kinases are typically upregulated at the level of mRNA or by post-translational control. No mutations or amplifications are identified in Aurora-A, and only limited mutation in Aurora-B (Figure 2B). We note that the analysis in Figure 2A indicates much higher mRNA levels of Aurora-A in tumor derived cell lines, including SCCHN cell lines, than in primary tumors. This striking difference suggests that selection of SCCHN and other tumors to survival on plastic may pose very significant selective pressure that alters tumor biology. Indeed, the recent study by Hennessey et al. notes profound differences in promoter methylation between SCCHN cell lines versus tumor and xenograft data, sounding a note of caution in extrapolating factors impacting mRNA expression from in vitro to clinical relevance.[55]
Increasingly, biological targets are studied as components of networks, rather than in isolation, to gain insight into coordinated or compensated changes that involve oncogenic changes in the target. For Aurora-A and –B, we initially compared profiles in the TCGA data set with the expression and mutational profiles of EGFR and p53 – both well-validated as linked to disease pathogenesis. No significant co-occurrence of alterations in p53 and aurora kinases was detected based on Fisher's exact test. We then performed similar analysis comparing expression of Aurora-A and –B with two well-characterized interacting partners, TPX2 (for Aurora-A) and survivin/BIRC5 (for Aurora-B). Here the same analysis identified significant co-occurrence of elevated expression levels of Aurora-A, Aurora-B, and TPX2 (p<0.05 in all cases). These may indicate specific functional interactions between this subgroup of proteins relevant to SCCHN; alternatively, this may reflect an underlying higher mitotic index of a subgroup of tumors, given the association of these three proteins with mitosis.
Taking this approach further, use of network-building tools in the TCGA resource highlights a subset of genes that physically and functionally interact with Aurora-A and –B, with a graphical representation in which intensity of node color represents the frequency of change in expression or mutation associated with SCCHN tumors (Figure 3). This immediately emphasizes proteins directly associated with the Auroras that are frequently affected in SCCHN: for instance, Aurora-A interactors GSK3β and AKT, which are well established as contributing to tumor pathology, or the Aurora-B interactors SMC4 and NSUN2/Misu. In silico analysis assessing whether these are positively or negatively correlated changes, coupled with functional analysis of phenotypic interactions, should seed a substantial number of testable hypotheses for new drug-drug combinations or prognostic biomarkers.
Finally, in all of these analyses, it is important to bear in mind that the Aurora kinases are subject to very extensive regulation post-translation, including phosphorylations that control their activity, and protein interactions that control rate of degradation. Some of the most important post-translational regulatory proteins, such as FBXW7 for Aurora-A, are emerging as tumor suppressors, and show variable expression in cancer; functional interactions require critical assessment at the protein level.[56] Given that the field of tumor proteomics has not yet caught up with genomics and transcriptomics, and faces technical hurdles that limit sensitivity, emphasizing the need for careful, low-throughput studies to evaluate mechanistic hypotheses.
Many studies have addressed the functional changes that accompany overexpression of Aurora-A in many types of tumor: results found in SCCHN cells generally are in accord with biological findings in other tumor types (reviewed in [21]). Overexpression is typically associated with elevated centrosome count and spindle abnormalities arising as a secondary consequence of defective cytokinesis, and associated with aneuploidy. In addition, elevated Aurora-A activity causes phosphorylation and increased activity of numerous proteins that are downstream effectors of EGFR and Ras, including RalA and AKT, as well as NF-kB, contributing to increased cell migration, invasion and survival. Cell cycle checkpoints are defective, in part because of negative regulation of p53 and p73 by Aurora-A. Cells overexpressing Aurora-A become addicted to its function, and susceptible to its inhibition. To emphasize results in SCCHN, one study found that HEp-2-S laryngeal cancer cells transfected with Aurora-A shRNA, knockdown of Aurora-A decreased the degree of aberrant chromosomal segregation. In addition, decreased expression of Aurora-A in these cells inhibited proliferation, migration and increased apoptosis.[57] This reduction in cell growth reflected accumulation in the G2-M phase, altered mitotic checkpoint response, and decreased expression of focal adhesion kinase (FAK), a tyrosine kinase that plays an important role in cell migration and the development of metastases. [57]
Recently, Chou and colleagues reported intriguing data which suggests that regulation of Aurora-A is in part mediated by the chromatin modifier protein, BMI1.[58] BMI1 supports cancer stem cell renewal and the epithelial-mesenchymal transition (EMT) that often precedes cancer invasion and metastasis. In hypopharyngeal cancer FaDu cells and other SCCHN cell lines, ectopic expression of BMI1 elevated Aurora-A mRNA and protein levels, while the reciprocal was seen following BMI1 knockdown. BMI1 regulation of Aurora-A required activation of the Akt-b-catenin pathway and was attenuated by a PI3K inhibitor. BMI1-induced EMT was mediated by Aurora-A interaction with GSK-3b and stabilization of Snail, an EMT regulator. In clinical SCCHN specimens, co-amplification of BMI1 and Aurora-A proteins was associated with a poor prognosis, suggesting that dual inhibition of these targets may be worthy of exploration.
