

Background: Bezlotoxumab is a neutralizing antibody targeting toxin B of Clostridium difficile.
Results: The structure of bezlotoxumab bound to a fragment of toxin B reveals its epitopes and mechanism of neutralization.
Conclusion: The epitopes overlap with two of the presumed carbohydrate binding pockets, preventing binding of the toxin to target host cells.
Significance: The data provide a molecular basis for neutralization by this clinically important antibody.
Keywords: Antibody, Bacterial Toxin, Crystal Structure, Drug Action, Epitope Mapping
Abstract
The symptoms of Clostridium difficile infections are caused by two exotoxins, TcdA and TcdB, which target host colonocytes by binding to unknown cell surface receptors, at least in part via their combined repetitive oligopeptide (CROP) domains. A combination of the anti-TcdA antibody actoxumab and the anti-TcdB antibody bezlotoxumab is currently under development for the prevention of recurrent C. difficile infections. We demonstrate here through various biophysical approaches that bezlotoxumab binds to specific regions within the N-terminal half of the TcdB CROP domain. Based on this information, we solved the x-ray structure of the N-terminal half of the TcdB CROP domain bound to Fab fragments of bezlotoxumab. The structure reveals that the TcdB CROP domain adopts a β-solenoid fold consisting of long and short repeats and that bezlotoxumab binds to two homologous sites within the CROP domain, partially occluding two of the four putative carbohydrate binding pockets located in TcdB. We also show that bezlotoxumab neutralizes TcdB by blocking binding of TcdB to mammalian cells. Overall, our data are consistent with a model wherein a single molecule of bezlotoxumab neutralizes TcdB by binding via its two Fab regions to two epitopes within the N-terminal half of the TcdB CROP domain, partially blocking the carbohydrate binding pockets of the toxin and preventing toxin binding to host cells.
Introduction
Clostridium difficile is an anaerobic Gram-positive bacillus that infects the colon of susceptible patients, mainly in hospital settings but also increasingly in the community. C. difficile infections (CDI)4 are typified by severe diarrhea, pseudomembranous colitis, and in extreme cases colonic rupture, sepsis, and death (1). Current treatments for CDI include vancomycin, metronidazole, and the recently approved antibiotic fidaxomicin (2). Despite excellent initial cure rates with these therapies, up to 30% of patients experience at least one recurrence and may require multiple rounds of treatment that can last several weeks to months, negatively impacting quality of life and costing the health care system at least $1 billion a year in the United States alone (2). For these reasons, the Centers for Disease Control have recently classified C. difficile as one of only three microorganisms that are “an immediate public health threat and that require urgent and aggressive action” (44). There is therefore a pressing need for new treatments against C. difficile, in particular ones that address the high rates of recurrence.
The pathogenic nature of C. difficile results from production of two exotoxins, toxin A (TcdA) and toxin B (TcdB), that are thought to target colonocytes via similar mechanisms that ultimately lead to cell death and disruption of the trans-epithelial resistance that normally exists across the gut wall (3). Damage to the gut epithelium results in fluid leakage into the gut lumen and release of proinflammatory mediators, such as IL-1β, TNFα, and IL-8, leading to an inflammatory response that includes recruitment of neutrophils and macrophages to the site of injury, further aggravating the disease (4). Extensive structural and functional work over the past several years has led to a basic understanding of the molecular events that lead to toxin-mediated cell death, as recently reviewed by Pruitt and Lacy (5). Following binding to specific receptors on the host cell, the toxins are internalized via endocytosis into clathrin-coated vesicles (6). Acidification of the endosome leads to conformational changes in the toxins (7, 8), allowing for transport of the glucosyltransferase domain (GTD) across the endosomal membrane via a poorly defined translocation process. The final steps of the cascade involve autocleavage of the toxin (catalyzed by a cysteine protease domain), leading to release of the GTD domain into the cytosol (9), where it glucosylates and inactivates small GTPases, such as Rac and Rho, which play a critical role in maintaining cellular morphology and in multiple other aspects of cellular homeostasis.
One aspect of toxin function that is still poorly understood is how TcdA and TcdB bind to host cells or, more specifically, what is the nature of the receptors to which the toxins bind. For TcdA, the receptor has been proposed to consist of a membrane-associated carbohydrate based on the following lines of evidence: (i) TcdA binds specifically to various galactose- and N-acetyl-glucose-containing carbohydrate molecules, including sugars known to be expressed on the extracellular surface of toxin-sensitive mammalian cells (10–16), and (ii) treatment with galactosidase abolishes TcdA binding (13–15). Evidence that the receptor for TcdB is also a carbohydrate is currently limited to the observation that this toxin is structurally highly homologous to TcdA (7, 17) and that it binds to carbohydrates that are structurally related to the TcdA ligands (10, 11). It is thought that toxin binding to receptor(s) on the cell surface occurs via the C-terminal domain of the toxins (known as the combined repetitive oligopeptide (CROP) domain) (12, 17–19), although recent data suggest that other domains within the toxins may also be involved in binding (20, 21). Analysis of the sequences of the TcdA and TcdB CROP domains shows that they consist of ∼20–30 short repeats (SRs) interspersed with a smaller number (seven in TcdA and four in TcdB) of homologous long repeats (LRs) (12, 17, 22, 23). Although a high-resolution structure of the intact toxin is still lacking, many of the individual toxin domains have been crystalized, including C-terminal fragments of the TcdA and TcdB CROP domains (12, 23–25). The structures of the TcdA/B CROPs display a recurring β-solenoid structural unit (defined herein as a “CROP unit”) containing one LR flanked by 2–3 SR β-hairpins on either side, consistent with the motifs uncovered through sequence analysis. Modeling based on a high-resolution structure of a peptide spanning the extreme C-terminal end of the TcdA CROP domain predicts an overall S-shaped elongated rodlike CROP domain for TcdA and a shorter horseshoe-shaped domain for TcdB (5, 23), supported by recent work from Lacy and co-workers using electron microscopy (7). A high resolution structure of a peptide spanning two CROP units of the TcdA CROP domain in complex with the carbohydrate α-Gal-(1,3)-β-Gal-(1,4)-β-GlcNAcO(CH2)8CO2CH3 was also obtained by the Ng group (12), demonstrating that the residues involved in carbohydrate binding are indeed located within the CROP domain, specifically centered around the LRs. More recently, the same group reported structures of C-terminal fragments of both TcdA and TcdB CROP domains bound to various camelid-neutralizing and non-neutralizing single-domain antibodies (nanobodies) (25).
The use of toxin-deficient strains of C. difficile in hamster models (26–28) and the observations that active and passive immunization against the C. difficile toxins is protective both in animal models (29–32) and in humans (33) demonstrate that TcdA and TcdB are indeed the primary contributors to disease. Based on this premise, a combination of the two monoclonal antibodies actoxumab (also known as MK-3415, GS-CDA1, and MDX-066) and bezlotoxumab (also known as MK-6072, MBL-CDB1, and MDX-1388), specific for TcdA and TcdB, respectively, is currently in phase III development for the treatment of recurrent CDI. In phase II trials, the actoxumab/bezlotoxumab combination reduced the rate of recurrence among CDI patients treated with standard of care therapy by 73% (33).
In this study, we have structurally characterized the interactions between the TcdB CROP domain and the neutralizing antibody bezlotoxumab. Following refinement of the bezlotoxumab epitopes using a number of biophysical approaches, we obtained the x-ray crystal structure of the N-terminal half of the TcdB CROP domain in complex with Fab fragments of bezlotoxumab, providing insight into the mechanism of neutralization of bezlotoxumab.
EXPERIMENTAL PROCEDURES
TcdB Protein Expression and Purification
The CROP domain fragments (B1 (residues 1834–2366), B2 (residues 1834–2101), B3 (residues 1949–2275) and B4 (residues 2102–2366)) and the catalytic domain (GTD) from C. difficile strain VPI 10463 were cloned into pET28a(+) vector (Invitrogen), which encodes a C-terminal His tag. TcdB fragments, including B2, were expressed in Escherichia coli BL21(DE3) (Novagen-EMD Millipore, Billerica, MA) in Terrific Broth supplemented with 50 μg ml−1 kanamycin for 18 h at 16 °C with 1 mm isopropyl 1-thio-β-d-galactopyranoside. The cells were harvested by centrifugation for 15 min at 6000 × g, and pellets were resuspended in 50 mm Tris, pH 8.0, 0.3 m NaCl, 1 mm DTT, 1 mg/ml protease inhibitor mixture III (EMD Millipore) and then lysed with a microfluidizer. The cell lysate was clarified by centrifugation at 100,000 × g for 1 h at 4 °C. The supernatant was filtered and loaded onto a Ni2+-immobilized metal ion affinity chromatography (Qiagen) column equilibrated with 50 mm Tris, pH 8.0, 0.3 m NaCl, 10% glycerol, and 1 mm DTT. The protein was eluted using an imidazole gradient (0–0.25 m) containing 50 mm Tris, pH 8.0, 0.3 m NaCl, 10% glycerol, and 1 mm DTT. Fractions containing TcdB proteins were pooled, diluted to 50 mm NaCl, and further purified by ion exchange chromatography using a Source 15Q column (GE Healthcare). The final storage buffer for TcdB proteins was 50 mm Hepes, pH 7.5, 0.15 m NaCl. Fractions with >95% pure peptides (as monitored by SDS-PAGE electrophoresis) were collected and concentrated to 5 mg/ml using a centrifugal concentrator. The identities of the peptides were confirmed by electrospray ionization-ion trap-MS using a LTQ-XL mass spectrometer and the Xcalibur software platform (Thermo Scientific). Purified TcdB peptides and Fab fragments were mixed in a 1:3 molar ratios, and excess Fab was separated by size exclusion chromatography (S200 26/60 column from GE Healthcare). The complex was concentrated to >15 mg/ml for crystallization trials.
