

Background: d-QuiNAc is found in many important bacteria, but its biosynthesis has not been defined.
Results: UDP-d-QuiNAc was synthesized in vitro from UDP-d-GlcNAc in reactions catalyzed by two cloned enzymes.
Conclusion: R. etli CE3 protein WreQ has UDP-2-acetamido-2,6-dideoxy-d-xylo-4-hexulose 4-reductase activity.
Significance: The defined pathway for UDP-d-QuiNAc biosynthesis provides this compound for in vitro biosynthetic studies and predicts organisms that contain d-QuiNAc.
Keywords: Bacteria; Carbohydrate Biosynthesis; Glycobiology; Nuclear Magnetic Resonance (NMR); Reductase; Rhizobium etli; UDP-2-acetamido-2,6-dideoxy-d-xylo-4-hexulose; UDP-d-GlcNAc; UDP-d-QuiNAc; WreQ
Abstract
N-acetyl-d-quinovosamine (2-acetamido-2,6-dideoxy-d-glucose, QuiNAc) occurs in the polysaccharide structures of many Gram-negative bacteria. In the biosynthesis of QuiNAc-containing polysaccharides, UDP-QuiNAc is the hypothetical donor of the QuiNAc residue. Biosynthesis of UDP-QuiNAc has been proposed to occur by 4,6-dehydration of UDP-N-acetyl-d-glucosamine (UDP-GlcNAc) to UDP-2-acetamido-2,6-dideoxy-d-xylo-4-hexulose followed by reduction of this 4-keto intermediate to UDP-QuiNAc. Several specific dehydratases are known to catalyze the first proposed step. A specific reductase for the last step has not been demonstrated in vitro, but previous mutant analysis suggested that Rhizobium etli gene wreQ might encode this reductase. Therefore, this gene was cloned and expressed in Escherichia coli, and the resulting His6-tagged WreQ protein was purified. It was tested for 4-reductase activity by adding it and NAD(P)H to reaction mixtures in which 4,6-dehydratase WbpM had acted on the precursor substrate UDP-GlcNAc. Thin layer chromatography of the nucleotide sugars in the mixture at various stages of the reaction showed that WbpM converted UDP-GlcNAc completely to what was shown to be its 4-keto-6-deoxy derivative by NMR and that addition of WreQ and NADH led to formation of a third compound. Combined gas chromatography-mass spectrometry analysis of acid hydrolysates of the final reaction mixture showed that a quinovosamine moiety had been synthesized after WreQ addition. The two-step reaction progress also was monitored in real time by NMR. The final UDP-sugar product after WreQ addition was purified and determined to be UDP-d-QuiNAc by one-dimensional and two-dimensional NMR experiments. These results confirmed that WreQ has UDP-2-acetamido-2,6-dideoxy-d-xylo-4-hexulose 4-reductase activity, completing a pathway for UDP-d-QuiNAc synthesis in vitro.
Introduction
Many rare sugars are found in the polysaccharide structures coating bacterial cell surfaces. A moderately rare example is N-acetyl-d-quinovosamine (QuiNAc),3 which so far has been found exclusively in the lipopolysaccharides (LPSs) or capsular polysaccharides of bacteria. Although confined to bacteria, it is found in numerous species, including species belonging to the genera Pseudomonas and Rhizobium (1, 2).
The QuiNAc residue of LPS in Rhizobium etli strain CE3 may play an important role in the biology of this bacterium. An R. etli mutant strain (CE166) carrying a Tn5 transposon in the wreQ gene locus (formerly designated lpsQ) fails in the infection stage of symbiosis with legume hosts. This mutant has much less O-antigen-containing LPS than normal, and its O-antigen lacks QuiNAc (3, 4). The QuiNAc is replaced in this mutant by its 4-keto derivative, 2-acetamido-2,6-dideoxy-d-xylo-4-hexulose (5). The deficiency in O-antigen amount can be suppressed by introducing into this mutant multiple copies of the other R. etli CE3 genes needed for O-antigen synthesis. Because QuiNAc is still replaced in the resulting strain by its 4-keto derivative, the effect of just this abnormality is thereby revealed. The mutant genetically suppressed in this way infects slowly, and resulting root nodules are widely dispersed (4). This result suggests that the symbiotic role of LPS may require a specific structural feature conferred by QuiNAc.
QuiNAc has been proposed to be the first sugar of the O-antigen of R. etli CE3 and the O-antigen-repeating unit in Pseudomonas aeruginosa O6 (6–8). However, these inferences lack evidence from biosynthesis of the polysaccharide in vitro. One obstacle to such studies is that the expected biosynthetic donor of the QuiNAc residue, UDP-QuiNAc, has never been produced in vitro.
The proposed biosynthetic pathway of UDP-QuiNAc, based on studies in R. etli CE3 and in P. aeruginosa O6 (5, 6), involves two steps. In the first step, the precursor UDP-GlcNAc is converted by a 4,6-dehydratase to its 4-keto-6-deoxy derivative. This reaction has been reproduced in vitro by use of several homologous enzymes (9–13). It is the common first step in synthesizing many UDP-sugars from UDP-GlcNAc (14).
In the proposed second step, the 4-keto intermediate is further converted to UDP-QuiNAc by a 4-reductase. This reaction and an enzyme to catalyze it have never been demonstrated in vitro. It has been proposed that the R. etli CE3 wreQ gene encodes the 4-reductase for this step. This gene was previously designated lpsQ (5), but all genes involved in R. etli CE3 O-antigen synthesis have been assigned the symbol wre to conform to bacterial polysaccharide genetic nomenclature (15). The predicted amino acid sequence suggests that WreQ belongs to the short chain dehydratase/reductase superfamily characterized by its GXXGXXG nucleotide-binding motif and the SYK catalytic triad (5, 16). As mentioned above, chemical analysis of O-antigen from the symbiosis-deficient wreQ-null mutant showed that QuiNAc was replaced by its 4-keto derivative at the same position (5), supporting that WreQ is the 4-reductase for the last step in QuiNAc synthesis.
P. aeruginosa serotype O6 contains QuiNAc in the O-antigen of its B-band LPS. A wreQ gene homolog, wbpV, was found in its B-band O-antigen gene cluster. Disruption of the wbpV gene abrogates O-antigen synthesis (6). P. aeruginosa O6 also contains the wbpM gene, which encodes a UDP-GlcNAc 4,6-dehydratase (6). The enzymatic activity of WbpM has been demonstrated in vitro biochemically (9, 13). The R. etli CE3 genome contains a wbpM gene homolog designated wreV. A logical prediction is that these gene pairs (wreV and wreQ in R. etli CE3 and wbpM and wbpV in P. aeruginosa O6) encode the enzymes needed to synthesize QuiNAc.
In the work presented here, the R. etli CE3 WreQ protein was purified, and its enzymatic activity in vitro was investigated. The nucleotide sugar product of the WreQ-catalyzed reaction was chemically identified to be UDP-d-QuiNAc. The results confirmed that WreQ can act as a 4-reductase to provide the first demonstration of UDP-d-QuiNAc synthesis in vitro.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Conditions
R. etli CE3 was derived from R. etli type strain CFN42 by a spontaneous mutation conferring resistance to streptomycin (17). As in all past studies of wreQ and almost all other studies of the LPS of R. etli CFN42, strain CE3 was the wild-type source of DNA and genotype for strain constructions. All R. etli strains were grown to stationary phase at 30 °C in TY liquid medium (0.5% tryptone (Difco), 0.3% yeast extract (Difco), and 10 mm CaCl2). Unless stated otherwise, all Escherichia coli strains were grown to stationary phase at 37 °C in Luria-Bertani (LB) liquid medium (1.0% tryptone, 0.5% yeast extract, and 0.5% NaCl). Agar medium contained 1.5% Bacto Agar (Difco).