Clinical Trials of Aurora inhibitors in SCCHN
Interest in targeting Aurora kinase has led to the development of multiple small molecule inhibitors of Aurora-A or B/C, as well as pan-Aurora inhibitors (Table 1).[59-63] Among a large number of agents evaluated preclinically, the pan-Aurora-Agents AMG900 and AT9283, the Aurora-A and -B targeting agents AZD1152, ZM447439, and ENMD2076, the Aurora-A-specific agent MLN8237 (alisertib), and the Aurora-B/C inhibitor GSK1070916A have progressed towards clinical trials.[21, 61, 64] The most advanced compound, MLN8237 (alisertib), is being evaluated in phase II trials for multiple types of cancer and phase III trials for the treatment of peripheral T-cell lymphoma [NCT01482962], and is generally well tolerated. [61, 62] Neutropenia, mucositis associated with nausea, fatigue and somnolence are the primary associated side effects, with neutropenia dose-limiting. [61, 62] These toxicities reflect the particular requirement for Aurora kinases in highly proliferative tissues.
Table 1.
| Agent | Target (IC50) |
|---|---|
| AMG900 | Aurora A (5 nM) |
| Aurora B (4 nM) | |
| Aurora C (1nM) | |
| MLN8237 | Aurora A (61 nM) |
| AZD1152 | Aurora A (1369 nM) |
| Aurora B (.36 nM) | |
| Aurora C (17 nM) | |
| PHA-739358 | Aurora A (13 nM) |
| Aurora B (79 nM0 | |
| Aurora C (61 nM) | |
| Tozasertib | Aurora A (0.6 nM) |
| Aurora B (18 nM) | |
| Aurora C (4.5 nM) | |
| AT9283 | Aurora A (3 nM) |
| Aurora B (3 nM) | |
| ENMD2076 | Aurora A (13 nM) |
| Aurora B (350 nM) |
The clinical data of Aurora inhibitors in SCCHN is currently limited. For alisertib, disease stability with over six cycles of treatment was observed in a limited number of patients with SCCHN. A follow up phase I study was conducted, which was followed by a planned phase 2 expansion which includes a cohort for head and neck cancer (NCT01045421).[61, 62] It seems likely that, as with many targeted therapies, development of future studies should focus on combinations. A number of current and proposed combinations are discussed below, and illustrated in Figure 4.
Figure 4. Aurora combination therapies and interrelation of drug targets.

Aurora-A (AURKA) and Aurora-B (AURKB) inhibitors (orange shading) block processes required for mitosis. Productive therapeutic combinations involve small molecules and antibodies targeting proteins influencing cell proliferation and survival signaling, or cytotoxics inducing DNA damage and mitotic checkpoints (drugs indicated in pink shading). See text for details.
Another ongoing study in SCCHN involves the combination of cetuximab, alisertib, and radiation for patients with newly diagnosed SCCHN. The concept for this study arose from a preclinical, functional genomics screen of a protein-interaction enriched EGFR signaling network [65] that revealed synthetic lethality between anti-EGFR agents and two related scaffolding proteins, NEDD9 (a pro-metastatic factor in SCCHN [66] and other tumor types) and its paralog BCAR1.[67] As a drug target closely interacting with NEDD9, Aurora-A was evaluated for functional interaction with EGFR; dual inhibition with small molecules showed synergistic effect in multiple cell lines. [26, 65] Of clinical benefit, cetuximab, targeting EGFR, and MLN8237, targeting Aurora-A, exhibit non-overlapping toxicities. A phase I study (NCT01540682) is the first in the clinical setting to study dual inhibition of Aurora-A and EGFR with these agents in the curative setting for SCCHN, while an additional phase I trial evaluates the effect of dual inhibition in non-small cell lung cancer (NSCLC), comprising adenocarcinoma, large cell undifferentiated cancer, and squamous cell carcinomas (NCT01471964).
A second independent study has shown benefit of dual inhibition of Aurora kinases and EGFR in SCCHN, in this case using the pan-Aurora inhibitor R763 and the EGFR inhibitor cetuximab. [68] This combination indicated additive inhibition of growth of SCCHN cell lines compared to monotherapy, and resulted in a disruption of cytokinesis and increased apoptosis. Interestingly, this study found additional benefit in dual inhibition of Aurora-A and -B versus Aurora-A over treatment with MLN8237, setting the stage for a new clinical trial proposal. Another interesting feature of this study was the observation that in a retrospective analysis of 180 cases, dual overexpression of EGFR and Aurora-A was a negative prognostic factor, compared to tumors with low expression of both of these biomarkers (p=0.024).[68] However, potency of the R763/cetuximab combination was independent of the degree of EGFR expression, suggesting translation of expression levels to patient selection for trials will not be straightforward.
Additional trials for Aurora inhibitor combinations now in progress for other cancer types suggest some potentially useful concepts for SCCHN. The idea of suppressing the RAS/RAF/MEK/ERK pathway in combination with an Aurora-A inhibitor is now being tested in a Phase I clinical trial (NCT01613261) in patients with non-hematological malignancies. In vitro studies [69] demonstrated that the combination of a MEK inhibitor, TAK-733, and alisertib/MLN8237 significantly reduced the ability of the cells to re-enter the cell cycle after one mitosis compared to either drug alone, thus supporting an idea for potential cooperation of the two pathways in driving cancer cell proliferation. Another study addressed pazopanib (Votrient) a VEGFR family inhibitor which also has a beneficial off-target activity against the Aurora-A and -B kinases.[70] A recent clinical trial in anaplastic thyroid carcinoma (ATC) demonstrated minimal activity of pazopanib monotherapy.[71] However, synergistic pazopanib combination with paclitaxel produced a durable response in one patient via a mechanism that involved an irreversible mitotic catastrophe in ATC cells, potentially due in part to Aurora-A inhibition.[70] A similar concept is now being tested in a Phase I clinical trial of the alisertib and pazopanib combination (NCT01639911) in solid tumors.
