

Abstract
The potential role of the posttranslational modification of proteins with O-linked N-acetyl-β-D-glucosamine (O-GlcNAc) in the pathogenesis of Alzheimer disease (AD) has been studied extensively, yet the exact function of O-GlcNAc in AD remains elusive. O-GlcNAc cycling is facilitated by only two highly conserved enzymes: O-GlcNAc transferase (OGT) catalyzes the addition, while O-GlcNAcase (OGA) catalyzes the removal of GlcNAc from proteins. Studies analyzing global O-GlcNAc levels in AD brain have produced inconsistent results and the reasons for altered O-GlcNAcylation in AD are still poorly understood. In this study, we show a 1.2 fold increase in cytosolic protein O-GlcNAc modification in AD brain when compared to age-matched controls. Interestingly, O-GlcNAc changes seem to be attributable to differential modification of a few individual proteins. While our finding of augmented O-GlcNAcylation concurs with some reports, it is contrary to others demonstrating decreased O-GlcNAc levels in AD brain. These conflicting results emphasize the need for further studies providing conclusive evidence on the subject of O-GlcNAcylation in AD. We further demonstrate that, while OGT protein levels are unaffected in AD, OGA protein levels are significantly decreased to 75 % of those in control samples. In addition, augmented protein O-GlcNAc modification correlates to decreased OGA protein levels in AD subjects. While OGA inhibitors are already being tested for AD treatment, our results provide a strong indication that the general subject of O-GlcNAcylation and specifically its regulation by OGA and OGT in AD need further investigation to conclusively elucidate its potential role in AD pathogenesis and treatment.
Keywords: Alzheimer disease, O-GlcNAc, O-GlcNAcase
1. Introduction
O-linked N-acetyl-β-D-glucosamine (O-GlcNAc) is a posttranslational modification in which a single N-acetylglucosamine is attached to a protein [1]. O-GlcNAc modification is localized to the nucleocytoplasmic compartment [1], it is dynamic and changes in response to various stimuli [2]. O-GlcNAc cycling is facilitated by only two enzymes. The addition of GlcNAc from its high energy donor substrate UDP-GlcNAc onto the hydroxyl groups of serine and threonine residues of target proteins is catalyzed by O-GlcNAc transferase (OGT) [3]. OGT activity is regulated over a broad range of UDP-GlcNAc concentrations [4] that are dependent on the flux of nutrients through the hexosamine biosynthetic pathway (HBP). The HBP converts about 2–3% of the intracellular glucose to UDP-GlcNAc [5], therefore, O-GlcNAc is often referred to as a nutrient sensor [6]. The enzyme β-N-acetylglucosaminidase (O-GlcNAcase or OGA), a member of the family of hexosaminidases, catalyzes the removal of GlcNAc from proteins. Unlike lysosomal hexosaminidases, OGA activity is highest at neutral pH and it localizes mainly to the cytosol [7]. Both OGT and OGA are transcribed by two highly conserved genes and are expressed ubiquitously with high levels in brain and pancreas [8,9]. The OGT gene on the X chromosome encodes three OGT isoforms with identical catalytic domains but a varying number of tetratricopeptide repeats: the longest and shortest isoforms, termed ncOGT and sOGT, respectively, are found in the nucleus and cytoplasm, while a third isoforms is localized to the mitochondria (mOGT) [10,11,12]. OGA is encoded by MGEA5 on chromosome 10 which gives rise to a full length OGA, FL-OGA, as well as a shorter nuclear variant termed NV-OGA [13,14]. OGT and OGA knockout studies have demonstrated that O-GlcNAc is essential for life as its absence leads to embryonic or neonatal lethality, respectively [10,15]. In accordance with its important role in development, perturbations in O-GlcNAc cycling are also associated with different diseases such as Alzheimer disease (AD), cancer, and type II diabetes (reviewed in [16]).
AD is clinically characterized by cognitive decline and memory impairment, and its histopathological hallmarks include extracellular senile plaques containing amyloid β-peptide, a toxic fragment of the amyloid precursor protein, and intracellular neurofibrillary tangles consisting of hyperphosphorylated tau protein [17]. In addition, positron emission tomography studies have demonstrated early impairment of cerebral glucose metabolism in AD [18]. Increasing evidence suggests that O-GlcNAc may play an important role in the pathogenesis of AD since both OGT and OGA are highly expressed in brain [8,9] and the OGA gene maps to a gene region that has been linked to late onset AD [19]. Furthermore, amyloid precursor protein and tau as well as other neuronal proteins are O-GlcNAc modified [20,21,22]. However, studies on O-GlcNAcylation in AD brain have revealed conflicting results. For example, one study reported increased protein O-GlcNAcylation, while another study reported decreased protein O-GlcNAcylation in AD brain [23,24]. In addition, the mechanisms behind altered protein O-GlcNAc modification in AD brain remain elusive.
With the exact role of O-GlcNAc in AD still unclear, the current study focuses on global O-GlcNAc levels as well as the enzymes controlling its cycling in samples from the inferior parietal lobule (IPL), a brain region known to be affected by AD [25].
2. Material and Methods
2.1 Chemicals
All chemicals and antibodies were from Sigma Aldrich (St. Louis, MO) unless noted otherwise. Precision Plus Protein Standard, 4–15% or 8–16% Criterion TGX Precast gels, 10x TGS running buffer, and 0.45 nm nitrocellulose membrane were from Bio-Rad (Hercules, CA) and 10x ReBlot Plus Strong Stripping Antibody Solution was from Millipore (Temecula, CA). Primary antibodies used in this study were monoclonal anti-O-GlcNAc antibody (clone CTD110.6), rabbit anti-actin antibody, monoclonal anti-β-actin antibody, monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody. Anti-OGT antibody clone AL28 was kindly provided by S. Arnold, Johns Hopkins University, Baltimore, MD, and anti-OGA antibody was kindly provided by G. Crawford, Mercer University, Macon, GA. Secondary antibodies used were anti-mouse IgG/IgM horseradish peroxidase (HRP) (Millipore, Temecula, CA), ECL™ anti-rabbit IgG HRP, ECL™ Plex goat-anti-mouse IgG, Cy™5, ECL™ Plex goat-anti-rabbit IgG, Cy™5 (all GE Healthcare, Pittsburgh, PA) and anti-rabbit IgG alkaline phosphatase.
