

Background: Two alleles of the mammalian Ras protein acyltransferase are associated with X-linked intellectual disabilities.
Results: Mutations of zDHHC9 isolated from XLID patients result in defects to steady state autopalmitoylation levels.
Conclusion: Although the zDHHC9 mutations produce similar phenotypes, they are guided by distinct mechanisms.
Significance: Elucidation of the catalytic mechanism of PATs is a critical step in designing pharmacological interventions.
Keywords: Enzyme Mechanism, Post-translational Modification (PTM), Protein Acylation, Protein Palmitoylation, Ras Protein, X-linked Intellectual Disability, zDHHC Proteins
Abstract
Early onset intellectual disabilities result in significant societal and economic costs and affect 1–3% of the population. The underlying genetic determinants are beginning to emerge and are interpreted in the context of years of work characterizing postsynaptic receptor and signaling functions of learning and memory. DNA sequence analysis of intellectual disability patients has revealed greater than 80 loci on the X-chromosome that are potentially linked to disease. One of the loci is zDHHC9, a gene encoding a Ras protein acyltransferase. Protein palmitoylation is a reversible modification that controls the subcellular localization and distribution of membrane receptors, scaffolds, and signaling proteins required for neuronal plasticity. Palmitoylation occurs in two steps. In the first step, autopalmitoylation, an enzyme-palmitoyl intermediate is formed. During the second step, the palmitoyl moiety is transferred to a protein substrate, or if no substrate is available, hydrolysis of the thioester linkage produces the enzyme and free palmitate. In this study, we demonstrate that two naturally occurring variants of zDHHC9, encoding R148W and P150S, affect the autopalmitoylation step of the reaction by lowering the steady state amount of the palmitoyl-zDHHC9 intermediate.
Introduction
Protein lipidation refers to the covalent modification of proteins with lipid moieties that include fatty acyl chains (myristoyl or palmitoyl), isoprenes (farnesyl or geranylgeranyl), or cholesterol. The major form of protein palmitoylation, S-palmitoylation, occurs via a thioester linkage on cysteine residues. Myristoylation and prenylation are irreversible modifications, whereas palmitoylation is reversible through proposed protein palmitoylthioesterases (1). The reversible nature of the thioester linkage provides for a dynamic acylation/deacylation cycle that is required for subcellular trafficking and function of palmitoylated proteins (2, 3). The addition of a palmitoyl moiety to a protein has at least three potential functions. First, palmitoylation facilitates membrane association and/or the micro-environmental distribution of proteins within membrane subdomains (4). Second, palmitoylation within transmembrane domains (TMD)2 causes the TMD to tilt within the membrane (5), thus affecting the protein's conformation. Third, palmitoylation of membrane proteins affects their ability to interact with other proteins as transient associations or to form complexes (6–12). Indeed, the dynamic nature of the thioester molecular switch adds an important aspect to the functional regulation of proteins.
Protein palmitoylation is catalyzed by protein acyltransferases (PATs), which belong to a conserved family of proteins referred to as zDHHC proteins for their DHHC (Asp-His-His-Cys) catalytic motif (13, 14). The zDHHC-catalyzed palmitoylation reaction utilizes a two-step mechanism. In the first step, a substrate-enzyme (PAT) intermediate is formed via a thioester linkage between palmitate from palmitoyl-CoA and the cysteine of the DHHC motif, a process referred to as autopalmitoylation (15). The proposed juxtaposition of the PAT catalytic active site with the cytosolic surface of the membrane implies that the enzyme can utilize membrane-associated palmitoyl-CoA as the acyl donor and suggests that after the formation of the palmitoyl-PAT intermediate, the active site of the enzyme will be tethered to the membrane through the palmitoyl moiety. In the second step, palmitate is transferred to the thiol of a cysteine of the protein substrate. In the absence of protein substrate, the thioester is hydrolyzed, releasing free palmitate and enzyme poised to react with another molecule of palmitoyl-CoA (15). In the Saccharomyces cerevisiae Erf2-Erf4 Ras PAT, the rate of the autopalmitoylation/hydrolysis cycle in the absence of Ras substrate is regulated by the Erf4 accessory subunit, although the molecular mechanism has not been elucidated (16).
Six of the 23 mammalian zDHHC PAT genes (HIP14, HIP14L, zDHHC8, zDHHC9, zDHHC12, and zDHHC15) have been implicated in brain disorders such as schizophrenia, Huntington disease, Alzheimer disease, infantile and adult onset forms of neuronal ceroid lipofuscinosis, and mental retardation (17). Like S. cerevisiae Erf2-Erf4, zDHHC9 is a member of a large evolutionarily conserved family of DHHC motifs containing enzymes that require the formation of a complex with an accessory protein to be functional. The accessory protein for zDHHC9 is the Golgi-localized membrane protein, GCP16 (18, 19). Intriguingly and again like the Erf2-Erf4 complex, zDHHC9 is a PAT for Ras proteins (19–21); however, it is suspected that it can palmitoylate other substrates. zDHHC9 expression has been linked to two forms of cancer, and loss-of-function alleles of zDHHC9 have been linked to X-linked intellectual disorders (XLIDs) (22). Currently, the substrate(s) of zDHHC9 in the context of these physiological processes are not known.
Although XLID is one of the most common causes of mental disorders, the term actually describes a broader set of developmental abnormalities and accounts for 10% of intellectual deficiencies in males (23). Identification of the genes responsible through linkage studies and sequencing of the X chromosome has led to the identification of ∼80 genes associated with XLID and includes more than 200 syndromes (24, 25). There are four alleles of zDHHC9 linked to XLID. The two nonsense mutations create a truncation and a frameshift, both deleting the essential DHHC catalytic domain of zDHHC9, and are therefore recessive. This study addresses the molecular defect manifested by the two missense genetic variants that map within the zDHHC9 gene and are linked to the Marfanoid features as well as the psychological disorders representative of syndromic XLIDs.
We provide genetic and biochemical evidence that zDHHC9 R148W and zDHHC9 P150S reduce the steady state level of autopalmitoylated zDHHC9 through different mechanisms. The R148W mutation increases the hydrolysis rate of the zDHHC9-palmitate thioester linkage, whereas the P150S mutation decreases the initial burst phase of the autopalmitoylation reaction. Both mutations affect the steady state level of the zDHHC9-palmitate intermediate, which in turn is predicted to affect palmitoylation target proteins involved in intellectual development.
EXPERIMENTAL PROCEDURES
Strains, Media, and Yeast Techniques
Yeast and bacteria growth media were prepared as described previously (26). Yeast cultures were grown in synthetic complete (SC) medium or YPD medium (1% yeast extract, 2% peptone, and 2% glucose) (26). Induction of GAL1,10 promoters was achieved by adding 4% galactose to SC medium. Yeast transformations were performed using the lithium acetate procedure (27). The yeast strains used in this study, RJY1842 (MATa/α ade2-1/ade2-1 leu2-3,112/leu2-3,112 ura3–1/ura3-1 trp1-1/trp1-1 his3-11,15/his3-11,15 can1-100/can1-100 erf4Δ::NATr/erf4Δ::NATr GAL+) (16) and RJY1330 (MATα leu2-3,112 ura3-52 ade2Δ ade8Δ lys2-801 ras1::HIS3 ras2::Ras2CS-Ext erf2::KANr 〈YCp52RAS2〉) have been previously described (20).