Concepts for future trials of combination strategies involving Aurora inhibitors
Docetaxel
Combination of Aurora kinase inhibitors with drugs interfering with mitotic spindle formation is mechanistically based on the established role of Aurora kinases A and B in the regulation of mitosis. Blocking both Aurora kinase and the microtubules assembly/disassembly process with microtubule poisons, taxanes and vinca alkaloids, provides an irreversible mitotic arrest. Inhibitors of aurora kinase, such as PF-03814735, have also shown to cause additive growth inhibition in mouse colorectal xenografts when given in combination with docetaxel.[63] This is consistent with results from Anand et al., in which overexpression of Aurora-A induces increased resistance to taxanes, such as paclitaxel in HeLa cells.[24] The interactions between taxanes and Aurora inhibitors have been demonstrated as synergistic in multiple cancer preclinical models including colon [72, 73], ovarian [15], B-cell lymphomas [74], esophageal and gastric cancer [75]. Early phase clinical trials exploring combination of Aurora inhibitors with taxanes are ongoing with myelosuppression being the main toxicity of the combination therapy ([76] and NCT01094288). As synergistic cytotoxic effects have been observed from combining siRNA depletion of Aurora-A with paclitaxel treatment of SCCHN cells[41], it is very reasonable to explore a MLN8237-docetaxel combination in SCCHN.
SRC
NEDD9 and BCAR1, noted above as physically and functionally interacting with Aurora-A, also physically interact directly with SRC [67], while functional analyses indicates that loss of NEDD9 sensitizes cells to loss or inhibition of Src family kinases [77, 78]. Subsequent direct testing of pharmacological inhibitors of SRC and Aurora kinases was strongly synergistic in multiple ovarian epithelial carcinoma cell lines, and potently killed cells entering mitosis [79] via interference with a post mitotic reattachment, and selective removal of aneuploid cell populations. Inhibition of SRC with dasatinib has already been shown to yield therapeutic benefit in SCCHN in treatment of cetuximab-resistant tumors.[80] Evaluation of Aurora-And SRC inhibition in this population of patients may yield enhanced therapeutic benefit.
HDACs
Although most studies of Aurora-A focus on its role at mitosis, a growing body of work supports additional functions in non-mitotic cells (reviewed in [21]). Significantly, a study identifying a role for Aurora-A kinase in regulating the cell cilium identified a direct interaction with HDAC6, a member of the histone deacetylase family that targets microtubules.[81] Inhibition of HDAC6 activity by the small molecule inhibitor tubacin or depletion of HDAC6 by siRNA in each case resulted in cilium stabilization.[81] Beyond these functions being relevant to cilia, entry into mitosis also involves acetylation of ciliary tubulin [82]. Reciprocally, in cancer cells, mRNA expression of Aurora-A and -B is influenced by the status of histone acetylation [83]. Combined pharmacological blockade of Aurora kinases and histone deacetylase has been shown as synergistic in preclinical models of B-cell lymphoma [84] and has led to the testing of this combination therapy in the patients with Hodgkin, B-Cell non-Hodgkin, or peripheral T-cell lymphoma (NCT01567709). It may be promising to explore this approach in SCCHN.
Survivin
As noted above, the function of Aurora-B involves close interactions with survivin, a nuclear and cytoplasmic localized protein which inhibits apoptosis and regulates mitosis. [34, 85] The survivin inhibitor YM155 is currently in clinical trials (e.g., with paclitaxel and carboplatin in patients with solid tumors (phase I) and advanced NSCLC (phase II) (NCT01100931)). A combination of YM155 with either a pan-Aurora targeted agent, or an Aurora-B-specific inhibitor such as GSK1070916A, may merit further exploration in SCCHN.
Potential strategies for biomarker development
Both early focused experiments and large datasets from the TCGA and other resources make it very clear that SCCHN exhibits heterogeneity, and that therapeutic gains may be realized by optimizing treatments based on the molecular characteristics of each tumor. As there is increasing clinical development of new targeted agents, and new combinations of existing agents, it will be essential to develop biomarkers to help determine which patients will benefit from inhibition of the Aurora kinases.[86] One current potential predictive biomarker includes Aurora-A expression levels. However, while increased Aurora-A mRNA and protein expression have been associated with decreased survival (Figure 3, and [39, 44]), it is not known if its overexpression is a biomarker of responsiveness to Aurora kinase inhibition. For example, EGFR expression and response to cetuximab has been extensively studied, and EGFR expression has not been validated as a predictive biomarker for EGFR inhibition.[87]
Since Aurora kinase inhibition results in increased cellular accumulation in mitosis, both the mitotic index and/or phosphorylated histone H3 levels have been studied as possible pharmacodynamic markers to assess biologic activity of novel Aurora inhibitors.[60-62] An increase in mitotic index, as measured in skin biopsies, was observed twenty-four hours after dosing with the Aurora-A-specific compound MLN8237. Histone H3 phosphorylation, which is associated with Aurora-B activity, has also been evaluated as a pharmacodynamic marker in phase I trials.[88-90] The degree of mitotic cell chromosome alignment and spindle bipolarity has been evaluated as well, and these are less apparent after treatment with MLN8054, a preclinical Aurora-A inhibitor.[59] Blockade of Aurora kinase activity triggers p53-mediated apoptosis in cells containing wild type p53, while loss of p53 upregulates Aurora-A. Given the frequent occurrence of p53 mutations in a large subset of SCCHN tumors (Figure 3), this immediately parses SCCHN tumors into groups with and without these signaling relationships. Prolonged mitosis following Aurora-B blockade has been shown to activate p53 in the subsequent G1 phase via a new mechanism involving telomeres de-protection [91], while Aurora-B directly phosphorylates p53 to induce its degradation [37]. Together, these findings suggest investigation of p53 status as a biomarker of response to Aurora-A inhibitors in SCCHN.
