

Abstract
McCune-Albright syndrome (polyostotic fibrous dysplasia, café-au-lait skin spots, and precocious puberty) is a genetically mosaic disorder with populations of mutant and normal cells in affected organs. Cushing syndrome, a rare feature of the condition, usually affects infants and is the result of corticotropin-independent primary bilateral adrenal disease, usually interpreted as nodular adrenocortical hyperplasia. In this study of 9 patients with Cushing syndrome and McCune-Albright syndrome, light microscopy revealed a characteristic bimorphic pattern of diffuse and nodular hyperplasia and a distinctive form of cortical atrophy with apparent zona glomerulosa hyperplasia in 8 patients, all very young. The pattern could be explained by the presence of a mosaic distribution of mutant and normal cells in the adrenal glands. The findings are different from those in inherited or other forms of genetically caused Cushing syndrome. The ninth patient, aged 17 years, had an adrenal adenoma and diffuse cortical hyperplasia in each adrenal gland.
Keywords: adrenal cortical atrophy, adrenal cortical hyperplasia, Cushing syndrome, GNAS1 mutation, McCune-Albright syndrome
In 1910, Cushing encountered a 17-year-old female patient with painful adiposity, hypertension, and other symptoms. In 1930, he saw another patient, a 30-year-old man, with the same findings. Cushing searched the literature for other patients with these unusual findings and found 10. He reported the 12 cases in 1932 in a landmark paper entitled “The Basophil Adenomas of the Pituitary Body and Their Clinical Manifestations (Pituitary Basophilism).”11 Today, we know that there are 2 distinct forms of the clinical conditions he described: one is pituitary dependent (Cushing disease) and the other is pituitary independent (Cushing syndrome). Cushing syndrome is almost always caused by excess secretion of cortisol either by a unilateral adrenal tumor (adenoma or carcinoma) or by 1 of several primary bilateral hyperplasias, some familial or syndrome related or both.
McCune-Albright syndrome is a rare, nonfamilial disorder reported almost simultaneously by McCune and Bruch,24 and Albright et al3 in 1937. Its principal features were osteitis fibrosa disseminata (later reclassified as polyostotic fibrous dysplasia), café-au-lait skin spots, and precocious puberty in female patients. Additional types of endocrine overactivity occurred occasionally, including gigantism, hyperthyroidism,15 and, infrequently, Cushing syndrome.20 Some cases featured hypophosphatemic rickets,2,22 hepatic dysfunction, cardiomegaly, and sudden death.33 Onset of the Cushing syndrome was usually in the first year of life and was very rarely congenital.26,31
There is considerable phenotypic variation in McCune-Albright syndrome. This led Happle16 to suggest that clinical variability and the absence of familial involvement could be explained by the presence of genetically different populations of cells existing side by side in the same organ (cellular mosaicism). In 1991, Weinstein et al35 discovered a constitutive activating mutation of the GNAS1 gene in adrenal nodules. Subsequently, the mutation was found in the pituitary, thyroid, testes, liver, and skin of affected patients.33
The anatomic basis of the Cushing syndrome has been attributed variously to bilateral nodular hyperplasia, 18 bilateral “atypical” adenomas,5 bilateral macronodular hyperplasia,14 and bilateral hyperplasia.21,29 In rare cases, the cause of hypercortisolism was apparently in 1 adrenal gland because Cushing syndrome resolved after unilateral adrenalectomy.14,32 Exceptionally, Cushing syndrome resolved spontaneously.13
In this study, we describe the adrenocortical histopathologic findings in 9 patients with McCune-Albright syndrome—associated Cushing syndrome. Eight patients had bilateral diffuse and nodular cortical hyperplasia and multifocal cortical atrophy; the ninth patient had a cortical adenoma and diffuse cortical hyperplasia in each adrenal gland.
PATIENTS AND METHODS
Patients
Nine patients with McCune-Albright syndrome and Cushing syndrome comprised the study group. The cases of 7 patients were previously reported.1,5,6,14,17,36 Additional clinical information, pathologic material, and follow-up in 6 of these cases were generously supplied by reporting investigators, the patients’ physicians, or the patients’ families. The tenth patient,23 a male infant with Cushing syndrome only, was included in the study because he had the same adrenal pathology as patients with McCune-Albright syndrome. He died suddenly at the age of 33 years; no explanation was found at autopsy.
Material for the study included (1) original pathology reports (6 cases), (2) hematoxylin and eosin-stained slides with 2 to 22 sections of the adrenal glands in the 4 patients who underwent bilateral adrenalectomy, 1 to 6 sections in 4 who had unilateral adrenalectomy, and 6 sections of the glands obtained at autopsy in 1 case (patient 6). One to 4 formalin-fixed, paraffin-embedded blocks were available in 3 cases. Seventeen sections of adrenal and 6 adrenal blocks were available in the tenth patient.
The following histochemical stains were obtained: hematoxylin and eosin, Masson trichrome, and reticulin. Immunostaining was performed with antibodies directed to the following: vimentin (Dual Env; PT link: 1/500 BRD; Dako; V9), synaptophysin (Ventana; CC1 mild; 1/50 BRD; Leica [Novocastro]; 27G12), inhibin-A (Advance; PT link; 1/60 BRD; AbD Serotec; R1), melan A (Advance; PT link; 1/500 BRD; Dako; A103), CD56 (Ventana; CC1 mild; 1/100 BRD; Dako; 123C3), and MIB1 (Ventana; CC1 mild; 1/100BRD; Dako; MIB-1). A polymer-based detection system was used.