Temperature-dependent Fluorescence (TdF)
Solutions of the various TcdB peptides (166 μm in 25 mm HEPES, pH 7.5, 0.15 m NaCl) were centrifuged for 5 min to remove insoluble material and diluted 100-fold. Sypro orange (Invitrogen) was added to the diluted peptide solution at a final concentration of 5×. For assessments in the presence of bezlotoxumab, the antibody was added at an antibody/peptide ratio of 1:1. 10-μl samples of TcdB peptide with and without bezlotoxumab were added to a white Abgene 384-well PCR plate (Thermo Scientific) and sealed with flat ultraclear caps (Bio-Rad). TdF measurements were conducted with a Roche PCR thermocycler (Roche Applied Science) equipped with a CCD camera for fluorescence detection. The temperature was increased from 25 to 95 °C in 0.2 °C increments using a 200-ms stabilization delay before reading. Fluorescence signals were acquired with excitation and emission wavelengths of 490 and 560 nm, respectively. A customized analysis program using a non-linear least square method based on the generalized reduced gradient algorithm was used to fit the protein unfolding model (34). The following parameters were floated during the fitting process: y intercepts for the intensity of Sypro Orange in both the native and denatured proteins (yn and yd); the associated slopes (Mn and Md); the midpoint of melting (Tm); and the enthalpy at the Tm (ΔHm).
Surface Plasmon Resonance
Binding of TcdB and TcdB fragments to the antibodies was studied by surface plasmon resonance using a ProteOn XPR36 instrument (Bio-Rad). Bezlotoxumab was immobilized to the sensor chip surface using an antibody capture method. Briefly, a ProteOn GLC Sensor chip was docked to the system, and after standard cleaning according to the manufacturer's recommendations, a mixture of 1× 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride + N-hydroxysulfosuccinimide was injected over the chip to activate the chip surface. A 25 μg/ml solution of goat anti-human IgG F(ab′)2 (Thermo Scientific) in ProteOn immobilization buffer (10 mm sodium acetate, pH 5.5) was injected over 2 min. 1 m ethanolamine-HCl was then injected over 5 min to block any unoccupied reactive sites on the chip surface. 20 μg/ml bezlotoxumab in ProteOn running buffer (PBS, pH 7.4, 0.005% Tween 20) was injected over 2 min. Toxin fragments were diluted at various concentrations in ProteOn running buffer and injected in horizontal orientation for 4 min (flow rate 25 μl/min). Association and dissociation were measured over time as changes in the refractive index. Data analyses were carried out using the ProteOn instrument software, and data were fitted using a two-site heterogeneous ligand model to determine kon, koff, and Kd.
Hydrogen Deuterium Exchange-Mass Spectrometry (HDX-MS)
HDX-MS was carried out largely as described previously (35). Briefly, pepsin digestion and fragment separation conditions were optimized to follow peptides spanning the entire TcdB CROP sequence. 41 μm TcdB B1 peptide (Fig. 1A) was acidified and passed over an immobilized pepsin column at 200 μl/min with buffer A (0.05% TFA in H2O). Peptic fragments were loaded onto a reverse phase trap column at 200 μl/min with buffer A, desalted with buffer A, and separated by a C18 column with a linear gradient of 13–35% buffer B (95% acetonitrile, 0.0025% TFA in water) over 23 min. Mass spectrometry data were acquired using a Thermo Fisher LTQ-Orbitrap. For digestion condition optimization experiments, MS/MS spectra were acquired in data-dependent mode. One MS1 full scan was carried out (15,000 resolving power, profile mode, from 380 to 2000 m/z, 500,000 target ions, and a 750-ms maximum ion injection time) followed by one MS/MS scan in the LTQ (10,000 target ions, 100-ms maximum ion injection time). Dynamic exclusion was enabled with a repeat count of 1 for 180 s, with a list size of 50. To determine the minimum amount of monoclonal antibody necessary to saturate the available binding sites on TcdB C1, 7.5 μm B1 peptide was titrated against various amounts of antibody in D2O at the following bezlotoxumab/B1 ratios: 0, 0.1, 0.25, 0.3, 0.4, 0.5, 0.6, 0.7, and 0.75. The titration mixtures were incubated for 300 s at 23 °C, quenched, and analyzed as above. On-exchange experiments of B1 in the presence and absence of bezlotoxumab were performed by mixing 14.7 μm B1 with 4.4 μm bezlotoxumab in D2O. The mixture was incubated at 23 °C for 30, 100, 300, or 1000 s, quenched, and analyzed as above. For the analysis of deuterium-labeled samples, the MS1 full scans were acquired as described above, and no MS/MS spectra were acquired. Percentage deuteration was averaged over all time points, and significant differences between deuteration levels in the presence versus the absence of bezlotoxumab are reported in Fig. 2B. Mapping of the HDX-protected regions onto TcdB, as shown in Fig. 5, was carried out by constructing a three-dimensional model based on the crystal structure of the C-terminal fragment of TcdA (23) and using the Molecular Operating Environment computer software (Chemical Computing Group Inc., Montreal, Canada).
FIGURE 1.

C. difficile toxins and toxin fragments. A, domain structures of TcdA and TcdB showing the putative receptor binding (CROP) domains at the C terminus. B, TcdB CROP domain showing SRs (green) and LRs (gray) and highlighting the four peptides used in this study, B1, B2, B3, and B4. The first of four CROP units, consisting of one LR flanked by three SRs on the N-terminal side and two SRs on the C-terminal side, is highlighted in red.
FIGURE 2.

Identification of the bezlotoxumab-binding regions within the TcdB CROP domain. A, Coomassie Blue-stained polyacrylamide gel and Western blot showing binding of bezlotoxumab to intact TcdB and to peptides B1, B2, and B3 but not to peptide B4 or to the TcdB catalytic domain (GTD). Molecular masses (kDa) of markers are indicated on the left. B, binding of bezlotoxumab to full-length TcdB, B1, B2, and B3, but not B4, as assessed by TdF and SPR. C, summary of HDX-MS experiments showing regions of the CROP domain that are protected from deuteration in the presence of bezlotoxumab and extent of protection. D, details of the HDX-MS analysis showing deuteration levels (color key at lower right) for peptide B1 alone (top, −B) and in the presence of bezlotoxumab (bottom, +B). Each box represents a distinct peptide identified by MS and is subdivided into four time points: 30, 100, 300, and 1000 s from top to bottom (see “Experimental Procedures”). The numbering used is that of full-length TcdB. The differences between deuteration levels in the presence versus the absence of bezlotoxumab were averaged over all time points, and significant differences are reported in Fig. 2C.
FIGURE 5.
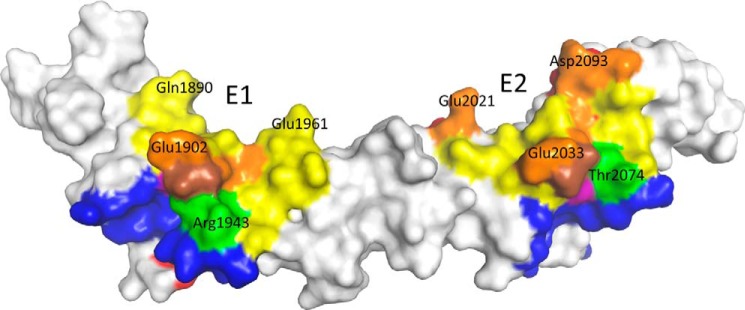
Surface representation of peptide B2 with bezlotoxumab epitopes and putative carbohydrate-interacting residues. Regions that interact with bezlotoxumab (as identified by x-ray crystallography) are shown in yellow on the surface of B2. Residues that are protected from deuteration in the HDX-MS analysis are shown in red. Residues putatively involved in carbohydrate binding are shown in blue. Overlapping regions are shown in orange (x-ray and HDX-MS), green (x-ray and carbohydrate binding), purple (HDX-MS and carbohydrate binding), or brown (x-ray, HDX-MS, and carbohydrate binding).