Finally, efforts towards personalized medicine are increasingly incorporating consideration of drug targets as components of linear signaling cascades, or dense signaling networks.[92] Mutational activation of a downstream effector predicts resistance to inhibition of an upstream activator, while mutational activation or overexpression of an upstream component suggests sensitivity to inhibition of a downstream effector. Concretely, in the signaling relationship in which EGFR signals through Ras to PI3K and to Raf and MEK, an activating mutation of Ras predicts resistance to EGFR targeting therapies [93],while tumors with overexpressed EGFR are sometimes responsive to inhibitors of PI3K or Raf. As shown in Figure 3, Aurora-A and -B are components of increasingly well studied signaling networks, with some of their direct interactive partners or upstream activators overexpressed or mutated in a subset of tumors. Study of such interactive maps should provide fertile ground for the generation of hypotheses regarding predictive biomarkers, which could then be evaluated in the context of a prospective clinical trial. Clear near-term candidates would include expression of TPX2, NEDD9, or other post-translational activators for Aurora-A, and survivin expression for Aurora-B.
Conclusions
The prognosis of patients with HPV- SCCHN and recurrent disease still remains poor, and there is a pressing need for new treatment options. The Aurora kinases are emerging as a potential therapeutic target in the treatment of SCCHN, with the first trials now underway for Aurora inhibitors. Evaluation of the combination of these agents with cytotoxic therapies or EGFR inhibitors is occurring in the near horizon. Studies in the middle distance will likely focus on the integration of Aurora kinase inhibitors into combinations involving EGFR inhibitors, taxanes, or radiotherapy for the treatment of SCCHN, and determine whether inhibition of Aurora-A, Aurora-B, or the combination is most effective. In subsequent years, the integration of genomic scale resources should yield considerable insight into whether integrated patterns of gene expression accompany sensitivity to Aurora inhibitors, and may yield robust biomarkers for response, and suggestions for effective therapeutic combinations involving these inhibitors.
Acknowledgments
We would like to acknowledge the following funding sources which have contributed to our work:
CA 06927 NIH core grant
Pew Charitable Fund
Concetta Greenberg donation to Fox Chase Cancer Center
Spore P50CA083638 (EA Golemis)
Tobacco Settlement Fund (EA Golemis and I Astsaturov)
NIHRO1 CA 50633
U54 CA 149147
NIH K22 CA 160725
R21 Ca 164205 (I Astsaturov)
Fox Chase Cancer Center Head and Neck Keystone
R01 CA63366
Footnotes
Search strategy and selection criteria: English language journals and articles were reviewed. Databases included Medline, PubMed, as well as those maintained by the TCGA, the Broad Institute, the Sanger Institute, Gene Expression Omnibus (GEO) and ArrayExpress.
Clinical trial information was obtained from clinicaltrials.gov and the American Society of Clinical Oncology abstract site. Trials included those that involved inhibition of aurora kinase pathways. Search terms included aurora and head and neck.
Contributors: RM – literature search and review, writing of manuscript
IGS – implementation and analysis of genomic database search, figures
BB – guidance with literature and manuscript review, interpretation of literature
IA - literature search and review, writing of manuscript, figures
EAG – manuscript writing, manuscript review, interpretation of literature
Author conflicts of interest: Mehra - spouse is employee of GlaxoSmithKline
Burtness - funding from Boehringer Ingelheim and Genentech
Golemis - no conflicts of interest
Astsaturov - no conflicts of interest
Serebriiskii – no conflicts of interest
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA: a cancer journal for clinicians. 2005 Mar-Apr;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Desantis C, Virgo K, Stein K, Mariotto A, Smith T, et al. Cancer treatment and survivorship statistics, 2012. CA: a cancer journal for clinicians. 2012 Jul;62(4):220–41. doi: 10.3322/caac.21149. [DOI] [PubMed] [Google Scholar]
- 3.D'Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, Koch WM, et al. Case-control study of human papillomavirus and oropharyngeal cancer. The New England journal of medicine. 2007 May 10;356(19):1944–56. doi: 10.1056/NEJMoa065497. [DOI] [PubMed] [Google Scholar]
- 4.Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011 Nov 10;29(32):4294–301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. Journal of the National Cancer Institute. 2008 Feb 20;100(4):261–9. doi: 10.1093/jnci/djn011. [DOI] [PubMed] [Google Scholar]
- 6.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. The New England journal of medicine. 2010 Jul 1;363(1):24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rischin D, Young RJ, Fisher R, Fox SB, Le QT, Peters LJ, et al. Prognostic significance of p16INK4A and human papillomavirus in patients with oropharyngeal cancer treated on TROG 02.02 phase III trial. J Clin Oncol. 2010 Sep 20;28(27):4142–8. doi: 10.1200/JCO.2010.29.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. The New England journal of medicine. 2008 Sep 11;359(11):1116–27. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 9.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006 Feb 9;354(6):567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 10.Herbst RS, Arquette M, Shin DM, Dicke K, Vokes EE, Azarnia N, et al. Phase II multicenter study of the epidermal growth factor receptor antibody cetuximab and cisplatin for recurrent and refractory squamous cell carcinoma of the head and neck. J Clin Oncol. 2005 Aug 20;23(24):5578–87. doi: 10.1200/JCO.2005.07.120. [DOI] [PubMed] [Google Scholar]