For measuring the thickness of the cortex, the sections were viewed in an M20 macroscope (Wild Heerbrugg, Switzerland) equipped with a digital camera (ProgRes C3; Jenoptik, Jena, Germany). The images were digitized using image capture software (Prog Res; Jenoptik) and the thickness of cortex measured interactively with image analysis software (KS 400; Carl Zeiss, Inc, MicroImaging, Inc, Thornwood, NY).
Normal Infant Adrenal Glands
Adrenal glands, obtained at autopsy from 2 female infants, 1 and 3 months of age, who succumbed to sudden infant death syndrome, were studied as indicated above.
DNA Studies
DNA was extracted from frozen tissue or paraffin blocks in cases 1,2, and 10 as described previously.18 The GNAS1 gene was sequenced as described by Shenker et al33 and Weinstein et al.35 Sequencing for the PRKAR1A gene was also obtained in selected cases as described by Kirschner et al.18
RESULTS
Patients
Demographic and clinical findings are presented in Table 1. Clinical findings in 2 patients are illustrated in Figure 1. Results of adrenal imaging and laboratory testing are shown in Table 2. Treatment, initial pathologic diagnosis, follow-up, and GNAS1 status are summarized in Table 3.
TABLE 1.
Age at Onset of Clinical Features in 9 Patients With McCune-Albright Syndrome*
| Patient | Sex | Cushing Syndrome | Café-au-lait Skin Patches | Polyostotic Fibrous Dysplasia | Precocious Puberty | Other | Reference |
|---|---|---|---|---|---|---|---|
| 1 | F | Neonatal | Neonatal | Neonatal | Absent | Hyperthyroidism (neonatal), cholestasis, nephrocalcinosis, cardiac arrhythmia, microcephaly | Not reported |
| 2 | F | 4 mo | Neonatal | 6 mo | 6 mo | Hyperthyroidism (4 y) | Not reported |
| 3 | M | 14 mo | Neonatal | 22 mo | Absent | Liver dysfunction (2 mo) | 14 |
| 4 | M | 17 y | Birth | 3 y | 6 y | Microcephaly, acanthosis nigricans, pituitary adenoma, nodular, thyroid, cardiomegaly | 5 |
| 5 | F | 2 mo (in utero in retrospect) | 2 mo | 18 mo | 7 mo | Growth failure, renal defect (1 wk), hyperthyroidism, hepatic dysfunction (2 wk) | Not reported |
| 6 | F | Birth | Birth | Birth | Absent | Hyperthyroidism (birth) | 36 |
| 7 | M | 3 mo | Absent | 3 y | Absent | Liver dysfunction (3 mo), hypopituitarism (14 y) | 6 |
| 8 | M | 4 mo | Birth | 4 mo | Absent | None | 17 |
| 9 | F | 2 mo | 2 mo | 2 y | 2 mo | Hypophosphatemic osteomalacia (9 y), cerebral thrombosis (13 y) | 32 |
| 10 | M | 4 mo | Absent | Absent | Absent | Acanthosis nigricans | 22 |
Patients 1 through 9 had Cushing syndrome and McCune-Albright syndrome. Patient 10 had Cushing syndrome, but not McCune-Albright syndrome.
F indicates female; M, male.
FIGURE 1.

A, Case 7. Moon-face and facial plethora in a 4-month-old male infant. B, Case 9. Multiple café-au-lait patches in a 2-month-old female infant.
TABLE 2.
Adrenal Imaging and Laboratory Study Findings*
| Patient | Adrenal Imaging | Plasma Corticotropin | Serum Cortisol | Urinary Free Cortisol | Serum Cortisol Circadian Rhythm | High-Dose Dexamethasone Sppression Test |
|---|---|---|---|---|---|---|
| 1 | MRI: bilateral nodular enlargement | ↓ | ↑ | ↑ | NA | NA |
| 2 | CT: R adrenal 15 × 9 mm; L gland not seen | ↓ | ↑ | NA | Absent | NA |
| 3† | CT and MRI: asymmetrical bilateral enlargement, L>R | ↓ | ↑ | ↑ | Present‡ | Cortisol not suppressed |
| 4 | Angiogram: L vascular mass | NA | Normal | NA | NA | Cortisol not suppressed |
| 5 | NA | NA | ↑ | ↑ | NA | Cortisol not suppressed |
| 6 | Scintigram: marked uptake bilaterally, L>R | ↓ | NA | ↑ | Absent | NA |
| 7 | CT: bilateral enlargement | ↑ | ↑ | NA | NA | Cortisol not suppressed |
| 8 | NA | ↓ | ↑ | Absent | Cortisol not suppressed | |
| 9 | US and MRI: R, multinodular enlargment; L, normal | ↓ | ↑ | NA | Absent | Cortisol not suppressed |
| 10 | Aortogram: no adrenal enlargement | ↓ | ↑ | ↑ | NA | Cortisol not suppressed |
Patients 1 through 10 had Cushing syndrome and McCune-Albright syndrome. Patient 10 had Cushing syndrome, but not McCune-Albright syndrome.
Urinary aldosterone secretion was normal.
Following unilateral adrenalectomy.
↓ indicates decreased or undetectable; ↑, increased; CT, computed tomography; L, left; MRI, magnetic resonance imaging; NA, not available; NMR, nuclear magnetic resonance; R, right; US, ultrasonography.
TABLE 3.
