

Background: CARF is a regulator of p53 pathway, cellular senescence, and apoptosis.
Results: Level of CARF expression decides the cell proliferation fate toward either senescence or tumorigenesis through regulation of DNA damage response and checkpoint signaling.
Conclusion: CARF regulates DNA damage response and cell proliferation in a dose-dependent manner.
Significance: CARF is an important target for cancer therapy.
Keywords: Cell Proliferation, Cellular Regulation, Cellular Senescence, DNA Damage Response, p53, CARF, Malignant
Abstract
Collaborator of ARF (CARF) has been shown to directly bind to and regulate p53, a central protein that controls tumor suppression via cellular senescence and apoptosis. However, the cellular functions of CARF and the mechanisms governing its effect on senescence, apoptosis, or proliferation are still unknown. Our previous studies have shown that (i) CARF is up-regulated during replicative and stress-induced senescence, and its exogenous overexpression caused senescence-like growth arrest of cells, and (ii) suppression of CARF induces aneuploidy, DNA damage, and mitotic catastrophe, resulting in apoptosis via the ATR/CHK1 pathway. In the present study, we dissected the cellular role of CARF by investigating the molecular pathways triggered by its overexpression in vitro and in vivo. We found that the dosage of CARF is a critical factor in determining the proliferation potential of cancer cells. Most surprisingly, although a moderate level of CARF overexpression induced senescence, a very high level of CARF resulted in increased cell proliferation. We demonstrate that the level of CARF is crucial for DNA damage and checkpoint response of cells through ATM/CHK1/CHK2, p53, and ERK pathways that in turn determine the proliferative fate of cancer cells toward growth arrest or proproliferative and malignant phenotypes. To the best of our knowledge, this is the first report that demonstrates the capability of a fundamental protein, CARF, in controlling cell proliferation in two opposite directions and hence may play a key role in tumor biology and cancer therapeutics.
Introduction
Although DNA integrity and genomic stability are the basis for organismal survival and determination of proliferative fate of cells in terms of senescence, apoptosis, or continued cell proliferation, they are constantly endangered by environmental agents, as well as endogenous metabolic processes, such as reactive species, and errors of cellular processes. Thus, the DNA damage response (DDR)7 pathways, which include DNA repair, cell cycle regulation, and cell fate determination, are of the utmost importance to prevent genomic instability and chromosomal abnormalities that may lead to genetic disorders, cancer, and aging (1).
The two major arms of DDR are epitomized by the two kinases, ATM (ataxia telangiectasia-mutated protein) and ATR (ataxia telangiectasia and Rad3-related protein), which are generally considered to sense double-stranded breaks and replication stress-related problems, respectively (2). These sensors provide extended time for adequate DNA repair by activating their downstream effectors, checkpoint kinase (CHK)1 and CHK2, which induce cell cycle checkpoints before entering the critical cell cycle stages. Recent developments have enlightened the roles of ATM and ATR and showed that they modulate a larger number of downstream effectors and coordinate a much wider variety of cellular activities, including DNA replication and repair, transcription, metabolic signaling, RNA splicing, and telomere maintenance, than initially anticipated, as reviewed earlier (3, 4).
The most well recognized molecule involved in DDR signaling and cell fate decision is p53, which acts in itself as a molecular sensor and regulator of cellular stress, senescence, and apoptosis (5). Notably, the ATM axis senses a DNA abnormality, mostly in the form of double-stranded breaks, which triggers CHK1/CHK2 phosphorylation leading to a cascade of events that involve p53 and p21WAF1 activation and ultimately cell cycle arrest while also phosphorylating γH2AX and inducing DNA repair processes (6). Recent studies have also found that the ERKs, which are components of the major survival pathway, are activated upon DNA damage through co-regulation with p53 to regulate apoptosis, cell cycle arrest, and proliferation (7).
Collaborator of ARF (CARF) was first discovered as a binding partner of ARF and has since been shown to have both ARF-dependent and -independent functions that converge to regulate the p53 pathway; CARF has been shown to directly bind to and regulate p53 to induce cellular senescence and apoptosis (8, 9). However, the cellular functions of CARF and the molecular mechanisms governing its effect on DNA damage, senescence, apoptosis, and proliferation remain elusive. We had previously shown that the overexpression of CARF in both normal and tumor cells (with or without ARF) results in up-regulation of p53 and its downstream effectors, such as p21WAF1, subsequently leading to senescence (10). It has also been found that CARF functions in a negative feedback loop involving p53 and HDM2, where it binds to and activates p53, and both get degraded by HDM2, whereas CARF transcriptionally represses HDM2. Recently, CARF has also been demonstrated to be regulated independently of p53, whereby suppression of CARF leads to aneuploidy, DNA damage, mitotic catastrophe, and apoptosis through the ATR/CHK1 pathway (11). Understanding the molecular regulations of these pathways is extremely important to address unsolved questions not only in cancer but also in aging studies. These findings support the role of CARF in tumor suppression through determination of cell fate between senescence and proliferation. In the present study, using in vitro and in vivo CARF overexpression systems, we further verify that CARF is fundamental to the proliferative fate of cells. Whereas its moderate overexpression induces senescence, a very high level, or superexpression, results in increased cell proliferation. We demonstrate that a critical level of CARF expression is crucial for genomic integrity. Deregulation of CARF leads to a loss of DNA damage response through the ATM/CHK1/CHK2, p53, and ERK pathways, causing either mitotic catastrophe and apoptosis (in case of CARF suppression) (11) or enhanced proliferation and malignant transformation (in the case of CARF superexpression) as demonstrated in this study. Because of such major control on the determination of cell proliferative fates from growth arrest/senescence to proproliferation and malignant transformation, CARF is proposed as a crucial player in carcinogenesis and its therapeutics.
EXPERIMENTAL PROCEDURES
Cell Culture
All cell lines were obtained from the America Type Culture Collection unless otherwise specified. The ATM-deficient cells FT/pEBS7 (hereby referred as FT vector or FTV) were derived from AT22IJE-T, an immortalized fibroblast line, and generously provided by Dr. Kum Kum Khanna (Queensland Institute of Medical Research, Herston, Australia) and AT5-BIVA cells were obtained from the Japanese Collection of Research Bioresources Cell Bank. All cell lines were cultured in DMEM supplemented with 5–10% FBS and 1% penicillin/streptomycin mix at 37 °C with 95% O2 and 5% CO2 in a humidified chamber. Cell culture reagents were purchased from Invitrogen, and all other chemical reagents were purchased from Sigma-Aldrich unless otherwise specified.
Retrovirus Infection
Exogenous expression of CARF was carried out using a retroviral carrier of GFP-tagged CARF, cloned into a pCX4neo vector (provided by Dr. Tsuyoshi Akagi, Osaka, Japan) as previously described (11). For the production of retroviruses, the Plat-E (Platinum-E) ecotropic murine leukemia virus packaging cell line (107 cells in 10-cm plates) was transfected with equal (6 μg) amounts of pVPack-GP (gag and pol), pVPack-VSV-G (vesicular stomatitis virus G) (both from Agilent, La Jolla, CA), and either pCX4neo empty vector or pCX4neo/GFP-CARF using FuGENE 6 (Roche), following the manufacturers' protocol. Fresh medium was replaced 24 h after transfection, and culture supernatant was collected at 60–72 h, passed through 0.45-μm filter, and used as viral stock for infection. The viral stock was diluted (1/1000–1/10), or undiluted stock was supplemented with 8 μg/ml polybrene and used to infect cells for the generation of CARF overexpressing (COE) and superexpressing (CSE) cell lines, respectively. After 18–24 h, fresh medium containing G418 (500–900 μg/ml) was added to select for positively infected cells to obtain stable GFP-CARF-expressing cell lines. To rule out the effect of retrovirus vector, per se, GFP-overexpressing and -superexpressing derivatives of U2OS were generated using the same retrovirus vector. Control cells for each experiment were infected with a concentrated pCX4neo vector virus dose corresponding to the CSE cells. The cells were expanded and maintained in 200 μg/ml G418-supplemented medium for 1–2 weeks. The cells were examined for expression of recombinant CARF by Western blotting as described below.