- 11.Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995 Apr 7;81(1):95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 12.Palazzo RE, Vogel JM, Schnackenberg BJ, Hull DR, Wu X. Centrosome maturation. Current topics in developmental biology. 2000;49:449–70. doi: 10.1016/s0070-2153(99)49021-0. [DOI] [PubMed] [Google Scholar]
- 13.Mahen R, Venkitaraman AR. Pattern formation in centrosome assembly. Curr Opin Cell Biol. 2012 Feb;24(1):14–23. doi: 10.1016/j.ceb.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 14.Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008 Jun 20;320(5883):1655–8. doi: 10.1126/science.1157425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008 Sep 4;455(7209):119–23. doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]
- 16.Chan EH, Santamaria A, Sillje HH, Nigg EA. Plk1 regulates mitotic Aurora A function through betaTrCP-dependent degradation of hBora. Chromosoma. 2008 Oct;117(5):457–69. doi: 10.1007/s00412-008-0165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, Dozier C, et al. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J Cell Sci. 2004 May 15;117(Pt 12):2523–31. doi: 10.1242/jcs.01108. [DOI] [PubMed] [Google Scholar]
- 18.Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. The Journal of cell biology. 2003 Apr 28;161(2):281–94. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nature reviews. 2003 Nov;4(11):842–54. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- 20.Kimmins S, Crosio C, Kotaja N, Hirayama J, Monaco L, Hoog C, et al. Differential functions of the Aurora-B and Aurora-C kinases in mammalian spermatogenesis. Molecular endocrinology (Baltimore, Md. 2007 Mar;21(3):726–39. doi: 10.1210/me.2006-0332. [DOI] [PubMed] [Google Scholar]
- 21.Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL, Jr, Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013 Feb;70(4):661–87. doi: 10.1007/s00018-012-1073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lampson MA, Cheeseman IM. Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends in cell biology. 2011 Mar;21(3):133–40. doi: 10.1016/j.tcb.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marumoto T, Zhang D, Saya H. Aurora-A - a guardian of poles. Nat Rev Cancer. 2005 Jan;5(1):42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- 24.Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer cell. 2003 Jan;3(1):51–62. doi: 10.1016/s1535-6108(02)00235-0. [DOI] [PubMed] [Google Scholar]
- 25.Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. The EMBO journal. 2002 Feb 15;21(4):483–92. doi: 10.1093/emboj/21.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pugacheva EN, Golemis EA. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nature cell biology. 2005 Oct;7(10):937–46. doi: 10.1038/ncb1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pugacheva EN, Golemis EA. HEF1-aurora A interactions: points of dialog between the cell cycle and cell attachment signaling networks. Cell Cycle. 2006 Feb;5(4):384–91. doi: 10.4161/cc.5.4.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL, Jr, Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013 Feb;70(4):661–87. doi: 10.1007/s00018-012-1073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Briassouli P, Chan F, Savage K, Reis-Filho JS, Linardopoulos S. Aurora-A regulation of nuclear factor-kappaB signaling by phosphorylation of IkappaBalpha. Cancer research. 2007 Feb 15;67(4):1689–95. doi: 10.1158/0008-5472.CAN-06-2272. [DOI] [PubMed] [Google Scholar]
- 30.Lim KH, Brady DC, Kashatus DF, Ancrile BB, Der CJ, Cox AD, et al. Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Molecular and cellular biology. 2010 Jan;30(2):508–23. doi: 10.1128/MCB.00916-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katayama H, Ota T, Jisaki F, Ueda Y, Tanaka T, Odashima S, et al. Mitotic kinase expression and colorectal cancer progression. Journal of the National Cancer Institute. 1999 Jul 7;91(13):1160–2. doi: 10.1093/jnci/91.13.1160. [DOI] [PubMed] [Google Scholar]
- 32.Sorrentino R, Libertini S, Pallante PL, Troncone G, Palombini L, Bavetsias V, et al. Aurora B overexpression associates with the thyroid carcinoma undifferentiated phenotype and is required for thyroid carcinoma cell proliferation. The Journal of clinical endocrinology and metabolism. 2005 Feb;90(2):928–35. doi: 10.1210/jc.2004-1518. [DOI] [PubMed] [Google Scholar]
- 33.Zeng WF, Navaratne K, Prayson RA, Weil RJ. Aurora B expression correlates with aggressive behaviour in glioblastoma multiforme. Journal of clinical pathology. 2007 Feb;60(2):218–21. doi: 10.1136/jcp.2006.036806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erpolat OP, Gocun PU, Akmansu M, Karakus E, Akyol G. High expression of nuclear survivin and Aurora B predicts poor overall survival in patients with head and neck squamous cell cancer. Strahlenther Onkol. 2012 Mar;188(3):248–54. doi: 10.1007/s00066-011-0042-7. [DOI] [PubMed] [Google Scholar]