Plasmid Construction
Plasmids used throughout this study are listed in Table 1. The sequences of the deoxyoligonucleotide primers used to construct zDHHC9 alleles are available upon request. The DNA sequences of all constructs were verified by DNA sequencing (GeneWiz, Plainfield, NJ). Yeast strain RJY1842 was used for homologous recombination-assisted ligation-independent cloning (21). B1250 was constructed using ligation-independent cloning by amplifying the ERF2 open reading frame via the polymerase chain reaction (PCR) utilizing forward and reverse primers whose 5′ sequences were homologous to the multicloning site (MCS1) of pESC(Trp), B1192, to allow the FLAG epitope to be in-frame with the ERF2 coding sequence.
TABLE 1.
Plasmids used in this study
| Bacterial strain | Plasmidsa |
|---|---|
| B1192 | pESC(Trp) |
| B1500 | pESC(Trp) DHHC9:His6:FLAG |
| B1856 | pESC(Trp) DHHC9:His6:FLAG/Myc:GCP16 |
| B1250 | pESC(Trp) FLAG:Erf2 |
| B1858 | pESC(Trp) DHHC9 R148W:His6:FLAG/Myc:GCP16 |
| B1860 | pESC(Trp) DHHC9 P150S:His6:FLAG/Myc:GCP16 |
| B1863 | pESC(Trp) DHHA9 C169A:His6:FLAG/Myc:GCP16 |
| B1859 | pESC(Trp) DHHC9 R148W:His6:FLAG |
| B1862 | pESC(Trp) DHHC9 P150S:His6:FLAG |
| B1910 | pESC(Trp) Myc:GCP16 |
| B1911 | pESC(Trp) DHHC9:His6:FLAG/Myc:GCP16 C69S,C72S |
a All plasmids were constructed for this study with the exception of pESC(Trp) (B1192) (Agilent, Santa Clara, CA).
zDHHC9 (accession number BC006200) and GCP16 (BC001227) were obtained from the I.M.A.G.E. Consortium (Image numbers 2964425 (zDHHC9) and 3456384 (GCP16)). The zDHHC9 open reading frame was amplified by PCR using primers that would encode an in-frame C-terminal His6-FLAG epitope flanked by sequences homologous to MCS1 of pESC-Leu, B1192 (Agilent Technologies, Santa Clara, CA, CA), to facilitate ligation-independent cloning, placing the zDHHC9 open reading frame immediately downstream of the divergent GAL1,10 promoter and producing B1500. B1856 was constructed by inserting the PCR-amplified GCP16 possessing the proper flanking sequences into MCS2 of B1500. This strategy allows GCP16 to also be under the control of the GAL1,10 promoter. Mutations zDHHC9 R148W, zDHHC9 P150S, and C169A (zDHHA9) were created using the QuikChange II mutagenesis kit (Agilent, Santa Clara, CA) as per the manufacturer's instructions, using B1500 (zDHHC9) and creating B1859 (zDHHC9 R148W) and B1862 (zDHHC9 P150S). Plasmids co-expressing the zDHHC9 alleles and GCP16 open reading frame from the divergent GAL1,10 promoter were generated from B1856 (zDHHC9/GCP16) using the QuikChange II system, creating B1858 (zDHHC9 R148W/GCP16), B1860 (zDHHC9 P150S/GCP16), and B1863 (zDHHA9/GCP16). All plasmid DNA sequences were verified by sequencing both DNA strands (GeneWiz, Plainfield, NJ).
Complementation Assay
The in vivo function of the zDHHC9 mutants along with the wild type protein was investigated using our previously described complementation assay (20) in yeast strain RJY1330. Briefly, strain RJY1330 was transformed with plasmids expressing the zDHHC9 alleles and was grown in liquid synthetic medium containing 2% glucose and lacking tryptophan at 30 °C until the culture reached an A600 between 0.8 and 1.2. Cells were serially diluted starting with 100,000 cells. The dilutions were spotted onto synthetic medium containing 2% glucose (lacking tryptophan) with and without 0.1% 5′-fluoroorotic acid to select against the presence of URA3-linked RAS2. The plates were incubated for up to 4 days at 30 °C.
Protein Expression and Purification
zDHHC9-GCP16 enzyme complexes were expressed from the pESC(Trp)-divergent GAL1,10 promoter in strain RJY1842 (which also harbored a second plasmid to enhance galactose induction, pMA210 (28)). Strain RJY1842 was transformed with zDHHC9-GCP16 and pMA210 and then grown in synthetic medium containing 2% glucose and lacking tryptophan and histidine overnight at 30 °C with shaking. Approximately 3 × 107 cells were inoculated into 50 ml of synthetic medium containing 2% glucose and lacking tryptophan and histidine, and the culture was grown at 30 °C until the cell density was between 1.6 × 107 cells/ml (A600 0.8) and 2.4 × 107 cells/ml (A600 1.2). To induce the expression of the zDHHC9-GCP16 enzyme complexes, ∼50 A600 of cells (1 × 109) were seeded into 1 liter of YEP containing 4% galactose, and the culture was incubated at 30 °C with shaking (230 rpm) for 18 h. The cells were harvested by centrifugation at 3000 × g for 15 min. The resulting pellet was resuspended in breaking buffer (50 mm Tris-Cl, pH 8, 500 ml NaCl, 1 mm EDTA, 1 mm dithiothreitol), 1× protease inhibitor mixture (PIC) (1 μm leupeptin, 2 μm pepstatin A, and 16 mm benzamidine), and the cells were lysed using glass beads (400–600 mesh, Sigma) for 40 min with 1-min pulses/1-min cooling (20 cycles). The resulting extract was spun at 3000 × g for 15 min to remove cellular debris and unbroken cells to yield a whole cell extract. Purification procedure of the zDHHC9-GCP16 enzyme complexes from the whole cell extract has been previously described for Erf2-Erf4 (15).
Immunoprecipitation and Western Blot Analysis
Immunoreactive zDHHC9-GCP16 enzyme complexes were isolated from 3 mg of whole cell extract using anti-FLAG (Sigma)-conjugated agarose beads (20 μl of 50% slurry, pre-equilibrated in 50 mm Tris-Cl, pH 8, 150 mm NaCl, 1.0% Triton X-100, 1× PIC). The extract was incubated with the beads for 2 h at 4 °C with agitation. The beads were allowed to settle (4 °C) and washed three times, allowing the beads to settle after each wash. Immunoprecipitates were removed from the beads by adding 1× protein loading buffer (nonreducing) and incubating the samples at 65 °C for 3 min. The beads were pelleted by centrifugation, 3500 × g for 10 s, and the sample was transferred to a new tube. DTT was added to a final concentration of 20 mm, and the sample was heated at 100 °C for 3 min. The proteins were separated by 12% SDS-PAGE, transferred to nitrocellulose, probed with an antibody to the c-Myc epitope (9E10) (Myc-GCP16 (Sigma)) at a 1:2000 dilution, and visualized using SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific, Waltham, MA), as per the manufacturer's instructions.