Size Exclusion Chromatography Coupled with Multiangle Laser Light Scattering (SEC-MALLS)
Bezlotoxumab was mixed at different molar ratios with peptide B1 at room temperature in PBS. The final antibody concentration was 0.17 mg/ml in the reaction mixture. SEC-MALLS was performed using a Dawn Heleos-II 18 angle light scattering detector and an Optilab T-rEX refractive index detector from Wyatt Technology (Santa Barbara, CA). Samples were separated on a YMC-pack Diol 200 column (300 × 8 mm, 5 μm) on an Agilent 1200 system at ambient temperature. The mobile phase was PBS (10 mm phosphate buffer, pH 7.4, with 137 mm NaCl and 2.7 mm KCl), and the flow rate was 0.5 ml/min. The running time was 30 min. Signals at 280 and 214 nm were recorded. Light scattering and refractive index signals were collected, and molecular masses of detected components were calculated, using the ASTRA VI software (Wyatt Technology).
Preparation of Fab Fragments
Bezlotoxumab Fab fragments were generated using the Pierce Fab Preparation Kit (Thermo Scientific), following the manufacturer's instructions. Briefly, antibody was incubated with immobilized papain resin at 37 °C for 5 h, following which undigested IgG and Fc fragments were removed using a protein A column. The Fab-containing flow-through was collected and further purified by gel filtration chromatography. N-terminal sequencing was used to confirm the Fab fragment identity.
X-ray Crystallography
Crystals of TcdB/Fab were obtained by sitting drop vapor diffusion, by mixing equal amounts of protein solution with the reservoir solution containing only 5% PEG 4000. Crystals appeared after 2 weeks and belonged to the space group P21 with unit cell dimensions of a = 79.41°, b = 134.66°, c = 102.58°, and β = 112.56°. The asymmetric unit had a solvent content of 69% and contained one TcdB and two Fab molecules. X-ray data were collected at 100 K using a Pilatus 6M detector at the Industrial Macromolecular Crystallography Association Collaborative Access Team beamline 17ID at the Advanced Photon Source (Argonne), at a wavelength of λ = 1.0000 Å to a resolution of 2.89 Å. Data set statistics are provided in Table 1. Data were processed with autoPROC (Global Phasing) (36). 2% of the reflections were set aside in the free R set. Molecular replacement was carried out with Molrep (CCP4 suite) (37) using a two-search model. The TcdB model was manually designed based on the TcdA crystal structure (12), using Maestro (Schrödinger, LLC, New York). The Fab molecules were derived from a previous Fab structure (PDB code 3QEG) based on sequence homology. The model was refined in real space interactively using Coot (38) and refined using autoBUSTER (Global Phasing). The final model contains 1132 of 1146 residues (271 TcdB residues, 427 Fab1, and 434 Fab2 residues) The final model was validated using MolProbity (39) and presented good stereochemistry, with over 95.9% of all residues in favored and additionally allowed regions of the Ramachandran plot.
TABLE 1.
Crystallographic data collection and refinement statistics
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 79.4, 134.6, 102.6 |
| α, β, γ (degrees) | 90, 112.56, 90 |
| Resolution (Å) | 42–2.89 (2.9–2.89)a |
| Rmerge | 0.056 (0.547) |
| I/σI | 17.3 (2.1) |
| Completeness (%) | 99.5 (100) |
| Redundancy | 3.4 (3.4l) |
| Refinement | |
| Resolution (Å) | 42–2.89 |
| No. of reflections | 44385 |
| Rwork/Rfree | 19.8/23.2 |
| No. of atoms | |
| Protein | 8673 |
| Ligand/ion | 0 |
| Water | 14 |
| B-factors | |
| Protein | 74 |
| Ligand/ion | n/a |
| Water | 49 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.010 |
| Bond angles (degrees) | 1.33 |
a Values in parentheses are for highest resolution shell.
Molecular Modeling
The model used to derive the crystal structure of B2 in complex with Fab fragments was constructed from a TcdA x-ray structure (PDB code 2G7C (12)), altering the CROP unit using Maestro (Schrödinger, LLC) to correspond to the 3SR-1LR-2SR fold of TcdB. The model of peptide B4 was built based on the B2 structure using Molecular Operating Environment (Chemical Computing Group Inc.). The model of the full TcdB CROP domain was built by fusing the B2 structure with the model of B4 (using Maestro), ensuring that the β-solenoid fold structure and rotation angles between β-hairpin repeats (120º between two SRs and 90º between LRs and flanking SRs, along 31 screw axes) were maintained.
Binding Free Energy Calculations
Binding free energies (ΔG) between Fab fragments and E1, E2, or E3 were based on crystal structures described herein and described previously (25) and calculated using Prime MMGBSA (Schrödinger, LLC), with atoms of Fab and TcdB held fixed.
TcdB Neutralization Assay
Vero cells were seeded at 2000 cells/well in 96-well dishes and incubated overnight. Purified TcdB toxin from strain VPI 10463 (tgcBIOMICS, Bingen, Germany) was diluted in culture medium to a final concentration of 10 pg/ml, previously demonstrated to result in a ∼95% decrease in cell viability (data not shown), in the absence or presence of various concentrations of bezlotoxumab, incubated at 37 °C for 2 h, and then added to cells. After 24 h at 37 °C, the medium was aspirated, plates were washed two times with phosphate-buffered saline, and 200 μl/well of complete medium was added, followed by incubation at 37 °C for 48 h. Medium was removed, and 100 μl/well of 10% cold TCA was added, followed by incubation for 60 min at 4 °C in order to fix the cells. TCA was removed, wells were washed four times with distilled water, and 100 μl/well of 100 μg/ml sulforhodamine B (Sigma) in 10% acetic acid was added. Plates were incubated for 15 min at room temperature and then washed four times with 10% acetic acid and air-dried. 150 μl/well of 10 mm Tris was added, and plates were incubated with shaking at room temperature for an additional 10 min. Plates were read in a SpectraMax plate reader (Molecular Biosystems) at an absorbance wavelength of 570 nm. The percentage of cell survival was calculated by defining A570 values in empty wells as representing 0% cell survival and wells containing cells incubated in the absence of toxin representing 100% cell survival.
TcdB Cell Surface Binding Assay
Vero cells (American Type Culture Collection) were grown at 37 °C in 5% CO2 in Eagle's minimum essential medium supplemented with 10% FBS, 100 units/ml penicillin, and 100 units/ml streptomycin. For cell surface binding, 10-cm dishes of confluent Vero cells were pretreated with a 14 μm concentration of the endocytosis inhibitor chlorpromazine (Enzo Life Sciences, Farmingdale, NY) in Vero cell culture medium for 1 h at 37 °C. 50 or 100 ng/ml TcdB (The Native Antigen Company, Oxfordshire, UK) were incubated with or without 200 μg/ml bezlotoxumab or actotoxumab at 37 °C in Vero cell culture medium for 30 min. Mixtures were then added to the Vero cells, keeping the chlorpromazine concentration constant at 14 μm. Plates were incubated at 37 °C in 5% CO2 to allow toxin binding. After 15 min, plates were chilled on ice and washed three times with cold PBS, and cells were harvested by scraping. Membrane proteins were isolated in the cold using the Mem-PER Plus membrane protein extraction kit (Thermo Scientific) according to the manufacturer's instructions. In the final step, membrane proteins were solubilized in a total volume of 100 μl of solubilization buffer. 33 μl of 4× Laemmli sample buffer was added, and samples were treated at 95 °C for 5 min and resolved on 4–12% SDS-polyacrylamide gels. Following transfer to nitrocellulose membrane, blots were probed with a 1:1 combination of bezlotoxumab and antibody 2A11 (29) and visualized as described below (“Western blotting”). Nitrocellulose membranes were also probed with rabbit anti-cadherin polyclonal antiserum (Cell Signaling, Beverly, MA) to ensure that equivalent amounts of protein were loaded in each lane. Band densities were quantitated using the Odyssey software application (LI-COR Biosciences, Lincoln, NE) and normalized to nanogram amounts based on the density of the control TcdB band (1 ng loaded in the well).
Western Blotting
Proteins were resuspended in Laemmli sample buffer, boiled, and separated by SDS-gel electrophoresis (3 μg of protein/well for the Coomassie-stained gel and 200 ng of protein/well for the Western blot). Gels were either stained with Coomassie Blue or transferred to nitrocellulose for Western blotting, according to standard methodology. The nitrocellulose membrane containing transferred protein was blocked in Odyssey blocking buffer (LI-COR Biosciences) followed by incubation with bezlotoxumab for 1 h at room temperature. After washing, the blot was incubated with a donkey anti-human IgG antibody coupled to IRDye 800CW (LI-COR Biosciences) for 30 min at room temperature. After additional washing, bands were visualized using the Odyssey Imaging System (LI-COR Biosciences).