DNA Techniques
R. etli genomic DNA was isolated using a GenElute Bacterial Genomic DNA kit (Sigma-Aldrich). Plasmid DNA isolation from E. coli cultures and purification from agarose gels were performed using the QIAprep Spin Miniprep kit (Qiagen) and Gel/PCR DNA Fragments Extraction kit (IBI Scientific), respectively. Amounts of DNA fragments were amplified using the Expand High Fidelity PCR System (Roche Applied Science) with custom primers (Eurofins MWG Operon) containing restriction sites to facilitate cloning. Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs. Vectors or recombinant plasmids were transformed into OmniMAX (Invitrogen) or 5-α (New England Biolabs) E. coli cells for cloning or into BL21(DE3) (Lucigen) E. coli cells for protein overexpression. DNA sequencing was performed by Functional Biosciences.
Construction of a pET Vector for WreQ Overexpression
The gene encoding WreQ was amplified from the genomic DNA of R. etli CE3 (NCBI accession number NC_007761; the wreQ gene sequence (nucleotide 2969313 to nucleotide 2970242) under this accession matches exactly with the lpsQ sequence under GenBankTM accession number AY391267 reported previously (5)). The primers designed based on this DNA sequence are 5′-CCGGCATATGCGATGCCTCGTCACC-3′ (forward) and 5′-GGCCGGATCCCTATTCCACTGCAAG-3′ (reverse). Restriction sites included in the primers are underlined. The wreQ gene was subcloned by insertion at the NdeI and BamHI sites of pET15b vector (Novagen), generating plasmid pLS7. The resulting construct was checked by restriction digestion and nucleotide sequence determination. It encodes the WreQ protein with additional amino acids at the amino terminus, MGSSHHHHHHSSGLVPRGSH (the His6 tag is underlined, and the thrombin cleavage site is in bold). The WreQ protein is referred to as His6-WreQ in this work.
Complementation of R. etli wreQ Mutant
For testing complementation of a wreQ mutant with his6-wreQ, the his6-wreQ gene together with the vector ribosome binding site was amplified from the constructed vector pLS7 with primers 5′-GCCGAATTCATACCCACGCCGAAACAAG-3′ (forward) and 5′-GCCGGTACCAGTTCCTCCTTTCAGCAAA-3′ (reverse). This fragment was subcloned by insertion into the EcoRI and KpnI sites of plasmid pFAJ1708 (18), generating plasmid pLS31. The pLS31 plasmid was introduced into the R. etli wreQ::Tn5 mutant CE166 by a triparental mating procedure described previously (8). LPS was prepared from 0.5 ml of full-grown cultures and analyzed by SDS-PAGE with periodate-silver stain (8). To determine whether QuiNAc was present, LPS was extracted from washed bacterial pellets (from 4.0 liters of culture) by the hot phenol-water method; dialyzed; treated with RNase A, DNase I, and protease K; and dialyzed again followed by lyophilization (8). For determination of neutral and amino sugars, LPS preparations were treated with 2 m trifluoroacetic acid for 2 h at 121 °C. After reduction with NaBD4 and acetylation, alditol acetate derivatives of the LPS sugars were analyzed by gas chromatography (GC) under conditions described previously (8).
To test complementation of a wreQ mutant with wbpV from P. aeruginosa O6, pFV611-26 plasmid (6) (provided by Dr. J. S. Lam, University of Guelph, Guelph, Canada) was digested with PstI and SacI to release a fragment containing the wbpV gene. This fragment was cloned into two pFAJ1700-based vectors (18), generating pJB7 and pJB8, respectively. Complementation was tested as described above.
Mutagenesis of R. etli wreV Gene and Its Complementation with wbpM Gene of P. aeruginosa
Site-directed mutagenesis of R. etli wreV gene was performed using the same method as described in the previous study (8). The gentamicin resistance cassette from plasmid pUCGm (19) was inserted in the R. etli CE3 wreV gene through the SalI sites, generating strain CE568.
To test complementation of the wreV mutant with wbpM from P. aeruginosa, the full-length wbpM gene was amplified from a pBAD-wbpM vector (provided by Dr. J. S. Lam) with primers 5′-GGCATATGTTGGATAATTTGAGG-3′ (forward) and 5′-TTAAGCTTTCAGGGTTCTCGCCGCC-3′ (reverse). This fragment was subcloned by insertion into the NdeI and HindIII sites of plasmid pJBC1 (20), generating plasmid pTL61. Complementation was tested as described above.
Overexpression and Purification of His6-WreQ
The pLS7 plasmid encoding His6-WreQ was transformed into electrocompetent BL21(DE3) E. coli cells by electroporation, and a purified transformant was grown overnight in LB medium containing ampicillin (100 μg/ml). A flask of 1 liter of LB medium was inoculated with 5 ml of overnight culture and grown at 37 °C with shaking until A600 reached 0.6. His6-WreQ expression was induced by adding isopropyl β-d-1-thiogalactopyranoside (Gold Biotechnology) to a final concentration of 0.1 mm, and the culture was shaken for a further 20 h at 16 °C. Cells were harvested by centrifugation at 5000 × g for 15 min at 4 °C, and the pellets were stored at −80 °C until needed.
The His6-WreQ protein was purified with Ni2+ affinity chromatography. Specifically, the frozen cell pellets were resuspended in lysis buffer (20 mm sodium phosphate, 300 mm NaCl, 5 mm imidazole, and 14.3 mm 2-mercaptoethanol, pH 7.0) and disrupted by sonication. Cell debris was removed by centrifugation (6000 × g for 20 min at 4 °C), and the supernatant was centrifuged at 65,000 × g for 120 min at 4 °C. The supernatant was passed through a column containing 1 ml of Ni2+ Profinity immobilized metal affinity chromatography resin (Bio-Rad). The column was washed with 10 ml of lysis buffer followed by washing with lysis buffer containing 20 mm imidazole. His6-WreQ protein was eluted with lysis buffer containing 300 mm imidazole. Triton X-100 was added to the combined elution fraction to a final concentration of 0.1%. Then the elution fraction was transferred to dialysis tubing (molecular mass cutoff, 12–14,000 Da) and dialyzed overnight at 4 °C in 1 liter of buffer containing 20 mm sodium phosphate, 300 mm NaCl, and 14.3 mm 2-mercaptoethanol, pH 7.0. The dialyzed protein sample was concentrated with an Amicon Ultra-15 (nominal molecular mass limit, 10 kDa) filter device. The concentrated protein was fast frozen on dry ice and preserved at −80 °C until needed. The typical yield of purified protein was 4.5 mg/liter of cell culture. Protein concentration was determined by the method of Bradford (21).
Overexpression, Purification, and Enzymatic Analysis of WbpM
The E. coli BL21(DE3)plysS strain carrying a pET vector derivative encoding a soluble truncated version of the WbpM protein (residues 262–665) (9) was generously provided by Dr. J. S. Lam. To express the WbpM-His-S262 protein, 1 liter of LB medium was inoculated with a 20-ml overnight culture of the BL21(DE3)plysS strain and grown at 37 °C. Protein expression was induced by addition of 0.5 mm isopropyl β-d-1-thiogalactopyranoside. After a 5-h induction at 37 °C, the cells were harvested by centrifugation at 5000 × g for 15 min at 4 °C. Purification of WbpM-His-S262 was carried out in the same manner as described above for His6-WreQ except that all the buffers did not contain 2-mercaptoethanol. Following purification, the elution fraction was dialyzed against 1 liter of buffer (20 mm sodium phosphate and 300 mm NaCl, pH 7.0) and concentrated with an Amicon Ultra-15 (nominal molecular mass limit, 10 kDa) filter device. Protein concentration was determined by the method of Bradford (21).
Radioactive enzyme assays were carried out in 10-μl reactions containing 20 mm sodium phosphate, pH 7.0, 0.5 mm UDP-GlcNAc (Sigma), 0.029 μm UDP-[3H]GlcNAc (0.1 mCi/mmol; PerkinElmer Life Sciences), and 9 μg of WbpM. The reaction was incubated at 30 °C and allowed to proceed for 30 min for complete substrate conversion.
WbpM-WreQ Coupled Enzyme Assay
The WreQ-catalyzed reaction was carried out by adding 15 μg of His6-WreQ protein and 1 mm NAD(P)H to the WbpM reaction mixture and allowed to proceed for another 4 h. In the control reactions where one or multiple components were omitted, the lost volume was compensated with buffer.