Coupled PAT Assay
The coupled PAT assay has been described previously (15). The 200-μl reaction contained 2 mm 2-oxoglutarate (α-ketoglutamic acid), 0.25 mm NAD+, 0.2 mm thiamine pyrophosphate, 2 μg of purified zDHHC9-GCP16 or zDHHC9-GCP16 mutant complex, 1 mm EDTA, 1 mm dithiothreitol, 32 milliunits of 2-oxoglutarate dehydrogenase (α-ketoglutarate dehydrogenase), in 50 mm sodium phosphate, pH 7.2. The production of NADH was monitored with a BioTek Mx fluorometer (BioTek, Winooski, VT) using 340 nm excitation/465 nm emission (29). The first 10 min of the reaction were analyzed to determine the initial rates of CoASH release. The PAT-specific activity was determined from a standard curve of NADH production using different amounts of CoASH, and the reaction was allowed to proceed to equilibrium before fluorescence was measured (340 nm excitation/465 nm emission).
Synthesis of BODIPY® C12:0-CoA
The synthesis of BODIPY® C12:0-CoA from BODIPY® C12:0 (Invitrogen) and CoASH (Sigma) was based on the procedure of Berthiaume et al. (30) and is described in Mitchell et al. (16).
Thin Layer Chromatography (TLC)
For performing the kinetics of autopalmitoylation reaction and measuring the initial rates of the consumption of substrates and the release of products, purified zDHHC9-GCP16 (or the zDHHC9-GCP16 mutant complexes) was added to 50 mm sodium phosphate, pH 7.4, with 0.5 mm DTT in a volume of 100 μl. BODIPY® C12:0-CoA was added to a final concentration of 40 μm, with constant stirring, to initiate the reaction, and aliquots were taken at various intervals ranging from 5 to 60 s. The components were separated using TLC on a Silica G60 plate using n-butyl alcohol/water/acetic acid (50:30:20) (v/v/v) as the mobile phase. Spotting the aliquots on a TLC plate effectively terminated the reaction (data not shown). The plate was allowed to dry, and the fluorescence was determined by fluorescence imaging of the plate using the Typhoon 9410 Variable Mode Imager (GE Healthcare) for BODIPY® fluorescence (488 nm excitation/532 nm emission). BODIPY® fluorescence located at the origin corresponded to BODIPY® C12:0-zDHHC9 (Rf = 0.0). BODIPY® C12:0-CoA had an approximate Rf = 0.5 and BODIPY® C12:0 an approximate Rf = 0.9. After allowing the samples to separate in the mobile phase, the plates were air-dried overnight in the dark. The amount of BODIPY® C12:0 transferred to zDHHC9-GCP16 was calculated from a standard curve of known amounts of BODIPY® C12:0-CoA.
BODIPY® C12:0 Transfer Assay
BODIPY® C12:0-CoA (40 μm final) was added to a 25-μl reaction containing ∼2.0 μg of enzyme (zDHHC9-GCP16) in 50 mm sodium phosphate buffer, pH 7.4, incubated at 30 °C for 5 min and then terminated by the addition of 5× nonreducing protein loading buffer (16). Each reaction was heated at 65 °C for 5 min and then subjected to SDS-PAGE (12%). The gel was washed three times in double distilled H2O and visualized on the Typhoon 9410 variable mode imager (GE Healthcare) for BODIPY® fluorescence (488 nm excitation/532 nm emission) to visualize transfer of the BODIPY® signal.
RESULTS
XLID-linked zDHHC9 Mutants (R148W and P150S) Are Conserved
The DHHC motif, along with six conserved cysteine residues, includes the presumed catalytic domain of all DHHC protein acyltransferases (Fig. 1A). Alignment of the Ras PAT subfamily that consists of yeast Erf2 and mammalian zDHHC9 reveals additional conserved residues that include a (R/K)PXR motif (denoted by arrows) juxtaposed to the catalytic DHHC motif (Fig. 1B). Two genetic variants of zDHHC9, R148W, and P150S, fall within the (R/K)XR motif and are associated with the Marfanoid features and psychological disorders characteristic of syndromic XLIDs. In a recent study, we screened the DHHC CRD of yeast Erf2 and found that the RPPR motif is involved in the catalytic step after the initial formation of the Erf2-palmitoyl thioester bond intermediate (15). This observation suggests that the palmitoylation defect in the XLID alleles of zDHHC9 might alter the kinetics of the autopalmitoylation step of the PAT reaction. Alternatively, the point mutations may affect the substrate transfer step. We initiated our studies by examining the autopalmitoylation reaction.
FIGURE 1.

Comparison of the consensus protein acyltransferase sequences with the amino acid sequence of zDHHC9. A, WebLogo sequence consensus representation of the 51-amino acid DHHC cysteine-rich domain using the 23 human protein acyltransferase protein sequences. The size of the amino acid letter code denotes the amount of conservation at that position; the boldface residues represent a minimum of 90% conservation. B, WebLogo sequence consensus representation of zDHHC9 and related cysteine-rich domains throughout evolution. The consensus is derived from the following: human zDHHC9, zDHHC14, and zDHHC18; S. cerevisiae Erf2; Ciona intestinalis zDHHC14 (GI: 198427890); Nematostella vectensis zDHHC9 (GI: 156377027); Drosophila melanogaster zDHHC9 (GI: 281366130); Strongylocentrotus purpuratis zDHHC14 (GI: 390334287), Amphimedon queenslandica zDHHC14 (GI: 340375495), Schistoma mansoni zDHHC9 (GI: 256084522), and Danio rerio zDHHC9 (GI: 158518002). An asterisk denotes the proposed catalytic cysteine residue of the DHHC motif (DHHC9 C169A). The arrows indicate the mutated positions of two naturally occurring isolates of human zDHHC9 (zDHHC9 R148W and zDHHC9 P150S).
zDHHC9 Mutants (R148W and P150S) Are Defective in in Vivo Ras Palmitoylation
We first tested the activity of zDHHC9 in a heterologous genetic assay. Given that Erf2 and zDHHC9 palmitoylate yeast and mammalian Ras proteins, respectively (19, 21), we tested whether zDHHC9 could functionally complement a loss of viability resulting from deletion of the yeast ERF2 gene (Fig. 2A). Expression of zDHHC9, but not the catalytically inactive zDHHA9 gene, suppressed the growth defect of a strain lacking Erf2 (erf2Δ) in the presence of GCP16, albeit to an extent that is ∼10% that of yeast ERF2. We next asked if zDHHC9 requires co-expression of GCP16, as observed previously for yeast Erf2 and Erf4 (16). The complementation of growth defect of the erf2Δ strain by zDHHC9 requires GCP16, even though ERF4 is expressed in the strain, indicating that the GCP16 and ERF4 genes are not functionally interchangeable. Interestingly, mutation of the two reported palmitoylation sites in GCP16 (C69S,C72S) failed to suppress the loss of ERF2 suggesting that palmitoylation of GCP16 is required for zDHHC9-GCP16 complex function and that palmitoylation of GCP16 occurs in S. cerevisiae. Based on the suppression results, zDHHC9 and GCP16 exhibit functionality in S. cerevisiae and are able, as a complex, to suppress the loss of the Erf2-associated PAT complex. In contrast to wild type zDHHC9-GCP16, zDHHC9 R148W-GCP16, and zDHHC9 P150S-GCP16 were not able to suppress the loss of ERF2. This was not due to a decrease in expression, as the mutant proteins were expressed to comparable levels as wild type. Furthermore, the interactions of wild type and mutant zDHHC9 proteins with GCP16 were indistinguishable (Fig. 2B). It is interesting to note that the interaction between inactive zDHHA9 and GCP16 is reduced, suggesting that palmitoylation of GCP16 by zDHHC9 may be required to increase the affinity of the interaction.