Plasmid Transfection
The cDNA encoding full-length GFP-CHK1 was a gift from Dr. Aziz Sancar from the University of North Carolina School of Medicine (Addgene plasmid no. 22888), GFP-CHK2 was a generous gift from Dr. Yasuhiro Minami from the University of Kobe (Japan), and GFP-ERK1 (Addgene plasmid no. 14747) was generously provided by Dr. Rony Seger from the Weizmann Institute of Science (Rehouot, Israel). The vectors, including a GFP control plasmid, were transiently transfected into control or CARF-expressing cells using FuGENE 6 (Roche) following the manufacturer's protocol. Briefly, the cells were plated into a 6-well plate, and 2–4 μg of each vector was transfected into cells at a ratio of 6:1 of transfection reagent to DNA in antibiotic-free medium with 10% FBS. After 48–72 h, the transfected cells were subjected to the various assays.
In parallel, CARF-expressing cells were also transfected with 50–100 nm siRNA against CHK1 and CHK2 (Cell Signaling Technology, Danvers, MA) using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. All the experiments were performed in triplicate and at least three times. For ERK inhibition, cells with exogenous CARF were treated with 20 μm of U0126, a MEK inhibitor (Cell Signaling), for 48 h.
Western Blot Analysis
Protein samples (10–20 μg) were harvested using Nonidet P-40 lysis buffer (20 mm Tris, 100 mm EDTA, 100 mm EGTA, 100 μm PMSF, 150 mm NaCl, and 1% Nonidet P-40), radioimmune precipitation assay buffer (Thermo Scientific, Waltham, MA) or 5× loading buffer, separated in SDS-polyacrylamide gels, and electroblotted onto PVDF membranes (Millipore, Billerica, MA) using a semidry transfer blotter (ATTO, Tokyo, Japan). Immunoblotting was performed with antibodies against ATM, p53, p21WAF1, total ERK1/2 and GFP (Santa Cruz, CA), and total CHK1, phospho-CHK1 (Ser317/345), total CHK2, phospho-CHK2 (Thr68), and phospho-ERK1/2 (Cell Signaling). The monoclonal anti-actin (Millipore), anti-phospho-ATM (Ser1981; Genetex, Irvine, CA), anti-γH2AX (Millipore), and polyclonal anti-CARF (8) antibodies were also used. The immunoblots were incubated with HRP-conjugated goat anti-mouse or anti-rabbit antibodies (Cell Signaling and Santa Cruz) and detected using ECL Prime substrate (Amersham Biosciences/GE Healthcare). Where applicable, densitometry quantitation of at least three immunoblots was carried out using ImageJ software (National Institute of Health), and the data are shown as relative units where control bands were given a value of 1, and CARF-overexpressing bands were calculated as fold change over control following normalization with actin. All the experiments were performed in triplicate and at least three times.
Cell Proliferation Assay
Five thousand cells were seeded into 96-well microtiter plates (E-plate 16; Roche). Cell morphology, attachment, spreading, and proliferation were monitored every 30 min for 48–72 h using an electrical impedance-based system, Real-Time Cell Analyzer DP (Roche), which was placed in a humidified incubator maintained at 37 °C with 95% O2 and 5% CO2. Cell sensor impedance is expressed as an arbitrary unit, referred to as Cell Index, which is defined at each time point as (Rn − Rb)/15, where Rn is the cell electrode impedance of a well that contains cells, and Rb is the background impedance of a vehicle-only well. Data analysis was carried out using Real-Time Cell Analyzer software 1.2 supplied with the instrument.
Clonogenic Survival Assays
To estimate the rate of cell proliferation and colony-forming efficiency for each cell line, 100 cells from each genotype were plated per well into 6-well plates or 500 cells into 10-cm dishes and allowed to grow for 6–12 days. Medium was replaced every 3–4 days. Termination of the experiment was performed by fixing the cells with equal amounts of ice-cold methanol and acetone for 10 min and staining with 0.1% crystal violet, followed by manual count of visible colonies.
Immunofluorescence
Cells were grown on glass coverslips or trypsinized after treatments and cytospun onto coated glass slides using the Cytospin 4 instrument (Thermo Scientific) and then fixed with equal ratio of ice-cold methanol and acetone for 10 min and permeabilized with PBS-Triton-X-100 (0.1%) for 10 min at room temperature, followed by a blocking step with 2% BSA for 10 min. The coverslips were incubated with antibodies against heterochromatin protein 1γ (HP1γ) (Cell Signaling Technology), p53, phospho-ATM (Ser1981), or γH2AX (antibodies as used for Western blots) at room temperature for 1 h or 4 °C overnight, probed with Alexa Fluor-conjugated secondary antibodies (Molecular Probes, Invitrogen), and finally counterstained with Hoechst 33258 (Sigma-Aldrich). The slides were viewed using a Zeiss Axioplan 2 microscope, and images were taken using a Zeiss AxioCam HRc camera.
Wound Healing Assay
A scratch wound was made in a monolayer of cells by passing a 200-μl pipette tip straight through the cell layer. Cells were washed with PBS to remove cell debris and fed with fresh medium. The time of wound creation was designated as 0 h. Cells were allowed to proliferate and migrate into the wound for at least 24 h, the process of which was recorded under a phase contrast microscope with a 10× phase objective lens. The images were analyzed with the Wimasis WimScratch software for quantitation of the scratch area.
Cell Invasion Assay
In vitro invasion assay was carried out by seeding 40,000 cells into the upper chambers of specially designed 16-well CIM-plates (Roche) with 8-μm pores, which are similar to conventional Transwells but with microelectrodes located on the underside of the membrane of the upper chamber. The upper wells were coated on the surface with 1/10 dilution of Matrigel (BD Biosciences). The number of cells that had spontaneously migrated (no chemoattractant was added to the lower chamber) from the upper chamber through the Matrigel and microporous membrane onto the underside of the membrane in the lower chamber was measured by the microelectrodes every 10 min (which avoids the effect of cell size and proliferation rate, respectively) up to 50 h using the Real-Time Cell Analyzer DP instrument (Roche) as described above. Data analysis was carried out using Real-Time Cell Analyzer software 1.2 supplied with the instrument.
In Vivo Study
Five-week-old nude mice were obtained from Charles River. Parental and CARF derivatives of HeLa cells (∼1 × 106) were injected subcutaneously into the abdomen. The mice were monitored for presence or absence of tumors for 2–3 weeks.
RT-PCR
RNA was extracted with Qiagen RNeasy kit, and cDNA was synthesized from 2 μg of RNA using the Thermoscript reverse transcriptase (Qiagen) following the manufacturer's protocol. Subsequently, PCR was performed using equal amounts of synthesized cDNA, and the primers sets are described in Table 1 with the Phusion high fidelity DNA polymerase system (Thermo Scientific). The PCR products were then resolved on a 1% agarose gel with ethidium bromide for visualization.
TABLE 1.