Separation of Sugar Nucleotides by Thin Layer Chromatography
Enzyme reactions were stopped by adding an equal volume of ice-cold 100% ethanol, and proteins were removed by centrifugation (16,000 × g for 10 min at 4 °C) after overnight storage at −20 °C. The supernatant was lyophilized to dryness and stored at −20 °C.
The dried residues of radioactive reactions were dissolved in 5 μl of glass-distilled water. The entire volume of each reaction sample was spotted on an aluminum-backed precoated Silica Gel 60 plate (EMD Chemicals) and developed in Solvent A (2-propanol/ammonium hydroxide/water, 6:3:1). Dried TLC plates were sprayed with EN3HANCE Autoradiography Enhancer (PerkinElmer Life Sciences) and exposed to film (Eastman Kodak Co. BioMax XAR film) for 3 days at −80 °C.
Gas Chromatography-Mass Spectrometry (GC-MS) Analysis of Glycosyl Residue
To detect the glycosyl residue of the final product of the WbpM-WreQ reaction, the reaction was carried out in a volume of 200 μl containing 1 mm UDP-GlcNAc, 20 μg of WbpM, 36 μg of His6-WreQ, and 1 mm NADH. The lyophilized reaction mixture was treated with 1 m hydrochloric acid for 15 min at 100 °C. After reduction with NaBD4 and acetylation, alditol acetate derivatives of sugars in the reaction were dissolved in dichloromethane and injected onto an HP-5MS (Agilent 19091S-433E) capillary column (0.25-mm inner diameter × 30 m, 0.25-μm film thickness) with helium as the carrier gas at a flow rate of 1.0 ml/min. The GC program was started with an oven temperature of 50 °C followed by an increase of 20 °C min−1 to 150 °C, then at 2 °C min−1 to 200 °C, then at 5 °C min−1 to 260 °C, and a hold at 260 °C for 10 min.
Purification of Nucleotide Sugars by Thin Layer Chromatography
To purify the non-radioactive WbpM reaction product for NMR analysis, the reaction was incubated at 30 °C for 1 h with a total volume of 500 μl consisting of 20 mm sodium phosphate buffer, pH 7.0, 1 mm UDP-GlcNAc, and 120 μg of WbpM. To purify the final product of the WbpM-WreQ reaction, 450 μg of His6-WreQ and 1 mm NADH were added to the WbpM reaction and further incubated for 4 h. Reactions were stopped and treated as described above, and after lyophilization, the dried residues were dissolved in a 50-μl volume in water. Of this solution, 24 μl (0.24 μmol) was spotted on a TLC plate (10 spots, 2 μl each) and developed in Solvent A as described above. One loaded lane of the TLC plate was excised from both sides of the TLC plate, sprayed with thymol reagent (5 mg/ml thymol in ethanol/concentrated sulfuric, 19:1 (v/v)), and incubated in a 110 °C oven until stained spots were observed to locate the position of the sugar nucleotide to be purified. At this position on the unstained part of the same TLC plate, the silica coating was scraped from the aluminum backing, and the sugar nucleotide candidates were eluted from the silica with Solvent A. The eluates were lyophilized and redissolved in 20 μl of water. The result of purification was analyzed by TLC with thymol staining, and the concentration was measured by UV absorbance at 260 nm.
NMR Spectroscopy
NMR experiments were performed at a static magnetic field strength of 14.1 teslas (proton Larmor frequency of 600 MHz) on an Agilent VNMRS instrument equipped with a ColdProbe. Standard TOCSY, gradient-enhanced COSY, and NOESY two-dimensional pulse sequences were used for assignments; the pulse sequences were obtained from the VNMRJ 2.0 software suite. Raw NMR data were processed using NMRPipe (22) and analyzed in Sparky (23). The NMR assignment experiments were performed at 30 °C to match conditions of the enzyme reaction directly monitored by NMR. In the assignment experiments, 99% D2O was used as a solvent with presaturation of the residual water signal. The chemical shifts measured at 30 °C were referenced to the water resonance because its temperature dependence is well known: the water chemical shift was calculated to be at 4.700 ppm at 30 °C (24, 25). As an additional reference, we recorded the chemical shift of the acetone methyl group in water at the same temperature; it was 2.201 ± 0.008 ppm.
NMR samples were prepared by dissolution of lyophilized powders in 99.8% D2O (Cambridge Isotope Laboratories). The final concentration of UDP-QuiNAc in the Shigemi tube sample was 0.23 mm. The one-dimensional proton spectrum was acquired with water presaturation, 16,384 points, and 128 transients (8 min of total acquisition time). All two-dimensional experiments were acquired with 16,384 points in direct and 512 points in indirect dimensions. The two-dimensional 1H-1H gradient-enhanced COSY was acquired in the real mode with 32 transients (5 h). The two-dimensional 1H-1H TOCSY was acquired with 16 transients (11 h). The two-dimensional 1H-1H NOESY was acquired with 16 transients in duplicate (total of 24 h). The final concentration of the 4-keto intermediate was significantly lower than that of UDP-QuiNAc, and this was partially compensated by increased acquisition time.
RESULTS
Cloning, Expression, and Purification of WreQ
The wreQ gene was amplified from chromosomal DNA of R. etli CE3 and cloned into the pET15b vector. The resultant plasmid encodes a WreQ protein with an amino-terminal six-histidine tag. Following overexpression, this His6-WreQ protein was purified from the soluble fraction by nickel affinity chromatography (Fig. 1A).
FIGURE 1.
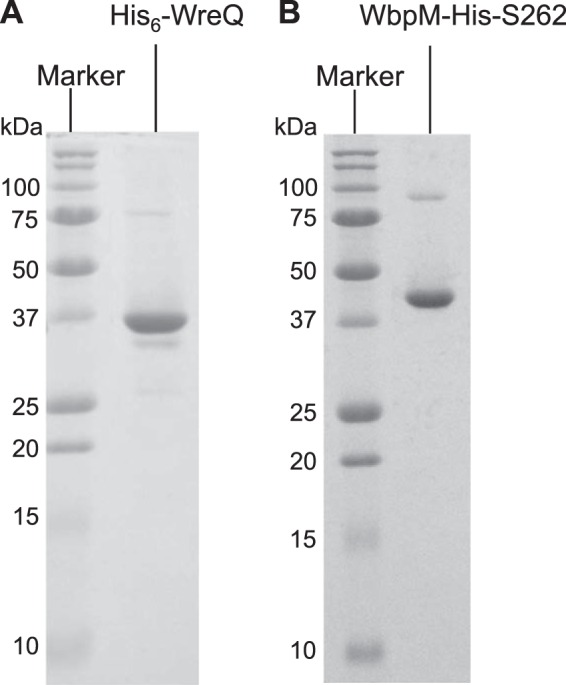
Coomassie Blue-stained SDS-PAGE (15% gel) showing purified proteins His6-WreQ (A) and WbpM-His-S262 (B) used for in vitro biosynthesis of UDP-QuiNAc. The apparent size of the purified His6-WreQ protein agreed with its predicted size of 35.3 kDa. The apparent size of the purified WbpM-His-S262 protein is consistent with the published result (9) (46.1 kDa).
The modified wreQ gene (his6-wreQ) was determined to be functional in vivo due to its ability to complement an R. etli wreQ mutant strain. In the complemented mutant, the level of smooth LPS (LPS I) was restored to normal (Fig. 2A), and the presence of quinovosamine in the LPS was confirmed by sugar composition analysis (Fig. 2B). Gene wbpV, the wreQ gene homolog from P. aeruginosa O6, also complemented the same mutant (Fig. 2, A and C). All of these results match those reported previously (5) when R. etli wild-type wreQ was used to complement this same wreQ mutant.
FIGURE 2.

Complementation of the R. etli wreQ mutant strain, CE166. A, silver-stained SDS-PAGE (18% gel) of LPS samples from whole-cell lysates. Lane 1, CE3; lane 2, CE166; lane 3, CE166/pLS31; lane 4, CE166/pJB7; lane 5, CE166/pJB8. LPS I, O-antigen-containing LPS; LPS II, core oligosaccharide-Lipid A lacking O-antigen. B and C, relative content of QuiNAc among the sugars of crude LPS as revealed by GC analysis after conversion of sugars to alditol acetates. CE3, wild-type R. etli strain; CE166, R. etli strain with a Tn5 insertion in wreQ; CE166/pLS31, CE166 strain carrying pLS31 vector, which expresses His6-WreQ; CE166/pJB7 and CE166/pJB8, CE166 strain carrying pJB7 or pJB8 plasmids, each carrying the wbpV gene. QuiN, quinovosamine.