FIGURE 2.

zDHHC9-GCP16 interaction is required to suppress the loss of ERF2. A, to determine the ability of different zDHHC9 alleles and zDHHC9-GCP16 constructs to suppress the loss of ERF2 in S. cerevisiae, we performed a functional plasmid shuffle assay (22). Strain RJY1330 harboring plasmids expressing zDHHC9, zDHHC9-GCP16, zDHHC9 R148W-GCP16, zDHHC9 P150S-GCP16, zDHHC9-GCP16 (C69S,C72S), zDHHA9-GCP16, or ERF2 from the GAL1,10 promoter, were monitored for RAS2-independent growth on synthetic medium lacking tryptophan and supplemented with 5′-fluoroorotic acid (5′-FOA) to select for those strains capable of losing the URA3-linked RAS2 plasmid. Strains were initially grown in liquid synthetic medium lacking tryptophan and were spotted in 10-fold dilutions (50 × 103 initial cfu) on solid medium lacking tryptophan (top panel) and medium lacking tryptophan supplemented with 0.1% 5′-fluoroorotic acid (bottom panel). The plates were incubated for 4 days at 30 °C. B, mutations at zDHHC9 amino acid positions R148W and P150S do not interfere with the GCP16 interaction. Immunoprecipitates (IP) from whole cell extracts isolated from strain RJY1330 expressing zDHHC9-GCP16, zDHHC9, GCP16, zDHHA9-GCP16, zDHHC9 R148W-GCP16, and zDHHC9 P150S-GCP16 proteins were isolated using anti-FLAG-conjugated agarose. The immunoprecipitates were separated using SDS-PAGE (12%), transferred to nitrocellulose, and probed with antibodies to the c-Myc epitope (to identify myc-GCP16) or antibodies to the FLAG epitope (to identify zDHHC9). Western blot (WB) analysis of the whole cell extract utilizing antibodies to c-Myc was performed to demonstrate the presence of myc-GCP16 in the extracts.
Autopalmitoylation Activity of Purified zDHHC9-GCP16
We previously demonstrated that the yeast Ras DHHC protein complex, Erf2-Erf4, undergoes autopalmitoylation and binds palmitate in a one-to-one complex (15). We performed a similar active-site titration reaction experiment using three concentrations of zDHHC9-GCP16 (20, 40, and 100 pmol). Partially purified zDHHC9-GCP16 complex was combined with BODIPY® C12:0 CoA and allowed to react for the indicated times to produce the zDHHC9-BODIPY® C12:0 intermediate, which in turn can be hydrolyzed to recycle zDHHC9 enzyme and release BODIPY® C12:0 (Fig. 3A). As a control, we performed the same experiment with the active site mutant protein, zDHHA9-GCP16. The values for the control reactions were subtracted from the values obtained for the reaction using the wild type enzyme. The reaction proceeded with a rapid burst of activity and then proceeded in a linear fashion over the next 30 s (Fig. 3B). This was similar to the observation using Erf2-Erf4, which proceeds by burst kinetics initially until steady state is reached (15), in this case between 0 and 5 s. Extrapolated to time 0 and plotted versus the amount of enzyme added, a linear response is observed with a slope of 0.58. Assuming a 1:1 complex between enzyme and the palmitoyl moiety, the enzyme is ∼58% active, which is similar to what we observed with Erf2-Erf4 (15).
FIGURE 3.

Active site titration of zDHHC9-GCP16 complexes. A, schematic representation of the protein acyltransferase autopalmitoylation reaction. The reactants and products were separated using TLC as follows: 1) BODIPY® C12:0-CoA (Rf = 0.5); 2) zDHHC9-BODIPY® C12:0 intermediate (Rf = 0.0), and 3) BODIPY® C12:0 (Rf = 0.9). B, active site analysis using thin layer chromatography separation of autopalmitoylation reactants and products. Active site titration analysis measuring the amount of CoASH production was performed at room temperature (24 °C) using 20, 40, and 100 pmol of zDHHC9-GCP16 complex (or zDHHA9-GCP16 complex) and BODIPY® C12:0-CoA as the acyl donor. Time points were taken at 0, 5, 10, 15, 20, 25, 30, 45, and 60 s. The t0 point was taken prior to enzyme addition. As a control for nonspecific activity, 20, 40, and 100 pmol of the inactive zDHHA9-GCP16 enzyme were assayed, and the values were subtracted from the values determined for the wild type complexes (n = 2). C, amounts of the products were determined using a standard curve of known amounts of BODIPY® C12:0 CoA (n = 2). D, graphical representation of active site titration data. The amount of CoASH is calculated as the amount of BODIPY® C12:0 formed plus the amount of zDHHC9-BODIPY® C12:0 intermediate at the TLC origin (Rf = 0). There is a nearly 1 to 1 correlation between the amount of zDHHC9-GCP16 used and the amount of CoASH produced (slope = 0.58) (inset). The data represent the average of two (n = 2) experiments.
zDHHC9 Mutants Alter the Kinetics of the Autopalmitoylation Reaction
The steady state autopalmitoylation/hydrolysis activity was measured by monitoring CoASH generated during the transfer of palmitate to the enzyme (autopalmitoylation reaction) using a coupled reaction in which α-ketoglutarate dehydrogenase (α-KDH) uses CoASH to reduce NADH, which is monitored in a fluorometer (340 nm excitation/460 nm emission) (Fig. 4A). Because there is a 1:1 correlation between the amount of autopalmitoylation and the amount of reduced NADH produced, the autopalmitoylation step of the zDHHC9 reaction can be monitored in real time. To determine Vmax and Km values for the zDHHC9 complexes, we determined the autopalmitoylation activity of the enzymes by varying the concentration of palmitoyl-CoA (Fig. 4B). The Vmax value of the zDHHC9-GCP16 complex was 47 pmol/min/μg, with a Km of 45 μm. In the absence of GCP16, zDHHC9 alone had a higher Vmax value (190.8 pmol/min/μg of enzyme) than zDHHC9-GCP16, but the apparent affinity was lower (Km of 84 μm). Thus, the kcat/Km value for zDHHC9 lacking GCP16 was higher than that of the wild type complex (43,867 min−1 m−1).
FIGURE 4.

zDHHC9 undergoes autopalmitoylation in the absence of GCP16. A, schematic representation of the coupled in vitro autopalmitoylation reaction (see under “Experimental Procedures”). B, graphical representation of the rate of CoASH production (pmol/min/μg) over the first 10 min of the reaction versus the concentration (μm) of acyl donor (palmitoyl-CoA). The ordinate axis is broken to accommodate the activity measurement of zDHHC9 R148W. Complexes are denoted as follows: zDHHA9-GCP16 (circle with X); zDHHC9 P150S-GCP16 (open square); zDHHC9-GCP16 (open triangle); zDHHC9 R148W-GCP16 (open circle); zDHHC9 (closed triangle); zDHHC9 R148W (closed circle), and zDHHC9 P150S (closed square). Curve fit was performed using Prizm™ software.