RT-PCR primer set sequences
| Sequence | |
|---|---|
| ATM | |
| Sense | 5′-TAGGGTGTACTAGTGGAGGA-3′ |
| Antisense | 5′-GTAGTAACTATTAGTTTCGTGCA-3′ |
| CHK1 | |
| Sense | 5′-GATGCAGACAAATCTTATCAATGC-3′ |
| Antisense | 5′-AGTTTGCAGGACAGGATAATCTTC-3′ |
| CHK2 | |
| Sense | 5′-TACAGGCGTGAGCCACTGTGCTGGG-3′ |
| Antisense | 5′-GTAGACATGATTTCTCCTGCAGAAC-3′ |
| p53 | |
| Sense | 5′-CTGCCCTCAACAAGATGTTTTG-3′ |
| Antisense | 5′-CTATCTGAGCAGCGCTCATGG-3′ |
| p21WAF1 | |
| Sense | 5′-ATGAAATTCACCCCCTTTCC-3′ |
| Antisense | 5′-CCCTAGGCTGTGCTCACTTC-3′ |
| GAPDH | |
| Sense | 5′-ACCTGACCTGCCGTCTAGAA-3′ |
| Antisense | 5′-TCCACCACCCTGTTGCTGTA-3′ |
MTT Assay
Five thousand cells of each genotype were seeded into 96-well microtiter plates following treatments, allowed to grow for 24–36 h before addition of the MTT reagent (Roche), and incubated for 2 h. Measurement was done with a plate reader.
Statistical Analysis
The data are reported as arithmetic means ± S.D. Statistical analyses were carried out using Student's t test or nonparametric Mann-Whitney U test, whichever was applicable, performed with the Prism software. Statistical significance was defined as p value ≤ 0.05.
RESULTS
COE and CSE Cells Exhibit Contrasting Cell Proliferation Phenotypes
To generate stable cells lines with varying levels of CARF expression, HeLa, U2OS, and HT1080 cells were infected with 1/1000 diluted or undiluted retrovirus carrying GFP-tagged CARF as described under “Experimental Procedures.” The expression level of CARF was detected by Western blotting (Fig. 1A). Morphology of control and CARF-expressing cells was captured, and differences could be observed between the control, COE (infected with 1/1000 diluted virus), and CSE (infected with undiluted virus) cells (Fig. 1B). Whereas COE cells appeared flat and spread out, CSE cells had more extensions and were less adherent. In contrast, the morphology of control GFP-overexpressing (GOE) and -superexpressing (GSE) derivatives of U2OS did not show any change (data not shown).
FIGURE 1.

Dose-dependent expression of CARF exerts opposite effects on cell growth. A, varying amounts of GFP-tagged and endogenous CARF levels in HeLa cells as detected by Western blotting is shown. Actin was used as loading control, and experiments were performed at least three times. B, morphology difference in control, COE, and CSE derivatives of HeLa cells. C, increased cell proliferation in CSE (blue line) and lower cell growth rate in COE (green line) as compared with the control (red line) cells. D, clonogenic survival assay showing more colonies in CSE and less in COE cells as compared with control cells. The data are presented as average numbers of colonies (from at least three experiments) ± S.D. *, p < 0.05. E and F, growth rate (E) and viability (F) of control, GOE, and GSE cells, respectively. G, senescence-related HP1γ (red) staining showing heterochromatin foci localization in COE cells. H, wound healing assay at 0, 18, and 36 h after scratch. I, images analyzed by the Wimasis WimScratch software quantitation. J, real time motility assay showing its increase in CSE cells and decrease in COE cells as compared with controls cells. K, control and CSE HeLa cells were subcutaneously injected into nude mice (n = 6/group). Two weeks later, only CSE mice showed big tumors. Details of the mice and experimental tumors are shown below the figure. Con, control.
The COE and CSE derivative cells were maintained in G418-supplemented media and confirmed the respective level of stable GFP-CARF expression by immunoblotting. Transduced GFP-CARF protein was high in CSE cells as compared with the COE cells at all time points analyzed (data not shown). The COE and CSE cells were subjected to quantitative growth and motility assays. Whereas growth assay by Real-Time Cell Analyzer revealed an increase in cell proliferation in CSE cells, the COE cells showed slower growth as compared with controls (Fig. 1C). This was also reflected in the clonogenic survival assay in which CSE cells produced statistically more colonies and COE had decreased colony-forming capacity, indicating an opposing growth effect with different levels of exogenous CARF (Fig. 1D). GOE and GSE control derivatives of U2OS when examined for growth characteristics showed comparable growth rate and viability (Fig. 1, E and F).
To further characterize the COE and CSE cells, we performed immunostaining of HP1γ, a senescence marker protein (12). As shown in Fig. 1G, COE cells exhibited focal heterochromatin localization of HP1γ as compared with the pan nuclear staining in control and CSE cells. Furthermore, the scratch wound healing assay (Fig. 1, H and I), in which phase contrast images were taken at 0 (just after scratch was made), 18, and 36 h after scratch, and quantitative motility assay by Real-Time Cell Analyzer in both HeLa and U2OS cells (Fig. 1J and data not shown) revealed higher motility of CSE cells and lower motility of COE derivatives as compared with the parent control cells. In in vivo nude mice tumor formation assays, we found that only the CSE cells formed big tumors in 3–4 weeks (Fig. 1K). Altogether, these data suggested that CARF might exert a dose-dependent regulation on proliferative capacity of cells. Whereas its overexpression caused senescence and decreased motile capacity of cells, the superexpressing derivatives experience an increase in cell proliferation and motility. Similar phenotypes were observed in three different cell lines, including HeLa, U2OS, and HT1080 (Fig. 1 and data not shown), demonstrating that the CARF-mediated dose-dependent regulation of cell growth and arrest is not specific to one cell line.
The p53 Pathway Is Activated in COE but Not in CSE Cells
Our previous study had shown that the overexpression of CARF induced senescence and was mediated by p53 activation (9). Considering the role of CARF as a positive regulator of p53, we first did immunoblotting to determine the expression of p53 and its downstream effector, p21WAF1 in U2OS cells (wild type and functional p53). We found that the p53 and p21WAF1 increased in COE but decreased in CSE cells (Fig. 2A), whereas HDM2 was inversely correlated (data not shown) and as reported previously (13). Furthermore, in HeLa cells, CARF overexpression was able to stabilize p53, despite its degradation by human papillomavirus (HPV) virus present in these cells (data not shown). Activation of p53 in HeLa-COE cells was also confirmed by up-regulation of p21WAF1. HeLa-CSE derivatives showed a decrease in p53 as compared with the control cells (quantitation from three independent experiments on two cell lines is shown in Fig. 2A. Immunostaining for p53 demonstrated its up-regulation and nuclear accumulation in COE cells only; CSE cells showed a decrease in comparison with the control (Fig. 2B, second row, and data not shown). GOE and GSE control derivatives of U2OS showed no difference in p53 expression examined by immunostaining (Fig. 2C) and Western blotting (data not shown). To characterize the differences in motility of COE and CSE cells, we examined the level of expression of matrix metalloproteinases (MMP-2, MMP-3, and MMP-9), established markers of cell migration (14). As shown in Fig. 2D (HeLa) and in U2OS (data not shown), we found a high level of expression of MMP-3 and MMP-9 in CSE derivatives. Because similar results were obtained in both U2OS and HeLa cells, we henceforth show only the results from HeLa cells.
FIGURE 2.