Generation of WreQ Substrate, the 4-Keto-6-deoxy Derivative of UDP-GlcNAc
A soluble truncated version of the WbpM protein, His-S262 (9), was used to synthesize the predicted substrate for WreQ. Purified His-S262 protein (Fig. 1B) converted UDP-GlcNAc to a new compound, which was separated by TLC and detected by autoradiography (Fig. 3, lanes 1 and 2). The same result was observed on a thymol-stained TLC plate. The new compound was purified from unstained TLC plates, and its one-dimensional and two-dimensional 1H NMR spectra (discussed below) agreed with the results of previous studies on the WbpM homolog Cj1120c (PglF) (13). The evidence that this product is the 4-keto-6-deoxy derivative of UDP-GlcNAc is presented below.
FIGURE 3.
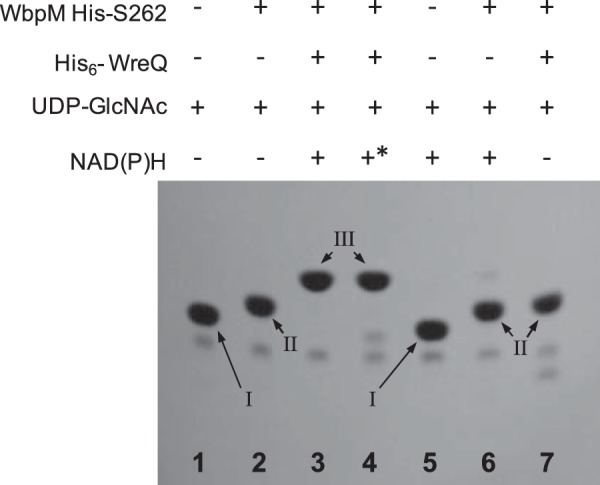
Autoradiograms of TLC-separated WbpM-WreQ reaction and control reactions. Lane 1, no enzymes were added to the nucleotide sugar substrate UDP-GlcNAc; lane 2, WbpM reaction for synthesizing the 4-keto intermediate; lane 3, WbpM-WreQ reaction for synthesizing UDP-QuiNAc; lane 4, NADPH in place of NADH; lane 5, negative control, no WbpM; lane 6, negative control, no WreQ; lane 7, no NAD(P)H. NMR analysis confirmed the identities of the spots as follows: Spot I, UDP-GlcNAc; Spot II, 4-keto intermediate; Spot III, UDP-QuiNAc. * indicates NADPH.
Substrate Conversion by WreQ
The strategy for assaying WreQ was to carry out a two-step reaction. Following the first step reaction catalyzed by WbpM, purified His6-WreQ protein and NAD(P)H were added to start the second step reaction. A new compound later shown to be UDP-QuiNAc was produced (Fig. 3, lanes 3 and 4). Both the WreQ enzyme and NAD(P)H were required to produce this new compound (Fig. 3, lanes 6 and 7). WreQ did not use UDP-GlcNAc as substrate; i.e. no conversion occurred in the absence of WbpM (Fig. 3, lane 5).
GC-MS Analysis of the WbpM-WreQ Reaction Product
To gain initial evidence that UDP-QuiNAc was synthesized in the WbpM-WreQ reaction, the reaction mixture was treated at 100 °C with acid to release the sugars from the nucleoside diphosphate linkages. The resulting compounds were reduced and acetylated to convert sugars into deuterated alditol acetates followed by combined GC-MS analysis.
A deuterated alditol acetate derivative eluted at 26.61 min (Fig. 4A). It fragmented into ions with m/z ratios 85, 103, and 145, which identified it as a 2-amino sugar, and ions with m/z ratios 201, 260, and 302 (Fig. 4B), which were 58 mass units smaller than the corresponding ions of the alditol acetate derived from glucosamine (data not shown). This difference agreed exactly with predicted differences at the C6 position where the glucosamine derivative is CH2OCOCH3 (molecular mass, 73 Da) and quinovosamine derivative is CH3 (molecular mass, 15 Da). These data supported the existence of the quinovosamine moiety in the reaction mixture as expected if UDP-QuiNAc is produced.
FIGURE 4.

GC-MS analysis of chemical derivatives after converting sugars to alditol acetates. A, GC profile of derivatives from the WbpM-WreQ reaction products. B, MS spectrum of the QuiNAc derivative peak in A. Inset, predicted MS fragmentation patterns of the alditol acetates derived from QuiNAc. MS peaks corresponding to the predicted fragmentation pattern are shown with numbers above. The peak designated with & in A is a common non-sugar peak. The peak designated with # may have resulted from other components in the enzyme reaction.
NMR Analysis of the WreQ Reaction Product UDP-d-QuiNAc
Compounds in a non-radioactive WbpM-WreQ reaction mixture were separated by TLC, and the UDP-QuiNAc candidate was purified from an unstained TLC plate. The purified compound was analyzed by one- and two-dimensional NMR spectroscopy (Figs. 5 and 6).
FIGURE 5.

Structural determination for UDP-QuiNAc. A, structure and proton NMR resonance assignments of the reaction product deduced to be UDP-d-QuiNAc. B–D, two-dimensional 1H-1H TOCSY, COSY, and NOESY analysis of UDP-QuiNAc. B, two-dimensional 1H-1H TOCSY correlation map for UDP-QuiNAc. C, two-dimensional 1H-1H COSY correlation map for UDP-QuiNAc. The experiment was acquired in real time mode; therefore, all resonances with chemical shifts beyond water appear mirrored. D, two-dimensional 1H-1H NOESY correlation map for UDP-QuiNAc.
FIGURE 6.
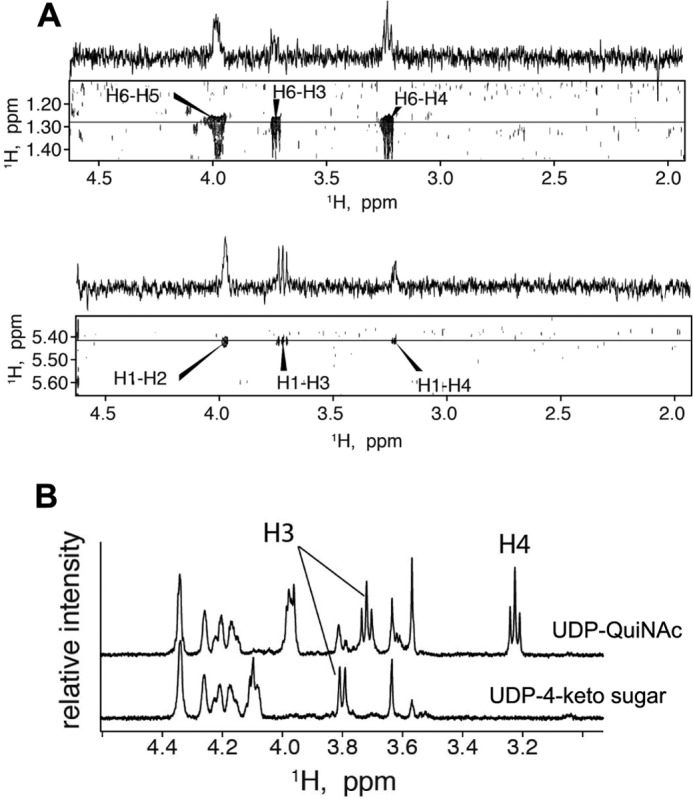
Assignment of proton chemical shifts for H3 and H4. A, strips of two-dimensional 1H-1H TOCSY correlation map for UDP-QuiNAc and one-dimensional slices taken at frequencies of H6 (top panel) and H1 (bottom panel) in indirect dimension demonstrating relative intensities of the cross-peaks. B, comparison of one-dimensional proton NMR spectra of purified UDP-QuiNAc (top trace) and UDP-2-acetamido-2,6-dideoxy-α-d-xylo-4-hexulose (UDP-4-keto sugar) (bottom trace).
