

Background: DspA/E is a bacterial type III effector toxic to eukaryotic cells.
Results: Ectopic expression of DspA/E in yeast decreases sphingolipid precursors long chain bases (LCBs) by altering regulation of serine palmitoyltransferase (SPT).
Conclusion: DspA/E toxicity is linked to LCB depletion.
Significance: Regulation of the sphingolipid pathway is a new cellular process manipulated by a bacterial type III effector.
Keywords: Cell Growth, Host-Pathogen Interaction, Phosphatase, Sphingolipid, Yeast Genetics, DspA/E, Long Chain Bases, Orm Regulation, Serine Palmitoyltransferase, Type III Effector
Abstract
Erwinia amylovora, the bacterium responsible for fire blight, relies on a type III secretion system and a single injected effector, DspA/E, to induce disease in host plants. DspA/E belongs to the widespread AvrE family of type III effectors that suppress plant defense responses and promote bacterial growth following infection. Ectopic expression of DspA/E in plant or in Saccharomyces cerevisiae is toxic, indicating that DspA/E likely targets a cellular process conserved between yeast and plant. To unravel the mode of action of DspA/E, we screened the Euroscarf S. cerevisiae library for mutants resistant to DspA/E-induced growth arrest. The most resistant mutants (Δsur4, Δfen1, Δipt1, Δskn1, Δcsg1, Δcsg2, Δorm1, and Δorm2) were impaired in the sphingolipid biosynthetic pathway. Exogenously supplied sphingolipid precursors such as the long chain bases (LCBs) phytosphingosine and dihydrosphingosine also suppressed the DspA/E-induced yeast growth defect. Expression of DspA/E in yeast down-regulated LCB biosynthesis and induced a rapid decrease in LCB levels, indicating that serine palmitoyltransferase (SPT), the first and rate-limiting enzyme of the sphingolipid biosynthetic pathway, was repressed. SPT down-regulation was mediated by dephosphorylation and activation of Orm proteins that negatively regulate SPT. A Δcdc55 mutation affecting Cdc55-PP2A protein phosphatase activity prevented Orm dephosphorylation and suppressed DspA/E-induced growth arrest.
Introduction
Sphingolipids comprise a major class of structural materials and lipid signaling molecules in all eukaryotic cells. Sphingolipid metabolism is generally conserved in animals, yeast, and plants (1). The major steps of the sphingolipid biosynthesis have been elucidated in yeast (Fig. 1). The sphingolipid biosynthesis pathway starts with the condensation of a serine with an acyl-CoA to give long chain bases (LCBs).3 This reaction is catalyzed by the enzyme serine palmitoyltransferase (SPT). The resulting LCBs are further modified to form ceramides, the backbone of more complex sphingolipids. Ceramides are then transported to the Golgi complex where they acquire a species-specific array of polar headgroups to form the complex sphingolipids. Sphingolipid synthesis is tightly regulated. Recent studies in yeast indicate that this regulation is driven by inhibition of SPT, the first and rate-limiting enzyme of the sphingolipid pathway. Two yeast homologous proteins, Orm1 and Orm2, associate with SPT to inhibit SPT activity. The activity of Orm proteins is regulated by phosphorylation (2). Orm proteins are inactivated via phosphorylation by the protein kinase Ypk1 (3, 4), and the Cdc55-PP2A phosphatase was recently identified as a key phosphatase that counteracts Ypk1-mediated phosphorylation of Orm proteins (5). This provides a mechanistic basis for homeostatic regulation of sphingolipid production and enables cells to respond rapidly to changing environment (6). When SPT is inhibited by myriocin, a potent inhibitor of SPT, yeast cells attempt to reinitiate sphingolipid synthesis through activation of the Ypk1 kinase, which phosphorylates and inactivates Orm proteins, thereby leading to increased SPT activity (3, 4).
FIGURE 1.
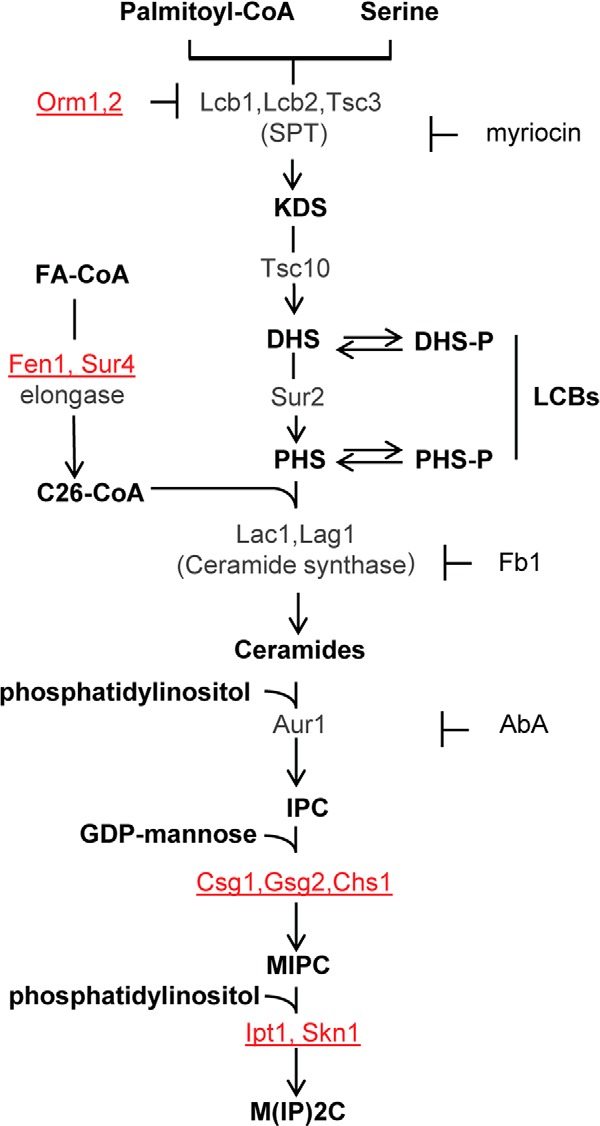
Schematic view of the sphingolipid metabolism in S. cerevisiae. Metabolic intermediates and complex sphingolipids are shown in bold; genes are indicated in gray or red. The genes indicated in red correspond to mutants resistant to DspA/E-mediated toxicity. Specific inhibitors of SPT (myriocin), ceramide synthase (Fb1), and IPC synthase (AbA) are also indicated. Sphingolipid biosynthesis starts in the endoplasmic reticulum with the condensation of serine and palmitoyl-CoA. This step is catalyzed by SPT to produce 3-ketodihydrosphingosine (KDS). Two related proteins, Lcb1 and Lcb2, heterodimerize to form the active SPT. A third protein, Tsc3, associates with the Lcb1/Lcb2 heterodimer and stimulates SPT activity several fold, whereas Orm1p and Orm2p proteins act as negative regulators of SPT. 3-Ketodihydrosphingosine is immediately reduced to DHS by an NADPH-dependent reaction catalyzed by an enzyme encoded by the TSC10 gene. DHS can then be hydroxylated by the Sur2 protein to give PHS. DHS and PHS are the yeast LCBs that are amide-linked to a very long chain fatty acid by the ceramide synthase to form ceramides. Ceramide synthase activity requires either of two redundant genes, LAC1 and LAG1. Ceramides are then modified in the Golgi apparatus to form complex sphingolipids. The first modification is the addition of myoinositol phosphate to form inositol-phosphoceramide (IPC). This reaction is catalyzed by IPC synthase whose activity requires the AUR1 gene. IPC is then mannosylated to yield mannose-inositol-phosphoceramide (MIPC), a reaction that requires the CSG1 and CSG2 genes. The terminal step in sphingolipid biosynthesis is the addition of inositol phosphate to MIPC to yield mannose-(inositol-P)2-phosphoceramide (M(IP)2C). The genes involved in this last reaction are IPT1 and SKN1. FA, fatty acyl.
To induce disease in their hosts, many pathogens inject virulence proteins called effectors, which function inside the host cells. Gram-negative bacteria, which are responsible for numerous diseases in plants and animals, possess three different syringe-like structures allowing injection of protein effectors into the host cells: the type III, type IV, and type VI secretion systems. Once inside host cells, the delivered effectors act as virulence factors modulating cellular processes and suppressing host defense for the benefit of the pathogen (7–9). The study of effector proteins delivered by these specialized machineries has provided remarkable insight not only into fundamental aspects of host-pathogen interactions but also into the basic biology of eukaryotic cells. Notably, type III effectors (T3Es) were shown to target components of the immune system, transcription, cell death, proteasome and ubiquitination systems, RNA metabolism, hormone pathways, and chloroplast and mitochondrion functions (10–12). A current challenge is to systematically determine the virulence functions, biochemical activities, and host targets of bacterial effectors.
Recently, surrogate hosts like the yeast Saccharomyces cerevisiae have become increasingly popular to study the function of effector proteins (13). This is based on the observation that effectors often target fundamental cellular processes that are conserved among eukaryotes. For example, a systematic screen of the yeast deletion strain collection for strains hypersensitive to the expression of the Shigella T3E OspF identified a role for this effector in innate immunity regulation (14).