2.2 Subjects
Frozen IPL and cerebellum samples of subjects with well-characterized AD and age-matched controls were obtained from Sanders-Brown Center on Aging of the University of Kentucky. Age, gender, post-mortem intervals (PMI) and Braak stages are listed in Table 1.
Table 1.
Demographic information of AD subjects and age-matched controls
| Age (years) Mean ± SD |
Sex | PMI (hours) Mean ± SD |
Braak stage | |
|---|---|---|---|---|
| Control | 85 ± 5.1 | 4M, 9F | 3.2 ± 2.2 | 0–IV |
| AD | 84 ± 5.8 | 4M, 9F | 3.2 ±0.8 | V–VI |
2.3 Sample preparation
Brain samples were homogenized in ice-cold isolation buffer (0.32 M sucrose, 20 mM HEPES, 2 mM EDTA, 2 mM EGTA, 0.1 M GlcNAc, pH 7.4 with 4 μg/ml leupeptin, 4 μg/ml pepstatin A, 5 μg/ml aprotinin, 0.2 mM PMSF) using a Wheaton glass homogenizer. One aliquot was set on ice and sonicated 2x 10s on 20% power using a 550 Sonic Dismembrator (Fisher Scientific, Rockford, IL) and centrifuged at 4°C for 10 minutes at 1000 x g to obtain homogenates. A different aliquot was subjected to subcellular fractionation. Samples were first centrifuged at 4°C for 10 minutes at 1000 x g, the supernatant (crude cytosolic fraction) was transferred into a new tube and centrifuged again at 4°C for 10 minutes at 16,090 x g. The resulting supernatant represents the cytosolic fraction. Protein estimation was performed using Pierce BCA Protein Assay (Thermo Scientific, Rockford, IL).
2.4 Western blot analysis
Samples (50 μg homogenate or 25 μg cytosolic fraction) were mixed with 4x sample buffer (0.2 M Tris HCl, pH 6.8, 40% glycerol, 20% β-mercaptoethanol, 8% sodium dodecyl sulfate (SDS), 0.01% bromophenolblue), heated 5 minutes at 95°C and subjected to SDS-polyacrylamide gel electrophoresis. Separated proteins were then transferred onto nitrocellulose membranes and membranes were stained with Ponceau S (0.1% Ponceau S, 7.5% acetic acid) to monitor transfer. After destaining in wash blot (150 mM NaCl, 3 mM NaH2PO4, 17 mM Na2HPO4, 0.04% Tween 20), membranes were blocked in blocking solution (3 % (w/v) bovine serum albumin (BSA) in wash blot). Primary antibody was diluted in blocking solution and membranes were incubated over night at 4°C. The membranes were washed three times in wash blot and then incubated with secondary antibody diluted in wash blot for 1h at room temperature. Membranes were washed three times with wash blot before signal detection. O-GlcNAc and OGA signals were detected chemiluminescently using Clarity™ Western ECL Substrate (Bio-Rad, Hercules, CA), OGT signals were developed colorimetrically using 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (Thermo Fisher Scientific, Rockford, IL) in alkaline phosphatase buffer (0.1 M Tris, 0.1 M NaCl, 5 mM MgCl2, pH 9.5) and loading controls were detected using CyDyes. All images were acquired with the ChemiDoc™ MP imaging system and image analysis was performed using Image Lab™ Software (Bio-Rad, Hercules, CA).
2.5 OGA activity assay
OGA activity assay was performed using the synthetic substrate p-nitrophenyl N-acetyl-β-D-glucosaminide (pNP-GlcNAc) as described by Zachara et al. (2011) [26]. Briefly, samples were homogenized in isolation buffer without EDTA, EGTA and GlcNAc, and crude cytosolic fractions were prepared as described above. The samples were incubated with Concanavalin A agarose for 30 minutes at 4°C to remove interfering hexosaminidases. Samples were then desalted using Zeba™ Desalt Spin Columns (Pierce Biotechnology, Rockford, IL) and protein estimation was performed. Sample (25 μg) was mixed with activity assay buffer (final concentrations: 50 mM sodium cacodylate, pH 6.4, 50 mM N-acetylgalactosamine, 0.3 % BSA, 2 mM pNP-GlcNAc). Reactions were incubated at 37°C for 2 h and stopped by the addition of 500 mM Na2CO3. Absorbance was read at 405 nm and OGA activity is reported as enzyme activity units where one unit catalyzes the release of 1 μmol pNP/min from pNP-GlcNAc.
2.6 Statistical analysis
Signal intensities from Western blot analyses were normalized to loading control (actin or GAPDH) and converted to % control. Data are shown as mean ± SEM. All statistical analyses were performed with GraphPad Prism 5 (GraphPad Software, San Diego, CA). For comparison of AD and age-matched controls Student’s t-test was performed where p < 0.05 was considered to be statistically significant. Possible relationship of two factors was assessed by Pearson correlation followed by computation of two-tailed p-value where p < 0.05 was considered to be statistically significant.