Assignment of proton resonances of the WreQ reaction product utilized a combination of NOE, TOCSY, and COSY correlations. The two-dimensional 1H-1H TOCSY experiment allowed establishing the resonance frequencies of all protons in the QuiNAc sugar ring (Fig. 5B). The UDP-ribose ring protons were observed between 4.174 and 4.344 ppm and at 5.959 ppm. The proton at 5.959 ppm was correlated to the uracil base proton at 7.935 ppm (not shown). The UDP resonances did not significantly change from UDP-GlcNAc to the 4-keto intermediate and to UDP-QuiNAc; they will not be discussed. Contaminants uncoupled from UDP-QuiNAc resonances were observed in small amounts in the UDP-QuiNAc sample as isolated spin systems at low intensity between 3.624 and 3.814 ppm, at 1.677 and 2.144 ppm, and at 1.221 ppm (Fig. 5B). The hexose protons assigned to the UDP-QuiNAc resonance chemical shifts (Table 1) are explained below.
TABLE 1.
Summary of the proton chemical shifts and multiplet structures for the hexose ring of UDP-d-QuiNAc and UDP-2-acetamido-2,6-dideoxy-d-xylo-4-hexulose at 30 °C
Standard deviation of the chemical shift values is on the order of 0.004 ppm (as reported by Sparky). Accuracy of J coupling constant estimates is ±0.2 Hz for UDP-d-QuiNAc and ±0.6 Hz for UDP-2-acetamido-2,6-dideoxy-d-xylo-4-hexulose.
| Compound | Proton | Chemical shift | Multiplet structure | J coupling constants |
|---|---|---|---|---|
| ppm | Hz | |||
| UDP-d-QuiNAc | H1 | 5.422 | 1:1:1:1 quadruplet | 3JH1,H2 = 3.6 |
| 3JH1,P = 7.2 | ||||
| H2 | 3.978 | Unresolved overlapped multiplet | ||
| H3 | 3.723 | 1:2:1 triplet | 3JH3,H4 = 9.8 | |
| 3JH2,H3 = 9.8 | ||||
| H4 | 3.231 | 1:2:1 triplet | 3JH3,H4 = 9.3 | |
| 3JH4,H5 = 9.3 | ||||
| H5 | 3.977 | Unresolved overlapped multiplet | ||
| H6 | 1.271 | 1:1 doublet | 3JH6,H5 = 6.0 | |
| UDP-2-acetamido-2,6-dideoxy-d-xylo-4-hexulosea | H1 | 5.433 | Poorly resolved multiplet | |
| H2 | 4.091 | Unresolved overlapped multiplet | 3JH3,H2 = 10.8 | |
| H3 | 3.803 | Doublet | ||
| H5 | 4.091 | Unresolved overlapped multiplet | ||
| H6 | 1.218 | Doublet | 3JH6,H5 = 5.4 |
a Due to a lower concentration of the latter sample, the spectral quality did not allow confident measurement of the smaller 3J coupling constants at H1 position.
The methyl proton doublet centered at 1.271 ppm was identified as H6 by comparison with the proton spectrum of UDP-GlcNAc that shows the only methyl resonance at 2.044 ppm originating from its N-acetyl group. Inspection of the two-dimensional 1H-1H COSY revealed that the neighbor of H6, the H5 proton, resonates at 3.977 ppm (Fig. 5C). The peak of H5 had significant asymmetry, indicating overlapping resonances, and showed four COSY cross-peaks.
The proton resonance at 5.442 ppm was assigned to be H1 by similarity to other sugar nucleotides (26). Inspection of the two-dimensional 1H-1H NOESY correlation map (Fig. 5D) reveals a strong (and the only) NOE cross-peak of H1 at 5.442 and 3.977 ppm, indicating that H5 must be overlapping with H2 at this chemical shift because H5 is too far away to produce NOE to H1. (In the COSY data, the H1 resonance was mirrored into the 4.0 ppm region, thus obscuring the expected cross-peak with H2.)
Of the two remaining resonances at 3.723 and 3.231 ppm, we assigned H4 to 3.231 according to the following considerations. 1) The TOCSY cross-peak of H6-H3 is weaker comparatively with the H6-H4 cross-peak in accord with the greater number of bonds separating H6 and H3 spins (Fig. 6A, top panel). 2) An opposite relationship is observed for TOCSY cross-peaks of H1-H3 and H1-H4, the latter being the weakest of the two, corresponding to the greater number of intervening bonds (Fig. 6A, bottom panel). 3) This resonance is absent in the 4-keto intermediate, which would not have a proton at position 4 (see Fig. 6B).
The configuration of the H4 and H5 in UDP-QuiNAc was determined to be axial based on analysis of 3J coupling constants. Both H3 and H4 are symmetrical triplets, indicating identical 3J coupling constants of 9–10 Hz for all H2-H3, H3-H4, and H4-H5 pairs (Fig. 6B, top trace). The large value of the 3J coupling constants is indicative of the axial orientation of coupled protons (27); therefore, we conclude that H4 and H5 are axial, which leads to the structure shown in Fig. 5A corresponding to the configuration of a d-QuiNAc ring.
NMR Analysis of the WreQ Substrate for UDP-d-QuiNAc Synthesis
The product of the first step of the reaction, the 4-keto intermediate, was purified using TLC from the reaction mixture containing the WbpM protein but not WreQ. Assignment of NMR signals was performed in a similar fashion as for the UDP-d-QuiNAc. The TOCSY correlation map demonstrated that the WbpM reaction product contains two isolated proton spin systems with signals of one assigned to H6 and H5, whereas the second spin system included H1, H2, and H3. The H2 and H5 resonances overlap in the same fashion as in the TOCSY spectra of UDP-d-QuiNAc (data not shown), but the absence of a H6-H3 cross-peak indicates a break in the continuous path for the TOCSY transfer from H6 to H3 due to the H4 proton missing in the 4-keto intermediate.
Fig. 6B compares the one-dimensional spectra of the UDP-d-QuiNAc and the WbpM reaction product, demonstrating the absence of the H4 signal in the 4-keto intermediate spectrum (bottom trace). Additionally, the H3 signal becomes a doublet, indicating that H3 has only one 3J coupling partner in this molecule, and it must be H2. These findings confirm that the WbpM reaction product and substrate of WreQ was, indeed, the 4-keto-6-deoxy derivative of UDP-GlcNAc.
Direct Observation of the Reaction Progress by NMR
Stability of the compounds in the NMR tube was directly assessed by comparison of one-dimensional proton spectra acquired before and after two-dimensional experiments (data not shown). Samples were found to be completely stable over the entire time of data acquisition.
To gain information about reaction kinetics, we performed the enzyme incubation in an NMR spectrometer and directly monitored the WbpM-WreQ reaction progress. Initial accumulation of the 4-keto intermediate and its subsequent conversion into UDP-QuiNAc were monitored by 1H NMR spectroscopy at 30 °C. Fig. 7 shows an overlay of one-dimensional proton spectra taken during the reaction course. The initial reaction mixture (black trace) contained UDP-GlcNAc, NADH, and purified WreQ but no WbpM. Addition of WbpM led to quick accumulation of the 4-keto-6-deoxy intermediate (reflecting 4,6-dehydratase activity of WbpM) as indicated by appearance of the new H6 methyl proton doublet centered at 1.217 ppm. Activity of WreQ led to subsequent conversion of the 4-keto-6-deoxy intermediate into UDP-QuiNAc that was detected by the appearance of its H6 methyl doublet centered at 1.271 ppm (and decreasing H6 methyl signal of the 4-keto-6-deoxy intermediate). Because the predicted product UDP-QuiNAc retains the methyl group in position 6, the chemical shift change must reflect the modification of the hexose ring at position 4. The WbpM-catalyzed reaction was significantly faster than that catalyzed by WreQ. Most of the 4-keto intermediate was produced within the first 10 min (black to blue traces), whereas subsequent slow conversion to UDP-QuiNAc continued during overnight incubation and even then was not complete (blue to red traces).
FIGURE 7.