Treatment, Initial Pathologic Diagnosis, GNAS1 Mutation, and Follow-up*
| Patient | Adrenalectomy (Age) | Initial Pathologic Diagnosis | GNAS1 Mutation | Follow-up |
|---|---|---|---|---|
| 1 | Bilateral (7 mo) | Nodular adrenocortical hyperplasia | R201H | Dead at 9 mo from complications of McCune-Albright and Cushing syndromes |
| 2 | Unilateral (7 mo) | NA | R201G | Thyrotoxicosis at 3 y;alive at 4 y; plasma cortisol elevated |
| 3 | Unilateral (25 mo) | Macronodular hyperplasia | R201H | Well at 5 y. Serum cortisol not suppressed by high-dose dexamethasone |
| 4 | Unilateral (17 y) | “Atypical” adenoma, 11 × 7 × 4 cm | Not tested | Sudden death at 18 y; unexplained at autopsy; cardiomegaly 870 g |
| 5 | Bilateral (3 mo). Preoperative ketaconazole | Nodular hyperplasia | Not tested | Sudden death at 2 y; cardiorespiratory arrest |
| 6 | Not done. Trilostane 30 mg/daily | Multinodular hyperplasia (autopsy) | Not tested | Dead at 4 mo; cardiac failure |
| 7 | Bilateral (6 mo) | Nonpigmented adrenocortical hyperplasia with nodular elements | R201C | Alive and well at 20 y |
| 8 | Bilateral (5 mo) | Bilateral nodular hyperplasia | No† | Sudden death at 5 mo; unexplained at autopsy |
| 9 | Unilateral (4 mo) | R: diffuse and nodular cortical hyperplasia without internodular atrophy. L (biopsy): no nodular cortical dysplasia | R201C | Unexplained death while in hospital for “tonsillitis” at 14 y |
| 10 | Bilateral (8 mo) | Diffuse and nodular hyperplasia | No | Sudden death at 33 y; unexplained at autopsy |
Patients 1 through 10 had Cushing syndrome and McCune-Albright syndrome. Patient 10 had Cushing syndrome but not McCune-Albright syndrome.
There was little nodular tissue in the specimen.
Pathology
Gross
The findings are presented in Table 4. With regard to the adrenal weights, a normal gland weighs about 4 gm at birth. Each gland loses up to 50% of its mass in the first month of life as a result of rapid involution of the fetal zone. Thereafter, the glands undergo further shrinkage, and weigh about 1 gm at 12 months of age.27 The gross appearance of the glands in 2 cases is illustrated in Figure 2.
TABLE 4.
Adrenocortical Pathologic Findings*
| Adrenal Weight, g
|
Microscopic Pathology
|
||||||
|---|---|---|---|---|---|---|---|
| Patient | Right | Left | Gross Pathology | Cortical Nodules |
Cortical Hyperplasia |
Cortical Atrophy |
Fetal Cortex |
| 1 | 4.2 | 4.7 | Slightly nodular, hyperplastic dark green parenchyma | Yes | Acidophilic; cortex up to 1 mm thick | Yes | Present |
| 2 | NA | NA | NA | No | Acidophilic | Yes | Present |
| 3 | NA | NA | Cortex thickened; yellow nodules, with darker center, some extra-adrenal | Yes | Clear cell >acidophilic | Yes | Absent |
| 4 | 9.5 | NA | Marked increase in cortical tissue | Absent. Adenoma bilaterally | Acidophilic | No | Absent |
| 5 | 3.5 | 4.0 | Smooth outline preserved; cut surface reddish-brown with alternating tan-to-green nodules | Yes, vague | Acidophilic; cortex up to 1.2 mm thick | Yes | Present |
| 6 | 3 | 3.2 | NA | Yes | Acidophilic | Yes | Present. Cytomegaly and cortical cysts |
| 7 | 1.5 | 2.0 | R: Tan nodule, 4 × 2 × 2 cm | Yes | Clear cell >Acidophilic | Yes | |
| 8 | 1.8 | 2.0 | Small yellow nodules, 2–3.5 mm. Residual cortex brown | Yes | Acidophilic | Yes | Present |
| 9 | 4 | Not resected | Yellow-gray cortex 0.05 cm thick | Yes | Acidophilic with cortex up to 0.6 mm thick† | Yes | Present |
| 10 | 2.6 | 2.3 | Cortices slightly thickened | Yes | Clear and acidophilic | Yes | Absent |
Patients 1 through 9 had Cushing syndrome and McCune-Albright syndrome. Patient 10 had Cushing syndrome, but not McCune-Albright syndrome.
Prominent accumulations of zona glomerulosa-type cells around periphery of gland.
NA indicates not available; R, right.
FIGURE 2.

Gross appearance of adrenal gland [case 3 (A) and case 5 (B)]. A, The external surface was yellow and partially covered with fat. Several nodules were visible. B, The reddish-brown thickened cortex (arrow) featured several “tan-to-green nodules.”
Light Microscopy
Hyperplastic cortex and nodules
The cortex ranged from 0.7 to 2.1 mm, mean 0.95 mm, in thickness. It was initially composed of outer clear and inner acidophilic layers. Eventually, the one or the other cell type predominated, and the cortex was completely replaced by enlarged cells with finely granular acidophilic cytoplasm or cells with vacuolated cytoplasm (Fig. 3). Occasionally, there was a mixture of the 2 cell types. Some of these hyperplastic zones bulged laterally pushing aside and compressing the adjacent cortex. Their continued expansion resulted in development of nodules, some of which had a very thin incomplete fibrous capsule connected with the adrenal capsule. There was no lipochrome in the hyperplastic cortex or in the nodules. The hyperplastic cells did not extend into the medulla.
FIGURE 3.

Cortical hyperplasia and cortical atrophy [A and B (case 7); C and D (case 8)]. A, Low-power micrograph showed diffuse clear cell hyperplasia with variable thickening of the cortex. Hyperplasia was punctuated by compressed and stretched zones of atrophy (arrows). B, Intermediate-power magnification area in box in panel A. The cortex lacked zonation and was composed of hyperplastic clear cells punctuated by 3 narrow tongues of atrophic cortex. Fetal cortex with vacuolar degeneration was present centrally. C, Cortex had a variegated appearance due to alternating areas of acidophilic cell hyperplasia interspersed in areas of cortical atrophy (outer light and inner dark layers). D, Intermediate-power magnification of area in box in panel C. Compact cell hyperplasia had 2 tinctorial appearances; moderately strong (arrows) and more intense (lower center).