The kinetic properties of the XLID mutant enzymes (zDHHC9 R148W-GCP16 and zDHHC9 P150S-GCP16) were similarly analyzed. The Vmax value for the generation of CoASH by the zDHHC9 R148W-GCP16 enzyme was 6.7-fold greater than wild type zDHHC9-GCP16, whereas the Vmax value for zDHHC9 P150S-GCP16 was similar to wild type. In the absence of the GCP16, the Vmax value for zDHHC9 R148W alone was 4-fold higher than wild type zDHHC9, and zDHHC9 P150S was lower (Fig. 4B). The Vmax and Km values for the autopalmitoylation reaction are summarized in Table 2. A different kinetic mechanism appears to underlie the zDHHC9 P150S mutant compared with zDHHC9 R148W. However, the ability of the mutant zDHHC9 enzymes to hydrolyze palmitoyl-CoA to generate palmitate and CoASH does not explain the defect observed in vivo.
TABLE 2.
Steady state autopalmitoylation/hydrolysis activity values for zDHHC9
NA means not applicable.
| Acyltransferase | Km | Vmax | kcat/Km |
|---|---|---|---|
| μm | pmol/min/μg | min−1 m−1 | |
| zDHHA9-GCP16 | NA | 8.2 ± 1.2 | NA |
| zDHHC9-GCP16 | 45 ± 4 | 55.2 ± 2.5 | 43.867 |
| zDHHC9 | 84 ± 6 | 199.0 ± 12 | 95,400 |
| zDHHC9 R148W-GCP16 | 32 ± 4 | 184.0 ± 11 | 230,738 |
| zDHHC9 R148W | 69 ± 7 | 1212 ± 45 | 732,748 |
| zDHHC9 P150S-GCP16 | 39 ± 6 | 60.8 ± 2.0 | 56,646 |
| zDHHC9 P150S | NA | 11.6 ± 3.0 | NA |
| Erf2-Erf4 | 43 ± 8 | 43.0 ± 3.0 | 66,667 |
XLID-linked zDHHC9 Mutants (R148W and P150S) Alter the Hydrolysis of zDHHC9-bound Palmitate
To understand the autopalmitoylation step of the palmitoyltransferase reactions, it is necessary to dissect the palmitoylation and hydrolysis reactions occurring at the level of the enzyme. To address this, we devised a TLC assay to monitor the levels of reaction intermediates (DHHC9-palmitoyl) and products (free BODIPY® C12:0 CoA and BODIPY® C12:0) versus time (Fig. 5). For this assay, zDHHC9-GCP16 enzyme complex was combined with BODIPY® C12:0 CoA, and serial aliquots were spotted over time onto silica TLC plates, which terminated the reaction. To determine the molecular mechanism regulating the formation and hydrolysis of the zDHHC9-palmitoyl thioester linkage, we used the TLC assay to compare the hydrolysis rates, CoASH production rates, and steady state amounts of zDHHC9-BODIPY® C12:0 over the first 30 s of the palmitoylation reaction (Fig. 5). Each reaction contained 50 pmol of the respective zDHHC9 enzyme and was initiated by the addition of 40 μm BODIPY® C12:0-CoA. We observed that, like the wild type zDHHC9-GCP16 enzyme complex and the yeast Erf2-Erf4 complex, the zDHHC9-GCP16 mutant complexes undergo a pre-steady state burst of activity to initiate the reaction. The steady state rates of CoASH production were linear over the assay time range with zDHHC9 R148W-GCP16 producing nearly 5-fold more CoASH (228.0 pmol/min) as wild type (44.4 pmol/min) and zDHHC9 P150S-GCP16 (50.7 pmol/min) (Fig. 5 and Table 3).
FIGURE 5.
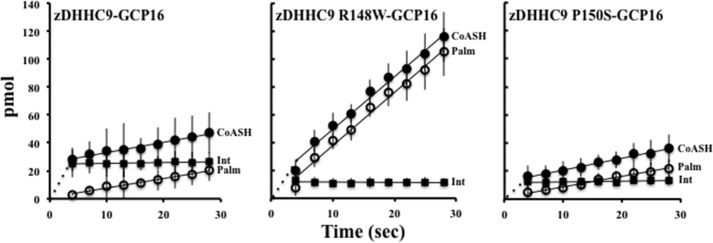
In vitro measurement of zDHHC9-BODIPY® C12:0 intermediate hydrolysis rate. The reaction highlighted in Fig. 3A was used to determine the rate of CoASH production, rate of zDHHC9-BODIPY® C12:0 hydrolysis, and the amount of zDHHC9-BODIPY® C12:0 intermediate formed. The reactions were terminated by spotting onto the TLC plate. The plates were developed using a mobile phase of n-butyl alcohol/water/acetic acid (50:30:20) (v/v/v). BODIPY® fluorescence was visualized using excitation 488-nm/emission 520-nm filters (Typhoon, GE Healthcare). The amount (in pmol) of CoASH (CoASH, closed circles), BODIPY® C12:0 (Palm, open circles), and zDHHC9-BODIPY® C12:0 intermediate (Int, closed squares) produced are graphically represented for the zDHHC9-GCP16, zDHHC9 R148W-GCP16, and zDHHC9 P150S-GCP16 complexes. The data represent three independent experiments.
TABLE 3.
Quantification of autopalmitoylation and hydrolysis activities (TLC)
| zDHHC9 protein | CoASH | CoASH t0 | Palmitate | Palmitoyl-zDHHC9 |
|---|---|---|---|---|
| pmol/min | pmol | pmol/min | pmol | |
| zDHHA9-GCP16 | 3.4 (0) ± 0.7 | 3.7 (0) ± 0.8 | 3.6 (0) ± 0.6 | 8.0 (0) ± 1.5 |
| zDHHC9-GCP16 | 47.8 (44.4) ± 5.6 | 29.3 (25.6) ± 7.6 | 44.4 (40.8) ± 12.9 | 32.7 (24.7) ± 9.6 |
| zDHHC9 R148W-GCP16 | 231.4 (228.0) ± 20.3 | 15.7 (12.0) ± 2.1 | 231.4 (228.0) ± 20.8 | 20.0 (12.0) ±3.3 |
| zDHHC9 P150S-GCP16 | 54.1 (50.7) ±7.8 | 15.7 (12.0) ± 6.6 | 48.6 (45.0) ± 5.9 | 20.0 (12.0) ± 3.0 |
The hydrolysis rate of the intermediate thioester bond was increased for zDHHC9 R148W-GCP16 (228.0 pmol/min) when compared with the wild type enzyme (40.8 pmol/min) and zDHHC9 P150S-GCP16 (45.0 pmol/min) beginning with an initial BODIPY® C12:0 production of 1.1 pmol (wild type), 1.0 pmol (zDHHC9 R148W-GCP16), and 1.0 pmol (zDHHC9 P150S-GCP16). From these data, it appears that the zDHHC9 R148W-GCP16 complex is defective at forming the BODIPY® C12:0-enzyme intermediate. However, once the intermediate is formed, the thioester is hydrolyzed at a rate that is ∼5-fold higher than that of the wild type complex. zDHHC9 P150S-GCP16, also defective at forming the first step of autopalmitoylation, hydrolyzes the thioester linkage at a rate similar to that of the wild type enzyme. This defect could reflect a change in the autopalmitoylation burst phase while not affecting the thioester hydrolysis rate. In addition, the amount of the intermediate is lower at steady state for both zDHHC9 R148W-GCP16 (12.0 pmol) and zDHHC9 P150S-GCP16 (12.0 pmol) compared with the wild type complex (24.7 pmol).