The bacterium Erwinia amylovora is the causal agent of fire blight disease of pear and apple trees (15). The ability of E. amylovora to promote disease depends on a type III secretion system and on a single injected T3E named DspA/E (16, 17). DspA/E belongs to the AvrE effector family of T3Es, and functional cross-complementation has been demonstrated between DspA/E of E. amylovora and AvrE of Pseudomonas syringae or WtsE of Pantoea stewartii subsp. stewartii (18, 19). T3Es of the AvrE family are widespread in plant-pathogenic bacteria and are among the few T3Es conserved in most analyzed bacterial genomes, suggesting that they provide basic virulence function (20, 21). They are important to promote bacterial growth following infection (17, 22–25) and to suppress callose deposition, a plant basal defense reaction that strengthens the plant cell wall (26, 27). Furthermore, when tested, their ectopic expression in plant and yeast is toxic (28–32). This indicates that they likely target a cellular process that is conserved in eukaryotic cells. However, effectors of the AvrE family are very large proteins of unknown function, and their mode of action once inside the plant cell remains unsolved. AvrE-like effectors are particularly amenable to study in yeast because these effectors are highly toxic when expressed in S. cerevisiae (30, 33).
We previously reported that the T3E DspA/E induces growth arrest and alters cellular trafficking in S. cerevisiae (33). To unravel the cellular processes targeted by DspA/E, in this work we performed a genetic screen to identify S. cerevisiae mutants resistant to DspA/E-mediated growth arrest. The best suppressors identified were mutants impaired in the sphingolipid biosynthetic pathway. Exogenously added sphingolipid precursors, LCBs, also suppressed the DspA/E-mediated growth defect. We further showed that expression of DspA/E led to a decrease of LCB levels. This LCB depletion was due to down-regulation of SPT activity. Interestingly, in contrast to the SPT inhibitor myriocin, DspA/E expression did not activate the Ypk1 kinase leading to hyperphosphorylation of Orm proteins but rather resulted in a dephosphorylation of Orm proteins via a functional Cdc55-PP2A protein phosphatase.
EXPERIMENTAL PROCEDURES
Media, Bacteria, and Yeast Strains
The bacterial strain used in this study was DH5-α. Bacterial cells were grown in Luria broth medium supplemented if required with 100 μg·ml−1 ampicillin. The wild-type yeast strain used for expression of DspA/E was BY4741 (MATa his3Δ1, leu2Δ0, met15Δ0, ura3Δ0; designated as BY) obtained from Euroscarf. Pools of the Euroscarf mutant library were kindly provided by Alain Jacquier (Pasteur Institute, Paris, France). Individual mutant strains used in this study were ordered at Euroscarf. To maintain the plasmid expressing DspA/E (hereafter DspA/E) or the pCMha189 empty vector (hereafter EV) (34), yeast cells were routinely grown at 30 °C in selective synthetic complete medium lacking uracil supplemented with 2% (w/v) glucose (designated as SD-Ura). To repress DspA/E expression, doxycycline (10 μg·ml−1) was added to the medium (designated as SD-Ura Doxy). When required, SD medium lacking both uracil and histidine (SD-Ura-His) was used. For transformation, yeast cells were grown overnight at 30 °C in YPD medium (1% (w/v) yeast extract, 1% (w/v) Bacto Peptone, 2% (w/v) glucose) and transformed with the appropriate plasmids selecting for prototrophy using the lithium acetate method.
Plasmid Construction
A fill-in was performed with T4 DNA polymerase on a DspA/E XbaI/SacI fragment issued from pTB4 (28). This fragment was then cloned into the yeast centromeric URA+ plasmid pCMha189 (34) previously digested with BamHI and filled with Klenow to give plasmid DspA/E. Other plasmids used in this study are described in Table 1.
TABLE 1.
Plasmids used in this study
aa, amino acids.
| Plasmids | Description | Source/Ref. |
|---|---|---|
| pDspA/E | pCMha189::dspA/E (CEN URA, XbaI-SacI) | This study |
| pRS413 | Centromeric HIS + vector | This study |
| pMS062 | pRS413::3xFLAG-ORM1 (CEN HIS, BamHI-EcoRI) | This study |
| pMS005 | pRS413::3xHA-ORM2 (CEN HIS, XbaI-SacI) | 46 |
| pMS064 | pRS413::3xFLAG-ORM1-3A S51A,S52A,S53A (CEN HIS) | This study |
| pMS040 | pGEX-6P-1::ORM1 [ORF aa 1–82] (BamHI-EcoRI) | 46 |
| pMS042 | pGEX-6P-1::ORM1 [ORF aa 1–82] S51A,S52A,S53A (BamHI-EcoRI) | 46 |
PCR Identification of the Yeast Deletion Mutants
For each of the 14 pools initially identified in our screen, 30 independent colonies grown on SD-Ura medium were resuspended in 5 μl of 0.07 n NaOH and incubated for 5 min at 97.5 °C. This template was then used for PCR amplification with two sets of primers: U1 (5′-GATGTCCACGAGGTCTCT-3′) and KD1 (5′-TCCTAACCTTTTATATTTCTC-3′) or U2 (5′-CGTACGCTGCAGGTCGAC-3′) and KU2 (5′-ATGGTATTGATAATCCTGATATG-3′). The amplified products were then sequenced to identify the 20-mer bar codes unique to each mutant.
Chemicals
6-Aminoquinolyl-N-hydroxysuccinimidyl carbamate reagent was purchased from Waters Corp. (Milford, MA) as an AccQ Fluor reagent kit and dissolved according to the manufacturer's instructions. C16-SPH was purchased from Matreya Inc. (Pleasant Gap, PA). Phytosphingosine (PHS) and dihydrosphingosine (DHS) were purchased from Enzo Biomol and dissolved in ethanol as 1 mg·ml−1 stock solutions. Phos-tagTM (Wako Chemicals) was prepared according to the manufacturer's instructions. For the thin-layer chromatography (TLC), fumonisin B1 (Fb1) (Sigma-Aldrich) and aureobasidin A (AbA) (Ozyme) were used at 100 μg·ml−1 (35) and 1 μg·ml−1 (36) concentrations, respectively. To induce Ypk1 kinase and Orm protein hyperphosphorylation and activation, myriocin (Sigma-Aldrich) was dissolved in 100% methanol to 500 μg·ml−1 and used at a final concentration of 0.5 μg·ml−1. The following antibodies were used: anti-Ypk1 at 1:1000 (37), monoclonal anti-Ypk1 Thr(P)-662 at 1:1000 (3) (kindly provided by R. Loewith), anti-FLAG at 1:1000 (M2, Sigma-Aldrich), anti-HA at 1:1000 (Cell Signaling Technology), anti-actin at 1:2000 (Millipore), and the appropriate HRP-conjugated anti-mouse IgG antibodies at 1:5000 (Pierce).
Free LCB Extraction
Strains were grown on SD-Ura Doxy plates for 48 h. Then the yeast cells were diluted to an A600 of 0.7 in 100 ml of SD-Ura and grown for 3 h to an A600 of 1. Cells were washed three times with ice-cold water, and pellets were suspended in 5 ml of methanol/chloroform (1:1) as described (38). At this stage, 10 ng of C16-SPH/A600 unit were added to each sample as a quantitative internal standard. Extraction was performed twice in a 50 °C bath with frequent mixing. Pooled lipid extracts were dried under nitrogen, desalted by butanol/water partitioning, dried under nitrogen, and resuspended in methanol. Then AccQ derivatization was done as described (39). Aliquots of derived lipids (20 μl) were subjected to HPLC as follows.
HPLC Separation and Analysis
Reversed phase chromatography was carried out on a C18 column (100 × 4.6 mm, C18, 100 Å, 2.6 μm, Kinetex Phenomenex) with a C18 guard column (4 × 3 mm, Gemini C18, 10/PK AFO-8497) on a Beckman Coulter System Gold 126 liquid chromatograph with 32 Karat software and with an RF-10A XL Shimadzu fluorescence detector. Elution was performed as described (39) except that elution was carried out for 35 min isocratically at a 0.3 ml·min−1 flow rate with acetonitrile/methanol (60:40) (solvent A). At the end of the run, the column was recycled using a 1-min linear gradient from 100% solvent A to 100% acetonitrile (solvent B), 5 min of solvent B at 0.5 ml·min−1, 0.5-min linear gradient to solvent A at 0.5 ml·min−1, and 5-min equilibration with solvent A at 0.3 ml·min−1. Elution of fluorescent compounds was monitored by excitation at 244 nm and emission at 398 nm. The data presented are mean values of three independent experiments Results are presented as means ± S.D. (n = 3; ***, p < 0.001; **, p < 0.05; *, p < 0.09; two-tailed Student's t test).
Cell Labeling and Thin-layer Chromatography
Cells were grown on SD-Ura Doxy plates for 48 h, diluted to an A600 of 0.7 in 10 ml of SD-Ura liquid medium, and grown for 90 min at 30 °C. When required, Fb1 and AbA were added 30 min prior to labeling. Yeast cells were then labeled at 25 °C for 40 min by adding 20 μCi of [14C]serine, diluted with 3 ml of fresh prewarmed SD-Ura medium, and further incubated for 80 min at 25 °C. Labeling was stopped by adding 40 μl of 3 mm NaN3, 10 mm NaF. Cells were then washed with water twice, suspended in methanol/chloroform (1:1), and disrupted with glass beads by vortexing. After 5 min of centrifugation at 3000 rpm, the supernatant was collected, and pellet was re-extracted twice with chloroform/methanol/water (10:10:3). Pooled supernatant were dried under nitrogen, and lipids were then desalted by butanol/water partitioning. Lipids were developed by ascending TLC using 0.2-mm TLC silica gel 60 with chloroform, methanol, 2 m NH4OH (40:10:1) as solvent. Radioactivity was detected and quantified using a Storm 840 (Amersham Biosciences).