Atrophic Cortex
The atrophic cortex was often visible on scanning microscopic examination because of its (1) thinness (0.2 to 0.5 mm, mean 3.5 mm in 7 cases), (2) histologic appearance (distinct outer light and inner dark layers), and (3) very sharp border with the acidophilic or clear cell hyperplastic cortex (Fig. 3). The amount of atrophic cortex varied considerably from case to case: the longest stretch found was 20 mm in length, but it usually occupied no >10% of the total non-nodular cortex.
The subcapsular outer clear zone of the atrophic cortex was composed of columns of cells several cells wide with vacuolated cytoplasm and a central hyperchromatic nucleus without a recognizable nucleolus. This layer was sharply delineated from an underlying layer of slightly smaller cells arranged in rows perpendicular to the gland capsule. The nuclei were similar to or slightly smaller than those in the outer layer, and occupied almost the entire cell, except for a barely discernible rim of finely granular eosinophilic cytoplasm. Frequently, the nuclei were molded in a direction parallel to the gland capsule (Fig. 4).
FIGURE 4.

Cortical atrophy (case 7). Cortex was 0.3-mm thick. There was a subcapsular layer of vacuolated cells with round dense nuclei and inner layer of rows of cells with molded nuclei and scarcely visible cytoplasm.
The atrophic zones were (1) flanked by hyperplastic cortex without transition between them, (2) in hyperplastic cortex as wedges or narrow tongues with the bases of the wedges at the gland capsule and the tapering end point deep in the cortex, and (3) in the nodules as isolated streaks (Fig. 3).
Fetal Cortex
Degenerating and viable fetal cortical remnants were found in 6 cases. The former featured vacuolar degeneration, ectatic vessels, and prominent hypocellular stroma that stained light blue with Masson trichrome. An oval area of cytomegaly (2 × 0.8 mm) and irregularly shaped subcapsular microcysts containing stringy mucoid material (Fig. 5) were present in 1 case (patient 1).
FIGURE 5.
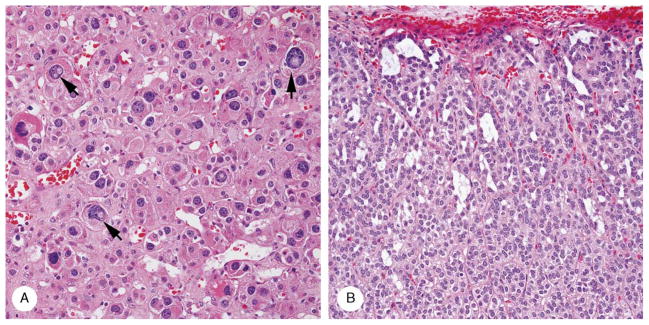
Cortical cytomegaly and microcysts (case 1). A, Cytomegaly featured large cells with eosinophilic cytoplasm and large to huge nuclei, several with intranuclear vacuoles (arrows). B, A series of polymorphic spaces (microcysts) separate the 2-cell columns of the subcapsular cortex.
Capsule
The capsule of the gland was usually intact. Commonly, it contained rows and little clusters of small cortical cells that differed cytologically from the intraglandular larger acidophilic and clear cells (Fig. 6). In some places, the capsule was deficient; this permitted cortical cells to extrude out of the gland into the periadrenal fat. Sometimes, the extraglandular protrusions formed demilunes with a thin, limiting capsule and base directly applied to the adrenal capsule. Larger extraadrenal cortical masses were often unencapsulated and in direct contact with retroperitoneal fat cells. In places, intraglandular nodules pushed the capsule outwards and adjacent extraglandular nodules pushed it inwards causing undulation of the capsule.
FIGURE 6.

Patterns of adrenal capsular involvement (case 6). A, Three intracapsular aggregates, 1 very small (arrow). The cytology and pattern of the cells are different in each of the aggregates. B, The capsule is distended by 2 nodules (asterisks), each having a thin fibrous capsule derived from the original capsule. In addition, rows of cortical cells are present in the capsule (arrow). The cells in the larger nodule (upper right) resemble those in the cortex proper (bottom right). Some of the cells in the smaller nodule (lower left) resemble those in the linear rows in the capsule. C, A roughly hemispherical mass of cortical cells with a very thin capsule is applied to the tip of a cortical ala in which the cortex is hyperplastic and lacks zonation. Fetal cortex is present at the center of the gland (F). D, A series of partially encapsulated nodules caused thickening of the cortex. The latter showed vague zonation, an outer clear zone with clear cytoplasm, and an inner acidophilic one with acidophilic cytoplasm (bottom half). The thickening of the cortex is increased by a series of partially encapsulated nodules (upper half). One nodule (arrow) is close to but separate from the adrenal gland. There is no infiltration of periadrenal fat.
Adenoma (Case 4)
Gross
The left adrenal gland was displaced by an irregularly shaped, yellow-gray mass composed of 2 lesions, 2 and 3.2 cm in diameter, respectively. The right-sided tumor measured 2 cm.
Light Microscopy
The findings in the 2 glands were generally similar (Fig. 7) and will be described together. The left tumor was unencapsulated and in direct contact on a broad front with periadrenal fat; the right lesion was partially surrounded by a thin fibrous capsule. The lesions were composed of uniform, large eosinophilic, and clear cells; the former were arranged in sheets and the latter in clusters. The cells were regular, but there was a scattering of larger cells. Most nuclei lacked a nucleolus. Myeloid metaplasia was present focally and there was lipochrome in the cells.