Preferential activation of p53 and DDR in COE and of MMPs in CSE cells. A, representative blots showing expression of GFP-tagged CARF, p53, and p21WAF1 in U2OS cells, indicating an increase and decrease of the p53 pathway in COE and CSE cells, respectively. Densitometric quantitation of bands from at least three experiments in U2OS and HeLa cells are shown with p53 and p21WAF1 expression levels as fold change over control (set as 1), and the data are shown as means ± standard deviation. B, HeLa cells with GFP-tagged CARF (green signal) were immunostained with anti-p53 (red) antibody. An increase in nuclear accumulation of p53 is observed only in COE cells (arrows). Immunostaining of pATM and γH2AX showed foci formation (middle panels, arrows) in COE, but not CSE cells (right panels). C, immunostaining showing p53 expression in control and GOE and GSE U20S cells. D, increased expression of MMPs in CSE cells, but not in COE cells. Densitometric quantitation of bands from three experiments, where MMP expression is shown as fold change over control. E, Western blot analysis showing activation of ATM (phosphorylated at serine 1981, pATM), as well as its downstream effectors, phosphorylated CHK1 (pCHK1), phosphorylated CHK2 (pCHK2), and BRCA1 in COE cells. CSE cells showing down-regulation of pCHK1 and up-regulation of pCHK2 and BRCA1. F, quantitation of signals from three independent experiments is shown. G and H, RT-PCR analysis for p53, p21WAF1, ATM, CHK1, and CHK2 showing increase in p53 and p21WAF1 transcripts in COE cells. ATM and CHK1 transcript levels remain unchanged in COE and CSE cells. CHK2 transcript increases in CSE cells. Actin and GAPDH were used as loading controls for the immunoblots and RT-PCR, respectively. All of the experiments were performed at least three times. Densitometric quantitation is from at least three experiments. *, p < 0.05; **, p < 0.01. Con, control.
DDR Pathway Is Activated Only in COE Cells
Based on the above findings that there is preferential up-regulation of p53 in COE cells and because p53 has a well established role as a major guardian of the genome, we investigated the DDR pathway in COE and CSE cells. The level of expression of ATM and its activated phosphorylated form (pATM) were examined and quantitated by Western blotting in several batches of COE and CSE cells generated in independent experiments. We found that similar to the increase in nuclear p53 in COE cells, pATM (phosphorylated at serine 1981, an established marker of initiation of DNA damage response) showed increase and distinct foci formation in these cells (Fig. 2B). Consistent with these results, γH2AX foci, an early response DDR component, were observed only in COE cells (Fig. 2B, bottom middle panel, arrows). Analyses of ATM, CHK1, and CHK2 proteins and their activated phosphorylated forms by Western blotting revealed that the levels of ATM (p = 0.05) and pATM (p = 0.008) increased in COE cells, and correspondingly, the expression and activation of CHK1 (CHK1, p = 0.04; pCHK1, p = 0.02; respectively) was up-regulated as well (Fig. 2E, lane 2). CSE cells, on the other hand, showed moderate but statistically significant decrease in the level of activated CHK1 (pCHK1; *, p = 0.007; Fig. 2E, lane 3). Quantitation from several independent experiments is shown in Fig. 2F.
Analysis of CHK2, a second downstream effector of ATM for DDR, revealed that its activated form, pCHK2, was increased not only in COE, but also in CSE cells. BRCA1, a downstream target of CHK2, was also found to be increased in both COE and CSE cells, suggesting that CHK2 is functionally triggered in both the cell types. However, pCHK2 increase in CSE was relatively low compared with COE cells. (Fig. 2, E and F), suggesting a differential regulation of DDR by serially increasing levels of CARF expression in COE and CSE cells. Taken together, these biochemical data suggested that (i) the induction of senescence in COE cells may be a DNA damage response mediated by activation of the ATM-CHK1-CHK-2 axis and (ii) differential regulation of CHK1 and CHK2 in CSE cells may enable these cells to dampen DDR resulting in escape from induction of senescence and acquisition of proproliferation fate. RT-PCR was performed to determine whether the expression of these proteins was altered at the transcript level in COE and CSE cells. As shown in Fig. 2G, p53 and p21WAF1 were up-regulated in COE cells as also reported earlier. ATM and CHK1 transcripts did not show any difference in their level of expression in control, COE, and CSE cells. CHK2, on the other hand, showed moderate increase in CSE cells (Fig. 2H). These data suggested that CARF regulates some, but not all, of these proteins at the transcriptional level. The proteins including ATM and CHK1 may be regulated at the post-translational level. Based on the above data, we next set out to determine the involvement of ATM, CHK1, and CHK2 in CARF-induced growth arrest and proproliferation phenotypes in COE and CSE cells, respectively.
ATM Deficiency Reverts the Growth Arrest in COE Cells, but CSE Cells Remain Unaffected
In view of the above findings that the overexpression of CARF may induce DDR by activation of the ATM-CHK1-CHK2 axis, we investigated the involvement of ATM using ATM-deficient cell lines (AT5-BIVA and FTV). COE derivatives of these cells, when examined for their growth characteristics (Fig. 3, A and B), did not show growth arrest. Their colony-forming capacity matched that of the control in the case of FTV cells and was even higher in AT5-BIVA cells, demonstrating that CARF overexpression did not induce senescence (Fig. 3, A–C) in these cells. On the other hand, a lack of ATM in CSE cells did not change cell proliferative phenotype. These cells still exhibited more colonies than the control cells. Based on these data that ATM-deficient COE cells did not show growth arrest and CSE cells showed increase in proliferation, it was concluded that whereas ATM is crucial to induce senescence in COE cells, it has no effect on the increase in proliferation in CSE cells.
FIGURE 3.
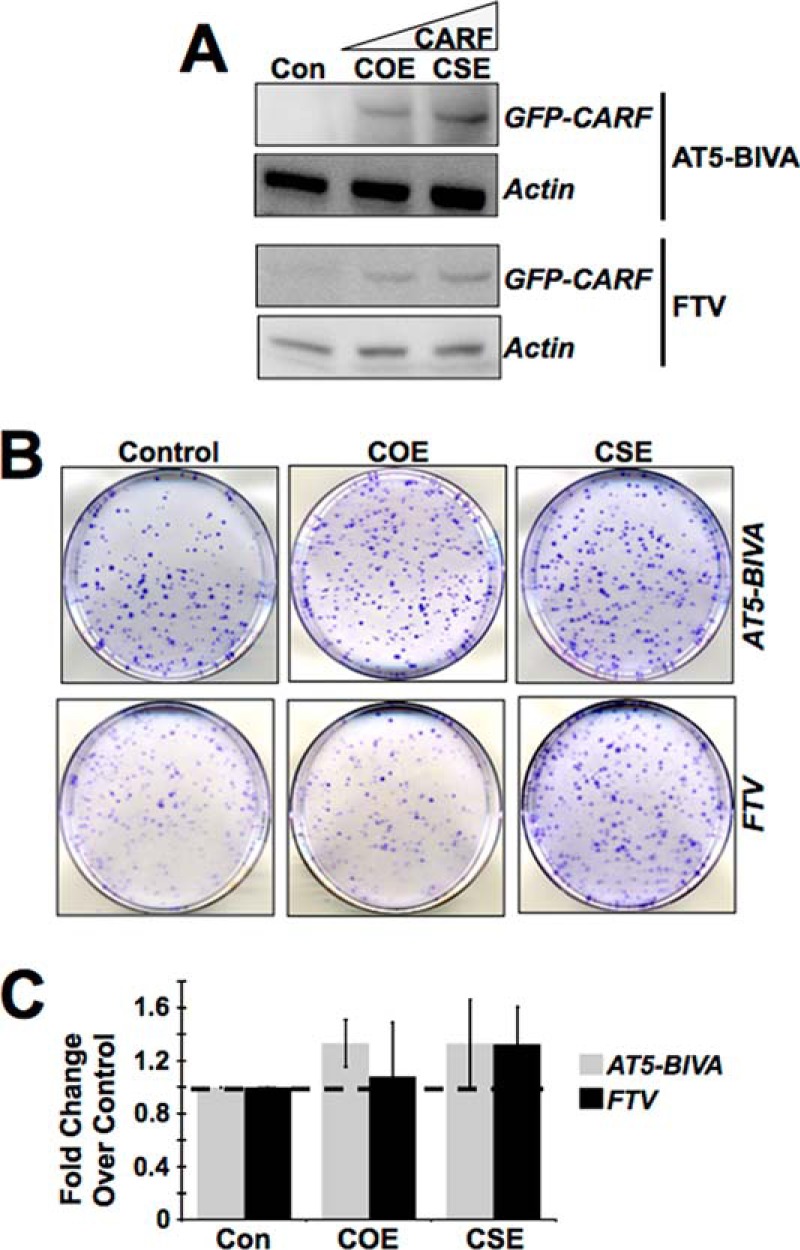
ATM is involved in CARF overexpression-induced growth arrest. A, in the ATM-null AT5-BIVA and FTV cell lines with moderate CARF overexpression, there is a reversion in the senescent phenotype, whereas the growth in CSE cells remains unaffected, as assessed by colony forming assay. B and C, colony number in ATM deficient COE and CSE cells are shown as fold change over control, which is set as 1. The results are from three independent experiments and shown as the means ± standard deviation. Con, control.