- 35.Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004 Jan;36(1):55–62. doi: 10.1038/ng1279. [DOI] [PubMed] [Google Scholar]
- 36.Chen SS, Chang PC, Cheng YW, Tang FM, Lin YS. Suppression of the STK15 oncogenic activity requires a transactivation-independent p53 function. The EMBO journal. 2002 Sep 2;21(17):4491–9. doi: 10.1093/emboj/cdf409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gully CP, Velazquez-Torres G, Shin JH, Fuentes-Mattei E, Wang E, Carlock C, et al. Aurora B kinase phosphorylates and instigates degradation of p53. Proc Natl Acad Sci U S A. 2012 Jun 12;109(24):E1513–22. doi: 10.1073/pnas.1110287109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tatsuka M, Sato S, Kanda A, Miki T, Kamata N, Kitajima S, et al. Oncogenic role of nuclear accumulated Aurora-A. Mol Carcinog. 2009 Sep;48(9):810–20. doi: 10.1002/mc.20525. [DOI] [PubMed] [Google Scholar]
- 39.Reiter R, Gais P, Jutting U, Steuer-Vogt MK, Pickhard A, Bink K, et al. Aurora kinase A messenger RNA overexpression is correlated with tumor progression and shortened survival in head and neck squamous cell carcinoma. Clin Cancer Res. 2006 Sep 1;12(17):5136–41. doi: 10.1158/1078-0432.CCR-05-1650. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Chen X, Jin Y, Liu B, Zhou L. Overexpression of Aurora-A promotes laryngeal cancer progression by enhancing invasive ability and chromosomal instability. Eur Arch Otorhinolaryngol. 2012 Feb;269(2):607–14. doi: 10.1007/s00405-011-1629-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mazumdar A, Henderson YC, El-Naggar AK, Sen S, Clayman GL. Aurora kinase A inhibition and paclitaxel as targeted combination therapy for head and neck squamous cell carcinoma. Head Neck. 2009 May;31(5):625–34. doi: 10.1002/hed.21007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tatsuka M, Sato S, Kitajima S, Suto S, Kawai H, Miyauchi M, et al. Overexpression of Aurora-A potentiates HRAS-mediated oncogenic transformation and is implicated in oral carcinogenesis. Oncogene. 2005 Feb 3;24(6):1122–7. doi: 10.1038/sj.onc.1208293. [DOI] [PubMed] [Google Scholar]
- 43.Kitajima S, Kudo Y, Ogawa I, Tatsuka M, Kawai H, Pagano M, et al. Constitutive phosphorylation of aurora-a on ser51 induces its stabilization and consequent overexpression in cancer. PloS one. 2007;2(9):e944. doi: 10.1371/journal.pone.0000944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fidler C, Yang D, Zhu F, Mehra R, Astsaturov I, Ridge R, et al. Expression of Aurora A and Phospho-Aurora-A is predictive of survival in patients with head and neck cancer. AACR. 2012;2012 [Google Scholar]
- 45.Moeder CB, Giltnane JM, Moulis SP, Rimm DL. Quantitative, fluorescence-based in-situ assessment of protein expression. Methods in molecular biology (Clifton, NJ. 2009;520:163–75. doi: 10.1007/978-1-60327-811-9_12. [DOI] [PubMed] [Google Scholar]
- 46.Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. Journal of the National Cancer Institute. 2000 May 3;92(9):709–20. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 47.Weinberger PM, Yu Z, Haffty BG, Kowalski D, Harigopal M, Brandsma J, et al. Molecular classification identifies a subset of human papillomavirus--associated oropharyngeal cancers with favorable prognosis. J Clin Oncol. 2006 Feb 10;24(5):736–47. doi: 10.1200/JCO.2004.00.3335. [DOI] [PubMed] [Google Scholar]
- 48.Weinberger PM, Yu Z, Haffty BG, Kowalski D, Harigopal M, Sasaki C, et al. Prognostic significance of p16 protein levels in oropharyngeal squamous cell cancer. Clin Cancer Res. 2004 Sep 1;10(17):5684–91. doi: 10.1158/1078-0432.CCR-04-0448. [DOI] [PubMed] [Google Scholar]
- 49.Qi G, Ogawa I, Kudo Y, Miyauchi M, Siriwardena BS, Shimamoto F, et al. Aurora-B expression and its correlation with cell proliferation and metastasis in oral cancer. Virchows Arch. 2007 Mar;450(3):297–302. doi: 10.1007/s00428-006-0360-9. [DOI] [PubMed] [Google Scholar]
- 50.Bolton MA, Lan W, Powers SE, McCleland ML, Kuang J, Stukenberg PT. Aurora B kinase exists in a complex with survivin and INCENP and its kinase activity is stimulated by survivin binding and phosphorylation. Molecular biology of the cell. 2002 Sep;13(9):3064–77. doi: 10.1091/mbc.E02-02-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barrett T, Troup DB, Wilhite SE, Ledoux P, Evangelista C, Kim IF, et al. NCBI GEO: archive for functional genomics data sets--10 years on. Nucleic acids research. 2011 Jan;39(Database issue):D1005–10. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hruz T, Laule O, Szabo G, Wessendorp F, Bleuler S, Oertle L, et al. Genevestigator v3: a reference expression database for the meta-analysis of transcriptomes. Advances in bioinformatics. 2008;2008:420747. doi: 10.1155/2008/420747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.TCGA [Google Scholar]