RESULTS
Bezlotoxumab-binding Regions of the TcdB CROP Domain
The two toxins of C. difficile, TcdA and TcdB, are structurally homologous, consisting of three distinct domains: a GTD, a translocation/cysteine protease domain, and a receptor binding CROP domain (Fig. 1A). In setting out to understand the nature of the interactions of bezlotoxumab with TcdB, we generated multiple overlapping peptides that together span the entire CROP domain (Fig. 1B), based on the fact that the antibody was raised against and is known to bind to this region of TcdB (29). Specifically, we expressed and purified the following constructs, all derived from the TcdB sequence of strain VPI 10463, against which bezlotoxumab was raised: B1 (residues 1834–2366 of TcdB), B2 (residues 1834–2101), B3 (residues 1949–2275), and B4 (residues 2102–2366). In order to facilitate the description of TcdB CROP domain fragments in this work, we define the term “CROP unit” as consisting of three SRs followed by one LR and two additional SRs (Fig. 1B). Using this nomenclature, B1 is predicted to contain four CROP units, whereas peptides B2, B3, and B4 are predicted to contain two CROP units each (Fig. 1B). Western blot analysis of these various peptides shows that bezlotoxumab binds to full-length TcdB, to peptide B1 (corresponding to the full CROP domain of TcdB), and to peptides B2 and B3 but not to peptide B4 or to a construct corresponding to the catalytic domain of TcdB (Fig. 2A). Binding of bezlotoxumab to full-length TcdB as well as to peptides B1, B2, and B3 (but not B4) was confirmed qualitatively by TdF and quantitatively by surface plasmon resonance (SPR) (Fig. 2B). The kinetics of bezlotoxumab binding to TcdB and its various fragments were determined by SPR, using a two-site binding model fit (see “Experimental Procedures”) because this model approximated the data more closely than a single-site model. As shown in Table 2, the SPR dissociation constants for the high (Kd1) and low affinity (Kd2) binding sites are similar for B1 (Kd1 = 41 ± 13 pm and Kd2 = 660 ± 13 pm), B2 (Kd1 = 46 ± 21 pm and Kd2 = 810 ± 56 pm), and full-length (Kd1 = 19 ± 5.1 pm and Kd2 = 370 ± 310 pm) TcdB proteins. The B3 fragment showed a 29-fold higher Kd2 compared with full-length TcdB, with no measurable high affinity binding site (Kd1). Peptide B4 and the TcdB catalytic domain showed no measurable affinity for bezlotoxumab, consistent with the Western blot and TdF data. The SPR data suggest that bezlotoxumab binds to two distinct epitopes within intact TcdB and within peptides B1 and B2 but that one epitope may be lost in B3 and that none are present in B4.
TABLE 2.
Association/dissociation rates and affinities of bezlotoxumab binding to TcdB and TcdB fragments, as determined by surface plasmon resonance
| Toxin fragment | High affinity binding site |
Low affinity binding site |
||||
|---|---|---|---|---|---|---|
| kona | koff | K′d | kon | koff | K′d | |
| m−1 s−1 | s−1 | pm | m−1 s−1 | s−1 | pm | |
| TcdB | 1.52 × 106 ± 0.45 × 106 | 2.83 × 10−5 ± 0.11 × 10−5 | 19 ± 5 | 1.57 × 107 ± 0.14 × 107 | 5.59 × 10−3 ± 4.36 × 10−3 | 370 ± 310 |
| B1 | 3.64 × 106 ± 0.11 × 106 | 1.41 × 10−4 ± 0.06 × 10−4 | 41 ± 13 | 2.09 × 107 ± 0.11 × 107 | 1.37 × 10−2 ± 0.07 × 10−2 | 660 ± 35 |
| B2 | 3.73 × 106 ± 0.11 × 106 | 1.61 × 10−4 ± 0.28 × 10−4 | 46 ± 21 | 1.35 × 107 ± 0.13 × 107 | 1.10 × 10−2 ± 0.18 × 10−2 | 810 ± 56 |
| B3 | NMb | NM | NM | 3.25 × 106 ± 1.52 × 106 | 3.03 × 10−2 ± 0.36 × 10−2 | 11,000 ± 6000 |
| B4 | NM | NM | NM | NM | NM | NM |
a All values are means ± S.D. of two independent determinations.
b NM, not measurable.
The binding sites of bezlotoxumab on TcdB were refined using HDX-MS, a method that relies on the measurement and comparison of the degree of deuterium incorporation by an antigen following incubation in D2O in the absence and presence of antibody (35). Characterization of peptide B1 (corresponding to the full CROP domain of TcdB) by HDX-MS (Fig. 2, C and D) reveals that fragments corresponding to residues 1902–1914 (EDGFKYFAPANTL), 2021–2025 (ENGEM), 2033–2038 (EDGFKY), and 2091–2096 (DEDTAE) of TcdB are significantly protected from deuteration (9, 7, 30, and 9%, respectively) upon preincubation with bezlotoxumab, suggesting that the antibody interacts with these regions of the CROP domain. Taken together with the Western blotting and biophysical data described above, these findings demonstrate that the bezlotoxumab epitopes lie in distinct regions within the N-terminal half of the TcdB CROP domain. We therefore chose peptide B2 (Fig. 1B) for crystal structure studies.
X-ray Structure of a TcdB CROP Fragment Bound to Bezlotoxumab Fab
The crystal structure of the B2-bezlotoxumab Fab complex was solved by molecular replacement. Although two Fab molecules were located in the asymmetric unit using a previous Fab structure (PDB code 3QEG) as the search model, molecule replacement was not successful in finding B2 using the three published TcdA x-ray structures (PDB entries 2G7C (12), 2F6E (23), and 2QJ6 (24)); the recently published structure of TcdB (25) was not available at the time these experiments were done. Attempts to solve the B2 structure using electron density calculated using only the two Fab molecules failed due to inadequate map quality. In order to create a better model for molecular replacement, a homology model of B2 was built using a TcdA structure (PDB code 2G7C (12)) as the template. With this model, one B2 solution was found, using the aromatic residues and the curvature of the backbone structure to validate the solution to the electron density. The structure refinement progressed smoothly with an R/Rfree of 20.2/23.8% at 2.89 Å (for complete data collection and refinement statistics, see Table 1). The final model (PDB entry 4NP4), contains residues 1834–2101 of TcdB, residues 1–213 of both bezlotoxumab light chains, residues 1–134 and 141–221 of the bezlotoxumab heavy chain of Fab1 (residues 135–140 are disordered), and residues 1–221 of Fab2 (Fig. 3).
FIGURE 3.

Crystal structure of the N-terminal half of the TcdB CROP domain bound to two bezlotoxumab Fab fragments. A, side view showing parallel binding of the two Fab fragments (Fab1 and Fab2) to their respective epitopes, E1 and E2. LRs are shown in gray and SRs in green. Fab heavy chains are shown in yellow (Fab1) and peach (Fab2) and light chains in pink (Fab1) and purple (Fab2). B, bottom-up view showing the Fab fragments bound perpendicularly to the curvature of the CROP domain. Residues of the CROP domain that directly interact with the heavy chains (yellow and peach) or light chains (pink and purple) of the Fab fragments are highlighted on the CROP surface. C, partial sequence of the light chains (Lc) and heavy chains (Hc) of bezlotoxumab, showing the six complementarity-determining regions (identified using the Molecular Operating Environment software from Chemical Computing Group); residues that interact with peptide B2 are highlighted in gray. D, sequence alignment of the two bezlotoxumab epitope regions (E1 and E2) and the two non-binding homologous regions (E3 and E4) of the TcdB CROP domain. Bezlotoxumab-interacting residues in E1 and E2 (and corresponding residues in E3 and E4) are highlighted in gray, with conserved and non-conserved substitutions (compared with E1) shown in green and red, respectively. Regions protected by bezlotoxumab in the HDX-MS experiment (Fig. 2c) are underlined, and residues putatively involved in carbohydrate binding are identified by asterisks. SRs and LRs are shown as green and gray boxes, respectively.
The structure of B2 confirms that this peptide spans two CROP units, each having a β-solenoid fold consisting of three SRs followed by an LR and two additional SR β hairpins, as predicted from the B2 sequence (Fig. 1B). The fold is characterized with few exceptions by a Tyr-Tyr-Phe repeat located on the second β strand of each β hairpin, and the hairpins themselves are stacked against each other with a 120° rotation between any two SRs and a 90° rotation between LRs and their flanking SRs. As a consequence, the CROP structure curves at the junction between LRs and SRs, causing kinks in what would be otherwise a stacked linear arrangement of SRs. Overall, the B2 fragment forms an elongated 115-Å-long rodlike superstructure with a ∼60° curvature. The two TcdB CROP units within B2 are structurally similar to each other, and Cα atoms can be superimposed with a root mean square deviation of 0.8 Å, which is only slightly above the experimental error of 0.4 Å calculated using the Luzatti plot (40). The structure of B2 is also highly analogous to those of the C-terminal fragments of the TcdA (12, 23) and TcdB (25) CROP domains, with root mean square deviation not exceeding 1.1 Å when comparing CROP units of each structure, demonstrating the conserved nature of the overall β-solenoid fold of toxin CROP domains. The major structural differences between CROP units occur within the first β hairpins and within the loops of the LR and result from specific crystal-packing contacts.