Actin Polarization State of Cells
Cells were grown to logarithmic phase, fixed by addition of 37% formaldehyde to a final concentration of 3.7%, and incubated for 30 min at room temperature. Cells were washed twice with PBS containing 1 mg·ml−1 BSA, and the cell pellet was resuspended in 25 μl of PBS containing 1 mg·ml−1 BSA. Then 5 μl of rhodamine-phalloidin (Molecular Probes; 3000 units/1.5 ml of MeOH) were added. After 1 h of incubation at room temperature in the dark, cells were washed three times and resuspended in 500 μl of PBS containing 1 mg·ml−1 BSA. The actin cytoskeleton was visualized using an Axioplan 2 fluorescence microscope from Zeiss. Quantification of polarized and depolarized cells was performed as described previously (33). At least 100 cells were counted for each condition. Data are presented as means ± S.D. of three independent experiments (n = 100; p < 0.001; two-tailed Student's t test).
Phosphoaffinity Gel Electrophoresis
To examine the phosphorylation state of FLAG-Orm1 and HA-Orm2, cells were grown for 6 h in SD-Ura-His medium and treated with 0.5 μg·ml−1 myriocin for 1 h when required. Cells were then harvested and resuspended in TBS lysis buffer containing 15% glycerol, 0.5% Tween 20, 10 mm NaF, 10 mm NaN3, 10 mm, p-nitrophenyl phosphate, 10 mm sodium pyrophosphate, and 10 mm glycerophosphate, 1 mm PMSF, and protease inhibitor mixtures (EDTA-free Complete, Roche Applied Science). Cells were broken by a FastPrep (45 s × 5 with 3-min intervals on ice) at 4 °C, and cell debris were removed by centrifugation at 500 × g for 10 min. 10 μg of proteins were then loaded onto 7.5% SDS-polyacrylamide gels containing 25 μm Phos-tag acrylamide and 10 μm MnCl2. The gels were run at 70-V constant voltage for 2.5 h and rinsed twice for 5 min in 1 mm EDTA-containing transfer buffer and then twice for 5 min in transfer buffer without EDTA before transfer onto nitrocellulose membrane. Immunodetection was performed using the anti-FLAG, anti-HA, and anti-actin antibodies described above. Gel to gel differences in band patterns are due to variability in phosphoaffinity gel resolution (2).
Ypk1 in Vitro Kinase Assay
Endogenous Ypk1 kinase was immunoprecipitated from total yeast cell lysates (200 μg) by Ypk1 antibodies (37). When needed, cells were treated with 0.5 μg·ml−1 myriocin for 1 h. Recombinant Orm proteins (GST-Orm1N1-82 and GST-Orm1N1-82-3A) were prepared as described (30). The kinase reaction (final volume, 100 μl) was performed in kinase buffer (25 mm Tris-HCl (pH 7.5), 5 mm β-glycerophosphate, 2 mm DTT, 0.1 mm Na3VO4, 10 mm MgCl2). Reactions were started by addition of 10 μl of ATP mixture (400 μm ATP and 3 μCi of [γ-32P]ATP (Amersham Biosciences)) and incubated for 30 min at 30 °C under vigorous shaking. Reactions were stopped by adding 5× SDS-PAGE sample buffer and incubated for 10 min at 65 °C. Proteins were separated on two SDS-polyacrylamide gels. One gel was transferred to nitrocellulose membrane for Western blotting, whereas the other gel was autoradiographed with an imaging plate (Fujifilm), and the images were captured by a Typhoon scanner (GE Healthcare).
Phosphorylation State of Ypk1 Kinase
Extracts were prepared as described previously for FLAG-Orm1 and HA-Orm2. Proteins were then resolved on a 7.5% phosphate affinity SDS-polyacrylamide gel and blotted on nitrocellulose membrane. Immunodetection was performed using anti-Ypk1Thr-662, anti-Ypk1, and anti-actin antibodies described above.
RESULTS
Yeast Mutants Affected in the Sphingolipid Biosynthetic Pathway Are Resistant to DspA/E-mediated Growth Defect
To unravel the cellular processes targeted by DspA/E in yeast, the DspA/E gene was cloned into the yeast centromeric plasmid pCMha189, which contains a doxycycline-regulatable promoter and URA3 as a selection marker (34). On SD-Ura medium, growth of the BY strain transformed with plasmid DspA/E was severely impaired, whereas growth of the BY strain transformed with the EV was not altered. On SD-Ura Doxy medium repressing DspA/E expression, the two strains grew equally well (Fig. 2A). To identify mutants resistant to DspA/E, we developed a screen based on transformation of strain BY with the plasmid DspA/E. When we transformed the strain BY with plasmid DspA/E and plated half of the transformed cells on SD-Ura medium and half of the transformed cells on SD-Ura Doxy medium, 2 days post-transformation the ratio of the number of colonies on SD-Ura versus the number of colonies on SD-Ura Doxy remained stable over 20 independent transformations (0.007 ± 0.002). We used this ratio to identify putative suppressors of the DspA/E-induced growth defect. Forty-five pools of 100 mutants of the Yeast Knock-out deletion collection (40, 41) were transformed with DspA/E. We reasoned that if a resistant mutant was present in a pool then the ratio of colonies grown on SD-Ura versus colonies grown on SD-Ura Doxy would be reproducibly higher than the ratio observed with the wild-type strain BY. This was indeed the case for 14 pools for which we observed a ratio above 0.03 2 days post-transformation following three independent transformations. From these 14 pools, 10 colonies were randomly picked from SD-Ura medium in each of the three independent transformations and further analyzed to identify the corresponding mutant. Sixty-three mutants, each found at least two times, were ordered from Euroscarf and transformed individually with plasmid DspA/E. Among nine mutants having a ratio following transformation superior to 0.5, six were affected in the sphingolipid biosynthetic pathway or in transport of metabolic precursors of this pathway (YPL057C/CSG1, YDR072C/IPT1, YDR497C/ITR1, YEL042W/GDA1, YDR372C/VPS74, and YPL056C/LCL1; Fig. 2B in red and Fig. 1). Because our screening procedure was not exhaustive, we decided to transform the available viable single mutants of the sphingolipid pathway with plasmid DspA/E. This allowed the identification of six new mutants (YBR036C/CSG2, YGR143W/SKN1, YCR034W/FEN1, YLR372W/SUR4, YGR038W/ORM1, YLR350W/ORM2, and YBR161W/CSH1) for which the calculated ratio of colonies grown on SD-Ura/colonies grown on SD-Ura Doxy is above 0.4 (Fig. 2B in blue and Fig. 1). When plated on SD-Ura medium, the 13 identified mutations were able to rescue the growth of the yeast strain expressing DspA/E, confirming their suppressor status (Fig. 2C).
FIGURE 2.
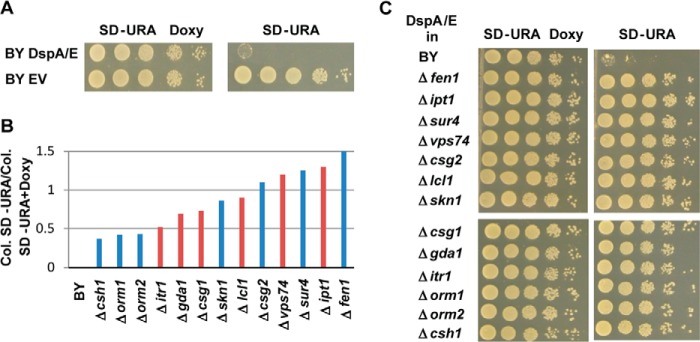
Sphingolipid mutants are suppressors of DspA/E-induced growth defect. A, serial dilutions of strain BY DspA/E or BY EV were spotted on SD-Ura Doxy or SD-Ura plates. Photographs were taken after 48 h growth. B, ratio of the number of colonies (Col.) observed on SD-Ura medium versus the number of colonies observed on SD-Ura Doxy medium for the indicated yeast strains transformed with DspA/E. The red bars represent the ratio observed with the yeast mutant strains identified through the general screening procedure, whereas the blue bars represent the ratio observed with the yeast mutant strains identified through systematic analysis of available viable single mutants of the sphingolipid pathway. C, serial dilutions of the indicated strains bearing plasmid DspA/E spotted on SD-Ura Doxy or SD-Ura. All photographs were taken after 48 h of growth.