Steady State Levels of zDHHC9 Autopalmitoylation
To examine the effect of the XLID mutation on steady state palmitoylation of zDHHC9, we used a gel-based assay and BODIPY® C12:0-CoA, a fluorescent analog of palmitoyl-CoA. Reactions containing zDHHC9, zDHHC9-GCP16, zDHHA9-GCP16, zDHHC9 R148W-GCP16, and zDHHC9 P150S-GCP16 were initiated with the addition of BODIPY® C12:0-CoA and allowed to equilibrate to steady state (Fig. 6). The wild type enzyme, zDHHC9-GCP16, resulted in the expected 46-kDa band and a second 15-kDa band, likely to be a degradation fragment of zDHHC9. Substituting the catalytically inactive zDHHA9-GCP16 abolished both bands. Expression of zDHHC9 in the absence of GCP16 resulted in a much lower steady state signal compared with the zDHHC9-GCP16 complex. Likewise, zDHHC9 R148W-GCP16 and zDHHC9 P150S-GCP16 also showed lower autopalmitoylation intensities for their steady state levels. Based on these results, we conclude that a combination of an unstable enzyme complex along with the reduction in the steady state level of the intermediate diminished the performance of the mutant enzyme complexes.
FIGURE 6.
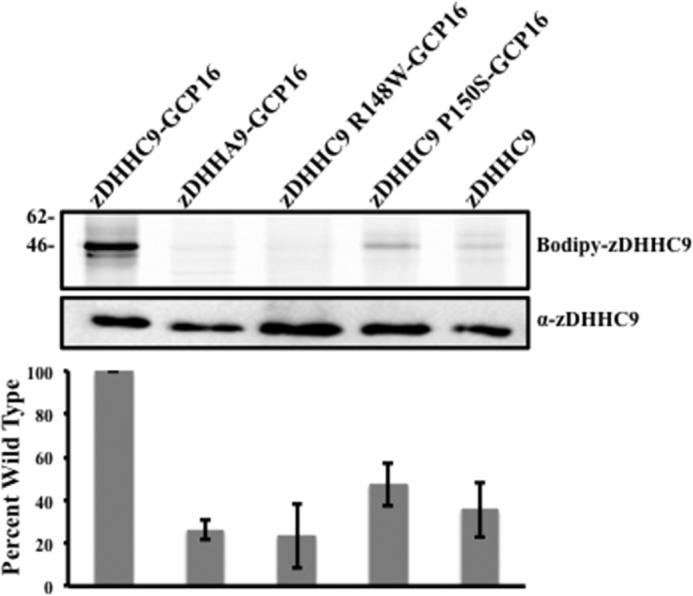
Mutations at positions Arg-148 and Pro-150 affect the autopalmitoylation activity of zDHHC9. In vitro autopalmitoylation reactions using BODIPY® C12:0-CoA as the acyl donor. Upper panel, in vitro steady state autopalmitoylation reactions were separated using SDS-PAGE under nonreducing conditions, and the fluorescence was visualized using excitation 488-nm/emission 520-nm filters (Typhoon, GE Healthcare). Lower panel, representative Western blot analysis used to quantify the amount of zDHHC9 and zDHHC9 mutants. The amount of BODIPY® C12:0 complexed to zDHHC9 was determined by using excitation 488-nm/emission 520-nm filters.
DISCUSSION
The identification of mutations in the zDHHC9 gene has implicated palmitoylation in the etiology of XLID (25). Of the four allelic variants, two were nonsense mutations within the zDHHC9 open reading frame. The nonsense mutations terminated the zDHHC9 open reading frame prior to the DHHC domain. The set of alleles also harbored missense mutations within the cysteine-rich region of the enzyme at residues Arg-148 and Pro-150. One hypothesis is that loss-of-function (or reduced function) alleles of zDHHC9 may affect the palmitoylation of Ras or another zDHHC9 substrate(s), which in turn may alter the spatial distribution and/or signaling in the synapse. Indeed, reduction in the amount of zDHHC9 by expression of the microRNA (miR-134) in somatostatin interneurons affects the palmitoylation status and localization of Ras in these cells (31). In addition, palmitoyltransferase activity levels impart a basic mechanism for regulating substrate recognition and specificity in Schizosaccharomyces pombe (32) as well as regulating Ras plasma membrane localization (33). It appears that the amount and activity of DHHC palmitoyltransferases play a direct role in the levels and types of palmitoylated substrates. However, it remains to be determined whether zDHHC9-dependent palmitoylation of Ras is directly involved in XLID or whether other, yet to be identified, zDHHC9 substrates are required.
In this study, we focused on two missense point mutations N-terminal to the DHHC motif within the cysteine-rich domain representing the first and third amino acid positions of the RPPRX2HC sequence found in 26% (6/23) of mammalian and 14% (1/7) of yeast DHHC proteins. These sequence variants were not detected in 445 normal control samples, representing 667 X chromosomes. The studies detailed above focus on the mutations within zDHHC9 at residues Arg-148 and Pro-150. A comparison of the zDHHC9 paralogs in humans reveals that Arg-148 and Pro-150 are conserved between zDHHC14, zDHHC5, zDHHC18, zDHHC8, and zDHHC19. In the remaining 18 human zDHHC proteins, only zDHHC1, zDHHC11, zDHHC12, zDHHC22, and zDHHC24 do not have an arginine or similar amino acid at position 148. Besides zDHHC9 and its paralogs, only zDHHC6, zDHHC22, and zDHHC24 have proline conserved at position 150. It would be interesting to compare the enzyme activities of zDHHC proteins with only one of two sites conserved with zDHHCs with both sites conserved (like zDHHC9) to determine the effect changes at these residues have on PAT activity. There are also variations of this sequence reflected in both mammals and yeast. The sequence RPXRX2HC is represented by 13% (3/23) of the mammalian sequences and none of the yeast sequences. KPXRX2HC is represented by 35% (8/23) of the mammalian sequences and 43% (3/7) of the yeast sequences. Interestingly, two other yeast sequences contain the KPXR sequence without the flanking XXHC sequence (Akr1 and Akr2). Finally, only one mammalian DHHC contains a KXPR sequence (DHHC6). These data are represented in WebLogo format in Fig. 1 with the consensus sequence of (R/K)PXRX2HC.
Previously, we examined the effects mutations within this domain have on the PAT activity of the S. cerevisiae Ras PAT enzyme, Erf2. We showed that Erf2 R185A (representing the fourth amino acid position of RPPRX2HC) increased the hydrolysis rate of the enzyme-palmitoyl intermediate thioester bond. In addition, the enzyme was able to transfer the palmitoyl moiety from the enzyme-palmitoyl intermediate to a Ras2 substrate. One possible molecular mechanism to explain these results is that the Erf2 R185A mutation increases the accessibility of water to the membrane-juxtaposed active site of the enzyme, therefore lowering the steady state levels of the enzyme-palmitoyl intermediate (15). Consistent with this interpretation, we were able to demonstrate palmitoyl transfer activity from the thioester linkage of the Erf2 R185A intermediate to the cysteine of the Ras substrate under conditions where the Ras substrate was near saturation. This finding implies that another interacting protein, in this case, Ras, could block water from entering the active site. We hypothesize that the relationship of the active site with the membrane is a mechanism for creating a hydrophobic environment for the transfer reaction and that the increase in thioester hydrolysis seen for Erf2 R185A was due, in part, to making the active site more accessible to water. This domain therefore appears to be important in the regulation of the enzyme's activity.