The Balance of CHK1 and CHK2 Activation Is Crucial to Induce Senescence in COE Cells and Proproliferation in CSE Cells
As shown in Fig. 2E, pCHK1 increased in COE cells. To address the contribution of pCHK1 in CARF overexpression-induced growth arrest, we knocked down CHK1 in COE cells using siRNA and found that these cells when compromised for CHK1 did not show growth arrest. Instead, it led to an increase in cell proliferation (1.97 ± 0.54-fold increase in CHK1 compromised COE cells compared with control siRNA-transfected COE cells; p = 0.0002) (Fig. 4A). The data were further confirmed by colony forming assay (Fig. 4B). Biochemical analysis of these cells revealed decreases in p53 and p21WAF1 (Fig. 4C, compare lanes 2), in accordance with their increased proliferative phenotype. As expected, the levels of ATM (upstream regulator of CHK1) did not change in control and CHK1-compromised cells (Fig. 4C).
FIGURE 4.

CHK1 is involved in growth arrest of COE cells, whereas it promotes proliferation of CSE cells. A, CHK1 knockdown by siRNA reverts the growth arrest in COE cells (2.48 ± 0.08-fold change in CHK1 siRNA-treated cells compared with control siRNA cells; p = 0.0002). B, colony forming assay showing CHK1 silencing produces more colonies than control siRNA treatment in COE cells. C, CHK1-compromised COE cells with increased proliferation show decreases in p53 and its downstream regulator p21WAF1, whereas ATM remains unchanged. D and E, decrease in cell growth in CHK1-overexpressing CSE cells as seen by colony-forming (D) and MTT assays (E) (0.78 ± 0.04-fold change in CHK1-CSE compared with control GFP cells; p = 0.0006). F, CHK1 overexpressing CSE cells with decreased proliferation show corresponding increases in p53 and p21WAF1. The cell viability graphs are shown as fold change over vehicle-transfected COE or CSE cells, which is set as 1, and the data are shown as means ± standard deviation. Actin was used as loading control for immunoblots, and each experiment was performed at least three times. *, average number of colonies ± S.D. from three independent experiments. Con, control.
To investigate the contribution of decreased level of activated CHK1 in proproliferation of CSE cells, we next restored its expression by introducing GFP-tagged CHK1 plasmid into these cells. Cell viability and clonogenic survival assays revealed that the proliferative phenotype of CSE cells was abolished by CHK1 overexpression. CHK1-overexpressing CSE cells produced fewer colonies (Fig. 4D), and their cell growth was substantially reduced (0.78 ± 0.04-fold change in CHK1-overexpressing CSE cells compared with control GFP cells; p = 0.0006) (Fig. 4E). Furthermore, the decrease in cell proliferation correlated with an increase in the expression level of p53 and p21WAF1 (Fig. 4F). The data suggested that the proproliferation caused by CARF superexpression was mediated by decrease in CHK1, and hence the exogenous overexpression was able to reinstate a checkpoint and growth arrest.
We next investigated the role of CHK2 protein in the phenotypes of COE and CSE cells. As shown in Fig. 2 (E and F), in contrast to CHK1, CHK2 was up-regulated both in COE and CSE cells. Thus, to evaluate the involvement of CHK2 increase in CARF-induced growth arrest in COE and proproliferation in CSE cells, we knocked down CHK2 by siRNA. Similar to CHK1 knockdown, compromise of CHK2 in COE cells caused an increase in proliferation (Fig. 5A, left panel; 4.01 ± 0.94-fold change in CHK2 siRNA-treated cells over control siRNA; p < 0.0001), suggesting that CHK2 up-regulation in COE cells was essential for proliferation inhibition/growth arrest. The data were also verified by colony-forming capacity of control and CHK2-compromised COE cells (Fig. 5B, left panel). Furthermore, consistent with the reversal of growth arrest by CHK2 silencing in COE cells, the levels of p53 and p21WAF1 also decreased (Fig. 5C, compare lane 2 of the control and CHK2 siRNA panels). These data suggested that the activation of CHK2 is involved in growth arrest of cells induced by overexpression of CARF.
FIGURE 5.

CHK2 is involved in cell growth arrest of COE cells. A, siRNA-mediated CHK2 suppression in COE and CSE cells results in substantial increase in cell viability in both cell types (MTT assay; p < 0.0001, p = 0.01; respectively). The graphs are shown as fold change over vehicle-transfected COE or CSE cells, which is set as 1. The data are shown as means ± standard deviation. B, colony forming assay of CHK2-silenced COE and CSE cells showing more colonies than the control siRNA-treated cells. C, Western blotting of CHK2 compromised COE and CSE cells showing decrease in CHK2 and corresponding decreases in p53 and p21WAF1 in COE cells only. Whereas CSE cells show higher levels of CHK2 after knockdown, p53 and p21WAF1 levels were also slightly increased as compared with control cells. D, clonogenic assay showing a decrease in proliferation in CHK2-overexpressing CSE cells. Quantitation from three independent experiments is included. E, CHK2-overexpressing CSE cells with lower proliferation rate show increases in p53 and p21WAF1. F, quantitation from three independent experiments. Actin was used as a loading control in immunoblots. Each experiment was performed at least three times. *, number of colonies ± S.D. from three independent experiments. Con, control.
Next, CHK2 was knocked down in CSE cells. Surprisingly, it was found that CHK2-compromised CSE cells showed a mild but statistically significant increase in proliferation as observed in multiple independent experiments including quantitation by MTT (Fig. 5A, right panel, 1.18 ± 0.10-fold increase in CHK2 compromised CSE cells over control, p = 0.01) and colony forming assays (Fig. 5B, right panel), suggesting that the increase in CHK2 induced by superexpression of CARF was not involved in the proproliferation effect and may instead reflect an adaptive tumor suppressor/checkpoint response, however insufficient, in these cells. To verify these data further, we overexpressed a GFP-tagged CHK2 in CSE cells. Cell proliferation and biochemical assays revealed the growth arrest of CHK2-overexpressing CSE cells as compared with the vector-transfected controls (Fig. 5D and data not shown). This was also accompanied by an increase in p53 and p21WAF1 (Fig. 5, E and F). These data endorsed that the up-regulation of CHK2 in CSE cells was not directly involved in enhanced proliferation of these cells and could be an adaptive tumor suppressor/proliferation checkpoint response and did not reach the level that could cause growth arrest. Taken together, it was concluded that CARF overexpression induced growth arrest and was mediated by activation of CHK1 and CHK2.