Exogenous LCBs Suppress DspA/E-mediated Growth Defect
We further investigated why mutations affecting the sphingolipid biosynthetic pathway are suppressors of DspA/E-induced growth arrest. Mutants blocked in complex sphingolipid biosynthesis downstream of SPT (Δipt1, Δskn1, Δcsg1, Δcsg2, and Δskn1) are known to accumulate precursors of the sphingolipid pathway such as ceramides, LCBs, or LCB-Ps (42). Mutants affected in acyl-CoA incorporation (Δsur4 and Δfen1) also result in LCB accumulation (36, 43). Finally, mutants affected in the regulation of SPT activity (Δorm1 and Δorm2) are no longer able to down-regulate SPT activity, which leads to accumulation of sphingolipid precursors (2). Overall, this suggests that a high level of LCBs may suppress DspA/E-mediated effects. If this is true, then exogenously added LCBs, which are rapidly incorporated in ceramides and complex sphingolipids (36), should also rescue the growth of yeast cells when DspA/E is expressed. To test this hypothesis, we grew the BY DspA/E and BY EV strains in the presence of 15 μm PHS or 15 μm DHS. On SD-Ura medium, both LCBs at 15 μm partially suppressed the growth defect of strain BY DspA/E (Fig. 3). Interestingly, careful observation of the growth assay showed that 15 μm PHS or 15 μm DHS was slightly toxic to BY EV yeast cells on medium supplemented with doxycycline, whereas growth of BY DspA/E cells was not affected. This suggests that on repressive medium DspA/E is not fully repressed and counteracts the toxicity induced by LCB addition to the growth medium.
FIGURE 3.
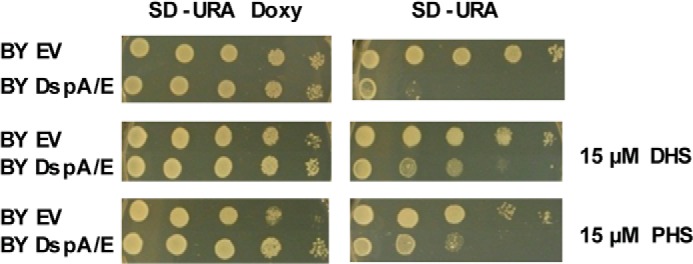
Exogenously added LCBs rescue DspA/E-mediated growth defect. Serial dilutions of strain BY DspA/E or BY EV were spotted on SD-Ura Doxy or SD-Ura plates supplemented or not with 15 μm DHS or 15 μm PHS. Photographs were taken after 48 h of growth.
DspA/E Expression Is Associated with Decreased LCB Levels
The above findings suggest that DspA/E-mediated toxicity may be linked to a decrease in LCB levels. To test this hypothesis, we examined the LCB levels (DHS and PHS) in BY cells expressing DspA/E. Because a dynamic balance exists between the LCBs and their phosphorylated forms (Fig. 1), we also monitored the levels of PHS-P and DHS-P. Overall, whatever the LCB species examined (DHS, PHS, DHS-P, or PHS-P), the level observed in BY DspA/E cells was lower than that observed in BY EV cells (Fig. 4A). We then examined the levels of LCB and LCB-P in the Δsur4 and Δfen1 cells with or without DspA/E. In Δsur4 cells carrying the empty vector, very high levels of PHS, DHS-P, and DHS were detected compared with those detected in BY EV cells, whereas the DHS-P level was lower than that observed in BY EV cells (Fig. 4B). In Δfen1 EV cells, the observed PHS-P, PHS, DHS-P, and DHS levels were all higher than those observed in BY EV cells and Δsur4 cells (Fig. 4D). These phenotypes are consistent with a previous publication (44). When DspA/E was expressed in Δsur4 or Δfen1 cells, the levels of LCBs and LCB-Ps were reduced compared with the control Δsur4 EV or Δfen1 EV cells (Fig. 4, B, C, and D). In Δsur4 DspA/E cells, the levels of LCBs and DHS-P were similar to those observed in the BY EV cells, whereas the PHS-P level was lower (Fig. 4, B and D). In Δfen1 DspA/E cells, the levels of LCBs and DHS-P remained higher than those observed in the BY EV cells, whereas the PHS-P level was similar (Fig. 4, C and D). Altogether, these results indicate that DspA/E expression induces LCBs and LCB-P depletion in BY, Δsur4, and Δfen1 cells. However, in the suppressor mutants Δsur4 and Δfen1, LCB levels were maintained at the same or above the level observed in BY EV cells.
FIGURE 4.

DspA/E-mediated growth defect is associated with depletion of LCB levels. A, histogram representing LCB (DHS and PHS) and LCB-P (DHS-P and PHS-P) levels observed in lipid extracts derived from BY EV or BY DspA/E cells 3 h after a shift from SD-Ura Doxy medium to SD-Ura medium. Quantities were calculated using C16-SPH as an internal standard. B and C, histograms representing LCB (DHS and PHS) and LCB-P (DHS-P and PHS-P) levels observed in extracts derived from BY DspA/E, Δsur4 EV, and Δsur4 DspA/E cells (B) and from BY DspA/E, Δfen1 EV, and Δfen1 DspA/E cells (C) 3 h after a shift from SD-Ura Doxy medium to SD-Ura medium. In B and C, results are expressed as -fold change (FC) compared with the BY EV control. In A, B, and C, results are presented as means, and error bars represent S.D. (n = 3; ***, p < 0.001; **, p < 0.05; *, p < 0.09; N.S, non-significant; two-tailed Student's t test). D, LCB and LCB-P levels observed (pmol·A600−1) for the above strains presented as means ± S.D.
DspA/E Expression Alters Sphingolipid Biosynthesis
DspA/E-induced LCB depletion suggests that the expression of DspA/E represses LCB synthesis. To examine this hypothesis, we performed in vivo metabolic labeling with [14C]serine of neosynthesized LCBs in yeast cells containing either the empty vector or plasmid DspA/E. As a control, we monitored the LCB level in the presence of AbA and Fb1, inhibitors of inositolphosphoceramide (IPC) synthesis and ceramide synthesis, respectively (Fig. 1). As expected, in BY EV cells, AbA caused ceramide accumulation (Fig. 5, A and C). Surprisingly, we also observed an LCB decrease in the presence of AbA, which could be interpreted as an attempt of the cell to overcome the inhibition of IPC synthesis by overproduction of ceramides (Fig. 5, A and B). The presence of Fb1, which blocks ceramide synthesis, caused a ceramide decrease and LCB accumulation as expected (Fig. 5, A, B, and C). Conversely, an uncharacterized lipid a characteristic of ceramide synthesis inhibition (36) and another uncharacterized lipid (denoted §) already observed in sphingolipid TLC profiles (45) appeared (Fig. 5A). Upon DspA/E expression, the amount of LCB observed was always reduced compared with the empty vector treatment (Fig. 5, A and B). The expression of DspA/E also counteracted the elevation of ceramides observed in the presence of AbA (Fig. 5, A and C), further indicating that the LCB decrease observed upon DspA/E expression cannot be explained by an accumulation of ceramides. Interestingly, a mild base-resistant uncharacterized lipid (denoted * in Fig. 5A) also appeared in cells expressing DspA/E. We could not detect this additional species in HPLC. It is therefore unlikely that this band could be 3-ketodihydrosphingosine. As a control, we also measured the uptake of [3H]serine by both BY EV and BY DspA/E cells and observed that expression of DspA/E in yeast did not affect the uptake (Fig. 5D). Altogether, these results indicate that LCB neosynthesis is reduced upon DspA/E expression. This reduction could be attributed to down-regulation of either Tsc10p or SPT activity (Fig. 1). However, because we could not detect 3-ketodihydrosphingosine accumulation, the reduction of LCB neosynthesis following DspA/E expression is likely due to down-regulation of SPT activity.
FIGURE 5.

DspA/E expression alters sphingolipid biosynthesis. A, TLC analysis of [14C]serine-labeled sphingolipids. 10 A600 units of BY cells harboring EV or expressing DspA/E were grown on SD-Ura medium supplemented or not with ceramide synthase inhibitor (Fb1; 100 μg·ml−1) or IPC synthase inhibitor (AbA; 1 μg·ml−1) before labeling. Lipids were extracted, O-deacetylated by NaOH treatment, and resolved by TLC in chloroform/methanol/NH4OH (40:10:1, v/v/v). Radiolabeled sphingolipids were revealed and quantified using a Storm phosphorimaging system. The same amount of cells (2.5 A600 units) was spotted for each sample. Assignments are supported by (I) the accumulation or absence of major bands upon specific inhibitor treatments, (II) comparison with cold PHS and DHS standards revealed by nihydrin and (iii) previous report (36, 45). Cer, ceramide; a, lipid a (36); §, uncharacterized Fb1-induced lipid (45); *, uncharacterized DspA/E-induced lipid; o, origin. The image presented is representative of three independent repetitions. B and C represent the quantification of LCBs and ceramides, respectively, shown in A in band intensity ×10−3. D, serine uptake assay in BY EV and BY DspA/E cells. Cells were labeled with [3H]serine, and incorporation of [3H]serine was measured by a scintillation counter. Results are represented as means, and error bars represent S.D. (n = 3; p > 0.1; n.s, not significant; two-tailed Student's t test).