The mammalian zDHHC protein with the highest degree of homology to Erf2 is zDHHC9. In addition to sequence identity, zDHHC9 is similar to Erf2 in at least two other ways. First, as with Erf2, zDHHC9 appears to have at least four TMDs based on hydropathy plot analysis (34) with the cytosolic localized catalytic domain placed between TMDs 2 and 3. Second, like the Erf2 interaction with Erf4, zDHHC9 interacts with an accessory protein, GCP16, which was identified on the basis of homology with Erf4 (19). The presence of an accessory subunit may not be exclusive to Erf2 and zDHHC9; however, to date, these enzymes are the only DHHCs shown to be heterodimers. Despite these similarities, zDHHC9 cannot substitute for ERF2 in vivo. This is most likely because zDHHC9 cannot interact or utilize Erf4 (Fig. 2A). Likewise, GCP16 cannot be utilized by Erf2 and therefore is not interchangeable with Erf4 (data not shown). However, the zDHHC9-GCP16 PAT complex can substitute for the loss of the Erf2-Erf4 complex. Although the accessory subunits are not interchangeable, they seem to have similar functions. Previously, we demonstrated that Erf4 stabilized Erf2 and protected the active site from water (16). Here, we show that GCP16 has a similar subset of functions. Loss of GCP16 does not abolish autopalmitoylation. zDHHC9 is still able to undergo autopalmitoylation, but with a higher rate of thioester hydrolysis (Figs. 5 and 6). However, unlike Erf4, GCP16 is dually palmitoylated on residues Cys-69 and Cys-72. These residues are critical for GCP16 to properly function as the accessory subunit by changing its ability to interact through mis-localization or compartmental proximity to zDHHC9 (35). We observed a similar phenomenon. Mutating GCP16 residues Cys-69 and Cys-72 to serine abolished the suppression we observe with wild type GCP16 (in the form of the zDHHC9-GCP16 complex). One possible explanation is that GCP16 requires palmitoylation to properly interact with zDHHC9. Indeed, we are unable to detect an interaction between zDHHC9 and GCP16 (C69S,C72S) (data not shown) and observe a reduction in the amount of interacting GCP16 in complexes utilizing zDHHA9 compared with those using zDHHC9 (Fig. 2B). In addition, it seems plausible that zDHHC9 may use GCP16 as a substrate. The partial interaction of GCP16 with zDHHA9 may reflect partial “promiscuous” palmitoylation of GCP16 by a yeast zDHHC enzyme(s). More experimentation is needed to address this issue.
In summary, we have demonstrated that zDHHC9 mutations (zDHHC9 R148W and zDHHC9 P150S) do not affect the steady state levels of the enzyme, do not affect the interaction with the accessory subunit GCP16, and do affect autopalmitoylation by causing a decrease in the steady state amount of the zDHHC9-palmitoyl intermediate. Therefore, it is the lack of palmitoylation stemming from these mutations that cause the disease. Although both of the mutant complexes demonstrate a lowering of the steady state level of the zDHHC9-palmitoyl intermediate, they appear to do so by distinct mechanisms (Fig. 7). Wild type zDHHC9 complexes undergo autopalmitoylation via burst kinetics to form the initial pool of zDHHC9-palmitoyl intermediate. This intermediate can be resolved to enzyme and palmitate through intrinsic hydrolysis of the thioester linkage. Likewise, zDHHC9 R148W can also undergo autopalmitoylation driven by burst kinetics, albeit to a lesser extent. However, the intrinsic hydrolysis rate of the thioester is much greater than for the wild type enzyme, leading to a reduction in the steady state amount of the intermediate. Finally, the burst of autopalmitoylation of zDHHC9 P150S appears to be 50% that of the wild type enzyme, while having an intrinsic hydrolysis rate similar to that of the wild type. This scenario also leads to a reduction in the steady state amount of the intermediate. These results provide a clear example of how mutations that affect the autopalmitoylation step of zDHHC9 can affect palmitoyltransferase activity. Furthermore, identification of zDHHC9 substrates in the brain will allow us to examine the transfer step of palmitoylation and should lead to a better understanding of the role of palmitoylation in XLID.
FIGURE 7.

Model of autopalmitoylation reaction for mutant zDHHC9 complexes. The thickness of the arrow indicates the relative amount of “burst” or rate of hydrolysis.
Acknowledgments
We thank Luc Berthiaume for the generous gift of α-zDHHC9. We also thank Jeremy Baker and Laura Pendleton for critical reading of the manuscript and many helpful and insightful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants GM073977 and CA050211 (to R. J. D.).
- TMD
- transmembrane domain
- XLID
- X-linked intellectual disorder
- PAT
- protein acyltransferase.
REFERENCES
- 1. Zhang M. M., Tsou L. K., Charron G., Raghavan A. S., Hang H. C. (2010) Tandem fluorescence imaging of dynamic S-acylation and protein turnover. Proc. Natl. Acad. Sci. U.S.A. 107, 8627–8632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rocks O., Gerauer M., Vartak N., Koch S., Huang Z. P., Pechlivanis M., Kuhlmann J., Brunsveld L., Chandra A., Ellinger B., Waldmann H., Bastiaens P. I. (2010) The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell 141, 458–471 [DOI] [PubMed] [Google Scholar]
- 3. Fukata Y., Fukata M. (2010) Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 11, 161–175 [DOI] [PubMed] [Google Scholar]
- 4. Zhang W., Trible R. P., Samelson L. E. (1998) LAT palmitoylation: its essential role in membrane microdomain targeting and tyrosine phosphorylation during T cell activation. Immunity 9, 239–246 [DOI] [PubMed] [Google Scholar]
- 5. Joseph M., Nagaraj R. (1995) Conformations of peptides corresponding to fatty acylation sites in proteins. A circular dichroism study. J. Biol. Chem. 270, 19439–19445 [DOI] [PubMed] [Google Scholar]
- 6. Zevian S., Winterwood N. E., Stipp C. S. (2011) Structure-function analysis of tetraspanin CD151 reveals distinct requirements for tumor cell behaviors mediated by α3β1 versus α6β4 integrin. J. Biol. Chem. 286, 7496–7506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Delandre C., Penabaz T. R., Passarelli A. L., Chapes S. K., Clem R. J. (2009) Mutation of juxtamembrane cysteines in the tetraspanin CD81 affects palmitoylation and alters interaction with other proteins at the cell surface. Exp. Cell Res. 315, 1953–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou B., Liu L., Reddivari M., Zhang X. A. (2004) The palmitoylation of metastasis suppressor KAI1/CD82 is important for its motility- and invasiveness-inhibitory activity. Cancer Res. 64, 7455–7463 [DOI] [PubMed] [Google Scholar]