The crystal structure also reveals that the two bezlotoxumab Fab fragments lie adjacent to each other and bind to distinct regions within a single B2 peptide, both roughly perpendicular to the curvature of the rodlike superstructure (Fig. 3) and rotated relative to each other by ∼50º due to the curved nature of the CROP domain. The two Fabs are highly similar to each other (0.7 Å root mean square deviation) and are oriented such that they could conceivably bind in a similar manner were they part of an intact monoclonal antibody molecule. The locations of the two epitopes are consistent with the binding data obtained by SPR (Table 2), namely the finding that only one of the two epitopes is present in peptide B3. Moreover, due to their proximity, the two Fab molecules interact with each other, with 204 Å2 of interacting surface area. Contacts between the two Fabs involve both the heavy and light chains of Fab1 and Fab2 (see supplemental Table 1). Among the interactions, there are several residue-specific contacts. The side chains of Glu-82Fab1/lc and Ser-85Fab2/hc form hydrogen bonds with each other, whereas the side chains of Thr-166Fab1/hc and Ser-167Fab1/hc and Lys-170Fab1/lc form hydrogen bonds with the peptide backbone of the Fab2 heavy chain.
Characterization of the Bezlotoxumab Epitopes
Extensive contacts are observed between B2 and the two Fab molecules (Fig. 3B). Fab1 and Fab2 cover areas on B2 of 825 and 906 Å2, respectively, and the heavy chains of each Fab cover more than 70% of that surface area. The complementarity-determining regions of both light and heavy chains, along with the nature of the residues that interact with B2, are shown in Fig. 3C. Residues having at least one atom within 4.0 Å of any Fab atom in the crystal structure are defined as being part of the two bezlotoxumab epitopes, referred to herein as E1 for Fab1 and E2 Fab2 (Fig. 3D; individual atomic interactions between TcdB and bezlotoxumab residues are shown in supplemental Table 2). The “anchor” points of the Fab/B2 interaction for both E1 and E2 are the β-hairpin loops of each LR, which protrude into the pocket between the heavy and light chains of the Fabs; additional contact points are provided by residues within the connecting loops between surrounding SRs and by a handful of residues within SR β-strands themselves (Fig. 3). The distance between the two anchor points is 50 Å.
The E1/Fab1 interface, although largely flat as is common for protein-protein interactions, contains mostly polar, charged, and/or aromatic residues that form extensive hydrogen bond networks and salt bridges (Fig. 4A). For example, within the anchoring LR β-hairpin loop, Glu-1902E1 forms a salt bridge with Arg-59Fab1/hc, whereas the Ser-1900E1 hydroxyl probably forms a hydrogen bond with the side chains of Trp-33Fab1/hc and Arg-99Fab1/hc. In addition, Gln-1890E1 and Tyr-1887E1 each form a hydrogen bond with one of the side chain oxygen atoms of Asp-55Fab1/hc. The Tyr-33Fab1/lc side chain interacts through hydrogen bonding with Asp-1903E1 and Asn-1941E1, the latter in turn forming a hydrogen bond with Trp-102Fab1/hc. The Glu-1947E1 side chain forms a salt bridge with Arg-109Fab1/hc and a hydrogen bond with Tyr-50Fab1/lc. Most of these interactions are conserved in E2 (Figs. 3D and 4B), as reflected by the high degree of overlap of the two binding sites (root mean square deviation of 0.8 Å, excluding solvent-exposed residues on the C-terminal side of the LR). Some key differences do exist between E1 and E2, including a different arrangement of the electrostatic interaction between Glu-1947E1 and Arg-100Fab1/hc; in E2, the structurally equivalent residue to Glu-1947E1 is Glu-2092E2 (which is a Pro residue at position 1960 in E1; see Fig. 4C). Other differences between the two epitopes are relatively minor and include a Ser-to-Asn substitution at position 2031 of E2 (Figs. 3D and 4B). Overall, differences between E1 and E2 are predicted to have little impact on binding of Fab, as demonstrated by similar binding free energies calculated for the two sites (Table 3).
FIGURE 4.

Bezlotoxumab epitopes and interactions with Fab residues. A, interacting residues within E1 (green) and Fab1 (yellow and pink for heavy and light chains, respectively). B, overlap of the Fab-interacting residues within E1 (green) and E2 (blue). C, structural conservation of a key salt bridge between Arg-100Fab/hc Glu residues in E1 (green) and E2 (blue). D, role of Gly-1963E1 (Ala-2095E2) in enabling key interactions between Ser-1900E1 (Asn-2031E2) and Trp-33Fab/hc, Tyr-52Fab/hc, and Arg-99Fab/hc.
TABLE 3.
Binding free energies for bezlotoxumab Fabs (and individual contributions by heavy and light chain residues) to the E1 and E2 regions of the TcdB CROP domain as well as to the whole B2 peptide
| Region | Binding free energy (ΔG) |
||
|---|---|---|---|
| Heavy chain | Light chain | Fab | |
| kcal/mol | |||
| E1 | −20.1 | −1.9 | −21.9 |
| E2 | −20.2 | −1.9 | −21.4 |
| E4 | 2340 | 246 | 2590 |
| B2 | NDa | ND | −43.3 |
a ND, not determined.
In addition to E1 and E2, two additional homologous regions, referred to herein as E3 and E4, are located in the C-terminal half of the domain (and therefore not present in peptide B2; see Fig. 3D). Based on the Western blotting, TdF, SPR, and HDX-MS data (Fig. 2), bezlotoxumab does not bind to these regions. In order to understand the lack of binding to E3 and E4, we constructed a homology model of peptide B4 (corresponding to the C-terminal half of the CROP domain and containing both E3 and E4; see Fig. 1B) based on the structure of B2. As expected, the model of B4 contains two CROP units consisting of 10 total SRs and 2 LRs, similar to B2. However, some key differences do exist between B2 and B4, specifically in the length of some of the connecting loops between SRs. Most significantly with regard to bezlotoxumab binding, the connecting loop between the two SRs that immediately follow each LR is longer in E3 than in E1 and E2, and this is predicted to sterically hinder Fab binding at this position (Fig. 3D). In addition, several substitutions at key positions within E3 and E4 may also explain the lack of bezlotoxumab binding at these regions. Most strikingly, the aforementioned salt bridges between Arg residues within the Fabs (at positions 59 and 100 of the heavy chain) and Glu residues within E1 (at positions 1902 and 1947) and E2 (at positions 2033 and 2092) are missing in E3 and E4 due to substitutions to non-Glu residues at the corresponding positions within these regions (Figs. 3D and 4C). As another example, Gly-1963E1 is substituted by a Lys at the equivalent position in E3, a change that probably perturbs the hydrogen bonding network between Ser-1900E1 (Asn-2031E2) and Trp-33Fab/hc/Tyr-52Fab/hc/Arg-99Fab/hc (Fig. 4D). Finally, significant van der Waals clashes would be expected from substitutions in E3 (relative to E1) at positions 2163 (Ser to Asp), 2265 (Glu to Ser), 2168 (Phe to Tyr), and 2229 (Gly to Lys) and in E4 (relative to E2) at positions 2336 (Asp to Thr), 2338 (Ser to Glu), and 2340 (Thr to Ile) (Fig. 3D). The recent publication of the structure of a C-terminal fragment of the TcdB CROP domain (25) provides additional insight into why bezlotoxumab does not bind to E4. The structure confirms the clashes predicted from the modeling described above and allows for the calculation of binding free energy values for binding of bezlotoxumab Fab to E4. As shown in Table 3, these values are in the high positive range, consistent with a very low binding affinity.