The Δsur4 and Δfen1 Mutants Restore Polarization of Actin Affected by DspA/E
In a previous study, we showed that the expression of DspA/E in yeast strongly affects the polarization of actin (33). This prompted us to examine whether the Δsur4 and Δfen1 mutants could also restore the polarization of actin affected by DspA/E. To test this hypothesis, cells were grown to exponential phase, and the actin cytoskeleton was visualized using rhodamine-phalloidin. We observed ∼75% polarized cells for the control strain BY EV (Fig. 6). When DspA/E was expressed, the strain BY DspA/E became depolarized as previously observed and contained only 20% polarized cells (Fig. 6). However, when DspA/E was expressed in Δsur4 or Δfen1 cells, we observed 58 and 82% polarized cells, respectively (Fig. 6). Based on these results, we conclude that the Δsur4 and Δfen1 mutations suppress the actin polarization defect observed in BY yeast cells following DspA/E expression.
FIGURE 6.
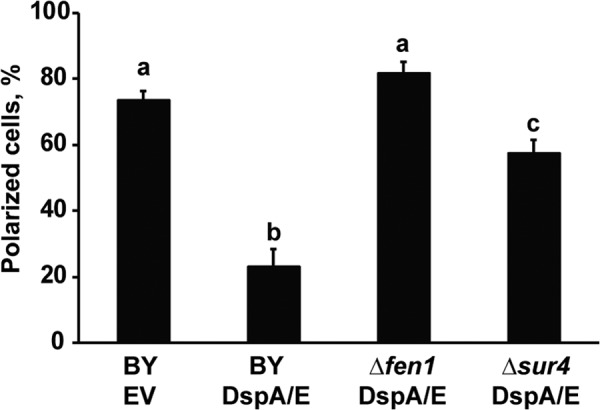
The Δsur4 and Δfen1 mutants restore actin polarization affected by DspA/E. Strains were grown in selective medium (SD-Ura) at 30 °C to exponential phase. Actin structure was then visualized using rhodamine-phalloidin and fluorescence microscopy. The histogram shows the average percentage of polarized cells versus the total number of cells. At least 100 cells were counted for each condition. Data are presented as means of three independent experiments, and conditions with different letters are statistically different (n = 100; p < 0.001; two-tailed Student's t test). Error bars represent S.D.
DspA/E Expression Induces Dephosphorylation of Orm Proteins
We next wondered how expression of DspA/E leads to SPT down-regulation. In yeast, Orm1 and Orm2 are dynamic negative regulators of SPT that are regulated by phosphorylation. We therefore tested whether DspA/E expression influences the phosphorylation status of Orm proteins. To test this, DspA/E was co-expressed in yeast cells with FLAG-Orm1 or HA-Orm2, and the phosphorylation status of both Orm proteins was checked by electrophoresis in SDS-polyacrylamide gels that incorporate a phosphate binding agent to improve the separation of phosphorylated species (Phos-tag). FLAG-Orm1 and HA-Orm2 appeared as multiple phosphorylated forms that collapsed to faster migrating species upon DspA/E expression, indicating that DspA/E expression decreased the phosphorylation status of Orm proteins (Fig. 7, A and B). Conversely, upon addition of the SPT inhibitor myriocin to BY cells, FLAG-Orm1 appeared hyperphosphorylated (Fig. 7A). As already described, this is an attempt for cells to restore a correct intracellular level of sphingolipids (4). Therefore, although both myriocin and DspA/E repress SPT activity, their mode of action is different.
FIGURE 7.

DspA/E expression induces dephosphorylation of Orm proteins without affecting Ypk1 activity. A, FLAG-Orm1 or FLAG-Orm1-3A proteins were expressed in wild-type BY cells harboring DspA/E or the EV. After 6 h on SD-Ura-His medium, the cells were lysed, and the resulting extracts were resolved by phosphate affinity SDS-PAGE (Phos-tag) gels and analyzed by immunoblotting with anti-FLAG. Loading controls on SDS-polyacrylamide gels were analyzed by anti-FLAG and anti-actin antibodies. As a control, myriocin-treated and untreated BY (EV) cells are shown. B, HA-Orm2 was expressed in wild-type BY cells harboring DspA/E or the EV and analyzed by anti-HA or anti-actin antibody. C, Ypk1 was immunopurified from BY yeast cells harboring DspA/E or EV, and its ability to phosphorylate wild-type GST-Orm1N1–82 protein (WT) or the GST-Orm1N1–82-3A phosphodeficient protein (3A) was tested. Controls of input Ypk1, wild-type GST-Orm1N1–82, and GST-Orm1N1–82-3A are shown below. D, Ypk1 was immunopurified from BY myriocin-treated or untreated cells, and its ability to phosphorylate wild-type GST-Orm1N1–82 was tested. Controls of input Ypk1 and wild-type GST-Orm1N1–82 are shown below. E, Ypk1 phosphorylation state was checked in vivo by using an antibody that specifically recognized phosphorylation of Thr-662 of Ypk1 (Ypk1T662). Two exposures, short (S.E) and long (L.E), are shown. Loading controls were analyzed by anti-Ypk1 and anti-actin antibody. BY cells harboring DspA/E or the EV are shown. Controls of myriocin-treated or solvent-treated (MeOH) BY cells are also shown.
DspA/E-mediated Dephosphorylation of Orm Proteins Requires the Cdc55-PP2A Phosphatase
To regulate SPT activity, phosphorylation status of Orm proteins is itself regulated by the Ypk1 kinase and the PP2A phosphatase (3–5). Two groups of phosphorylation sites have been described on Orm1. The first is exclusively phosphorylated by the Ypk1 kinase at serines 51, 52, and 53 and has been described as important for SPT activity (4), whereas the second (serines 29, 32, 34, 35, and 36) is not involved in this regulation (46). To examine whether DspA/E affects the phosphorylation triggered by Ypk1, we checked the phosphorylation status of the FLAG-Orm1-WT and FLAG-Orm1-3A devoid of the Ypk1 phosphorylation site (serines 51, 52, and 53) in BY DspA/E and BY EV cells. The expression of DspA/E had only a slight impact on the phosphorylation status of FLAG-Orm1-3A, indicating that expression of DspA/E mostly impacts phosphorylation of the Ypk1 site (Fig. 7A). We therefore examined the impact of DspA/E expression on Ypk1 activity both in vitro and in vivo. Ypk1 was immunoprecipitated from yeast cells expressing DspA/E and assayed for kinase activity using wild-type GST-Orm1N1–82 protein (Fig. 7C, WT) as a substrate in vitro. We also used GST-Orm1N1–82-3A phosphodeficient protein (Fig. 7C, 3A) devoid of Ypk1 phosphorylation sites as a control substrate. GST-Orm1N1–82 wild-type protein was phosphorylated to the same extent by Ypk1 immunopurified from either BY DspA/E or BY EV cells, whereas no phosphorylation was detected with the GST-Orm1N1–82-3A protein (Fig. 7C). As a control, we checked GST-Orm1N1–82 hyperphosphorylation by Ypk1 purified from myriocin-treated BY EV cells (Fig. 7D). We further checked the phosphorylation status of Ypk1 following DspA/E expression in vivo using an antibody specifically raised against the phosphorylated Thr-662 residue of Ypk1 (3). Although an increased level of Ypk1Thr-662 phosphorylation was observed with the myriocin-treated control, expression of DspA/E did not affect Ypk1Thr-662 phosphorylation (Fig. 7E). Therefore, expression of DspA/E, contrary to treatment with myriocin, does not activate Ypk1.
We then tested whether DspA/E-induced Orm dephosphorylation requires the PP2A phosphatase. PP2A is a heterotrimeric enzyme composed of three distinct subunits: a scaffold subunit, a catalytic subunit, and a regulatory subunit encoded by two distinct genes, CDC55 and RTS1 (47). Stability of the heterotrimeric complex requires a methylation of the C terminus of the catalytic subunit by the carboxymethyltransferase Ppm1 protein (48, 49). Interestingly, we identified Δppm1 in our screen as a good suppressor of DspA/E-mediated growth arrest (Fig. 8A). A major function of the regulatory subunit is to target substrate phosphoproteins to the PP2A phosphatase (47). Orm proteins were described as hyperphosphorylated in a Δcdc55 mutant, whereas their phosphorylation status was not modified in a Δrts1 mutant, indicating that Cdc55 is the only PP2A regulatory subunit involved in Orm-dependent sphingolipid regulation (5). We therefore checked suppression of DspA/E-mediated growth arrest in a Δcdc55 mutant (Fig. 8A) and compared the phosphorylation status of FLAG-Orm1 protein upon DspA/E expression in either Δcdc55 or BY cells (Fig. 8B). Control myriocin-treated Δcdc55 and BY cells showed hyperphosphorylation of FLAG-Orm1. As already observed in Fig. 6A, DspA/E expression had a severe impact on FLAG-Orm1 phosphorylation in BY cells; however, in Δcdc55 cells, FLAG-Orm1 phosphorylation remained mostly unchanged (Fig. 8B). This indicates that DspA/E-mediated dephosphorylation of Orm proteins requires a functional Cdc55-PP2A phosphatase.
FIGURE 8.

DspA/E-induced growth arrest and Orm protein dephosphorylation require a functional Cdc55-PP2A phosphatase. A, serial dilutions of strains BY, Δppm1, and Δcdc55 harboring DspA/E or the EV were spotted on SD-Ura Doxy or SD-Ura plates. Photographs were taken after 48 h of growth. B, FLAG-Orm1 was expressed in BY and Δcdc55 mutant cells expressing DspA/E or not (EV). After 6 h on SD-Ura medium, the cells were lysed, and the resulting extracts were resolved by Phos-tag gels and analyzed by immunoblotting with anti-FLAG. Loading controls on SDS-polyacrylamide gels were analyzed by anti-FLAG or anti-actin antibodies. Controls of BY and Δcdc55 mutant treated with myriocin are also shown.