ERKs Are Involved in CARF-mediated Effects
It is well known that the ERKs play an important role in cell cycle regulation and are the downstream effectors of the ATM-CHK axis in DDR. Thus, we set out to determine whether ERKs contributed to the CARF-mediated contrasting proliferative phenotypes. As shown in Fig. 6A, the CSE cells exhibited a substantial increase in ERK1/2 and its activated phosphorylated forms (pERK1/2). To determine whether ERK1/2 is involved in the proliferation of these cells, they were inhibited by pharmacological intervention (U0126). We found that the proliferation of CSE cells diminished (Fig. 6B; 0.80 ± 0.13-fold change over vehicle control, p = 0.02), corroborated by increases in p53 and p21WAF1 (Fig. 6C). The data were also supported by increase in the levels of pERK1/2 in proproliferative derivative cells, such as COE + CHK1 siRNA, COE + CHK2 siRNA, and CSE + CHK2 siRNA (Fig. 6D). In contrast to CSE cells, COE cells exhibited a low level of expression and activation of ERK1/2 (Fig. 6A), suggesting that such a decrease might be involved in growth arrest of COE cells. We overexpressed GFP-tagged ERK1 in COE cells and found that the clonogenic capacity of these cells was highly increased as compared with GFP transfected control COE cells (Fig. 6E), indicating an abolishment of the CARF overexpression-induced growth arrest, which was also accompanied by decrease in p53 and p21WAF1 (Fig. 6F).
FIGURE 6.
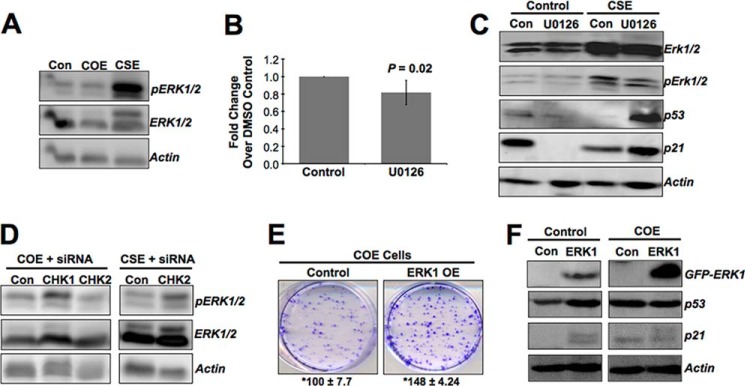
ERKs are involved in CARF-mediated up-regulation of proliferation in CSE cells. A, an elevated level of ERK phosphorylation in CSE cells, whereas it decreases in COE cells. B and C, HeLa cells when treated with U0126, an inhibitor of ERKs, lead to decrease in proliferation of CSE cells (B), as assessed by MTT assay (p = 0.02), and correspond with increases in p53 and p21WAF1 by Western blot (C). D, phosphorylated ERK increases in COE cells compromised for CHK1 or CHK2, and CSE cells compromised for CHK2 (all these cells show higher proliferation as compared with their respective controls). E and F, in parallel experiments, ERK1 overexpression in COE cells lead to an augmentation in cell growth (E), as shown by clonogenic assay, which is corroborated by decreases in p53 and p21WAF1 (F). The graph is shown as fold change over vehicle-treated CSE cells, which is set as 1, and the data are shown as means ± standard deviation. Actin was used as a loading control in immunoblots. Each experiment was performed at least three times. The mean number of colonies ± S.D. from three independent experiments is shown. Con, control.
Dual Regulation of DDR and Cell Proliferation by CARF Involves Its Interaction with CHK2 and ERK1
The above data demonstrated that CARF poses a dose-dependent control on cell proliferation. Whereas its overexpression caused growth arrest/senescence of cells (by activation of DDR components including ATM, CHK1, and CHK2 leading to decrease in activated ERKs and increase in growth arrest proteins, p53 and p21WAF1), its superexpression caused increase in CHK2, but not in CHK1 transcript, accounting for the dampening of DDR resulting in a proproliferation effect mediated by an increase in ERK activation and a decrease in the growth-arresting proteins p53 and p21WAF1 (Fig. 7A). It has been earlier shown that CARF interacts with ARF, p53, and HDM2 and regulates the stability and activity of these proteins. RT-PCR data as shown in Fig. 2G suggested that CARF also regulates these proteins at the transcriptional level in a dose-dependent manner. In light of the present findings that CARF regulates DDR by the ATM-CHK1-CHK2-ERK axis in a dose-dependent manner, we investigated whether such regulation of CHK1, CHK2, and ERK involves direct interaction with CARF. Classic co-immunoprecipitation assay using anti-CARF antibody to pull down its binding partners demonstrated the binding of CARF with CHK2 and ERK1 (Fig. 7B).
FIGURE 7.

CARF binds to and interacts with CHK2 and ERK1. A, schematic diagram showing the pathways involved in CARF overexpression-induced growth arrest and superexpression-induced proproliferation phenotypes as predicted from the data in Figs. 1–6. CARF overexpression induces DNA damage and activates DDR via up-regulation of pATM, pCHK1/CHK2, p53, and p21WAF1 (shown by gray boxes and upward arrows). On the other hand, superexpression of CARF inhibits DDR by up-regulation of CHK2 and ERKs (shown by black boxes and upward arrows). Increase in CHK2 shifts the balance to its unphosphorylated form, resulting in p53 and p21WAF1 decreases (shown by white boxes and downward arrows), leading to an increase in cell proliferation. An increase in ERKs results in down-regulation of p53 and p21WAF1 and up-regulation of ERK targets involved in mitotic signaling. B, immunoprecipitation (IP) assay with anti-CARF antibody showing the pulldown of CHK2 and ERK1 by CARF. The data suggest that CARF binds directly to these proteins. C, HeLa cells were cultured in the presence of adriamycin for 48 h to 2 weeks. Immunoblotting revealed increases in CARF, p53, and p21WAF1 in cells undergoing growth arrest in the presence of adriamycin at 48 h. Cells escape growth arrest under prolonged (2 weeks) treatment with adriamycin and exhibit further increases in the level of CARF but decreases in p53 and p21WAF1 proteins. Actin was used as a loading control for immunoblots. The experiments were performed at least three times.
To finally confirm the dose-dependent regulation of proliferative fate of cells by CARF and its physiological relevance, we used an adriamycin-induced growth arrest as a model. Adriamycin treatment for 48 h led to senescence, associated with an increase in endogenous CARF expression as compared with the control cells (Fig. 7C). Of note, the cells that escaped the effect of adriamycin and formed colonies during 2 weeks of incubation showed further increase in the expression of CARF. This was correlated with increases in p53 and p21WAF1 at 48 h treatment time, both of which were decreased in adriamycin-resistant colonies at 2 weeks. Taken together with the above data, it is concluded that (i) CARF imposes a dose-dependent regulation on cell proliferation, whereas its overexpression causes growth arrest, and CARF superexpression has a proproliferative impact, and (ii) this occurs by dose-dependent differential regulation of DDR proteins, ATM, CHK1, and CHK2.