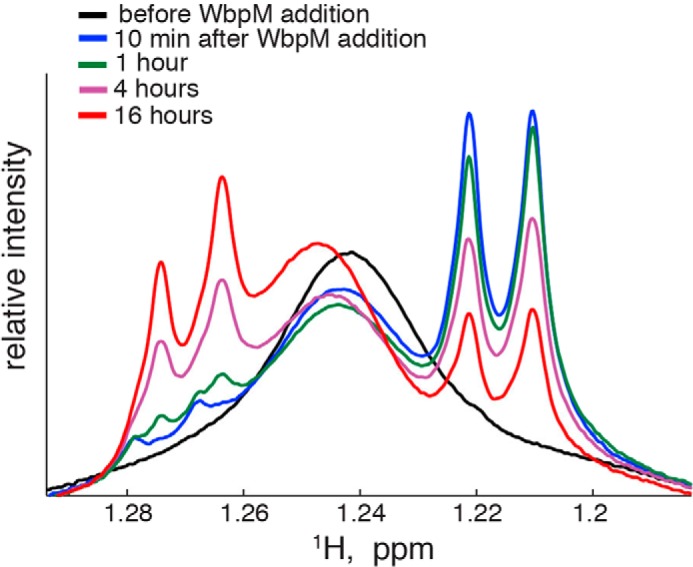
One-dimensional proton NMR spectra obtained from the reaction mixture at different incubation times. A broad resonance at 1.241 ppm is due to methyl groups of the detergent Triton X-100 present in the reaction mixture. This reaction was performed with 1 mm UDP-GlcNAc, 1 mm NADH, 48 μg of WbpM, and 54 μg of WreQ in a total volume of 300 μl. WbpM was added last to start the reaction.
DISCUSSION
Quinovosamine was first identified in bacterial polysaccharides at least 50 years ago (28, 29). Attempts to establish the pathway of its biosynthesis foundered on the use of crude extracts and the problem of demonstrating a specific reaction by adding NADH to a crude extract (30). Only now, with the use of a genetic approach leading to a facile enzyme purification, has an enzymatic pathway for its synthesis been demonstrated in vitro. In this report, we present evidence that UDP-QuiNAc can be synthesized in vitro from UDP-GlcNAc via the two steps proposed previously (5) (Fig. 8).
FIGURE 8.
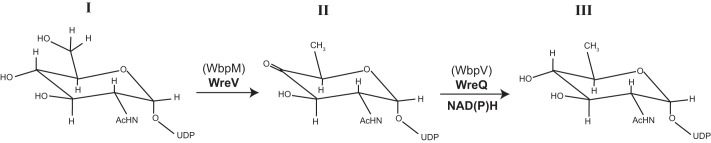
The proposed biosynthesis pathway of UDP-d-QuiNAc in R. etli CE3 and P. aeruginosa O6. The P. aeruginosa O6 proteins are indicated in parentheses. I, UDP-d-GlcNAc; II, UDP-2-acetamido-2,6-dideoxy-α-d-xylo-4-hexulose; III, UDP-d-QuiNAc.
The first step is catalyzed by UDP-GlcNAc 4,6-dehydratase, resulting in the 4-keto intermediate UDP-2-acetamido-2,6-dideoxy-d-xylo-4-hexulose. A soluble truncated version of the WbpM protein from P. aeruginosa was used in this study to catalyze this reaction in vitro (9). The R. etli CE3 wreV gene encodes a protein that is homologous to WbpM. An insertion mutation in wreV abolished O-antigen synthesis in R. etli, and wbpM complemented this mutant (Fig. 9). Hence, WreV is inferred to be the 4,6-dehydratase responsible in R. etli for the first step of UDP-QuiNAc synthesis.
FIGURE 9.
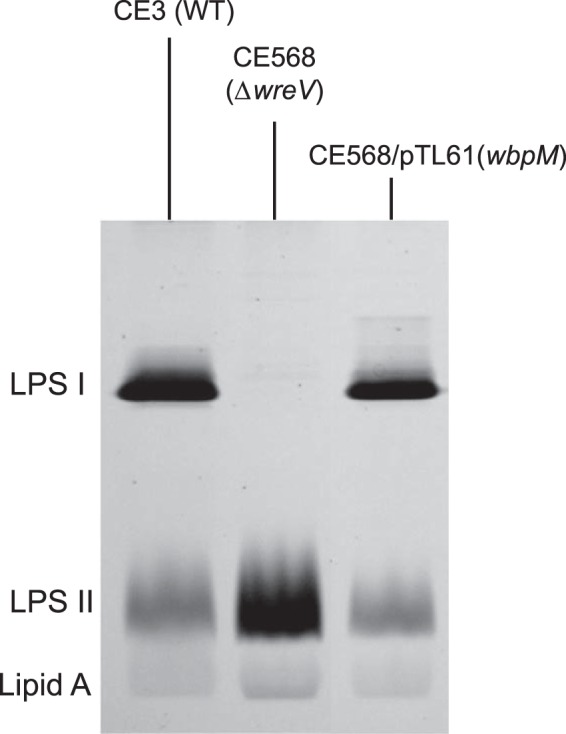
P. aeruginosa wbpM gene can complement R. etli wreV mutant (CE568, ΔwreV). Shown is silver-stained SDS-PAGE (18% gel) of LPS samples from whole-cell lysates. LPS I, O-antigen-containing LPS; LPS II, core oligosaccharide-Lipid A lacking O-antigen. CE3, wild-type R. etli strain; CE568, R. etli strain with a gentamicin resistance cassette insertion in wreV (ΔwreV); CE568/pTL61, CE568 strain carrying plasmid pTL61, which expresses the full-length WbpM.
The second step in Fig. 8 is catalyzed by a 4-reductase. Previous studies (4, 5) suggested that wreQ encodes this enzyme in R. etli CE3. The in vitro reaction followed in this study confirmed that WreQ is a 4-reductase. The stereospecificity of WreQ determines that the 4-keto intermediate is reduced to UDP-d-QuiNAc but not to its C4 epimer UDP-N-acetyl-d-fucosamine. A homolog of WreQ in P. aeruginosa O6 encoded by wbpV has been proposed to catalyze the same reaction (6). As predicted, the wbpV gene can complement the R. etli wreQ mutant (Fig. 2, A and C).
In surveying bacterial strains shown to contain d-QuiNAc and having a sequenced genome, wreV and wreQ gene homologs were always found and usually were closely linked on the genomes (Table 2). Thus, biosynthesis via WreV and WreQ homologs may be conserved in all bacteria containing d-QuiNAc. Conversely, the presence of d-QuiNAc in bacterial strains could be predicted based on whether they contain the WreV and WreQ homologs. For instance, we predict that Chlorobium phaeobacteroides DSM 266, Herbaspirillum seropedicae SmR1, and Polaromonas naphthalenivorans CJ2 as well as several Rhizobium and Pseudomonas strains not listed in Table 2 produce d-QuiNAc that would be found if the compositions of their polysaccharides were to be analyzed. More examples are given in Table 3.
TABLE 2.
Bacterial strains that have been reported to contain d-QuiNAc and have a sequenced genome revealed R. etli CE3 WreV and WreQ protein homologs
| Bacterial strain | WreV homolog accession no.a/E-value | WreQ homolog accession no.a/E-value |
|---|---|---|
| R. etli CE3 (1)b | YP_471773/0.0 | YP_470339/0.0 |
| P. aeruginosa O6 (34) | AAF23989/0.0 | AAF23991/2e−98 |
| Francisella tularensis subsp. tularensis SCHU S4 (35) | YP_170401/2e−113 | YP_170399/2e−28 |
| Legionella pneumophila subsp. pneumophila str. Philadelphia 1 (36) | YP_095000/2e−118 | YP_094797/6e−74 |
| Pseudomonas tolaasii NCPPB2192 (37) | WP_016970514/0.0 | WP_016971490/5e−96 |
| Vibrio vulnificus M06-24 (38) | YP_004189989/0.0 | YP_004189991/2e−100 |
a Accession numbers are from genome sequencing projects except P. aeruginosa O6 for which the accession numbers were associated with Belanger et al. (6).
b References following the name of bacterial strains are studies that identified d-QuiNAc in that strain.
TABLE 3.