DISCUSSION
To unravel the mode of action of the bacterial effector DspA/E, we performed a genetic screen and identified several S. cerevisiae mutants resistant to DspA/E-induced growth arrest. Our screen, based on transformation efficiency on inducing medium relative to the repressive medium, was simple, efficient, and particularly suited to study highly toxic effectors. Furthermore, it allowed the classification of suppressor strength, a feat not possible using a simple serial dilution plate assay (Fig. 2, compare B and C).
Using this screen, we discovered that mutants affected in the sphingolipid biosynthesis pathway are the most resistant to the DspA/E-induced growth defect. Further analysis showed that expression of DspA/E decreased LCB levels both in wild type and sphingolipid mutants. However, in the sphingolipid mutants, LCB levels were maintained at the same or above the level observed in the wild-type strain when DspA/E was expressed. Interestingly, Δfen1, the best suppressor identified in our screening procedure (Fig. 1B), also showed very high intracellular LCB levels (Fig. 4D). Exogenously added LCBs also partially suppressed the DspA/E-mediated growth defect. Altogether, these results indicate that when LCB levels are maintained above the threshold observed in the wild-type yeast strain cell growth is possible, whereas below this threshold, cell growth is stopped. This highlights the importance of LCB levels in DspA/E-induced growth arrest.
We previously described that expression of DspA/E in S. cerevisiae impairs endocytosis and actin polarization (33). This alteration of cellular trafficking can now be attributed to SPT repression and LCB depletion because we showed in this work that the Δsur4 and Δfen1 mutations, for which LCB levels are maintained at the same or above the level observed in the wild-type strain expressing DspA/E, suppressed the DspA/E-induced actin polarization defect. Our results are consistent with a previous study showing that repression of SPT activity, induced at 37 °C with the lcb1-100 thermosensitive mutation, blocks LCB synthesis and impairs endocytosis and actin organization (50).
SPT repression is not directly driven by DspA/E, and we have shown that DspA/E expression led to dephosphorylation and activation of Orm proteins, the negative regulators of SPT. Interestingly, myriocin, a direct inhibitor of SPT, has an opposite effect on Orm phosphorylation. Myriocin induces Ypk1-mediated hyperphosphorylation and inactivation of Orm proteins (2). This is an attempt of the cell to restore sphingolipid production. Contrarily, DspA/E, by decreasing Orm phosphorylation, prevents any attempt of the yeast cell to restore sphingolipid production.
The phosphorylation status of Orm proteins is driven by the Ypk1 kinase and the Cdc55-PP2A phosphatase (4, 5). We showed that, upon DspA/E expression, Ypk1 activity was not affected, suggesting that Orm dephosphorylation is mainly driven by the Cdc55-PP2A phosphatase. Actually, we observed that the Δcdc55 mutation affecting the regulatory unit of the Cdc55-PP2A phosphatase suppressed DspA/E-mediated growth arrest and dephosphorylation of Orm proteins. This confirms that DspA/E-mediated effects require the Cdc55-PP2A phosphatase. At this stage, we cannot determine whether DspA/E directly activates Cdc55-PP2A. A previous study also proposed the Cdc55-PP2A phosphatase to be involved in sphingolipid synthesis repression (5), but it is currently unknown how Cdc55-PP2A is activated.
Overall, our results led to the following model explaining DspA/E toxicity (Fig. 9). DspA/E-mediated growth arrest requires a functional Cdc55-PP2A phosphatase, which drives Orm dephosphorylation, thereby resulting in SPT repression and LCB depletion, which lead to the observed toxicity. This is a new way for a bacterial effector to perturb the eukaryotic cell. The sphingolipid pathway was shown previously to be perturbed by several plant fungal pathogens producing mycotoxins such as Fb1 and Alternaria alternata f. sp. lycopersici toxin that competitively inhibit ceramide biosynthesis (51, 52), but until now, this pathway was not described as being perturbed by plant bacterial pathogens. As DspA/E belongs to a widespread family of T3Es conserved in plant-pathogenic bacteria (53), the mode of action that we unraveled in this work is likely important for most plant-bacterium interactions.
FIGURE 9.
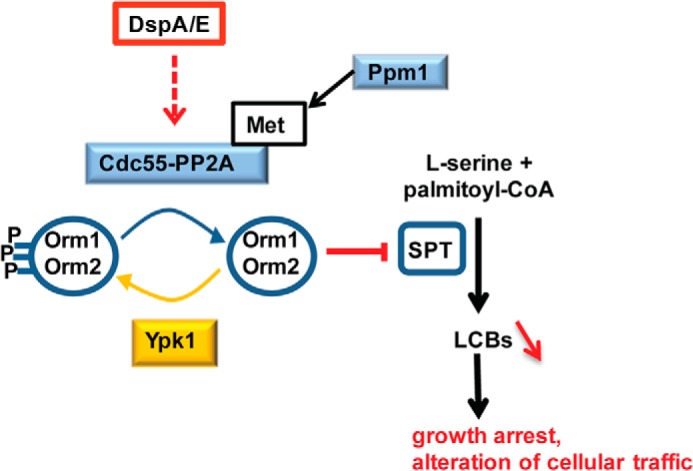
Proposed model for DspA/E-induced toxicity. DspA/E requires Cdc55-PP2A activity to induce dephosphorylation of Orm proteins as shown by analysis of the Δcdc55 mutant. Upon Orm dephosphorylation, SPT inhibition is enhanced, which in turn leads to LCB depletion. DspA/E-induced growth arrest and alteration of cellular traffic are counteracted in sphingolipid mutants that compensate DspA/E-induced LCB depletion, indicating that it is the LCB depletion that triggers growth arrest and perturbation of cellular traffic.
The sphingolipid biosynthetic pathway is highly conserved in all eukaryotes. In particular, ORM genes are evolutionarily conserved, and ORM homologs At1G01230 and At5G42000 are found in the genome of the model plant Arabidopsis thaliana (54). It is therefore likely that Orm proteins function similarly in plants as key regulator of sphingolipid homeostasis. Hence, it has been reported in several plant-pathogen interactions that the sphingolipid biosynthetic pathway is induced during the hypersensitive response, an immunity-related form of programmed cell death that blocks pathogen attack at the site of infection (54). Interestingly, induction of SPT and accumulation of LCBs in planta during the early stage of the hypersensitive response has been reported and linked to plant cell death (55–57). Therefore, inhibition of SPT could be seen as a mechanism that delays hypersensitive response cell death and allows bacterial development in planta. As observed in yeast, perturbation of sphingolipid metabolism is also deleterious to plant vesicular trafficking (58, 59). This may explain why DspA/E, following infection, is required to block basal defense, which require active cellular traffic, such as callose deposition at the cell wall surface (26, 28). Perturbation of the sphingolipid pathway could also explain why DspA/E induces plant cell death (28, 30, 32) because inhibition of the sphingolipid pathway ultimately leads to plant cell death (60–62). Our results in yeast pave the way for future plant studies.
Acknowledgments
We thank Alain Jacquier for the gift of the pooled yeast mutant library and Naïma Belgareh-Touzé, Emmanuel Baudouin, Jacques Pédron, and Jonas Körner for critical reading of the manuscript and helpful discussions throughout this work. We also thank Robbie Loewith (University of Geneva, Geneva, Switzerland) for phospho-Ypk1Thr-662 antibody and Yasunori Kozutsumi (University of Kyoto, Kyoto, Japan) for Ypk1 total antibody.
This work was supported in part by the Agence Nationale de la Recherche Jeune Chercheur DspCellDeath grant.
- LCB
- long chain base
- TE3
- type III effector
- DHS
- dihydrosphingosine
- PHS
- phytosphingosine
- SPT
- serine palmitoyltransferase
- PP2A
- protein phosphatase 2A
- Fb1
- fumonisin B1
- AbA
- aureobasidin A
- BY
- BY4741
- EV
- empty vector
- Doxy
- doxycycline
- SD
- synthetic defined
- PHS-P
- PHS phosphate
- DHS-P
- DHS phosphate
- LCB-P
- LCB phosphate
- IPC
- Inositol-phosphoceramide.