3. Results
3.1 Increased O-GlcNAc levels in AD brain
Total O-GlcNAc modification in cytosolic IPL samples from AD subjects is increased when compared to age-matched controls (100±3 vs. 119±7; p=0.03; n=13; Fig. 1A,B). As shown in Fig. 1A, O-GlcNAc signals within the groups demonstrate great variations. Especially the O-GlcNAc signals at approximately 75, 50 and 27 kDa (as indicated by the arrows) differ greatly between the samples and were therefore analyzed separately (Fig. 1C). The 75 kDa O-GlcNAc band is decreased to less than half of that of the control samples (100±11 vs. 43±6; p=0.0002; n=13) while the O-GlcNAc signals at 50 kDa and 27 kDa are both increased in AD (100±7 vs. 143±21; p=0.06 and 100±11 vs. 165±29; p=0.04; n=13). Proteomics analyses are underway to discover the identity of differentially O-GlcNAc-modified proteins in AD. Our preliminary results indicate the 27 kDa band may represent O-GlcNAc-modified triose phosphate isomerase, a glycolytic enzyme which has been previously described as O-GlcNAc modified [27]. Pre-incubation of anti-O-GlcNAc antibody with 0.1 M GlcNAc prevented signal detection on a replicate blot indicating antibody specificity. Only after long exposure times a signal at approximately 35 kDa could be detected (Fig. 1F). Analysis of cerebellum samples demonstrated no significant difference in cytosolic protein O-GlcNAcylation between AD and age-matched controls (100±2 vs. 91±6; p=0.20; n=8; Fig. 1D,E).
Fig. 1. O-GlcNAc levels in cytosolic fraction of IPL and cerebellar samples of subjects with AD and age-matched controls.

(A) Representative Western blot of control and AD IPL cytosolic fractions probed for O-GlcNAc and actin. (B) Densitometric quantification of total O-GlcNAc signals in IPL cytosolic fractions of control and AD samples (n=13). (C) Densitometric quantification of O-GlcNAc-positive bands at 75, 50 and 27 kDa, as indicated by the arrows in (A), in IPL cytosolic fractions of control and AD samples (n=13). (D) Representative Western blot of control and AD cerebellar cytosolic fractions probed for O-GlcNAc and GAPDH. (E) Densitometric quantification of O-GlcNAc signals in cerebellar cytosolic fraction of control and AD samples (n=8). (F) O-GlcNAc detection in IPL homogenate (1) and cytosolic fraction (2) after preincubation of CTD110.6 with 0.1M GlcNAc. The bar indicates the 37 kDa band of the molecular weight marker (M). For densitometric analyses, normalized O-GlcNAc signals were converted to % control and results are shown as mean ± SEM; *p < 0.05; ***p < 0.001.
3.2 No change in OGT levels in AD brain
OGT protein levels show no difference in IPL or cerebellar samples from AD subjects when compared to age-matched controls (IPL: 100±8 vs. 101±4; p=0.89; n=13; cerebellum: 100±7 vs. 82±7; p=0.09; n=8; Fig. 2). Only the largest of the three OGT isoforms, ncOGT with an apparent molecular weight of ~110 kDa, was analyzed as additional lower molecular weight signals could only be detected after sustained incubation with developing agents and seemed to result from cross-reaction rather than from a specific antibody reaction.
Fig. 2. OGT protein levels in IPL and cerebellar samples of subjects with AD and age-matched controls.
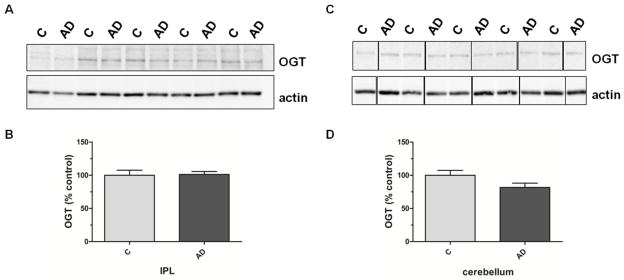
(A) Representative Western blot of control and AD IPL samples probed with specific antibodies for OGT and actin. (B) Densitometric quantification of OGT signals in IPL homogenates of control and AD samples (n=13). (C) Representative Western blot of control and AD cerebellar samples probed with specific antibodies for OGT and actin. (D) Densitometric quantification of OGT signals in cerebellar homogenates of control and AD samples (n=8). For densitometric analyses, normalized O-GlcNAc signals were converted to % control and results are shown as mean ± SEM.
3.3 Decreased OGA protein levels in AD brain
Both OGA isoforms, cytosolic full-length OGA (FL-OGA) with an apparent molecular weight of ~130 kDa as well as the nuclear variant of OGA (NV-OGA) at ~75 kDa, could be detected. FL-OGA protein levels are significantly decreased in IPL samples from AD subjects when compared to age-matched controls, while NV-OGA protein levels are unaffected (100±8 vs. 75±8; p=0.04 and 100±8 vs. 108±6; p=0.43 n=13; Fig. 3A,B). There is no significant difference in OGA levels in the cerebellum of AD and age-matched controls (FL-OGA: 100±10 vs. 82±12; p=0.28; n=8; NV-OGA: 100±5 vs. 91±9; p=0.40; n=8; Fig. 3C,D). Analysis of OGA activity using a synthetic substrate revealed decreased OGA activity in crude cytosolic fractions from AD samples when compared to age-matched controls (0.025±0.0025 vs. 0.015±0.002; p=0.01; n=5; Fig. 4A). However, after normalization of OGA activity to the corresponding FL-OGA protein levels no differences in OGA activity could be detected between AD and control samples (Fig. 4B).
Fig. 3. OGA protein levels in IPL and cerebellar samples of subjects with AD and age-matched controls.

(A) Representative Western blot of control and AD IPL samples probed for OGA and actin. (B) Densitometric quantification of OGA signals in IPL of control and AD samples (n=13). (C) Representative Western blot of control and AD cerebellar samples probed for OGA and actin. (D) Densitometric quantification of OGA signals in cerebellar homogenates of control and AD samples (n=8). For densitometric analyses, normalized O-GlcNAc signals were converted to % control and results are shown as mean ± SEM; *p < 0.05.