- 54.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012 May;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hennessey PT, Ochs MF, Mydlarz WW, Hsueh W, Cope L, Yu W, et al. Promoter methylation in head and neck squamous cell carcinoma cell lines is significantly different than methylation in primary tumors and xenografts. PloS one. 2011;6(5):e20584. doi: 10.1371/journal.pone.0020584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lau AW, Fukushima H, Wei W. The Fbw7 and betaTRCP E3 ubiquitin ligases and their roles in tumorigenesis. Front Biosci. 2012 Jun;17:2197–212. doi: 10.2741/4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang H, Chen X, Liu B, Zhou L. Effects of stable knockdown of Aurora kinase A on proliferation, migration, chromosomal instability, and expression of focal adhesion kinase and matrix metalloproteinase-2 in HEp-2 cells. Mol Cell Biochem. 2011 Nov;357(1-2):95–106. doi: 10.1007/s11010-011-0879-1. [DOI] [PubMed] [Google Scholar]
- 58.Chou CH, Yang NK, Liu TY, Tai SK, Hsu DS, Chen YW, et al. Chromosome Instability Modulated by BMI1-AURKA Signaling Drives Progression in Head and Neck Cancer. Cancer research. 2013 Jan 15;73(2):953–66. doi: 10.1158/0008-5472.CAN-12-2397. [DOI] [PubMed] [Google Scholar]
- 59.Chakravarty A, Shinde V, Tabernero J, Cervantes A, Cohen RB, Dees EC, et al. Phase I assessment of new mechanism-based pharmacodynamic biomarkers for MLN8054, a small-molecule inhibitor of Aurora A kinase. Cancer research. 2011 Feb 1;71(3):675–85. doi: 10.1158/0008-5472.CAN-10-1030. [DOI] [PubMed] [Google Scholar]
- 60.Cohen RB, Jones SF, Aggarwal C, von Mehren M, Cheng J, Spigel DR, et al. A phase I dose-escalation study of danusertib (PHA-739358) administered as a 24-hour infusion with and without granulocyte colony-stimulating factor in a 14-day cycle in patients with advanced solid tumors. Clin Cancer Res. 2009 Nov 1;15(21):6694–701. doi: 10.1158/1078-0432.CCR-09-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dees EC, Cohen RB, von Mehren M, Stinchcombe TE, Liu H, Venkatakrishnan K, et al. Phase I Study of Aurora A Kinase Inhibitor MLN8237 in Advanced Solid Tumors: Safety, Pharmacokinetics, Pharmacodynamics, and Bioavailability of Two Oral Formulations. Clin Cancer Res. Aug 2; doi: 10.1158/1078-0432.CCR-12-0589. [DOI] [PubMed] [Google Scholar]
- 62.Dees EC, Infante JR, Cohen RB, O'Neil BH, Jones S, von Mehren M, et al. Phase 1 study of MLN8054, a selective inhibitor of Aurora A kinase in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2011 Apr;67(4):945–54. doi: 10.1007/s00280-010-1377-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jani JP, Arcari J, Bernardo V, Bhattacharya SK, Briere D, Cohen BD, et al. PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Molecular cancer therapeutics. 2010 Apr;9(4):883–94. doi: 10.1158/1535-7163.MCT-09-0915. [DOI] [PubMed] [Google Scholar]
- 64.Hardwicke MA, Oleykowski CA, Plant R, Wang J, Liao Q, Moss K, et al. GSK1070916, a potent Aurora B/C kinase inhibitor with broad antitumor activity in tissue culture cells and human tumor xenograft models. Molecular cancer therapeutics. 2009 Jul;8(7):1808–17. doi: 10.1158/1535-7163.MCT-09-0041. [DOI] [PubMed] [Google Scholar]
- 65.Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci Signal. 2010;3(140):ra67. doi: 10.1126/scisignal.2001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lucas JT, Jr, Salimath BP, Slomiany MG, Rosenzweig SA. Regulation of invasive behavior by vascular endothelial growth factor is HEF1-dependent. Oncogene. 2010 Aug 5;29(31):4449–59. doi: 10.1038/onc.2010.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tikhmyanova N, Little JL, Golemis EA. CAS proteins in normal and pathological cell growth control. Cell Mol Life Sci. 2010 Apr;67(7):1025–48. doi: 10.1007/s00018-009-0213-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoellein A, Pickhard A, von Keitz F, Schoeffmann S, Piontek G, Rudelius M, et al. Aurora kinase inhibition overcomes cetuximab resistance in squamous cell cancer of the head and neck. Oncotarget. 2011 Aug;2(8):599–609. doi: 10.18632/oncotarget.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Collins S, Blair D, Zarycki J, Szynal C, Gangolli E, Vincent P, et al. A rationale for combining the targeted investigational agents TAK-733, a MEK1/2 inhibitor, with alisertib (MLN8237), an Aurora A kinase inhibitor, for cancer therapy Cancer research. 2012 Apr 15;72(8) 2012. [Google Scholar]
- 70.Isham CR, Bossou AR, Negron V, Fisher KE, Kumar R, Marlow L, et al. Pazopanib enhances paclitaxel-induced mitotic catastrophe in anaplastic thyroid cancer. Science translational medicine. 2012 Jan;5(166):166ra3. doi: 10.1126/scitranslmed.3004358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bible KC, Suman VJ, Menefee ME, Smallridge RC, Molina JR, Maples WJ, et al. A multiinstitutional phase 2 trial of pazopanib monotherapy in advanced anaplastic thyroid cancer. The Journal of clinical endocrinology and metabolism. 2012 Sep;97(9):3179–84. doi: 10.1210/jc.2012-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arbitrario JP, Belmont BJ, Evanchik MJ, Flanagan WM, Fucini RV, Hansen SK, et al. SNS-314, a pan-Aurora kinase inhibitor, shows potent anti-tumor activity and dosing flexibility in vivo. Cancer Chemother Pharmacol. 2010 Mar;65(4):707–17. doi: 10.1007/s00280-009-1076-8. [DOI] [PubMed] [Google Scholar]
- 73.VanderPorten EC, Taverna P, Hogan JN, Ballinger MD, Flanagan WM, Fucini RV. The Aurora kinase inhibitor SNS-314 shows broad therapeutic potential with chemotherapeutics and synergy with microtubule-targeted agents in a colon carcinoma model. Mol Cancer Ther. 2009 Apr;8(4):930–9. doi: 10.1158/1535-7163.MCT-08-0754. [DOI] [PubMed] [Google Scholar]