REFERENCES
- 1. Hannun Y. A., Obeid L. M. (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 2. Breslow D. K., Collins S. R., Bodenmiller B., Aebersold R., Simons K., Shevchenko A., Ejsing C. S., Weissman J. S. (2010) Orm family proteins mediate sphingolipid homeostasis. Nature 463, 1048–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berchtold D., Piccolis M., Chiaruttini N., Riezman I., Riezman H., Roux A., Walther T. C., Loewith R. (2012) Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat. Cell Biol. 14, 542–547 [DOI] [PubMed] [Google Scholar]
- 4. Roelants F. M., Breslow D. K., Muir A., Weissman J. S., Thorner J. (2011) Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 108, 19222–19227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun Y., Miao Y., Yamane Y., Zhang C., Shokat K. M., Takematsu H., Kozutsumi Y., Drubin D. G. (2012) Orm protein phosphoregulation mediates transient sphingolipid biosynthesis response to heat stress via the Pkh-Ypk and Cdc55-PP2A pathways. Mol. Biol. Cell 23, 2388–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Breslow D. K., Weissman J. S. (2010) Membranes in balance: mechanisms of sphingolipid homeostasis. Mol. Cell 40, 267–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Block A., Li G., Fu Z. Q., Alfano J. R. (2008) Phytopathogen type III effector weaponry and their plant targets. Curr. Opin. Plant Biol. 11, 396–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen L.-Q., Hou B.-H., Lalonde S., Takanaga H., Hartung M. L., Qu X.-Q., Guo W.-J., Kim J.-G., Underwood W., Chaudhuri B., Chermak D., Antony G., White F. F., Somerville S. C., Mudgett M. B., Frommer W. B. (2010) Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cui H., Xiang T., Zhou J. (2009) Plant immunity: a lesson from pathogenic bacterial effector proteins. Cell. Microbiol. 11, 1453–1461 [DOI] [PubMed] [Google Scholar]
- 10. Dean P. (2011) Functional domains and motifs of bacterial type III effector proteins and their roles in infection. FEMS Microbiol. Rev. 35, 1100–1125 [DOI] [PubMed] [Google Scholar]
- 11. Galán J. E. (2009) Common themes in the design and function of bacterial effectors. Cell Host Microbe 5, 571–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Angot A., Vergunst A., Genin S., Peeters N. (2007) Exploitation of eukaryotic ubiquitin signaling pathways by effectors translocated by bacterial type III and type IV secretion systems. PLoS Pathog. 3, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Curak J., Rohde J., Stagljar I. (2009) Yeast as a tool to study bacterial effectors. Curr. Opin. Microbiol. 12, 18–23 [DOI] [PubMed] [Google Scholar]
- 14. Kramer R. W., Slagowski N. L., Eze N. A., Giddings K. S., Morrison M. F., Siggers K. A., Starnbach M. N., Lesser C. F. (2007) Yeast functional genomic screens lead to identification of a role for a bacterial effector in innate immunity regulation. PLoS Pathog. 3, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Malnoy M., Martens S., Norelli J. L., Barny M. A., Sundin G. W., Smits T. H., Duffy B. (2012) Fire blight: applied genomic insights of the pathogen and host. Annu. Rev. Phytopathol. 50, 475–494 [DOI] [PubMed] [Google Scholar]
- 16. Barny M. A., Guinebretière M. H., Marçais B., Coissac E., Paulin J. P., Laurent J. (1990) Cloning of a large gene cluster involved in Erwinia amylovora CFBP1430 virulence. Mol. Microbiol. 4, 777–786 [DOI] [PubMed] [Google Scholar]
- 17. Gaudriault S., Malandrin L., Paulin J. P., Barny M. A. (1997) DspA, an essential pathogenicity factor of Erwinia amylovora showing homology with AvrE of Pseudomonas syringae, is secreted via the Hrp secretion pathway in a DspB-dependent way. Mol. Microbiol. 26, 1057–1069 [DOI] [PubMed] [Google Scholar]
- 18. Bogdanove A. J., Kim J. F., Wei Z., Kolchinsky P., Charkowski A. O., Conlin A. K., Collmer A., Beer S. V. (1998) Homology and functional similarity of an hrp-linked pathogenicity locus, dspEF, of Erwinia amylovora and the avirulence locus avrE of Pseudomonas syringae pathovar tomato. Proc. Natl. Acad. Sci. U.S.A. 95, 1325–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ham J. H., Majerczak D. R., Arroyo-Rodriguez A. S., Mackey D. M., Coplin D. L. (2006) WtsE, an AvrE-family effector protein from Pantoea stewartii subsp stewartii, causes disease-associated cell death in corn and requires a chaperone protein for stability. Mol. Plant Microbe Interact. 19, 1092–1102 [DOI] [PubMed] [Google Scholar]
- 20. Kvitko B. H., Park D. H., Velásquez A. C., Wei C.-F., Russell A. B., Martin G. B., Schneider D. J., Collmer A. (2009) Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 type III secretion effector genes reveal functional overlap among effectors. PLoS Pathog. 5, e1000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baltrus D. A., Nishimura M. T., Romanchuk A., Chang J. H., Mukhtar M. S., Cherkis K., Roach J., Grant S. R., Jones C. D., Dangl J. L. (2011) Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Pathog. 7, e1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frederick R. D., Ahmad M., Majerczak D. R., Arroyo-Rodríguez A. S., Manulis S., Coplin D. L. (2001) Genetic organization of the Pantoea stewartii subsp stewartii hrp gene cluster and sequence analysis of the hrpA, hrpC, hrpN, and wtsE operons. Mol. Plant Microbe Interact. 14, 1213–1222 [DOI] [PubMed] [Google Scholar]
- 23. Mor H., Manulis S., Zuck M., Nizan R., Coplin D. L., Barash I. (2001) Genetic organization of the hrp gene cluster and dspAE/BF operon in Erwinia herbicola pv. gypsophilae. Mol. Plant Microbe Interact. 14, 431–436 [DOI] [PubMed] [Google Scholar]
- 24. Badel J. L., Shimizu R., Oh H. S., Collmer A. (2006) A Pseudomonas syringae pv. tomato avrE1/hopM1 mutant is severely reduced in growth and lesion formation in tomato. Mol. Plant Microbe Interact. 19, 99–111 [DOI] [PubMed] [Google Scholar]
- 25. Holeva M. C., Bell K. S., Hyman L. J., Avrova A. O., Whisson S. C., Birch P. R., Toth I. K. (2004) Use of a pooled transposon mutation grid to demonstrate roles in disease development for Erwinia carotovora subsp atroseptica putative type III secreted effector (DspE/A) and helper (HrpN) proteins. Mol. Plant Microbe Interact. 17, 943–950 [DOI] [PubMed] [Google Scholar]
- 26. DebRoy S., Thilmony R., Kwack Y. B., Nomura K., He S. Y. (2004) A family of conserved bacterial effectors inhibits salicylic acid-mediated basal immunity and promotes disease necrosis in plants. Proc. Natl. Acad. Sci. U.S.A. 101, 9927–9932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boureau T., Siamer S., Perino C., Gaubert S., Patrit O., Degrave A., Fagard M., Chevreau E., Barny M.-A. (2011) The HrpN effector of Erwinia amylovora, which is involved in type III translocation, contributes directly or indirectly to callose elicitation on apple leaves. Mol. Plant Microbe Interact. 24, 577–584 [DOI] [PubMed] [Google Scholar]
- 28. Boureau T., ElMaarouf-Bouteau H., Garnier A., Brisset M. N., Perino C., Pucheu I., Barny M. A. (2006) DspA/E, a type III effector essential for Erwinia amylovora pathogenicity and growth in planta, induces cell death in host apple and nonhost tobacco plants. Mol. Plant Microbe Interact. 19, 16–24 [DOI] [PubMed] [Google Scholar]
- 29. Ham J. H., Majerczak D., Ewert S., Sreerekha M.-V., Mackey D., Coplin D. (2008) WtsE, an AvrE-family type III effector protein of Pantoea stewartii subsp. stewartii, causes cell death in non-host plants. Mol. Plant Pathol. 9, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oh C. S., Martin G. B., Beer S. V. (2007) DspA/E, a type III effector of Erwinia amylovora, is required for early rapid growth in Nicotiana benthamiana and causes NbSGT1-dependent cell death. Mol. Plant Pathol. 8, 255–265 [DOI] [PubMed] [Google Scholar]
- 31. Degrave A., Fagard M., Perino C., Brisset M. N., Gaubert S., Laroche S., Patrit O., Barny M. A. (2008) Erwinia amylovora type three-secreted proteins trigger cell death and defense responses in Arabidopsis thaliana. Mol. Plant Microbe Interact. 21, 1076–1086 [DOI] [PubMed] [Google Scholar]