FIGURE 7.

Bilateral adrenocortical adenoma (case 4). A, A circumscribed, lobulated tumor (autopsy specimen) protruded from the hyperplastic right adrenal cortex that lacked normal zonation. B, The left adrenal tumor (surgical specimen) was composed of acidophilic and clear cells and lacked a capsule. C, Tumor was composed of a sheet of cells with eosinophilic cells (upper right) and clustered clear cells (lower left). Scattered nuclei were enlarged (arrows). D, An unencapsulated 1.5-mm nodule of adrenocortical cells with eosinophilic cytoplasm and lymphocytic infiltrates in the periadrenal fat. The region of the zona fasciculata in the cortex proper lacked the normal clear appearance.
The residual adrenal cortex, 1 to 1.3 mm thick, was composed of large acidophilic cells with lipochrome in the deepest cells. Smaller vacuolated cells formed a very narrow subcapsular band on the left side. There was also a focus of disorganized acidophilic and clear cells, the former having lipochrome.
On both sides, there was an extra-adrenal unencapsulated focus of enlarged acidophilic cortical cells in the periadrenal fat adjacent to but separate from the tumor masses and from the adrenal gland capsule. The lesions measured 0.9 mm and 1.5 mm in diameter.
Immunocytochemistry
The results are presented in Table 5 and are illustrated in Figure 8. The hyperplastic cortex stained strongly with vimentin, synaptophysin, and inhibin, and less strongly with melan A. Staining was absent to minimal with CD56. The atrophic cortex stained strongly with CD56 and did not stain or stained minimally with synaptophysin, inhibin-A, and melan A. The gross, microscopic, and immunostaining findings in case 10 (Fig. 9) were similar to those just described.
TABLE 5.
Adrenocortical Immunocytochemical Findings*
| Vimentin
|
Synaptophysin
|
Inhibin-A
|
Melan A
|
CD56
|
MIB-1
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Hyperplasia and Nodules | Atrophy | Hyperplasia and Nodules | Atrophy | Hyperplasia and Nodules | Atropy | Hyperplasia and Nodules | Atropy | Hyperplasia and Nodules | Atrophy | Hyperplasia and Nodules | Atropy |
| 1 | 3+, outer cortex, fine gran | Neg | 2+, cyto body | Neg | 3+, fine gran | Superficial : neg. Deep: 1+ | 3+, coarse gran | Neg to 1+, fine gran | Neg | 1+, cell membrane, fine gran | Occ stained nuclei | Many stained nuclei |
| 2 | 3+, fine gran | Neg | 3+, large cyto body | 1+, outer | 3+2 2+, coarse grain |
1+, deep, cell membrane | 3+, fine gran | 1+, fine gran | 1+, patchy, cell membrane | 3+, cell membrane | Occ stained nuclei | Occ stained nuclei |
| 3 | 3+, patchy, cell membrane and cyto body, fine gran | Occ cells + | 3+, patchy, fine gran, cell membrane and cyto body | Neg | 3+ to 2+, coarse gran | Neg | 3+, coarse bran | 2+ to 1+, fine gran | 1+, cell membrane, patchy to neg | 3+, cell membrane | 1+ | 2+ to neg |
| 5 | Neg | Neg | 3+, fine gran, cell membrane and cyto body | Neg to 1+ | 3+ to 2+, fine and coarse gran | Neg to 1+ | 2+ to 1+, patchy, fine and coarse gran | Neg | 1+, subcapsular | 2+, cell membrane, coarse gran, focal | NA | NA |
| 8 | 3+, patchy, fine and coarse gran | Neg | 3+, cell membrane and cyto body, fine gran | Neg | 3+ to +2, coarse gran | Neg | 1+, fine gran | Neg | Neg | 3+, cell membrane, cyto, fine gran | 1+ | 1+ |
| 10 | 3+, coarse gran | Neg | 1+, cell membrane and cyto body, fine gran | Neg | 3+, coarse gran | Neg | 3+ to 2+, coarse gran | Neg | Neg | 3+, cell membrane, fine gran | 1+ | 1+ |
Cyto indicates cytoplasmic; gran, granularity; NA, not available; occ, occasional.
Patients 1,2, 3, 5, and 8 had Cushing syndrome and McCune-Albright syndrome. Patient 10 had Cushing syndrome but not McCune-Albright syndrome.
Staining, negative 3+, heavy ; 2+, moderate; 1+, light.
FIGURE 8.

Immunostaining of hyperplastic and atrophic cortex (case 8). The hyperplastic cortex stained strongly with synaptophysin (A) and inhibin-A (B), less strongly with melan A (C), and did not stain or stained minimally with CD56 (D). The atrophic cortex (asterisks) stained strongly with CD56 and minimally with melan A.
FIGURE 9.

Cortical hyperplasia and cortical atrophy (case 10). A, Areas of hyperplasia with mixed clear and acidophilic cells (arrows) are juxtaposed with zones of atrophic cortex with outer clear and inner dark cells (asterisks). B, Mixed clear and acidophilic cell hyperplasia with cortical atrophy (arrows). C, CD56-stained zones of cortical atrophy. Zones of mixed clear and acidophilic cell hyperplasia were unstained.
Normal Infant Adrenal Glands
Gross and Light Microscopy
The cortex was thin, and yellow-tan in color. Gland weights were not available. The cortex had a mean thickness of 0.47 mm, based on 608 measurements. Beneath the capsule, there was a narrow band of aggregates of very small cells with finely acidophilic, minimally vacuolated cytoplasm and a high nuclear-to-cytoplasmic ratio. Deep to these, the cortex lacked the distinct zonation present in the normal adult gland, although there was a subtle change from superficial to deep cortex (Fig. 10A). Vacuolated weakly acidophilic cells formed columns that became more acidophilic and less vacuolated deeper in the cortex. Fetal cortical remnants were present. It connection with the foregoing, the cortical zonation present in adults is not fully developed until adrenarche, and the zona glomerulosa, discontinuous in adults, is continuous in infants and children.28
FIGURE 10.