Fig. 4. OGA activity in IPL of subjects with AD and age-matched controls.
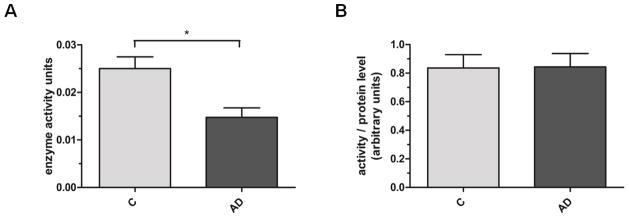
(A) OGA activity in crude cytosolic fractions of control and AD IPL samples was measured as cleavage of pNP-GlcNAc and absorbance of free pNP was detected at 405nm (n=5). Results are shown as enzyme activity units where one unit represents the amount of enzyme catalyzing the release of 1 μmol/min of pNP from pNP-GlcNAc. (B) OGA enzyme activity units were normalized to the corresponding FL-OGA protein levels. Results are shown as mean ± SEM; *p < 0.05.
3.4 Increased O-GlcNAc levels correlate with decreased OGA protein levels
In IPL samples, increased cytosolic protein O-GlcNAcylation correlates with decreased FL-OGA levels (p=0.002). No significant correlation could be detected between O-GlcNAc and NV-OGA or OGT protein levels (p=0.71 and p=0.25, respectively). In addition, O-GlcNAcylation did not correlate with age or PMI (p=0.60 and p=0.78, respectively) While there is no statistically significant correlation between O-GlcNAc and Braak stage, a potential, albeit weak, link between increasing O-GlcNAcylation and higher Braak stage may exist (p=0.10). Graphs and data are included in supplemental Fig. S1 and Table S2.
4. Discussion
Accumulating evidence indicates that O-GlcNAc may be implicated in the pathogenesis of AD. In this study, we observed significantly augmented O-GlcNAc modification of proteins in the cytosolic fraction of IPL samples from AD subjects. In accordance with previous results, protein O-GlcNAc modification in the cerebellum was unaltered when comparing AD subjects to age-matched controls [23,24]. However, our finding in IPL stands in contrast to results from Liu and colleagues, who demonstrated a decline in protein O-GlcNAcylation in AD brain [24,28]. Study of different brain areas, different sample preparation and analysis methods as well as the use of different anti-O-GlcNAc antibodies may explain the observed discrepancies between different studies. Liu et al. have analyzed O-GlcNAc modification in frontal cortex by radioimmuno-dot-blot assay or quantitative immuno-dot-blot assay [24,28] while we have examined parietal cortex samples by Western blot analysis. Additionally, we have used a different antibody for O-GlcNAc detection. Our detection methods also differed from the O-GlcNAc-specific ELISA applied by Griffith and Schmitz, who reported increased O-GlcNAc modification in the detergent insoluble cytoskeletal fraction of different AD-affected brain areas including the parietal cortex [23]. In addition, O-GlcNAcylation levels have been reported to decline with postmortem delay [28], but we do not find significant correlation between O-GlcNAcylation and PMI which may be due to the relatively short PMI of averagely 3.2 hours for both control and AD subjects.
Due to the conflicting findings of O-GlcNAc levels in AD, the development and application of a standardized method for O-GlcNAc analysis in brain samples would be useful in order to find a conclusive answer in this matter. While antibody CTD110.6 is commonly used for O-GlcNAc detection, repetition of our experiments, maybe using a different antibody, would have strengthened our data significantly. Due to the limited amount of human brain sample available for this study, experiments could only be repeated in few individual cases. However, independent verification would have been especially meaningful with respect to our findings of great variations in O-GlcNAc signal patterns within the analyzed samples. Intriguingly, the overall observed changes in O-GlcNAcylation appear to be dependent on changes of a few individual bands. Whether these changes are based solely on differential O-GlcNAcylation or may additionally be explained in part by differential expression levels of certain proteins will be the subject of further investigation in our group.
Liu et al. explain decreased O-GlcNAcylation with reduced neuronal glucose availability due to down regulation of glucose transporters GLUT1 and GLUT3 in the AD brain [29]. Reduced glucose availability in AD brain has indeed been reported by various groups. Kalaria and Harik report reduced glucose transporters at the blood-brain barrier as well as in the cerebral neocortex and hippocampus but not the cerebellum of subjects with AD [30]. Harr et al. also report a large reduction of neuron-specific GLUT3 in AD [31]. In addition, positron emission tomography studies have shown decreased glucose utilization in AD brain [18]. In accordance, reduction of brain glucose supply by starvation of mice lead to reduced protein O-GlcNAcylation in brain [28]. Paradoxically, in cell culture studies glucose deprivation induced a strong increase in O-GlcNAcylation [32,33,34]. Augmented O-GlcNAc modification despite lower UDP-GlcNAc concentrations was linked to increased OGT mRNA and protein levels and/or decreased OGA protein levels, and the authors explain the observed effects with the induction of different stress signaling pathways [32,33,34]. It is highly probable that glucose deprivation will have different effects on O-GlcNAc cycling enzymes and the HBP as only the mere decrease in glucose availability that has been described in AD brain. However, with the exception of this paper, no analyses of OGT and OGA enzymes and/or their activities have been reported in AD. In addition, glucose deprivation in cell culture studies and chronically reduced glucose availability in AD brain may induce different stress signaling pathways.