- 9. Scheffer K. D., Gawlitza A., Spoden G. A., Zhang X. A., Lambert C., Berditchevski F., Florin L. (2013) Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J. Virol. 87, 3435–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharma C., Rabinovitz I., Hemler M. E. (2012) Palmitoylation by DHHC3 is critical for the function, expression, and stability of integrin α6β4. Cell. Mol. Life Sci. 69, 2233–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gagnoux-Palacios L., Dans M., van't Hof W., Mariotti A., Pepe A., Meneguzzi G., Resh M. D., Giancotti F. G. (2003) Compartmentalization of integrin α6β4 signaling in lipid rafts. J. Cell Biol. 162, 1189–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang X., Kovalenko O. V., Tang W., Claas C., Stipp C. S., Hemler M. E. (2004) Palmitoylation supports assembly and function of integrin-tetraspanin complexes. J. Cell Biol. 167, 1231–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mitchell D. A., Vasudevan A., Linder M. E., Deschenes R. J. (2006) Protein palmitoylation by a family of DHHC protein S-acyltransferases. J. Lipid Res. 47, 1118–1127 [DOI] [PubMed] [Google Scholar]
- 14. Tsutsumi R., Fukata Y., Fukata M. (2008) Discovery of protein-palmitoylating enzymes. Pflugers Arch. 456, 1199–1206 [DOI] [PubMed] [Google Scholar]
- 15. Mitchell D. A., Mitchell G., Ling Y., Budde C., Deschenes R. J. (2010) Mutational analysis of Saccharomyces cerevisiae Erf2 reveals a two-step reaction mechanism for protein palmitoylation by DHHC enzymes. J. Biol. Chem. 285, 38104–38114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mitchell D. A., Hamel L. D., Ishizuka K., Mitchell G., Schaefer L. M., Deschenes R. J. (2012) The Erf4 subunit of the yeast Ras palmitoyl acyltransferase is required for stability of the Acyl-Erf2 intermediate and palmitoyl transfer to a Ras2 substrate. J. Biol. Chem. 287, 34337–34348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Young F. B., Butland S. L., Sanders S. S., Sutton L. M., Hayden M. R. (2012) Putting proteins in their place: palmitoylation in Huntington disease and other neuropsychiatric diseases. Prog. Neurobiol. 97, 220–238 [DOI] [PubMed] [Google Scholar]
- 18. Mansilla F., Birkenkamp-Demtroder K., Kruhøffer M., Sørensen F. B., Andersen C. L., Laiho P., Aaltonen L. A., Verspaget H. W., Orntoft T. F. (2007) Differential expression of DHHC9 in microsatellite stable and instable human colorectal cancer subgroups. Br. J. Cancer 96, 1896–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Swarthout J. T., Lobo S., Farh L., Croke M. R., Greentree W. K., Deschenes R. J., Linder M. E. (2005) DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H- and N-Ras. J. Biol. Chem. 280, 31141–31148 [DOI] [PubMed] [Google Scholar]
- 20. Bartels D. J., Mitchell D. A., Dong X., Deschenes R. J. (1999) Erf2, a novel gene product that affects the localization and palmitoylation of Ras2 in Saccharomyces cerevisiae. Mol. Cell. Biol. 19, 6775–6787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dong X., Mitchell D. A., Lobo S., Zhao L., Bartels D. J., Deschenes R. J. (2003) Palmitoylation and plasma membrane localization of Ras2p by a nonclassical trafficking pathway in Saccharomyces cerevisiae. Mol. Cell. Biol. 23, 6574–6584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., Stevens R. C. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raymond F. L. (2006) X linked mental retardation: a clinical guide. J. Med. Genet. 43, 193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tarpey P. S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O'Meara S., Latimer C., Dicks E., Menzies A., Stephens P., Blow M., Greenman C., Xue Y., Tyler-Smith C., Thompson D., Gray K., Andrews J., Barthorpe S., Buck G., Cole J., Dunmore R., Jones D., Maddison M., Mironenko T., Turner R., Turrell K., Varian J., West S., Widaa S., Wray P., Teague J., Butler A., Jenkinson A., Jia M., Richardson D., Shepherd R., Wooster R., Tejada M. I., Martinez F., Carvill G., Goliath R., de Brouwer A. P., van Bokhoven H., Van Esch H., Chelly J., Raynaud M., Ropers H. H., Abidi F. E., Srivastava A. K., Cox J., Luo Y., Mallya U., Moon J., Parnau J., Mohammed S., Tolmie J. L., Shoubridge C., Corbett M., Gardner A., Haan E., Rujirabanjerd S., Shaw M., Vandeleur L., Fullston T., Easton D. F., Boyle J., Partington M., Hackett A., Field M., Skinner C., Stevenson R. E., Bobrow M., Turner G., Schwartz C. E., Gecz J., Raymond F. L., Futreal P. A., Stratton M. R. (2009) A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 41, 535–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raymond F. L., Tarpey P. S., Edkins S., Tofts C., O'Meara S., Teague J., Butler A., Stevens C., Barthorpe S., Buck G., Cole J., Dicks E., Gray K., Halliday K., Hills K., Hinton J., Jones D., Menzies A., Perry J., Raine K., Shepherd R., Small A., Varian J., Widaa S., Mallya U., Moon J., Luo Y., Shaw M., Boyle J., Kerr B., Turner G., Quarrell O., Cole T., Easton D. F., Wooster R., Bobrow M., Schwartz C. E., Gecz J., Stratton M. R., Futreal P. A. (2007) Mutations in ZDHHC9, which encodes a palmitoyltransferase of NRAS and HRAS, cause X-linked mental retardation associated with a Marfanoid habitus. Am. J. Hum. Genet. 80, 982–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sherman F., Fink G. R., Hicks J. B. (1986) Laboratory Course Manual: Methods in Yeast Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 27. Ito H., Fukuda Y., Murata K., Kimura A. (1983) Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153, 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma J., Ptashne M. (1987) Deletion analysis of GAL4 defines two transcriptional activating segments. Cell 48, 847–853 [DOI] [PubMed] [Google Scholar]
- 29. Massey V. (1960) The composition of the ketoglutarate dehydrogenase complex. Biochim. Biophys. Acta 38, 447–460 [DOI] [PubMed] [Google Scholar]
- 30. Berthiaume L., Peseckis S. M., Resh M. D. (1995) Synthesis and use of iodo-fatty acid analogs. Methods Enzymol. 250, 454–466 [DOI] [PubMed] [Google Scholar]
- 31. Maak S., Boettcher D., Tetens J., Swalve H. H., Wimmers K., Thaller G. (2010) Expression of microRNAs is not related to increased expression of ZDHHC9 in hind leg muscles of splay leg piglets. Mol. Cell. Probes 24, 32–37 [DOI] [PubMed] [Google Scholar]
- 32. Zhang M. M., Wu P. Y., Kelly F. D., Nurse P., Hang H. C. (2013) Quantitative control of protein S-palmitoylation regulates meiotic entry in fission yeast. PLoS Biol. 11, e1001597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Young E., Zheng Z. Y., Wilkins A. D., Jeong H. T., Li M., Lichtarge O., Chang E. C. (2014) Regulation of Ras localization and cell transformation by evolutionarily conserved palmitoyltransferases. Mol. Cell. Biol. 34, 374–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Politis E. G., Roth A. F., Davis N. G. (2005) Transmembrane topology of the protein palmitoyl transferase Akr1. J. Biol. Chem. 280, 10156–10163 [DOI] [PubMed] [Google Scholar]
- 35. Ohta E., Misumi Y., Sohda M., Fujiwara T., Yano A., Ikehara Y. (2003) Identification and characterization of GCP16, a novel acylated Golgi protein that interacts with GCP170. J. Biol. Chem. 278, 51957–51967 [DOI] [PubMed] [Google Scholar]