Definitive and residual fetal adrenal cortex (3-month-old male infant). A, Definitive cortex lacked distinct zonation, was 0.5-mm thick, and surrounded the fetal cortex (asterisk). B, Synaptophysin stained superficial and deep bands of the definitive cortex leaving a central zone unstained. Medullary cells were heavily stained. Fetal cortical cells were less heavily stained. C, Inhibin-A stained a band of deep definitive cortical and fetal cells. D, Melan A stained the entire cortex and also fetal cortical cells. E, CD56 stained a continuous subcapsular band of cortical cells. F, MIB1 stained scattered nuclei in the atrophic cortex (arrows).
Immunocytochemistry
Vimentin: no staining. Synaptophysin: weak cytoplasmic and cytoplasmic globular body staining in upper and lower one thirds. Inhibin-A: strong fine granular staining in the deeper one half, patchy staining of middle cortex. Melan A: very strong coarsely granular staining of the subcapsular cell aggregates. CD56: membrane staining of the subcapsular cell aggregates (Fig. 10D). Medullary pheochromocytes were also stained.
DISCUSSION
The Cushing syndrome in this study of patients with McCune-Albright syndrome had several unusual features: early age at onset, life-threatening severity, occasional spontaneous remission and remission after unilateral adrenalectomy, and very unusual adrenocortical histopathology.
The anatomic cause of the Cushing syndrome has generally been attributed to bilateral nodular adrenocortical hyperplasia.7,15 Our findings showed that in addition to nodular hyperplasia there were sharply defined zones of cortical atrophy that gave the cortex a bimorphic appearance. In 8 patients, the microscopic appearance was similar, a combination compact or clear cell hyperplasia with tinctorially similar nodules, and cortical atrophy. The ninth patient had adenoma(s) and diffuse compact cell hyperplasia in each adrenal gland. The findings in the tenth patient who did not have McCune-Albright syndrome will be discussed later.
In the 8 patients just mentioned, there was (1) hypertrophy and hyperplasia of compact or clear cells, occasionally a mixture of both, (2) formation of distinct or vague compact and clear cell nodules, some partially or completely surrounded by a very thin fibrous capsule, (3) small intracapsular cortical cell aggregates and larger spheroidal cell “masses” that intruded on the cortex proper and protruded into the periadrenal fat, and (4) an unusual histologic type of thin cortex.
Thickened hyperplastic cortex was juxtaposed to thin atrophic cortex without transition between them creating an unusual and characteristic appearance. The atrophic cortex was usually recognizable even at scanning microscopic examination because of its sharply separated outer light and inner dark layers, an appearance due to vacuolated cells in the former and to the basophilic nuclei in the latter. Sometimes, the amounts of the atrophic cortex were inconspicuously scattered in zones of hyperplasia and in the cortical nodules and could easily escape attention or be disregarded as inconsequential because of their minuteness.
Histologically, the atrophic zones were different from the type of extratumoral cortex seen in association with cortisol-secreting cortical adenomas in adult patients and in primary pigmented nodular adrenocortical disease. In the former, the thin cortex is composed of large vacuolated cells.25 In the latter, it features small cells with weakly eosinophilic cytoplasm without distinct zonation. 34 The vacuolated cell zone in the cases we describe might correspond to that in the extratumoral cortex in association with cortisol-secreting adenomas, although the cells are much smaller. However, the accompanying apparent hyperplasia of the zona glomerulosa (see later) is unexplained. Urinary excretion of aldosterone was normal in 1 patient in the study (case 3).
The prominence of cortical cells in the adrenal capsule and on its external surface was an exaggeration of normal findings8—but the intracapsular adrenal cells were different cytologically from those within the gland. Thus, they did not obviously appear to be an extension of them into the capsule. The manner in which some nodules acquired a capsule was not evident. Possibly, some nodules originated intracapsularly and acquired a capsule in this way.
The immunostaining results provided good distinction between cortical hyperplasia and the nodules, on one hand, and cortical atrophy, on the other. The former stained with vimentin, synaptophysin, inhibin-A, and melan A, and not with CD56. The atrophic zones stained consistently with CD56 and not with the other markers. CD56, a surface binding glycoprotein, is expressed in normal zona glomerulosa cells and in aldosteromas.10 The CD56 findings revealed an apparent hyperplasia of the zona glomerulosa that paradoxically occurred in zones of cortical atrophy.
The study provided other unexpected findings, first, MIB1-stained nuclei in the atrophic cortex, second, cytomegaly and cortical superficial microcysts in 1 patient (case 2) and, third, age-dependent pathologic findings (hyperplasia in the young and adenoma in the late second decade). The cytomegaly and superficial cortical microcysts, findings typical of Beckwith-Wiedemann syndrome, 4 are unexplained. We have also observed both lesions in a premature infant (27-week gestation), without stigmata of McCune-Albright syndrome who died 3 days after birth following repair of a patent ductus arteriosus. Her monozygotic twin was normal. The apparent age-dependent pathologic findings are unexplained. It did not seem that the hyperplasia and atrophy seen in infancy could over time evolve into the bilateral adenomas found in case 4 (the patient had café-au-lait spots since birth).