- 32. Degrave A., Moreau M., Launay A., Barny M.-A., Brisset M.-N., Patrit O., Taconnat L., Vedel R., Fagard M. (2013) The bacterial effector DspA/E is toxic in Arabidopsis thaliana and is required for multiplication and survival of fire blight pathogen. Mol. Plant Pathol. 14, 506–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Siamer S., Patrit O., Fagard M., Belgareh-Touzé N., Barny M.-A. (2011) Expressing the Erwinia amylovora type III effector DspA/E in the yeast Saccharomyces cerevisiae strongly alters cellular trafficking. FEBS Open Bio 1, 23–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boyer J., Badis G., Fairhead C., Talla E., Hantraye F., Fabre E., Fischer G., Hennequin C., Koszul R., Lafontaine I., Ozier-Kalogeropoulos O., Ricchetti M., Richard G. F., Thierry A., Dujon B. (2004) Large-scale exploration of growth inhibition caused by overexpression of genomic fragments in Saccharomyces cerevisiae. Genome Biol. 5, R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vallée B., Riezman H. (2005) Lip1p: a novel subunit of acyl-CoA ceramide synthase. EMBO J. 24, 730–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guillas I., Kirchman P. A., Chuard R., Pfefferli M., Jiang J. C., Jazwinski S. M., Conzelmann A. (2001) C26-CoA-dependent ceramide synthesis of Saccharomyces cerevisiae is operated by Lag1p and Lac1p. EMBO J. 20, 2655–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tanoue D., Kobayashi T., Sun Y., Fujita T., Takematsu H., Kozutsumi Y. (2005) The requirement for the hydrophobic motif phosphorylation of Ypk1 in yeast differs depending on the downstream events, including endocytosis, cell growth, and resistance to a sphingolipid biosynthesis inhibitor, ISP-1. Arch. Biochem. Biophys. 437, 29–41 [DOI] [PubMed] [Google Scholar]
- 38. Nagiec M. M., Nagiec E. E., Baltisberger J. A., Wells G. B., Lester R. L., Dickson R. C. (1997) Sphingolipid synthesis as a target for antifungal drugs. Complementation of the inositol phosphorylceramide synthase defect in a mutant strain of Saccharomyces cerevisiae by the AUR1 gene. J. Biol. Chem. 272, 9809–9817 [DOI] [PubMed] [Google Scholar]
- 39. Lester R. L., Dickson R. C. (2001) High-performance liquid chromatography analysis of molecular species of sphingolipid-related long chain bases and long chain base phosphates in Saccharomyces cerevisiae after derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate. Anal. Biochem. 298, 283–292 [DOI] [PubMed] [Google Scholar]
- 40. Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., Arkin A. P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., Brachat S., Campanaro S., Curtiss M., Davis K., Deutschbauer A., Entian K. D., Flaherty P., Foury F., Garfinkel D. J., Gerstein M., Gotte D., Güldener U., Hegemann J. H., Hempel S., Herman Z., Jaramillo D. F., Kelly D. E., Kelly S. L., Kötter P., LaBonte D., Lamb D. C., Lan N., Liang H., Liao H., Liu L., Luo C., Lussier M., Mao R., Menard P., Ooi S. L., Revuelta J. L., Roberts C. J., Rose M., Ross-Macdonald P., Scherens B., Schimmack G., Shafer B., Shoemaker D. D., Sookhai-Mahadeo S., Storms R. K., Strathern J. N., Valle G., Voet M., Volckaert G., Wang C. Y., Ward T. R., Wilhelmy J., Winzeler E. A., Yang Y. H., Yen G., Youngman E., Yu K. X., Bussey H., Boeke J. D., Snyder M., Philippsen P., Davis R. W., Johnston M. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 [DOI] [PubMed] [Google Scholar]
- 41. Winzeler E. A., Shoemaker D. D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J. D., Bussey H., Chu A. M., Connelly C., Davis K., Dietrich F., Dow S. W., El Bakkoury M., Foury F., Friend S. H., Gentalen E., Giaever G., Hegemann J. H., Jones T., Laub M., Liao H., Liebundguth N., Lockhart D. J., Lucau-Danila A., Lussier M., M'Rabet N., Menard P., Mittmann M., Pai C., Rebischung C., Revuelta J. L., Riles L., Roberts C. J., Ross-MacDonald P., Scherens B., Snyder M., Sookhai-Mahadeo S., Storms R. K., Véronneau S., Voet M., Volckaert G., Ward T. R., Wysocki R., Yen G. S., Yu K., Zimmermann K., Philippsen P., Johnston M., Davis R. W. (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906 [DOI] [PubMed] [Google Scholar]
- 42. Dickson R. C., Sumanasekera C., Lester R. L. (2006) Functions and metabolism of sphingolipids in Saccharomyces cerevisiae. Prog. Lipid Res. 45, 447–465 [DOI] [PubMed] [Google Scholar]
- 43. Guillas I., Jiang J. C., Vionnet C., Roubaty C., Uldry D., Chuard R., Wang J., Jazwinski S. M., Conzelmann A. (2003) Human homologues of LAG1 reconstitute acyl-CoA-dependent ceramide synthesis in yeast. J. Biol. Chem. 278, 37083–37091 [DOI] [PubMed] [Google Scholar]
- 44. Oh C. S., Toke D. A., Mandala S., Martin C. E. (1997) ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. J. Biol. Chem. 272, 17376–17384 [DOI] [PubMed] [Google Scholar]
- 45. Voynova N. S., Vionnet C., Ejsing C. S., Conzelmann A. (2012) A novel pathway of ceramide metabolism in Saccharomyces cerevisiae. Biochem. J. 447, 103–114 [DOI] [PubMed] [Google Scholar]
- 46. Shimobayashi M., Oppliger W., Moes S., Jenö P., Hall M. N. (2013) TORC1-regulated protein kinase Npr1 phosphorylates Orm to stimulate complex sphingolipid synthesis. Mol. Biol. Cell 24, 870–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shi Y. (2009) Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484 [DOI] [PubMed] [Google Scholar]
- 48. Wu J., Tolstykh T., Lee J., Boyd K., Stock J. B., Broach J. R. (2000) Carboxyl methylation of the phosphoprotein phosphatase 2A catalytic subunit promotes its functional association with regulatory subunits in vivo. EMBO J. 19, 5672–5681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wei H., Ashby D. G., Moreno C. S., Ogris E., Yeong F. M., Corbett A. H., Pallas D. C. (2001) Carboxymethylation of the PP2A catalytic subunit in Saccharomyces cerevisiae is required for efficient interaction with the B-type subunits CDC55p and RTS1p. J. Biol. Chem. 276, 1570–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zanolari B., Friant S., Funato K., Sütterlin C., Stevenson B. J., Riezman H. (2000) Sphingoid base synthesis requirement for endocytosis in Saccharomyces cerevisiae. EMBO J. 19, 2824–2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dutton M. F. (1996) Fumonisins, mycotoxins of increasing importance: their nature and their effects. Pharmacol. Ther. 70, 137–161 [DOI] [PubMed] [Google Scholar]
- 52. Brandwagt B. F., Mesbah L. A., Takken F. L., Laurent P. L., Kneppers T. J., Hille J., Nijkamp H. J. (2000) A longevity assurance gene homolog of tomato mediates resistance to Alternaria alternata f. sp. lycopersici toxins and fumonisin B-1. Proc. Natl. Acad. Sci. U.S.A. 97, 4961–4966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Siamer S., Gaubert S., Boureau T., Brisset M.-N., Barny M.-A. (2013) Mutational analysis of a predicted double-propeller domain of the DspA/E effector of Erwinia amylovora. FEMS Microbiol. Lett. 342, 54–61 [DOI] [PubMed] [Google Scholar]
- 54. Berkey R., Bendigeri D., Xiao S. (2012) Sphingolipids and plant defense/disease: the “death” connection and beyond. Front. Plant Sci. 3, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peer M., Stegmann M., Mueller M. J., Waller F. (2010) Pseudomonas syringae infection triggers de novo synthesis of phytosphingosine from sphinganine in Arabidopsis thaliana. FEBS Lett. 584, 4053–4056 [DOI] [PubMed] [Google Scholar]
- 56. Birch P. R. J., Avrova A. O., Duncan J. M., Lyon G. D., Toth R. L. (1999) Isolation of potato genes that are induced during an early stage of the hypersensitive response to Phytophthora infestans. Mol. Plant Microbe Interact. 12, 356–361 [Google Scholar]
- 57. Shi L., Bielawski J., Mu J., Dong H., Teng C., Zhang J., Yang X., Tomishige N., Hanada K., Hannun Y. A., Zuo J. (2007) Involvement of sphingoid bases in mediating reactive oxygen intermediate production and programmed cell death in Arabidopsis. Cell Res. 17, 1030–1040 [DOI] [PubMed] [Google Scholar]
- 58. Bach L., Gissot L., Marion J., Tellier F., Moreau P., Satiat-Jeunemaître B., Palauqui J.-C., Napier J. A., Faure J.-D. (2011) Very-long-chain fatty acids are required for cell plate formation during cytokinesis in Arabidopsis thaliana. J. Cell Sci. 124, 3223–3234 [DOI] [PubMed] [Google Scholar]
- 59. Markham J. E., Molino D., Gissot L., Bellec Y., Hématy K., Marion J., Belcram K., Palauqui J.-C., Satiat-Jeunemaître B., Faure J.-D. (2011) Sphingolipids containing very-long-chain fatty acids define a secretory pathway for specific polar plasma membrane protein targeting in Arabidopsis. Plant Cell 23, 2362–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen M., Han G., Dietrich C. R., Dunn T. M., Cahoon E. B. (2006) The essential nature of sphingolipids in plants as revealed by the functional identification and characterization of the Arabidopsis LCB1 subunit of serine palmitoyl transferase. Plant Cell 18, 3576–3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dietrich C. R., Han G., Chen M., Berg R. H., Dunn T. M., Cahoon E. B. (2008) Loss-of-function mutations and inducible RNAi suppression of Arabidopsis LCB2 genes reveal the critical role of sphingolipids in gametophytic and sporophytic cell viability. Plant J. 54, 284–298 [DOI] [PubMed] [Google Scholar]
- 62. Saucedo-García M., Guevara-García A., González-Solís A., Cruz-García F., Vázquez-Santana S., Markham J. E., Lozano-Rosas M. G., Dietrich C. R., Ramos-Vega M., Cahoon E. B., Gavilanes-Ruíz M. (2011) MPK6, sphinganine and the LCB2a gene from serine palmitoyltransferase are required in the signaling pathway that mediates cell death induced by long chain bases in Arabidopsis. New Phytol. 191, 943–957 [DOI] [PubMed] [Google Scholar]