Bacterial strains predicted to contain d-QuiNAc based on the presence of genes encoding homologs of R. etli CE3 WreV and WreQ in their genome
WreQ was used as the query sequence in a BLAST search against the non-redundant protein sequence (nr) database using the blastp algorithm. Bacterial strains selected all meet the following criteria: 1) encode a WreQ homolog with lower E-value than that of WbpV, 7e−91 (the WreQ homolog of P. aeruginosa O6), 2) have a fully sequenced genome, and 3) encode a WreV homolog (40 of 41 strains contain both WreV and WreQ homologs).
| Genus | Species | Strain |
|---|---|---|
| Chlorobium | phaeobacteroides | DSM 266 |
| Colwellia | psychrerythraea | 34H |
| Cycloclasticus | sp. | P1 |
| Gallionella | capsiferriformans | ES-2 |
| Geobacter | bemidjiensis | Bem |
| sp. | M21 | |
| Herbaspirillum | seropedicae | SmR1 |
| Herminiimonas | arsenicoxydans | |
| Marinobacter | aquaeolei | VT8 |
| Methylobacillus | flagellatus | KT |
| Methylomicrobium | alcaliphilum | 20Z |
| Methylotenera | mobilis | JLW8 |
| versatilis | 301 | |
| Nitrosomonas | eutropha | C91 |
| Pelodictyon | phaeoclathratiforme | BU-1 |
| Polaromonas | naphthalenivorans | CJ2 |
| sp. | JS666 | |
| Pseudomonas | entomophila | L48 |
| fluorescens | Pf0-1 | |
| mendocina | ymp | |
| monteilii | SB3078 | |
| protegens | CHA0, Pf-5 | |
| putida | BIRD-1, GB-1, HB3267, W619 | |
| stutzeri | DSM 10701, RCH2 | |
| Rhizobium | etli | CIAT 652, bv. mimosae str. Mim1 |
| leguminosarum | bv. trifolii WSM1325, bv. trifolii WSM1689, bv. trifolii WSM2304, bv. viciae 3841 | |
| Sideroxydans | lithotrophicus | ES-1 |
| Thioalkalivibrio | sp. | K90mix |
| Vibrio | cholerae | O1 biovar El Tor str. N16961 |
| Variovorax | paradoxus | EPS |
The second reaction catalyzed by WreQ in Fig. 8 was relatively slow. The low activity may be an artifact of one or more features of the in vitro reaction. The His6-WreQ protein used in vitro has additional amino acids at the amino terminus that could affect structure at the active site and slow the catalysis. Another consideration is that during dialysis to reduce the concentration of imidazole used in purification of the His-tagged protein the His6-WreQ protein precipitated in the dialysis tubing. It was found that adding the detergent Triton X-100 alleviated the precipitation, but the presence of detergent (0.1% of reaction volume) may have affected the reaction rate. It is unclear why His6-WreQ precipitated during dialysis. One possible explanation is that WreQ normally associates with the membrane or other proteins in R. etli CE3 and that they are required for its stability. If that is the case, overproduction of WreQ in E. coli, which is devoid of its naturally associated partner, may render it unstable.
The pathway intermediate, UDP-2-acetamido-2,6-dideoxy-d-xylo-4-hexulose, was relatively stable. Although other 4-keto-6-deoxy sugars have been reported to be unstable (26, 31–33), this 4-keto-6-deoxy derivative of UDP-GlcNAc was able to withstand the purification by TLC and was stable for several days during NMR data acquisition. Thus, we were able to obtain both this intermediate and UDP-d-QuiNAc in stable, relatively pure form.
Analysis of O-antigen mutants and the sequences of the wre genes required for O-antigen have prompted speculation that O-antigen synthesis in R. etli CE3 begins with reactions that result in 1-pyrophospho-QuiNAc attached to a lipid carrier (8). As noted in the Introduction, a previous study showed that QuiNAc was replaced by its 4-keto derivative in the LPS of the wreQ mutant strain, CE166 (5). This observation leads to questions regarding the substrate specificities of WreQ and the putative glycosyltransferase that catalyzes initial sugar-lipid linkage. It is conceivable that the initiator glycosyltransferase works better with the 4-keto intermediate as substrate. If so, it may be that the resulting lipidated sugar intermediate is a better substrate for WreQ than the 4-keto intermediate with a UDP moiety. This speculation provides another possible explanation for the relatively slow catalysis of WreQ noted in this study. The compounds generated in vitro in this study provide an essential starting point for answering these speculative questions.
Acknowledgments
We acknowledge the technical assistance of Jodie Box in studies with wbpV. We thank Dr. J. S. Lam for providing the WbpM-His-S262 expression vector and the pBAD-wbpM and pFV611-26 plasmids.
This work was supported, in whole or in part, by National Institutes of Health Grant 1 R15 GM087699-01A1 (to K. D. N.).
- QuiNAc
- N-acetyl-d-quinovosamine
- UDP-GlcNAc
- UDP-2-acetamido-2-deoxy-α-d-glucose
- 4-keto intermediate and 4-keto-6-deoxy intermediate
- UDP-2-acetamido-2,6-dideoxy-α-d-xylo-4-hexulose
- UDP-QuiNAc
- UDP-2-acetamido-2,6-dideoxy-d-glucose
- TOCSY
- total correlation spectroscopy
- wre
- designation for genes specifically devoted to the R. etli CE3 O-antigen (15) (previous symbols had been lps and lpe).
REFERENCES
- 1. Forsberg L. S., Bhat U. R., Carlson R. W. (2000) Structural characterization of the O-antigenic polysaccharide of the lipopolysaccharide from Rhizobium etli strain CE3. A unique O-acetylated glycan of discrete size, containing 3-O-methyl-6-deoxy-l-talose and 2,3,4-tri-O-methyl-l fucose. J. Biol. Chem. 275, 18851–18863 [DOI] [PubMed] [Google Scholar]
- 2. Knirel Y. A., Bystrova O. V., Kocharova N. A., Zähringer U., Pier G. B. (2006) Conserved and variable structural features in the lipopolysaccharide of Pseudomonas aeruginosa. J. Endotoxin Res 12, 324–336 [DOI] [PubMed] [Google Scholar]
- 3. Cava J. R., Elias P. M., Turowski D. A., Noel K. D. (1989) Rhizobium leguminosarum CFN42 genetic regions encoding lipopolysaccharide structures essential for complete nodule development on bean plants. J. Bacteriol. 171, 8–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Noel K. D., Forsberg L. S., Carlson R. W. (2000) Varying the abundance of O antigen in Rhizobium etli and its effect on symbiosis with Phaseolus vulgaris. J. Bacteriol. 182, 5317–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Forsberg L. S., Noel K. D., Box J., Carlson R. W. (2003) Genetic locus and structural characterization of the biochemical defect in the O-antigenic polysaccharide of the symbiotically deficient Rhizobium etli mutant, CE166. Replacement of N-acetylquinovosamine with its hexosyl-4-ulose precursor. J. Biol. Chem. 278, 51347–51359 [DOI] [PubMed] [Google Scholar]
- 6. Bélanger M., Burrows L. L., Lam J. S. (1999) Functional analysis of genes responsible for the synthesis of the B-band O antigen of Pseudomonas aeruginosa serotype O6 lipopolysaccharide. Microbiology 145, 3505–3521 [DOI] [PubMed] [Google Scholar]
- 7. Bystrova O. V., Shashkov A. S., Kocharova N. A., Knirel Y. A., Lindner B., Zähringer U., Pier G. B. (2002) Structural studies on the core and the O-polysaccharide repeating unit of Pseudomonas aeruginosa immunotype 1 lipopolysaccharide. Eur. J. Biochem. 269, 2194–2203 [DOI] [PubMed] [Google Scholar]
- 8. Ojeda K. J., Simonds L., Noel K. D. (2013) Roles of predicted glycosyltransferases in the biosynthesis of the Rhizobium etli CE3 O antigen. J. Bacteriol. 195, 1949–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Creuzenet C., Lam J. S. (2001) Topological and functional characterization of WbpM, an inner membrane UDP-GlcNAc C6 dehydratase essential for lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Mol. Microbiol. 41, 1295–1310 [DOI] [PubMed] [Google Scholar]