A growing body of evidence indicates oxidative stress as another important player in AD [35]. Intriguingly, O-GlcNAc has been shown to increase in response to various stressors including oxidative stress [2], and UDP-GlcNAc pools are also susceptible to cellular stress [36]. Hyperglycemia-induced overproduction of superoxide has been shown to inhibit GAPDH activity and to increase UDP-GlcNAc levels probably by rerouting glucose and its metabolites from glycolysis towards the HBP [36]. These effects are independent of glucose availability as reduced GAPDH activity and increased UPD-GlcNAc levels can be completely prevented by inhibition of mitochondrial superoxide overproduction [36]. This mechanism may also apply in AD as GAPDH and other glycolytic enzymes are oxidized in AD [37,38] and their malfunction may divert glucose and its downstream metabolite fructose-6-phosphate to favor UDP-GlcNAc synthesis via the HBP. However, further studies are needed to investigate this possible link.
As noted above, O-GlcNAc cycling is mediated by only two enzymes, OGT and OGA. The OGT gene encodes for three isoforms, the 110 kDa ncOGT, the 103 kDa mOGT, and the 78 kDa sOGT [11,12]. In our study only one OGT isoform, ncOGT, could be detected and analysis of IPL and cerebellar samples revealed that ncOGT protein levels were unaffected in AD. In rat brain sOGT expression declined after maturation and no mOGT expression could be detected in adult brain [39] which may explain why no mOGT and sOGT were detected in our study. Investigation of OGT activity is necessary to allow for a meaningful discussion of the influence of OGT on O-GlcNAcylation in AD as altered or unaltered protein levels may not reflect changes in enzyme activity. Unfortunately, current OGT activity assays are based on the measurement of [3H]GlcNAc incorporation into peptides. Furthermore, the exact mechanisms of substrate specificity and regulation of OGT are still being researched. OGT substrate specificity and activity are dependent on UDP-GlcNAc concentrations [4] and OGT activity is potently inhibited by UDP [3]. In addition, OGT is tyrosine phosphorylated and O-GlcNAc-modified [40], indicating possible regulation by posttranslational modification. A more detailed analysis of OGT activity and regulation is essential for the understanding of its role in differential protein O-GlcNAcylation in AD.
Alternative splicing of the MGEA5 gene results in of two OGA isoforms, FL- and NV-OGA with an apparent molecular weight of approximately 130 kDa and 75 kDa, respectively. Both isoforms contain the N-terminal O-GlcNAcase domain, however, the truncated NV-OGA lacks the C-terminal domain of FL-OGA [14]. Consistent with nucleocytoplasmic localization of OGT and O-GlcNAcylated proteins, OGA activity has been reported in cytosol and nucleus [7], however, no mitochondrial OGA has yet been found. FL-OGA resides predominantly in the cytoplasm whereas NV-OGA was thought to be located mainly to the nucleus [14]. Recently, NV-OGA has been found in lipid droplets as well [41]. Initially, no O-GlcNAcase activity of NV-OGA could be detected using pNP-GlcNAc as a substrate, however, assays using a more sensitive fluorogenic substrate proved that NV-OGA was active too albeit less than FL-OGA [42]. Recently, this finding was confirmed with pNP-GlcNAc showing 400-fold lower catalytic efficiency of NV-OGA when compared to FL-OGA [43]. Little is yet known about OGA regulation. OGA has been shown to interact with and to be a substrate of OGT [44, 45] suggesting possible feed-back mechanisms for O-GlcNAc regulation. In our study, probing for OGA revealed two OGA bands corresponding to the molecular weight of FL- and NV-OGA, respectively. Detection of NV-OGA was unexpected as is has been shown to be quickly down regulated during embryonic development and was undetectable in rat brain after birth [39]. FL-OGA protein levels were significantly decreased in IPL samples from subjects with AD while NV-OGA levels were unaffected when compared to controls. Since altered protein levels might not correspond directly to a change in protein activity, we used a common protocol to examine OGA activity. In the current study a significant decline in OGA activity in AD samples was observed. Despite our finding of decreased FL-OGA protein levels, the same amount of total protein was used in the OGA activity assay. While Macauley and Vocadlo demonstrated activity of NV-OGA towards pNP-GlcNAc, extremely high concentrations of recombinantly expressed, purified and concentrated enzyme were applied in this study and NV-OGA activity was shown to be significantly impaired when compared to FL-OGA [43]. Based on these findings, OGA activity was normalized to FL-OGA protein levels revealing no difference in OGA activity between AD and age-matched controls indicating that diminished OGA activity in AD may be due to the reduction of cytosolic OGA protein but not to its malfunction.
However, the cause for the observed FL-OGA protein decrease is yet unknown. Transcription and/or translation of FL-OGA could be reduced or isoforms-specific degradation could be increased. A recent whole transcriptome sequencing analysis revealed that while the MGEA5 gene encoding for OGA is not differentially expressed in AD, it shows alternative splicing in AD [46]. Furthermore, OGA is a substrate for caspase-3 upon induction of apoptosis. However, its cleavage does not affect its activity as the OGA fragments remain associated [47]. Finding out how exactly decreased OGA protein levels and O-GlcNAc increase as seen in this study may contribute to the progression of AD will require further investigation. Finding answers to these questions will be highly interesting especially in light of recent animal studies demonstrating that OGA inhibition rescued memory impairment in a mouse model of familial AD [48]. In mice that show early amyloid β deposition and memory deficits, long-term OGA inhibition was initiated well before cognitive impairment could be detected when amyloid plaque deposition begins. In addition to its positive effect on memory, OGA inhibition also decreased amyloid β levels and plaque load and reduced neuroinflammation in brain [48]. In a different mouse model focusing on the tau-related AD pathology, increased tau O-GlcNAc modification by OGA inhibition correlated with decreased numbers of neurofibrillary tangles and reduced neuronal loss [49]. As in the Aβ mouse model, long-term OGA inhibition was initiated at an early time point before tau pathology was apparent [49]. In contrast to these studies, we observe OGA decline at a late AD stage which may have differential effects than early OGA inhibition. It would therefore be highly interesting to analyze the effects of OGA inhibition initiated well after the onset of AD as this is more likely to be the time point when AD treatment in humans will be implemented. Interestingly, in a mouse model of pre-clinical tauopathy OGA inhibition was initiated at a later time point leading to increased O-GlcNAcylation of brain proteins and mitigation of several symptoms including reduced body weight and motor deficits and was beneficial for overall clinical condition of transgenic mice [50].