Insights into the Mechanism of TcdB Neutralization by Bezlotoxumab
To gain a better understanding of how bezlotoxumab might impact the activity of TcdB on its target host cells, we mapped the residues putatively involved in carbohydrate binding (residues that are structurally analogous to those of TcdA that interact with α-Gal-(1,3)-β-Gal-(1,4)-β-GlcNAcO(CH2)8CO2CH3 in the structure by Greco et al. (12)) onto the B2 structure (Fig. 5) and compared these regions with the epitopes identified by x-ray crystallography (E1 and E2) and those predicted by HDX-MS as described above (Fig. 2C). As expected, the crystal structure epitopes overlap significantly with the areas identified as being protected from deuteration in the HDX-MS studies. Interestingly, there is also significant overlap between bezlotoxumab-binding residues and putative carbohydrate-binding regions, suggesting that bound antibody may directly impact the ability of TcdB to bind to carbohydrates. Based on this information, and because it has been proposed, at least for TcdA, that toxin mediates its biological activities via binding to its target host cells through specific carbohydrate moieties on the cell surface (10–16), we assessed the ability of bezlotoxumab to neutralize the cytotoxic effects of TcdB on Vero cells (considered the gold standard for assessing TcdB activity due to their high sensitivity to this toxin). As shown in Fig. 6A, bezlotoxumab potently neutralizes the activity of TcdB in these cells. Furthermore, we observed (Fig. 6, B and C) that bezlotoxumab blocks binding of TcdB to the surface of Vero cells by measuring membrane-bound TcdB following incubation of cells with the toxin in the absence or presence of antibody. As expected, the isotype control anti-TcdA antibody actoxumab has no effect on TcdB binding in this assay. The binding experiments were carried out at 37 °C in the presence of the endocytosis inhibitor chlorpromazine, at a concentration (14 μm) previously shown to inhibit endocytosis in Vero cells (41). Similar results (not shown) were obtained when binding was carried out (i) at 4 °C in the absence of chlorpromazine and (ii) at 37 °C with Vero cell membranes rather than live cells.
FIGURE 6.
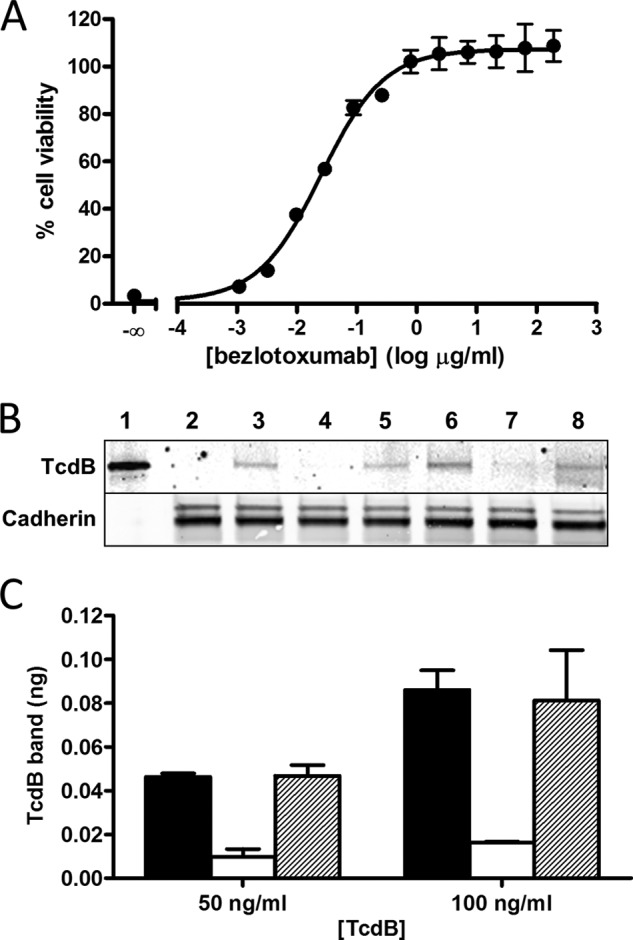
Inhibitory effect of bezlotoxumab on activity and binding of TcdB in Vero cells. A, neutralization of the cytotoxic effects of purified TcdB by bezlotoxumab. The concentration of TcdB used (10 pg/ml) was previously determined to cause a ∼95% decrease in cell viability in this assay. B, Western blots of Vero cell membranes following incubation of cells with 50 ng/ml (lanes 3–5) or 100 ng/ml (lanes 6–8) TcdB, in the absence and presence of bezlotoxumab or actoxumab (200 μg/ml). Binding experiments were carried out at 37 °C in the presence of the endocytosis inhibitor chlorpromazine fixed at 14 μm. Lane 1, 1 ng of purified TcdB (no membranes); lane 2, membranes from cells incubated without TcdB; lanes 3 and 6, membranes from cells incubated with TcdB in the absence of bezlotoxumab; lanes 4 and 7, membranes from cells incubated with TcdB in the presence of bezlotoxumab; lanes 5 and 8, membranes from cells incubated with TcdB in the presence of actoxumab. Levels of TcdB are shown in the top panel, and levels of cadherin (loading control) are shown in the bottom panel. C, quantification of membrane-bound TcdB (normalized to the 1 ng of TcdB control lane and to cadherin band densities) following incubation of Vero cells in the presence of 50 or 100 ng/ml TcdB alone (black bars) or in the presence of bezlotoxumab (white bars) or actoxumab (hatched bars). Values are averages ± S.D. of two independent determinations.
Model of Bezlotoxumab Bound to Full-length TcdB CROP Domain
Taken together, the biophysical and crystal structure data demonstrate that two Fab molecules bind to a single TcdB CROP domain, raising the question of whether one or two intact IgG molecules of bezlotoxumab can bind to TcdB. Studies using size exclusion chromatography (Fig. 7) demonstrate that the stoichiometry of bezlotoxumab to TcdB CROP domain (B1 peptide) is 1:1, regardless of the ratio of bezlotoxumab to B1, consistent with a model wherein the two distinct TcdB epitopes are occupied by each Fab region of a single bezlotoxumab IgG molecule. We therefore constructed a model of the full-length TcdB CROP domain (residues 1836–2367) bound to an intact molecule (IgG1) of bezlotoxumab (Fig. 8) by fusing the crystal structure of the B2 peptide to a model of the B4 peptide (based itself on the structure of B2). The Fc domain of bezlotoxumab was modeled on a published IgG1 structure (PDB entry 1HZH (42)). The TcdB CROP domain forms a horseshoe-shaped elongated structure, consistent with previous models (5, 23, 25). The bezlotoxumab antibody binds perpendicularly to the curvature of the CROP domain at sites that partially overlap with two of the four putative carbohydrate-interacting pockets, which are located largely along the convex surface of the CROP domain.
FIGURE 7.

SEC-MALLS analysis of bezlotoxumab, B1, and bezlotoxumab-B1 immune complexes. A, light scattering and molecular mass distributions of complexes formed at various bezlotoxumab/B1 molar ratios (1:0 (magenta), 0:1 (olive green), 1:5 (bright green), 1:1 (red), and 5:1 (navy blue)) as a function of elution time. B, average detected molecular mass of each complex. The molecular masses of the B1-bezlotoxumab complexes are approximately equal to the sum of B1 alone and bezlotoxumab alone, indicating a 1:1 stoichiometry.
FIGURE 8.
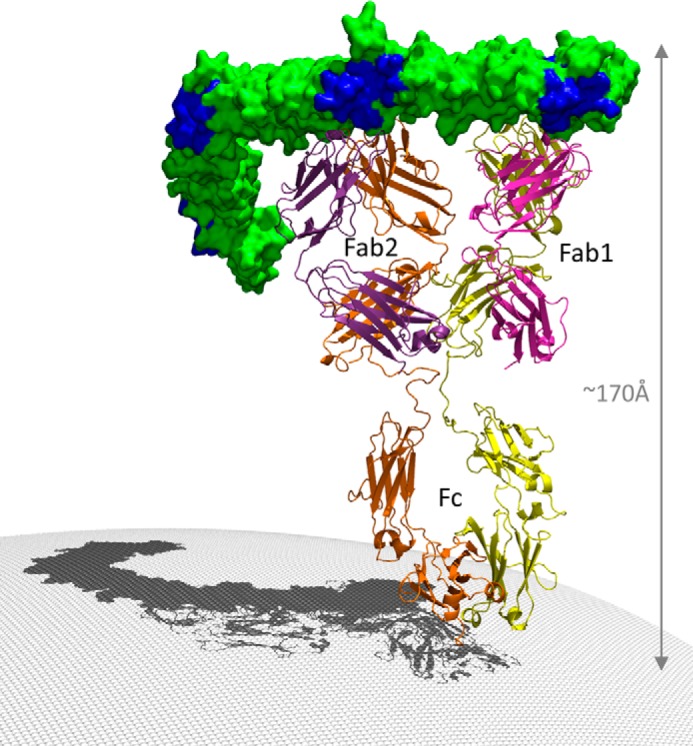
Model of the TcdB CROP domain bound to a full-length molecule of bezlotoxumab. The C-terminal (left) half of the CROP domain was modeled based on the structure of the B2 peptide. The Fc region of bezlotoxumab is based on a published high-resolution structure of human IgG1. The four putative carbohydrate binding regions are shown in dark blue within the otherwise green CROP domain.