DISCUSSION
In the present study, we demonstrated that CARF, based on its level of expression, regulates the proliferation fate of cells in opposite directions. A moderate level of overexpression induced growth arrest/senescence, and its very high level (superexpression) led to increased proliferation. To confirm that the high level of virus used for generating CARF superexpressing cells was not responsible for observed effects in this study, we generated GFP-overexpressing and -superexpressing cells using the low and high amount of virus vehicle with GFP encoding cDNA. Cell phenotypes and p53 level were examined in these cells. As shown in the data in Fig. 1 (E and F), we did not find any difference in the growth rate of cells. p53 (examined by immunostaining and Western blotting; Fig. 2C and data not shown) did not show any difference in GOE and GSE cells, implying that the effects observed for COE and CSE were due to the different level of CARF expression and not due to the virus vector.
By knockdown and overexpression of specific DDR proteins, it was further found that these phenotypes were a result of deregulation of the DDR and checkpoint pathway. Activation of DDR by CARF overexpression was evident by increased activation of (i) ATM, CHK1, CHK2, p53, and its downstream effector, p21WAF1, and (ii) down-regulation of ERK1/2 that resulted in growth arrest of cells. In contrast, superexpression of CARF (i) caused up-regulation of CHK2, (ii) inhibited the activation of CHK1 and p53 pathway, and (iii) activated ERKs to increase cell proliferation. Together with our previous findings, these data demonstrate that CARF plays a role in maintaining genome stability, and a delicate balance in the level of CARF is essential for its regulation. We propose that the physiological function of CARF is a DNA damage sensor that regulates and activates checkpoint arrest upon detection of genomic insults. A low or moderately high level of CARF expression induces DDR leading to apoptosis or growth arrest, respectively, but its very high level drives the cells toward proproliferation, higher migration capacity, and malignancy.
DDR is activated by sensing double-stranded breaks generated through exogenous and endogenous insults to the genome, the latter of which include disrupted replication, telomere shortening/degradation, as well as other metabolic processes (3, 6). Briefly, double-stranded breaks are first sensed by a complex consisting of MRE11, RAD50, and NBS1, leading to the recruitment and activation of the ATM kinase, which exists as an inactive dimer and is activated by autophosphorylation at serine 1981 (referred to as pATM here) to its monomeric form. Activated ATM in turn phosphorylates H2AX (γH2AX), as well as other critical effectors, such as BRCA1, an essential mediator of DNA repair through homologous recombination, and the two checkpoint kinases (CHK1 and CHK2), which induce cell cycle arrest via activation of p53 and p21WAF1 for DNA repair to progress. We found that the moderate overexpression of CARF induces the classic DDR response culminating into cell cycle arrest and/or senescence. Because overexpression of CARF was also found in cells undergoing replicative or stress-induced senescence (10), it is suggested that CARF functions in DDR including the one associated with replication errors, telomere shortening, and cellular metabolism. Alternatively, recent work by Bonilla et al. (15) in yeast and Soutoglou and Misteli (16) in mammalian cells has shown that just tethering certain DNA damage sensors and repair machinery components to the chromatin renders the chromatin capable of triggering the DDR without apparent DNA damage. Thus, CARF may be a potential recruiter and anchor that directly binds to and tethers DNA response machinery components to the chromatin. Indeed, we found that CARF forms physical complex with CHK2 (Fig. 7B); the stoichiometry of such complexes and the involvement of other DDR proteins warrant further investigation.
We had previously reported that CARF mediates senescence-associated growth arrest by activation of p53 and p21WAF1. The present study verified these findings and provided evidence that there is an involvement of the ATM-CHK1-CHK2-p53-p21WAF1 axis in CARF-mediated growth arrest of cells. ATM or CHK1/CHK2-compromised COE cells did not show growth arrest, demonstrating that these DDR proteins are crucial for CARF-induced growth arrest by the p53-p21WAF1 axis (Figs. 2–4). At the same time, the data endorsed that ATM and CHK1 are not involved in the proproliferative phenotype of CSE cells. Instead, superexpression of CARF was seen to cause transcriptional activation of CHK2 (Fig. 2H), shifting its balance to unphosphorylated and inactive form, resulting in an inactivation of p53-p21WAF1 and proproliferative fate of cells. Such an increase in CHK2 may trigger down-regulation of CHK1 as an compensatory/adaptive response and may further add to the proproliferation phenotype (17). This hypothesis was supported by data in Fig. 4D that showed partial reversal of proliferation by CHK1 reconstitution in CSE cells. These data demonstrated that CARF could be a major regulator of the checkpoint kinases, CHK1 and CHK2, and its level of expression decides the cell proliferation fate by regulating the balance of these two kinases.
CARF was previously shown to stabilize and activate wild type p53 in U2OS cells (9). This occurs in HeLa cells also and was able to overcome the inhibitory effects of human papillomavirus E6 protein on p53 in these cells. Thus, in addition to disrupting the CHK balance as discussed above, superexpression of CARF may result into excessive increase in p53 and its downstream effector and antagonist, HDM2, that in turn may trigger a negative feedback through p53 degradation, resulting into a proproliferation phenotype of cells (Fig. 6A). Interestingly, we found that HDM2 was up-regulated at the transcript level in CSE cells (data not shown), and that might also contribute to the decreased level of p53. The roles of mutant p53 and HDM2 in CARF-mediated regulation of cell proliferation warrant further study.
Furthermore, ERKs have been shown to function in DDR, although its effects are cell type- and genomic insult-specific (18–20). ERKs have recently been established in mediating ATM activation and promoting cell cycle arrest or apoptosis in both p53-dependent and -independent manners (7, 21, 22). On the other hand, p53 was found to inhibit ERK2 through caspase-mediated cleavage following DNA damage to decrease its prosurvival functions (23). Moreover, ERKs have been shown to be activated upon CHK1 or CHK2 inhibition as a prosurvival and proproliferation mechanism, although the underlying mechanisms as to how they interact are unknown (24, 25). In light of this information and the data in Fig. 6, the proliferative phenotype of CSE cells seemed to be promoted by increase in ERKs and was confirmed by decreased proliferation by inhibition of ERK by a specific inhibitor U0126. On the other hand, ERK overexpressing COE derivatives showed enhanced proliferation (Fig. 6, E and F) that was mediated by decrease in p53 and p21WAF1 (Fig. 6F) and may also involve other ERK effectors for mitotic signaling (Fig. 7A). Involvement of such effectors in proproliferation of CSE cells warrant further study. As a mechanism of up-regulation of ERK1 by CARF superexpression, we found that CARF can directly bind to ERK1 (Fig. 7B). In similar assays, a CARF-CHK2 complex was also detected (Fig. 7B), suggesting that CARF may modulate the activity of these proteins at the post-translational level, possibly by stabilization.
Finally, adriamycin-treated arrested and growing cells provided evidence to the physiological relevance of CARF dosage with the proliferation phenotypes. Cells that showed growth arrest in response to short term treatment of adriamycin (48 h) showed an increase in endogenous CARF expression, supporting the induction of DDR and increases in p53 and p21WAF1 (Fig. 7C). On the other hand, long term (2 weeks) treated cells that escaped the growth arrest and formed colonies exhibited further increase in the expression of CARF, supportive of abscission of DDR and a decrease in p53 and p21WAF1. Taken together with the above data, it is concluded that (i) CARF imposes a dose-dependent regulation on cell proliferation, whereas its overexpression causes growth arrest, and CARF superexpression has a proproliferative impact; and (ii) this occurs by dose-dependent differential regulation of DDR proteins ATM, CHK1, and CHK2. To the best of our knowledge, this is the first report that demonstrates the capability of a protein, based on the level of expression, to regulate contrasting proliferative fates of cells. The present study might also offer clues to its role in shift of growth-arrested senescent cells to proliferating cancer cells caused by deregulation of DDR. We have recently shown that CARF is up-regulated during replicative senescence, in response to DNA-damaging drugs, telomere unprotection by TRF2 siRNA, and oncogenic Ras-induced stress (26) and hence may represent an essential element of DNA damage response originating from either telomere shortening or direct DNA damage. Exogenous expression of CARF, without apparent DNA damage, was also able to induce senescence in cells, which further suggested that CARF is not only a DNA damage sensor but also is involved in execution of growth arrest by activation of DDR proteins. Superexpression of CARF seems to disrupt the regulation of DDR control so that cells not only escape growth arrest but also proliferate extensively as shown in the present study. Further study to investigate the physiological role of CARF and its interactions with its effector proteins, including ERKs and CHKs (therapeutic targets), is warranted for better understanding of CARF biology and its role in cancer.