Other observations were also intriguing—the juxta-position of the hyperplastic and atrophic zones of cortex and the apparent involvement of only 1 gland in some cases. The explanation for both may lie in the mechanism proposed by Happle16 in 1986 to explain the distribution of lesions within an organ or organ system. Happle16 suggested that the McCune-Albright syndrome could be the result of an autosomal-dominant lethal gene that was compatible with viability of the fetus when it occurred in the mosaic state, having arisen by somatic mutation. In support of this suggestion is a finding that, among monozygotic twins, 1 of whom had McCune-Albright syndrome, the second did not.12,30 The bimorphic cortex we report is likely explained by the presence of 2 types of adrenocortical cells; one genetically altered and hyperplastic and nodular, and the other putatively normal but morphologically atrophic because of lack of corticotropin stimulation.
Patient 10 in our study did not have McCune-Albright syndrome, clinically or molecularly, but he had Cushing syndrome and the same bimorphic adrenal pathology as in 8 patients who did have the syndrome. The bimorphic adrenal findings we report, thus, are not restricted to the McCune-Albright syndrome, and suggest the existence on another mosaic type disorder. In this connection, it will be recalled that mutations of different genes may result in the same pathologic phenotype, as with primary pigmented nodular adrenocortical disease.9,19
In summary, we have described the clinical, imaging, laboratory, and adrenal pathologic findings in 9 patients with McCune-Albright syndrome and Cushing syndrome. One patient had cortical adenoma(s). The remaining 8 had similar pathology, diffuse, and nodular cortical hyperplasia, and multifocal cortical atrophy, constituting a bimorphic cortical pathology. These findings also occurred in a tenth patient, an infant apparently with sporadic Cushing syndrome. As this Cushing syndrome occurred in early infancy and was caused by a bilateral adrenal disorder, it is likely that the syndrome was ultimately the result of a yet undiscovered genetic mutation.
Acknowledgments
The authors thank the following colleagues who provided information and materials used in this study: B.A. Boston, MD, D.J. Carson, MD, M. J. Finegold, MD, M. Ito, MD, S. Kolouskova, MD, S. LaFranchi, MD, W. Lebl, MD, W. Rabl, MD, B. Sherrington, MD, and C. Thornton, MB. The authors thank Monalisa Azevdo, MD and Anelia Horvath, PhD (NICHD, NIH) for some of the genetic (DNA) studies. The authors also thank the families of patients 5 and 10 for the information they provided.
Footnotes
Conflicts of Interest and Sources of Funding: J.A.C. received funding from the Mayo Foundation and the Intramural Program, NIH project Z01HD00064204.
References
- 1.Aarskog D, Tveteraas E. McCune-Albright’s syndrome following adrenalectomy for Cushing syndrome in infancy. J Pediatr. 1968;73:89–96. doi: 10.1016/s0022-3476(68)80043-5. [DOI] [PubMed] [Google Scholar]
- 2.Albright F, Butler AM, Bloomberg E. Rickets resistant to vitamin D therapy. Am J Dis Child. 1937;54:529–547. [Google Scholar]
- 3.Albright F, Butler AM, Hampton AO, et al. Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation and endocrine dysfunction, with precocious puberty in females: report of five cases. N Engl J Med. 1937;216:727–746. [Google Scholar]
- 4.Beckwith JB. Macroglossia, omphalocele, adrenal cytomegaly, gigantism, and hyperplastic visceromegaly. Birth Defects. 1969;5:188–196. [Google Scholar]
- 5.Benjamin DR, McRoberts JW. Polyostotic fibrous dysplasia associated with Cushing syndrome. Arch Pathol. 1973;96:175–178. [PubMed] [Google Scholar]
- 6.Boston BA, Mandel S, LaFranchi S, et al. Activating mutation in the stimulatory guanidine nucleotide-binding protein in an infant with Cushing’s syndrome and nodular adrenal hyperplasia. J Clin Endocrinol Metab. 1994;79:890–993. doi: 10.1210/jcem.79.3.8077378. [DOI] [PubMed] [Google Scholar]
- 7.Brown RJ, Kelly MH, Collins MT. Cushing syndrome in McCune-Albright syndrome. J Clin Endocrinol Metab. 2010;95:1508–1515. doi: 10.1210/jc.2009-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carney JA, Lloyd RV. Adrenal. In: Mills SE, editor. Histology for pathologists. PA: Lippincott: Williams and Wilkins; 2007. pp. 1167–1188. [Google Scholar]
- 9.Carney JA, Gaillard RC, Bertherat J, et al. Familial micronodular adrenocortical disease, Cushing syndrome, and mutations of the gene encoding phosphodiesterase 11A4 (PDE 11A) Am J Surg Pathol. 2010;34:547–555. doi: 10.1097/PAS.0b013e3181d31f49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caroccia B, Fassina A, Seccia TM, et al. Isolation of human adrenocortical aldosterone-producing cells by a novel immunomagnetic beads method. Endocrinology. 2010;151:1375–1380. doi: 10.1210/en.2009-1243. [DOI] [PubMed] [Google Scholar]
- 11.Cushing H. The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism) Bull Johns Hopkins Hosp. 1932;1:137–195. [Google Scholar]