Investigating the time frame at which OGA decline and augmented protein O-GlcNAcylation occur in AD and what role impaired glucose metabolism may play in these events will be the subject of further studies in our laboratory. Whether the observed changes are downstream consequences of altered metabolism or whether changes in OGA and O-GlcNAc may contribute to the progression from healthy to pathologic states could influence potential therapeutic strategies. In conclusion, this paper adds to the ongoing discussion of the potential role of O-GlcNAcylation in AD. Herein we present the first report of decreased OGA protein level in AD and furthermore, demonstrate a significant correlation between reduced OGA levels and increased cytosolic O-GlcNAcylation. However, conflicting reports on O-GlcNAcylation in AD emphasize the need for further, more detailed studies on this topic. In addition, comprehensive studies of the activity and complex regulation of O-GlcNAc cycling enzymes in AD are crucial for the implementation of potential future AD therapeutic strategies involving O-GlcNAc modulation.
Supplementary Material
Fig. S1: Increased O-GlcNAcylation correlates with decreased OGA protein levels
Correlation analyses were performed between O-GlcNAcylation and various factors in IPL samples from subjects with AD and age-matched controls. Global cytosolic O-GlcNAc levels as well as OGA and OGT protein levels were determined by Western blot analysis (n=13). OGT and OGA levels as well as age, Braak stage and post-mortem interval (PMI) were plotted against the corresponding O-GlcNAc signals. A linear regression line is included in the graph where Pearson correlation was significant (p < 0.05).
Fig. S3: Additional Western blots of cytosolic O-GlcNAc and OGT or OGA protein levels in homogenates of IPL and cerebellar samples of subjects with AD and age-matched controls
(A) Western blots of IPL cytosolic fractions probed for O-GlcNAc and actin. (B) Western blot of cerebellar cytosolic fractions probed for O-GlcNAc and GAPDH. Western blots of IPL (C) and cerebellar (D) samples probed for OGT and actin. Western blots of IPL (E) and cerebellar (F) samples probed for OGA and actin.
Table S2: Data for correlation analyses.
Global cytosolic O-GlcNAc levels as well as FL-OGA, NV-OGA and OGT protein levels were determined by Western blot analysis, normalized to loading controls and are represented as % control values. In addition, age at death (in years), Braak stage and post-mortem interval (PMI; in hours) are listed for each sample.
Acknowledgments
The authors thank the University of Kentucky ADC Clinical Neuropathology Core faculty for providing the brain specimen used for this study. We thank Dr. Garland Crawford and Dr. Shane Arnold for providing antibodies and Dr. Simone Diestel for critical reading of the manuscript. This work was supported in part by a NIH grant to D.A.B. [AG-05119].
Abbreviations
- AD
Alzheimer disease
- O-GlcNAc
O-linked N-acetyl-β-D-glucosamine
- OGT
O-GlcNAc transferase
- OGA
O-GlcNAcase
- HBP
Hexosamine biosynthetic pathway
Footnotes
The authors state no conflict of interest associated with this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Sarah Förster, Email: forster@uni-bonn.de.
Andrew S. Welleford, Email: andrew.welleford@uky.edu.
Judy C. Triplett, Email: judy.triplett@uky.edu.
Rukhsana Sultana, Email: Rukhsana.Sultana@UTSouthwestern.edu.
Brigitte Schmitz, Email: schmitz@uni-bonn.de.
D. Allan Butterfield, Email: dabcns@uky.edu.
References
- 1.Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes, Evidence for O-linked GlcNAc. J Biol Chem. 1984;250:3308–3317. [PubMed] [Google Scholar]
- 2.Zachara NE, O’Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem. 2004;279:30133–30142. doi: 10.1074/jbc.M403773200. [DOI] [PubMed] [Google Scholar]
- 3.Haltiwanger RS, Holt GD, Hart GW. Enzymatic addition of O-GlcNAc to nuclear and cytoplasmic proteins. Identification of a uridine diphospho-N-acetylglucosamine:peptide beta-N-acetylglucosaminyltransferase. J Biol Chem. 1990;265:2563–2568. [PubMed] [Google Scholar]
- 4.Kreppel LK, Hart GW. Regulation of a cytosolic and nuclear O-GlcNAc transferase. Role of the tetratricopeptide repeats. J Biol Chem. 1999;274:32015–32022. doi: 10.1074/jbc.274.45.32015. [DOI] [PubMed] [Google Scholar]
- 5.Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem. 1991;266:4706–4712. [PubMed] [Google Scholar]
- 6.Wells L, Vosseller K, Hart GW. A role for N-acetylglucosamine as a nutrient sensor and mediator of insulin resistance. Cell Mol Life Sci. 2003;60:222–228. doi: 10.1007/s000180300017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong DL, Hart GW. Purification and characterization of an O-GlcNAc selective N-acetyl-beta-D-glucosaminidase from rat spleen cytosol. J Biol Chem. 1994;269:19321–19330. [PubMed] [Google Scholar]
- 8.Okuyama R, Marshall S. UDP-N-acetylglucosaminyl transferase (OGT) in brain tissue: temperature sensitivity and subcellular distribution of cytosolic and nuclear enzyme. J Neurochem. 2003;86:1271–1280. doi: 10.1046/j.1471-4159.2003.01939.x. [DOI] [PubMed] [Google Scholar]