Acknowledgments
We thank Ran Gao, Navjot Shah, Kamrul Hasan, and Tomoko Yaguchi for help.
This work was supported by grants from the Japan Society for Promotion of Science, New Energy & Industrial Technology Development Organization of Japan.
- DDR
- DNA damage response
- CARF
- collaborator of ARF
- CHK
- checkpoint kinase
- COE
- CARF overexpressing
- CSE
- CARF superexpressing
- HP
- heterochromatin protein
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- GOE
- GFP-overexpressing
- GSE
- GFP-superexpressing
- MMP
- matrix metalloproteinase.
REFERENCES
- 1. Bartek J., Bartkova J., Lukas J. (2007) DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 26, 7773–7779 [DOI] [PubMed] [Google Scholar]
- 2. Papazoglu C., Mills A. A. (2007) p53: at the crossroad between cancer and ageing. J. Pathol. 211, 124–133 [DOI] [PubMed] [Google Scholar]
- 3. Ciccia A., Elledge S. J. (2010) The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shiloh Y. (2003) ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3, 155–168 [DOI] [PubMed] [Google Scholar]
- 5. Kracikova M., Akiri G., George A., Sachidanandam R., Aaronson S. A. (2013) A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 20, 576–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ljungman M. (2005) Activation of DNA damage signaling. Mutat. Res. 577, 203–216 [DOI] [PubMed] [Google Scholar]
- 7. Khalil A., Morgan R. N., Adams B. R., Golding S. E., Dever S. M., Rosenberg E., Povirk L. F., Valerie K. (2011) ATM-dependent ERK signaling via AKT in response to DNA double-strand breaks. Cell Cycle 10, 481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hasan M. K., Yaguchi T., Sugihara T., Kumar P. K., Taira K., Reddel R. R., Kaul S. C., Wadhwa R. (2002) CARF is a novel protein that cooperates with mouse p19ARF (human p14ARF) in activating p53. J. Biol. Chem. 277, 37765–37770 [DOI] [PubMed] [Google Scholar]
- 9. Hasan M. K., Yaguchi T., Minoda Y., Hirano T., Taira K., Wadhwa R., Kaul S. C. (2004) Alternative reading frame protein (ARF)-independent function of CARF (collaborator of ARF) involves its interactions with p53: evidence for a novel p53-activation pathway and its negative feedback control. Biochem. J. 380, 605–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hasan K., Cheung C., Kaul Z., Shah N., Sakaushi S., Sugimoto K., Oka S., Kaul S. C., Wadhwa R. (2009) CARF Is a vital dual regulator of cellular senescence and apoptosis. J. Biol. Chem. 284, 1664–1672 [DOI] [PubMed] [Google Scholar]
- 11. Cheung C. T., Singh R., Yoon A. R., Hasan M. K., Yaguchi T., Kaul S. C., Yun C. O., Wadhwa R. (2011) Molecular characterization of apoptosis induced by CARF silencing in human cancer cells. Cell Death Differ. 18, 589–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Collado M., Serrano M. (2010) Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer 10, 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hasan M. K., Yaguchi T., Harada J. I., Hirano T., Wadhwa R., Kaul S. C. (2008) CARF (collaborator of ARF) interacts with HDM2: evidence for a novel regulatory feedback regulation of CARF-p53-HDM2-p21WAF1 pathway. Int. J. Oncol. 32, 663–671 [PubMed] [Google Scholar]
- 14. Kessenbrock K., Plaks V., Werb Z. (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bonilla C. Y., Melo J. A., Toczyski D. P. (2008) Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol. Cell 30, 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Soutoglou E., Misteli T. (2008) Activation of the cellular DNA damage response in the absence of DNA lesions. Science 320, 1507–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zaugg K., Su Y. W., Reilly PT, Moolani Y, Cheung CC, Hakem R, Hirao A, Liu Q, Elledge SJ, Mak TW. (2007) Cross-talk between Chk1 and Chk2 in double-mutant thymocytes. Proc. Natl. Acad. Sci. U.S.A. 104, 3805–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wei F., Yan J., Tang D. (2011) Extracellular signal-regulated kinases modulate DNA damage response: a contributing factor to using MEK inhibitors in cancer therapy. Curr. Med. Chem. 18, 5476–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee S. J., Lee S. H., Yoon M. H., Park B. J. (2013) A new p53 target gene, RKIP, is essential for DNA damage-induced cellular senescence and suppression of ERK activation. Neoplasia 15, 727–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tentner A. R., Lee M. J., Ostheimer G. J., Samson L. D., Lauffenburger D. A., Yaffe M. B. (2012) Combined experimental and computational analysis of DNA damage signaling reveals context-dependent roles for Erk in apoptosis and G1/S arrest after genotoxic stress. Mol. Syst. Biol. 8, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heo J. I., Oh S. J., Kho Y. J., Kim J. H., Kang H. J., Park S. H., Kim H. S., Shin J. Y., Kim M. J., Kim M., Kim S. C., Park J. B., Kim J., Lee J. Y. (2012) ATM mediates interdependent activation of p53 and ERK through formation of a ternary complex with p-p53 and p-ERK in response to DNA damage. Mol. Biol. Rep. 39, 8007–8014 [DOI] [PubMed] [Google Scholar]
- 22. Tang D., Wu D., Hirao A., Lahti J. M., Liu L., Mazza B., Kidd V. J., Mak T. W., Ingram A. J. (2002) ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J. Biol. Chem. 277, 12710–12717 [DOI] [PubMed] [Google Scholar]
- 23. Marchetti A., Cecchinelli B., D'Angelo M., D'Orazi G., Crescenzi M., Sacchi A., Soddu S. (2004) p53 can inhibit cell proliferation through caspase-mediated cleavage of ERK2/MAPK. Cell Death Differ. 11, 596–607 [DOI] [PubMed] [Google Scholar]
- 24. Dai Y., Chen S., Pei X. Y., Almenara J. A., Kramer L. B., Venditti C. A., Dent P., Grant S. (2008) Interruption of the Ras/MEK/ERK signaling cascade enhances Chk1 inhibitor-induced DNA damage in vitro and in vivo in human multiple myeloma cells. Blood 112, 2439–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dai B., Zhao X. F., Mazan-Mamczarz K., Hagner P., Corl S., Bahassi el M., Lu S., Stambrook P. J., Shapiro P., Gartenhaus R. B. (2011) Functional and molecular interactions between ERK and CHK2 in diffuse large B-cell lymphoma. Nat. Commun. 2, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh R., Kalra R. S., Hasan K., Kaul Z., Cheung C. T., Huschtscha L., Reddel R. R., Kaul S. C., Wadhwa R. (2014) Molecular characterization of collaborator of ARF (CARF) as a DNA damage response and cell cycle checkpoint regulatory protein. Exp. Cell Res. 322, 324–334 [DOI] [PubMed] [Google Scholar]