- 10. Creuzenet C., Schur M. J., Li J., Wakarchuk W. W., Lam J. S. (2000) FlaA1, a new bifunctional UDP-GlcNAc C6 dehydratase/C4 reductase from Helicobacter pylori. J. Biol. Chem. 275, 34873–34880 [DOI] [PubMed] [Google Scholar]
- 11. Olivier N. B., Chen M. M., Behr J. R., Imperiali B. (2006) In vitro biosynthesis of UDP-N,N′-diacetylbacillosamine by enzymes of the Campylobacter jejuni general protein glycosylation system. Biochemistry 45, 13659–13669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pinta E., Duda K. A., Hanuszkiewicz A., Kaczyński Z., Lindner B., Miller W. L., Hyytiäinen H., Vogel C., Borowski S., Kasperkiewicz K., Lam J. S., Radziejewska-Lebrecht J., Skurnik M., Holst O. (2009) Identification and role of a 6-deoxy-4-keto-hexosamine in the lipopolysaccharide outer core of Yersinia enterocolitica serotype O:3. Chemistry 15, 9747–9754 [DOI] [PubMed] [Google Scholar]
- 13. Schoenhofen I. C., McNally D. J., Vinogradov E., Whitfield D., Young N. M., Dick S., Wakarchuk W. W., Brisson J. R., Logan S. M. (2006) Functional characterization of dehydratase/aminotransferase pairs from Helicobacter and Campylobacter: enzymes distinguishing the pseudaminic acid and bacillosamine biosynthetic pathways. J. Biol. Chem. 281, 723–732 [DOI] [PubMed] [Google Scholar]
- 14. Hao Y., Lam J. S. (2011) in Bacterial Lipopolysaccharides (Knirel Y. A., Valvano M. A., eds) pp. 195–235, Springer; Vienna [Google Scholar]
- 15. Ojeda K. J., Box J. M., Noel K. D. (2010) Genetic basis for Rhizobium etli CE3 O-antigen O-methylated residues that vary according to growth conditions. J. Bacteriol. 192, 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kavanagh K. L., Jörnvall H., Persson B., Oppermann U. (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell. Mol. Life Sci. 65, 3895–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Noel K. D., Sanchez A., Fernandez L., Leemans J., Cevallos M. A. (1984) Rhizobium phaseoli symbiotic mutants with transposon Tn5 insertions. J. Bacteriol. 158, 148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dombrecht B., Vanderleyden J., Michiels J. (2001) Stable RK2-derived cloning vectors for the analysis of gene expression and gene function in Gram-negative bacteria. Mol. Plant Microbe Interact. 14, 426–430 [DOI] [PubMed] [Google Scholar]
- 19. Schweizer H. D. (1993) Small broad-host-range gentamycin resistance gene cassettes for site-specific insertion and deletion mutagenesis. BioTechniques 15, 831–834 [PubMed] [Google Scholar]
- 20. Bouhenni R. A., Vora G. J., Biffinger J. C., Shirodkar S., Brockman K., Ray R., Wu P., Johnson B. J., Biddle E. M., Marshall M. J., Fitzgerald L. A., Little B. J., Fredrickson J. K., Beliaev A. S., Ringeisen B. R., Saffarini D. A. (2010) The Role of Shewanella oneidensis MR-1 outer surface structures in extracellular electron transfer. Electroanalysis 22, 856–864 [Google Scholar]
- 21. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 22. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 23. Goddard T. D., Kneller D. G. (2008) SPARKY 3, University of California, San Francisco [Google Scholar]
- 24. Hartel A. J., Lankhorst P. P., Altona C. (1982) Thermodynamics of stacking and of self-association of the dinucleoside monophosphate m2(6)A-U from proton NMR chemical shifts: differential concentration temperature profile method. Eur. J. Biochem. 129, 343–357 [DOI] [PubMed] [Google Scholar]
- 25. Orbons L. P., van der Marel G. A., van Boom J. H., Altona C. (1987) An NMR study of the polymorphous behavior of the mismatched DNA octamer d(m5C-G-m5C-G-T-G-m5C-G) in solution. The B, Z, and hairpin forms. J. Biomol. Struct. Dyn. 4, 939–963 [DOI] [PubMed] [Google Scholar]
- 26. King J. D., Poon K. K., Webb N. A., Anderson E. M., McNally D. J., Brisson J. R., Messner P., Garavito R. M., Lam J. S. (2009) The structural basis for catalytic function of GMD and RMD, two closely related enzymes from the GDP-D-rhamnose biosynthesis pathway. FEBS J. 276, 2686–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Claridge T. D. W. (2009) High-Resolution NMR Techniques in Organic Chemistry, Elsevier, Amsterdam [Google Scholar]
- 28. Smith E. J. (1964) The isolation and characterization of 2-amino-2:6-dideoxy d-glucose (d-quinovosamine) from a bacterial polysaccharide. Biochem. Biophys. Res. Commun. 15, 593–597 [Google Scholar]
- 29. Lüderitz O., Gmeiner J., Kickhöfen B., Mayer H., Westphal O., Wheat R. W. (1968) Identification of d-mannosamine and quinovosamine in Salmonella and related bacteria. J. Bacteriol. 95, 490–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Daniel A., Raff R. A., Wheat R. W. (1972) Identification of products of the uridinediphospho-N-acetyl-d-glucosamine oxidoreductase system from Citrobacter freundii ATCC 10053. J. Bacteriol. 110, 110–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bonin C. P., Potter I., Vanzin G. F., Reiter W. D. (1997) The MUR1 gene of Arabidopsis thaliana encodes an isoform of GDP-d-mannose-4,6-dehydratase, catalyzing the first step in the de novo synthesis of GDP-l-fucose. Proc. Natl. Acad. Sci. U.S.A. 94, 2085–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ohyama C., Smith P. L., Angata K., Fukuda M. N., Lowe J. B., Fukuda M. (1998) Molecular cloning and expression of GDP-d-mannose-4,6-dehydratase, a key enzyme for fucose metabolism defective in Lec13 cells. J. Biol. Chem. 273, 14582–14587 [DOI] [PubMed] [Google Scholar]
- 33. McNally D. J., Schoenhofen I. C., Mulrooney E. F., Whitfield D. M., Vinogradov E., Lam J. S., Logan S. M., Brisson J. R. (2006) Identification of labile UDP-ketosugars in Helicobacter pylori, Campylobacter jejuni and Pseudomonas aeruginosa: key metabolites used to make glycan virulence factors. Chembiochem 7, 1865–1868 [DOI] [PubMed] [Google Scholar]
- 34. Dmitriev B. A., Kocharova N. A., Knirel Y. A., Shashkov A. S., Kochetkov N. K., Stanislavsky E. S., Mashilova G. M. (1982) Somatic antigens of Pseudomonas aeruginosa. The structure of the polysaccharide chain of Ps.aeruginosa O:6 (Lanyi) lipopolysaccharide. Eur. J. Biochem. 125, 229–237 [DOI] [PubMed] [Google Scholar]
- 35. Wang Q., Shi X., Leymarie N., Madico G., Sharon J., Costello C. E., Zaia J. (2011) A typical preparation of Francisella tularensis O-antigen yields a mixture of three types of saccharides. Biochemistry 50, 10941–10950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Knirel Y. A., Moll H., Zähringer U. (1996) Structural study of a highly O-acetylated core of Legionella pneumophila serogroup 1 lipopolysaccharide. Carbohydr. Res. 293, 223–234 [DOI] [PubMed] [Google Scholar]
- 37. Molinaro A., Bedini E., Ferrara R., Lanzetta R., Parrilli M., Evidente A., Lo Cantore P., Iacobellis N. S. (2003) Structural determination of the O-specific chain of the lipopolysaccharide from the mushrooms pathogenic bacterium Pseudomonas tolaasii. Carbohydr. Res. 338, 1251–1257 [DOI] [PubMed] [Google Scholar]
- 38. Reddy G. P., Hayat U., Abeygunawardana C., Fox C., Wright A. C., Maneval D. R., Jr., Bush C. A., Morris J. G., Jr. (1992) Purification and determination of the structure of capsular polysaccharide of Vibrio vulnificus M06-24. J. Bacteriol. 174, 2620–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]