- 9.Gao Y, Wells L, Comer FI, Parker GJ, Hart GW. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J Biol Chem. 2001;276:9838–9845. doi: 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- 10.Shafi R, Iyer SP, Ellies LG, O’Donnell N, Marek KW, Chui D, Hart GW, Marth JD. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci U S A. 2000;97:5735–5739. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lubas WA, Hanover JA. Functional expression of O-linked GlcNAc transferase. Domain structure and substrate specificity. J Biol Chem. 2000;275:10983–10988. doi: 10.1074/jbc.275.15.10983. [DOI] [PubMed] [Google Scholar]
- 12.Hanover JA, Yu S, Lubas WB, Shin SH, Ragano-Caracciola M, Kochran J, Love DC. Mitochondrial and nucleocytoplasmic isoforms of O-linked GlcNAc transferase encoded by a single mammalian gene. Arch Biochem Biophys. 2003;409:287–297. doi: 10.1016/s0003-9861(02)00578-7. [DOI] [PubMed] [Google Scholar]
- 13.Heckel D, Comtesse N, Brass N, Blin N, Zang KD, Meese E. Novel immunogenic antigen homologous to hyaluronidase in meningioma. Hum Mol Genet. 1998;7:1859–1872. doi: 10.1093/hmg/7.12.1859. [DOI] [PubMed] [Google Scholar]
- 14.Comtesse N, Maldener E, Meese E. Identification of a nuclear variant of MGEA5, a cytoplasmic hyaluronidase and a beta-N-acetylglucosaminidase. Biochem Biophys Res Commun. 2001;283:634–640. doi: 10.1006/bbrc.2001.4815. [DOI] [PubMed] [Google Scholar]
- 15.Yang YR, Song M, Lee H, Jeon Y, Choi EJ, Jang HJ, Moon HY, Byun HY, Kim EK, Kim DH, Lee MN, Koh A, Ghim J, Choi JH, Lee-Kwon W, Kim KT, Ryu SH, Suh PG. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell. 2012;11:439–448. doi: 10.1111/j.1474-9726.2012.00801.x. [DOI] [PubMed] [Google Scholar]
- 16.Bond MR, Hanover JA. O-GlcNAc cycling: a link between metabolism and chronic disease. Annu Rev Nutr. 2013;33:205–229. doi: 10.1146/annurev-nutr-071812-161240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 18.Heiss WD, Szelies B, Kessler J, Herholz K. Abnormalities of energy metabolism in Alzheimer’s disease studied with PET. Ann NY Acad Sci. 1991;640:65–71. doi: 10.1111/j.1749-6632.1991.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 19.Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, Yhu S, McInnis MG, Go RC, Vekrellis K, Selkoe DJ, Saunders AJ, Tanzi RE. Evidence for genetic linkage of Alzheimer’s disease to chromosome 10q. Science. 2000;290:2302–2303. doi: 10.1126/science.290.5500.2302. [DOI] [PubMed] [Google Scholar]
- 20.Griffith LS, Mathes M, Schmitz B. Beta-amyloid precursor protein is modified with O-linked N-acetylglucosamine. J Neurosci Res. 1995;41:270–278. doi: 10.1002/jnr.490410214. [DOI] [PubMed] [Google Scholar]
- 21.Arnold CS, Johnson GV, Cole RN, Dong DL, Lee M, Hart GW. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J Biol Chem. 1996;271:28741–28744. doi: 10.1074/jbc.271.46.28741. [DOI] [PubMed] [Google Scholar]
- 22.Dong DL, Xu ZS, Chevrier MR, Cotter RJ, Cleveland DW, Hart GW. Glycosylation of mammalian neurofilaments. Localization of multiple O-linked N-acetylglucosamine moieties on neurofilament polypeptides L and M. J Biol Chem. 1993;268:16679–16687. [PubMed] [Google Scholar]
- 23.Griffith LS, Schmitz B. O-linked N-acetylglucosamine is upregulated in Alzheimer brains. Biochem Biophys Res Commun. 1995;213:424–431. doi: 10.1006/bbrc.1995.2149. [DOI] [PubMed] [Google Scholar]
- 24.Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, Iqbal K, Gong CX. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain. 2009;132:1820–1832. doi: 10.1093/brain/awp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacobs HD, von Boxtel MPJ, Jolles J, Verhey FRJ, Uylings HBM. Parietal cortex matters in Alzheimer’s disease: An overview of structural, funcational and metabolic findings. Neurosci Biobehav Rev. 2012;36:297–309. doi: 10.1016/j.neubiorev.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 26.Zachara NE, Vosseller K, Hart GW. Detection and analysis of proteins modified by O-linked N-acetylglucosamine. Curr Protoc Protein Sci. 2011;66:12.8.1–12.8.33. doi: 10.1002/0471140864.ps1208s66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cieniewski-Bernard C, Bastides B, Lefebvre T, Lemoine J, Mounier Y, Michalski JC. Identification of O-linked N-Acetylglucosamine proteins in rat skeletal muscle using two-dimensional gel electrophoresis and mass spectrometry. Mol Cell Proteomics. 2004;3:577–585. doi: 10.1074/mcp.M400024-MCP200. [DOI] [PubMed] [Google Scholar]
- 28.Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:10804–10809. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008;582:359–364. doi: 10.1016/j.febslet.2007.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalaria RN, Harik SI. Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem. 1989;53:1083–1038. doi: 10.1111/j.1471-4159.1989.tb07399.x. [DOI] [PubMed] [Google Scholar]
- 31.Harr SD, Simonian NA, Hyman BT. Functional alterations in Alzheimer’s disease:decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J Neuropathol Exp Neurol. 1995;54:38–41. [PubMed] [Google Scholar]
- 32.Cheung WD, Hart GW. AMP-activated protein kinase and p38 MAPK activate O-GlcNAcylation of neuronal proteins during glucose deprivation. J Biol Chem. 2008;283:13009–13020. doi: 10.1074/jbc.M801222200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor RP, Parker GJ, Hazel MW, Soesanto Y, Fuller W, Yazzie MJ, McClain DA. Glucose deprivation stimulates O-GlcNAc modification of proteins through up-regulation of O-linked N-acetylglucosaminyltransferase. J Biol Chem. 2008;283:6050–6057. doi: 10.1074/jbc.M707328200. [DOI] [PubMed] [Google Scholar]