DISCUSSION
We present here the structural characterization of the TcdB CROP domain in complex with the Fab fragments from the neutralizing antibody bezlotoxumab, not only providing important insight into the mechanism of neutralization of this important clinical antibody but also furthering our understanding of the TcdB CROP domain structure. The overall β-solenoid fold of the N-terminal half of the TcdB CROP domain is in large part consistent with the previously published model based on the TcdA crystal structure (7, 23) and with the recent structure of a C-terminal fragment of the TcdB CROP (25), but the structure described herein provides additional information, including the precise orientations of the β-hairpins/loops and side chain conformations of the N-terminal half of the CROP domain. Combined with the structure of the C-terminal peptide TcdB-B1 (slightly less than one full CROP unit encompassing four SRs and one LR) published recently (25), the structure of the N-terminal peptide B2 described herein (two full CROP units encompassing 10 SRs and 2 LRs) provides structural information for nearly 73% of the entire TcdB CROP domain.
The structure of the B2-Fab complex also reveals how bezlotoxumab interacts with the TcdB CROP domain. The SPR data (Table 2) provided a first clue that bezlotoxumab can bind to TcdB at two distinct binding sites, and this was confirmed by the crystal structure. The identification by SPR of high affinity and low affinity sites for bezlotoxumab binding to peptides B1 and B2 (with a single low affinity site in B3 and no binding to B4; see Table 2) suggests one of two distinct possibilities: (i) bezlotoxumab has distinct affinities for the two epitopes E1 and E2, with E2 corresponding to the low affinity site (because peptide B3 contains only this epitope), or (ii) the antibody binds to the two epitopes cooperatively, with the low affinity state identified by SPR corresponding to bezlotoxumab binding at a single epitope and the high affinity state corresponding to binding at both epitopes. The similar binding free energies calculated for the two epitopes (Table 3) supports the latter possibility.
Taken together, the Western blotting, temperature-dependent fluorescence, SPR, HDX-MS (Fig. 2), and crystal structure (Fig. 3) data provide insight into the mechanism of neutralization of TcdB by bezlotoxumab. The observation that the stoichiometry of bezlotoxumab to TcdB CROP is 1:1 even in the presence of a 5-fold excess of antibody (Fig. 7) suggests that bezlotoxumab binding to TcdB does not lead to antibody-mediated cross-linking, supporting the notion that bezlotoxumab directly neutralizes the toxin rather than decreasing its effective concentration via the formation of large immune complexes. As noted previously, the preponderance of existing data suggests that TcdA binds to its target host cells through interactions of its CROP domain with specific carbohydrate moieties on the cell surface. Indeed, binding of specific α-galactose-containing carbohydrate ligands to the CROP domain of TcdA has been demonstrated (10–16), and it has been shown that toxin binding is abolished upon treatment with α-galactosidase (13–15). Furthermore, TcdA-induced cell rounding is significantly reduced in the presence of intact TcdA CROP domain (18). Although a formal demonstration that TcdB binds to cell surface carbohydrates via its CROP domain is still lacking, it is tempting to ascribe a similar mode of binding to TcdB based on the high level of structural and functional homology between the two toxins and on the observation that TcdB binds to carbohydrates that are structurally related to the known TcdA ligands. As shown in Figs. 3D and 5, a significant overlap exists between the bezlotoxumab epitopes and the carbohydrate binding pockets of TcdB. Furthermore, bezlotoxumab directly blocks binding of TcdB to Vero cells (Fig. 6). These observations support a mechanism whereby bezlotoxumab neutralizes TcdB through direct blockade of receptor binding at the sites located near E1 and E2. Because additional putative carbohydrate binding pockets (to which bezlotoxumab does not bind) are located near E3 and E4, binding to the cell surface at these latter sites may be prevented (or significantly reduced) either through steric hindrance provided by bezlotoxumab bound at E1 and E2 or because high-affinity binding of TcdB to the cell surface requires at least three intact carbohydrate binding pockets. The fact that TcdA and TcdB have evolved to contain seven and four such pockets, respectively, supports the notion that multiple interactions with the cell surface are necessary for full binding/biological activity of the toxins (10). In further support of this, Just and co-workers (18) have previously shown that a peptide spanning the entire TcdA CROP domain efficiently competes with intact TcdA in a cell cytotoxicity assay, whereas a peptide spanning only the N-terminal half (three putative carbohydrate binding pockets) is unable to compete even at a high molar ratio.
The binding motif of bezlotoxumab described here is distinct from those of the camelid single-domain antibodies A20.1, A26.8, and B39, recently described by Murase et al. (25). To better understand these differences, we overlapped the structures of (i) A20.1 bound to a TcdA CROP fragment, (ii) B39 bound to a TcdB CROP fragment, and (iii) bezlotoxumab bound to peptide B2, aligning the highly homologous β-solenoid backbones of the three CROP fragments (Fig. 9). We also included the α-Gal-(1,3)-β-Gal-(1,4)-β-GlcNAcO(CH2)8CO2CH3 nestled within its binding pocket in TcdA, as described by Greco et al. (12). We excluded the neutralizing antibody A26.8, which binds to the extreme C terminus of TcdA and for which no homologous epitope exists within peptide B2. The model reveals the distinct epitopes of each antibody and suggests that binding of bezlotoxumab is more likely to interfere with carbohydrate binding than binding of A20.1 and B39. This is in line with the fact that bezlotoxumab is a fully neutralizing antibody, whereas B39 is non-neutralizing (25) and A20.1 is poorly neutralizing (43).
FIGURE 9.

Alignment of the structures of various CROP-antibody complexes. The backbones of the CROP domains of TcdA and TcdB were used for the alignment. Binding of nanobodies A20.1 (green) and B39 (white) as well as of Fab2 (yellow and cyan) and Fab1 (pink and purple) is shown, as is the TcdA carbohydrate ligand α-Gal-(1,3)-β-Gal-(1,4)-β-GlcNAcO(CH2)8CO2CH3.
Although the data presented in Fig. 6 show that bezlotoxumab interferes with TcdB binding to Vero cells and that this is the likely mechanism of toxin neutralization in this system, they do not entirely rule out the possibility that bezlotoxumab binding to the TcdB CROP domain neutralizes toxin by a mechanism other than direct occlusion of the receptor binding pockets of TcdB. As noted above, carbohydrate binding has not been unequivocally demonstrated to be the basis for TcdB binding to the cell surface, nor has the CROP domain itself been confirmed to be the receptor binding domain of TcdB; indeed, recent data have challenged the notion that the CROP domain is the sole determinant of cell binding in both TcdA and TcdB (20, 21). Furthermore, the residues of TcdB defined herein as being involved in carbohydrate binding are based on the structure of TcdA bound to α-Gal-(1,3)-β-Gal-(1,4)-β-GlcNAcO(CH2)8CO2CH3, and it is possible that the TcdB ligand interacts with different residues (encompassing a smaller or larger surface area) within TcdB than are highlighted here in Figs. 3 and 5. Therefore, the possibility exists that bezlotoxumab prevents TcdB binding to cells through conformational alterations in TcdB that mask its receptor binding regions or through steric hindrance by preventing cell binding via a distal receptor pocket (located on a domain other than the CROP). Another possibility is that bezlotoxumab does indeed occlude the receptor binding pockets of TcdB but that these pockets are distinct from the carbohydrate-binding domains, perhaps overlapping to an even larger extent with the epitopes of bezlotoxumab.
As mentioned above, the two Fab regions of a single bezlotoxumab molecule bind to TcdB at two distinct sites, E1 and E2. Of the 18 bezlotoxumab-interacting residues in common between E1 and E2, only 10 are identical, although 6 of the 8 amino acids substitutions are conservative (Fig. 3D). The structural tolerance for binding exhibited by bezlotoxumab suggests that this antibody should bind to and neutralize toxins from various C. difficile strains, although this remains to be demonstrated. Notably, the TcdB sequence of the hypervirulent strain NAP1/BI/027 is 70% identical and 90% homologous compared with that of VPI 10463 (against which bezlotoxumab was raised) within E1 (90% identical within E2), yet bezlotoxumab is fully protective against this strain in a piglet model of infection (31). Importantly, the flexibility inherent in binding of bezlotoxumab to its epitopes is not unlimited because the antibody does not bind to the E1/E2-homologous regions E3 and E4, nor does it bind to TcdA, despite the overall similar β-solenoid fold of the TcdA CROP domain.
The data presented here not only enhance our understanding of the structure of the TcdB CROP domain but also provide a possible structural basis for neutralization of TcdB by the clinically important antibody bezlotoxumab. Future studies should focus on elucidating structure-function relationships involved in carbohydrate binding for TcdB and how/whether such binding is truly the basis for toxin binding to host cells, as well as exploring the structural basis for binding/neutralization of TcdA to actoxumab, the other half of the important novel C. difficile therapy that is currently in phase III development for the prevention of recurrent CDI.
Supplementary Material
Acknowledgment
We thank Dr. Rumin Zhang for help with the interpretation of the SPR data.