- 12.Endo M, Yamada Y, Matsuura N, et al. Monozygotic twins discordant for the major signs of McCune-Albright syndrome. Am J Med Genet. 1991;41:216–220. doi: 10.1002/ajmg.1320410217. [DOI] [PubMed] [Google Scholar]
- 13.Gillis D, Rosler A, Hannon TS, et al. Prolonged remission of severe Cushing syndrome without adrenalectomy in an infant with McCune-Albright syndrome. J Pediatr. 2008;152:882–884. doi: 10.1016/j.jpeds.2008.01.037. [DOI] [PubMed] [Google Scholar]
- 14.Hamajima T, Maruwaka K, Homma K, et al. Unilateral adrenalectomy can be an alternative therapy for infantile onset of Cushing’s syndrome caused by ACTH-independent macronodular adrenal hyperplasia with McCune-Albright syndrome. Endocrin J. 2010;57:819–824. doi: 10.1507/endocrj.k10e-003. [DOI] [PubMed] [Google Scholar]
- 15.Hamilton CR, Jr, Maloof T. Unusual types of hyperthyroidism. Medicine. 1973;52:195–213. doi: 10.1097/00005792-197305000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Happle R. The McCune-Albright syndrome: a lethal gene surviving by mosaicism. Clin Genet. 1986;29:321–324. doi: 10.1111/j.1399-0004.1986.tb01261.x. [DOI] [PubMed] [Google Scholar]
- 17.Kirk JMW, Brain CE, Carson DJ, et al. Cushing’s syndrome caused by nodular adrenal hyperplasia in children with McCune-Albright syndrome. J Pediatr. 1999;134:789–992. doi: 10.1016/s0022-3476(99)70302-1. [DOI] [PubMed] [Google Scholar]
- 18.Kirschner LS, Carney JA, Pack SD, et al. Mutations of the gene encoding the protein kinase A type 1-α regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 19.Landis CA, Masters SB, Spada A, et al. GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumors. Nature. 1989;340:692–696. doi: 10.1038/340692a0. [DOI] [PubMed] [Google Scholar]
- 20.Lumbrosa S, Paris F, Sultan C. Activating Gs-alpha mutations: analysis of 113 patients with signs of McCune-Albright syndrome—a European collaborative study. J Clin Endocrinol Metab. 2004;89:2107–2113. doi: 10.1210/jc.2003-031225. [DOI] [PubMed] [Google Scholar]
- 21.Mauras N, Blizzard RM. The McCune-Albright syndrome. Acta Endocrinol Suppl (Copenh) 1986;279:207–217. doi: 10.1530/acta.0.112s207. [DOI] [PubMed] [Google Scholar]
- 22.McArthur RG, Hayles AB, Lambert PW. Albright’s syndrome with rickets. Mayo Clin Proc. 1979;54:313–320. [PubMed] [Google Scholar]
- 23.McArthur RG, Bahn RC, Hayles AB. Primary adrenocortical nodular dysplasia as a cause of Cushing’s syndrome in infants and children. Mayo Clin Proc. 1982;57:58–63. [PubMed] [Google Scholar]
- 24.McCune DJ, Bruch H. Progress in pediatrics: osteodystrophia fibrosa. Am J Dis Child. 1937;54:806–848. [Google Scholar]
- 25.Neville AM, Symington T. The pathology of the adrenal gland in Cushing’s syndrome. J Pathol Bacteriol. 1967;93:19–35. doi: 10.1002/path.1700930103. [DOI] [PubMed] [Google Scholar]
- 26.O’Bryan RM, Smith RW, Jr, Fine G, et al. Congenital adrenocortical hyperplasia with Cushing’s syndrome. JAMA. 1964;187:257–261. doi: 10.1001/jama.1964.03060170011002. [DOI] [PubMed] [Google Scholar]
- 27.O’Hare MJ. Origin and development of the adrenal gland. In: O’Hare MJ, editor. The human adrenal cortex. Pathology and biology—An integrated approach. Berlin, Heidelberg, New York: Springer-Verlag; 1982. pp. 11–15. [Google Scholar]
- 28.O’Hare MJ. Morphological changes in the adrenal cortex with age. In: O’Hare MJ, editor. The Human Adrenal Cortex. Pathology and Biology—An Integrated Approach. Berlin, Heidelberg, New York: Springer-Verlag; 1982. pp. 41–60. [Google Scholar]
- 29.Paris F, Philibert P, Lumbroso S, et al. Isolated Cushing’s syndrome: an unusual presentation of McCune-Albright syndrome in the neonatal period. Horm Res. 2009;72:315–319. doi: 10.1159/000245934. [DOI] [PubMed] [Google Scholar]
- 30.Peleg R, Avizou L, Eliakin A, et al. McCune-Albright syndrome in discordant twins. Isr Med Assoc J. 2009;11:343–347. [PubMed] [Google Scholar]
- 31.Post EM, Consenstein L, Hitch D, et al. Congenital Cushing syndrome with polyostotic fibrous dysplasia (PFD) Pediatr Res. 1983;17:169A. [Google Scholar]
- 32.Rabl W, Hahn H, Chi P, et al. Congenital Cushing syndrome (Cs) due to unilateral multinodular adrenocortical hyperplasia in McCune-Albright syndrome. Exp Clin Endocrinol. 1994;102(Suppl 1):74. [Google Scholar]
- 33.Shenker A, Weinstein LS, Moran A, et al. Severe endocrine and nonnendocrine manifestations of the McCune-Albright syndrome associated with activating mutations of the stimulatory G protein GS. J Pediatr. 1993;123:509–518. doi: 10.1016/s0022-3476(05)80943-6. [DOI] [PubMed] [Google Scholar]
- 34.Shenoy BV, Carpenter PC, Carney JA. Bilateral primary pigmented nodular adrenocortical disease: rare cause of the Cushing syndrome. Am J Surg Pathol. 1984;8:335–344. doi: 10.1097/00000478-198405000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Weinstein LS, Shenker A, Gejman PV, et al. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991;325:1688–1695. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 36.Yoshimoto M, Kakayama M, Baba T, et al. A case of neonatal McCune-Albright syndrome with Cushing syndrome and hyperthyroidism. Acta Paediatr Scand. 1991;80:984–987. doi: 10.1111/j.1651-2227.1991.tb11769.x. [DOI] [PubMed] [Google Scholar]