- 74.Mahadevan D, Stejskal A, Cooke LS, Manziello A, Morales C, Persky DO, et al. Aurora A inhibitor (MLN8237) plus vincristine plus rituximab is synthetic lethal and a potential curative therapy in aggressive B-cell non-Hodgkin lymphoma. Clin Cancer Res. 2012 Apr 15;18(8):2210–9. doi: 10.1158/1078-0432.CCR-11-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sehdev V, Katsha A, Ecsedy J, Zaika A, Belkhiri A, El-Rifai W. The combination of alisertib, an investigational Aurora kinase A inhibitor, and docetaxel promotes cell death and reduces tumor growth in preclinical cell models of upper gastrointestinal adenocarcinomas. Cancer. 2013 Feb 15;119(4):904–14. doi: 10.1002/cncr.27801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Minton SE, LoRusso P, Lockhart AC, Saif M, Krishnamurthi SS, Pickett-Gies CA, et al. A phase I study of MK-5108, an oral aurora A kinase inhibitor, in both monotherapy and in combination with docetaxel in patients with advanced solid tumors. ASCO Meeting Abstracts. 2010 Jun 14;28(15_suppl):e13026. 2010. [Google Scholar]
- 77.Singh MK, Izumchenko E, Klein-Szanto AJ, Egleston BL, Wolfson M, Golemis EA. Enhanced genetic instability and dasatinib sensitivity in mammary tumor cells lacking NEDD9. Cancer research. 2010 Nov 1;70(21):8907–16. doi: 10.1158/0008-5472.CAN-10-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tikhmyanova N, Golemis EA. NEDD9 and BCAR1 negatively regulate E-cadherin membrane localization, and promote E-cadherin degradation. PloS one. 2011;6(7):e22102. doi: 10.1371/journal.pone.0022102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ratushny V, Pathak HB, Beeharry N, Tikhmyanova N, Xiao F, Li T, et al. Dual inhibition of SRC and Aurora kinases induces postmitotic attachment defects and cell death. Oncogene. 2012 Mar 8;31(10):1217–27. doi: 10.1038/onc.2011.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wheeler DL, Iida M, Kruser TJ, Nechrebecki MM, Dunn EF, Armstrong EA, et al. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer biology & therapy. 2009 Apr;8(8):696–703. doi: 10.4161/cbt.8.8.7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007 Jun 29;129(7):1351–63. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jackman M, Lindon C, Nigg EA, Pines J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat Cell Biol. 2003 Feb;5(2):143–8. doi: 10.1038/ncb918. [DOI] [PubMed] [Google Scholar]
- 83.Zhang XH, Rao M, Loprieato JA, Hong JA, Zhao M, Chen GZ, et al. Aurora A, Aurora B and survivin are novel targets of transcriptional regulation by histone deacetylase inhibitors in non-small cell lung cancer. Cancer biology & therapy. 2008 Sep;7(9):1388–97. doi: 10.4161/cbt.7.9.6415. [DOI] [PubMed] [Google Scholar]
- 84.Kretzner L, Scuto A, Dino PM, Kowolik CM, Wu J, Ventura P, et al. Combining histone deacetylase inhibitor vorinostat with aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer research. 2011 Jun 1;71(11):3912–20. doi: 10.1158/0008-5472.CAN-10-2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qi G, Kudo Y, Ando T, Tsunematsu T, Shimizu N, Siriwardena SB, et al. Nuclear Survivin expression is correlated with malignant behaviors of head and neck cancer together with Aurora-B. Oral Oncol. 2010 Apr;46(4):263–70. doi: 10.1016/j.oraloncology.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 86.Carpinelli P, Moll J. Aurora kinase inhibitors: identification and preclinical validation of their biomarkers. Expert opinion on therapeutic targets. 2008 Jan;12(1):69–80. doi: 10.1517/14728222.12.1.69. [DOI] [PubMed] [Google Scholar]
- 87.Licitra L, Storkel S, Kerr KM, Van Cutsem E, Pirker R, Hirsch FR, et al. Predictive value of epidermal growth factor receptor expression for first-line chemotherapy plus cetuximab in patients with head and neck and colorectal cancer: Analysis of data from the EXTREME and CRYSTAL studies. Eur J Cancer. 2012 Dec 19; doi: 10.1016/j.ejca.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 88.Arkenau HT, Plummer R, Molife LR, Olmos D, Yap TA, Squires M, et al. A phase I dose escalation study of AT9283, a small molecule inhibitor of aurora kinases, in patients with advanced solid malignancies. Ann Oncol. 2012 May;23(5):1307–13. doi: 10.1093/annonc/mdr451. [DOI] [PubMed] [Google Scholar]
- 89.Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, et al. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000 Aug 4;102(3):279–91. doi: 10.1016/s0092-8674(00)00034-9. [DOI] [PubMed] [Google Scholar]
- 90.Schoffski P, Jones SF, Dumez H, Infante JR, Van Mieghem E, Fowst C, et al. Phase I, open-label, multicentre, dose-escalation, pharmacokinetic and pharmacodynamic trial of the oral aurora kinase inhibitor PF-03814735 in advanced solid tumours. Eur J Cancer. 2011 Oct;47(15):2256–64. doi: 10.1016/j.ejca.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 91.Hayashi MT, Cesare AJ, Fitzpatrick JA, Lazzerini-Denchi E, Karlseder J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol. 2012 Apr;19(4):387–94. doi: 10.1038/nsmb.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bagnyukova TV, Serebriiskii IG, Zhou Y, Hopper-Borge EA, Golemis EA, Astsaturov I. Chemotherapy and signaling: How can targeted therapies supercharge cytotoxic agents? Cancer biology & therapy. 2010 Nov 1;10(9):839–53. doi: 10.4161/cbt.10.9.13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011 May 20;29(15):2011–9. doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]