- 34.Zou L, Zhu-Mauldin X, Marchase RB, Paterson AJ, Liu J, Yang Q, Chatham JC. Glucose deprivation-induced increase in protein O-GlcNAcylation in cardiomyocytes is calcium-dependent. J Biol Chem. 2012;287:34419–34431. doi: 10.1074/jbc.M112.393207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sultana R, Perluigi M, Butterfield DA. Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer’s disease: insights into mechanism of neurodegeneration from redox proteomics. Antioxid Redox Signal. 2006;8:2021–2037. doi: 10.1089/ars.2006.8.2021. [DOI] [PubMed] [Google Scholar]
- 36.Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A. 2000;97:12222–12226. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 38.Butterfield DA, Hardas SS, Lange ML. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: many pathways to neurodegeneration. J Alzheimers Dis. 2010;20:369–393. doi: 10.3233/JAD-2010-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Li X, Yu Y, Shi J, Liang Z, Run X, Li Y, Dai CL, Grundke-Iqbal I, Iqbal K, Liu F, Gong CX. Developmental regulation of protein O-GlcNAcylation, O-GlcNAc transferase, and O-GlcNAcase in mammalian brain. PLoS One. 2012;7:e43724. doi: 10.1371/journal.pone.0043724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem. 1997;272:9308–9315. doi: 10.1074/jbc.272.14.9308. [DOI] [PubMed] [Google Scholar]
- 41.Keembiyehetty CN, Krzeslak A, Love DC, Hanover JA. A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J Cell Sci. 2011;124:2851–2860. doi: 10.1242/jcs.083287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim EJ, Kang DO, Love DC, Hanover JA. Enzymatic characterization of O-GlcNAcase isoforms using a fluorogenic GlcNAc substrate. Carbohydr Res. 2006;341:971–982. doi: 10.1016/j.carres.2006.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Macauley MS, Vocadlo DJ. Enzymatic characterization and inhibition of the nuclear variant of human O-GlcNAcase. Carbohydr Res. 2009;344:1079–1084. doi: 10.1016/j.carres.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 44.Whisenhunt TR, Yang X, Bowe DB, Paterson AJ, van Tine BA, Kudlow JE. Disrupting the enzyme complex regulating O-GlcNAcylation blocks signaling and development. Glycobiology. 2006;16:551–563. doi: 10.1093/glycob/cwj096. [DOI] [PubMed] [Google Scholar]
- 45.Lazarus BD, Love DC, Hanover JA. Recombinant O-GlcNAc transferase isoforms: identification of O-GlcNAcase, yes tyrosine kinase, and tau as isoform-specific substrates. Glycobiology. 2006;16:415–421. doi: 10.1093/glycob/cwj078. [DOI] [PubMed] [Google Scholar]
- 46.Twine NA, Janitz K, Wilkins MR, Janitz M. Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer’s disease. PLoS One. 2011;6:e16266. doi: 10.1371/journal.pone.0016266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butkinaree C, Cheung WD, Park S, Park K, Barber M, Hart GW. Characterization of beta-N-acetylglucosaminidase cleavage by caspase-3 during apoptosis. J Biol Chem. 2008;283:23557–23566. doi: 10.1074/jbc.M804116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim C, Nam DW, Park SY, Song H, Hong HS, Boo JH, Jung ES, Kim Y, Baek JY, Kim KS, Cho JW, Mook-Jung I. O-linked β-N-acetylglucosaminidase inhibitor attenuates β-amyloid plaque and rescues memory impairment. Neurobiol Aging. 2013;34:275–285. doi: 10.1016/j.neurobiolaging.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 49.Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol. 2012;8:393–399. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- 50.Borghgraef P, Menuet C, Theunis C, Louis JV, Devijver H, Marin H, Smet-Nocca C, Lippens G, Hillaire G, Gijsen H, Moechars D, van Leuven F. Increasing Brain Protein O-GlcNAcylation Mitigates Breathing Defects and Mortality of Tau.P301L Mice. PLoS One. 2013;8:e84442. doi: 10.1371/journal.pone.0084442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1: Increased O-GlcNAcylation correlates with decreased OGA protein levels
Correlation analyses were performed between O-GlcNAcylation and various factors in IPL samples from subjects with AD and age-matched controls. Global cytosolic O-GlcNAc levels as well as OGA and OGT protein levels were determined by Western blot analysis (n=13). OGT and OGA levels as well as age, Braak stage and post-mortem interval (PMI) were plotted against the corresponding O-GlcNAc signals. A linear regression line is included in the graph where Pearson correlation was significant (p < 0.05).
Fig. S3: Additional Western blots of cytosolic O-GlcNAc and OGT or OGA protein levels in homogenates of IPL and cerebellar samples of subjects with AD and age-matched controls
(A) Western blots of IPL cytosolic fractions probed for O-GlcNAc and actin. (B) Western blot of cerebellar cytosolic fractions probed for O-GlcNAc and GAPDH. Western blots of IPL (C) and cerebellar (D) samples probed for OGT and actin. Western blots of IPL (E) and cerebellar (F) samples probed for OGA and actin.
Table S2: Data for correlation analyses.
Global cytosolic O-GlcNAc levels as well as FL-OGA, NV-OGA and OGT protein levels were determined by Western blot analysis, normalized to loading controls and are represented as % control values. In addition, age at death (in years), Braak stage and post-mortem interval (PMI; in hours) are listed for each sample.