This article contains supplemental Tables 1 and 2.
The atomic coordinates and structure factors (code 4NP4) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CDI
- C. difficile infection(s)
- CROP
- combined repetitive oligopeptide
- GTD
- glucosyl transferase domain
- HDX
- hydrogen/deuterium exchange
- LR
- long repeat
- SEC-MALLS
- size exclusion chromatography coupled with multiangle laser light scattering
- SPR
- surface plasmon resonance
- SR
- short repeat
- TdF
- temperature-dependent fluorescence
- PDB
- Protein Data Bank.
REFERENCES
- 1. Rupnik M., Wilcox M. H., Gerding D. N. (2009) Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 7, 526–536 [DOI] [PubMed] [Google Scholar]
- 2. Bassetti M., Villa G., Pecori D., Arzese A., Wilcox M. (2012) Epidemiology, diagnosis and treatment of Clostridium difficile infection. Expert Rev. Anti. Infect. Ther. 10, 1405–1423 [DOI] [PubMed] [Google Scholar]
- 3. Carter G. P., Rood J. I., Lyras D. (2012) The role of toxin A and toxin B in the virulence of Clostridium difficile. Trends Microbiol. 20, 21–29 [DOI] [PubMed] [Google Scholar]
- 4. Shen A. (2012) Clostridium difficile toxins: mediators of inflammation. J. Innate Immun. 4, 149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pruitt R. N., Lacy D. B. (2012) Toward a structural understanding of Clostridium difficile toxins A and B. Front. Cell Infect. Microbiol. 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Papatheodorou P., Zamboglou C., Genisyuerek S., Guttenberg G., Aktories K. (2010) Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS One 5, e10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pruitt R. N., Chambers M. G., Ng K. K., Ohi M. D., Lacy D. B. (2010) Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc. Natl. Acad. Sci. U.S.A. 107, 13467–13472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qa'Dan M., Spyres L. M., Ballard J. D. (2000) pH-induced conformational changes in Clostridium difficile toxin B. Infect. Immun. 68, 2470–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pfeifer G., Schirmer J., Leemhuis J., Busch C., Meyer D. K., Aktories K., Barth H. (2003) Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J. Biol. Chem. 278, 44535–44541 [DOI] [PubMed] [Google Scholar]
- 10. Dingle T., Wee S., Mulvey G. L., Greco A., Kitova E. N., Sun J., Lin S., Klassen J. S., Palcic M. M., Ng K. K., Armstrong G. D. (2008) Functional properties of the carboxy-terminal host cell-binding domains of the two toxins, TcdA and TcdB, expressed by Clostridium difficile. Glycobiology 18, 698–706 [DOI] [PubMed] [Google Scholar]
- 11. El-Hawiet A., Kitova E. N., Kitov P. I., Eugenio L., Ng K. K., Mulvey G. L., Dingle T. C., Szpacenko A., Armstrong G. D., Klassen J. S. (2011) Binding of Clostridium difficile toxins to human milk oligosaccharides. Glycobiology 21, 1217–1227 [DOI] [PubMed] [Google Scholar]
- 12. Greco A., Ho J. G., Lin S. J., Palcic M. M., Rupnik M., Ng K. K. (2006) Carbohydrate recognition by Clostridium difficile toxin A. Nat. Struct. Mol. Biol. 13, 460–461 [DOI] [PubMed] [Google Scholar]
- 13. Krivan H. C., Clark G. F., Smith D. F., Wilkins T. D. (1986) Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Galα1–3Galβ1–4GlcNAc. Infect. Immun. 53, 573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pothoulakis C., Gilbert R. J., Cladaras C., Castagliuolo I., Semenza G., Hitti Y., Montcrief J. S., Linevsky J., Kelly C. P., Nikulasson S., Desai H. P., Wilkins T. D., LaMont J. T. (1996) Rabbit sucrase-isomaltase contains a functional intestinal receptor for Clostridium difficile toxin A. J. Clin. Invest. 98, 641–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pothoulakis C., LaMont J. T., Eglow R., Gao N., Rubins J. B., Theoharides T. C., Dickey B. F. (1991) Characterization of rabbit ileal receptors for Clostridium difficile toxin A. Evidence for a receptor-coupled G protein. J. Clin. Invest. 88, 119–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tucker K. D., Wilkins T. D. (1991) Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect. Immun. 59, 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. von Eichel-Streiber C., Laufenberg-Feldmann R., Sartingen S., Schulze J., Sauerborn M. (1992) Comparative sequence analysis of the Clostridium difficile toxins A and B. Mol. Gen. Genet. 233, 260–268 [DOI] [PubMed] [Google Scholar]
- 18. Frisch C., Gerhard R., Aktories K., Hofmann F., Just I. (2003) The complete receptor-binding domain of Clostridium difficile toxin A is required for endocytosis. Biochem. Biophys. Res. Commun. 300, 706–711 [DOI] [PubMed] [Google Scholar]
- 19. von Eichel-Streiber C., Sauerborn M., Kuramitsu H. K. (1992) Evidence for a modular structure of the homologous repetitive C-terminal carbohydrate-binding sites of Clostridium difficile toxins and Streptococcus mutans glucosyltransferases. J. Bacteriol. 174, 6707–6710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Olling A., Goy S., Hoffmann F., Tatge H., Just I., Gerhard R. (2011) The repetitive oligopeptide sequences modulate cytopathic potency but are not crucial for cellular uptake of Clostridium difficile toxin A. PLoS One 6, e17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schorch B., Song S., van Diemen F. R., Bock H. H., May P., Herz J., Brummelkamp T. R., Papatheodorou P., Aktories K. (2014) LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. Proc. Natl. Acad. Sci. U.S.A. 10.1073/pnas.1323790111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. von Eichel-Streiber C., Sauerborn M. (1990) Clostridium difficile toxin A carries a C-terminal repetitive structure homologous to the carbohydrate binding region of streptococcal glycosyltransferases. Gene 96, 107–113 [DOI] [PubMed] [Google Scholar]
- 23. Ho J. G., Greco A., Rupnik M., Ng K. K. (2005) Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. Proc. Natl. Acad. Sci. U.S.A. 102, 18373–18378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Albesa-Jové D., Bertrand T., Carpenter E. P., Swain G. V., Lim J., Zhang J., Haire L. F., Vasisht N., Braun V., Lange A., von Eichel-Streiber C., Svergun D. I., Fairweather N. F., Brown K. A. (2010) Four distinct structural domains in Clostridium difficile toxin B visualized using SAXS. J. Mol. Biol. 396, 1260–1270 [DOI] [PubMed] [Google Scholar]
- 25. Murase T., Eugenio L., Schorr M., Hussack G., Tanha J., Kitova E. N., Klassen J. S., Ng K. K. (2014) Structural basis for antibody recognition in the receptor-binding domains of toxins A and B from Clostridium difficile. J. Biol. Chem. 289, 2331–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kuehne S. A., Cartman S. T., Heap J. T., Kelly M. L., Cockayne A., Minton N. P. (2010) The role of toxin A and toxin B in Clostridium difficile infection. Nature 467, 711–713 [DOI] [PubMed] [Google Scholar]
- 27. Kuehne S. A., Collery M. M., Kelly M. L., Cartman S. T., Cockayne A., Minton N. P. (2014) The importance of toxin A, toxin B and CDT in virulence of an epidemic Clostridium difficile strain. J. Infect. Dis. 209, 83–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lyras D., O'Connor J. R., Howarth P. M., Sambol S. P., Carter G. P., Phumoonna T., Poon R., Adams V., Vedantam G., Johnson S., Gerding D. N., Rood J. I. (2009) Toxin B is essential for virulence of Clostridium difficile. Nature 458, 1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Babcock G. J., Broering T. J., Hernandez H. J., Mandell R. B., Donahue K., Boatright N., Stack A. M., Lowy I., Graziano R., Molrine D., Ambrosino D. M., Thomas W. D., Jr. (2006) Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect. Immun. 74, 6339–6347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kink J. A., Williams J. A. (1998) Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect. Immun. 66, 2018–2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Steele J., Mukherjee J., Parry N., Tzipori S. (2013) Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J. Infect. Dis. 207, 323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Torres J. F., Lyerly D. M., Hill J. E., Monath T. P. (1995) Evaluation of formalin-inactivated Clostridium difficile vaccines administered by parenteral and mucosal routes of immunization in hamsters. Infect. Immun. 63, 4619–4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lowy I., Molrine D. C., Leav B. A., Blair B. M., Baxter R., Gerding D. N., Nichol G., Thomas W. D., Jr., Leney M., Sloan S., Hay C. A., Ambrosino D. M. (2010) Treatment with monoclonal antibodies against Clostridium difficile toxins. N. Engl. J. Med. 362, 197–205 [DOI] [PubMed] [Google Scholar]
- 34. Sheth P. R., Liu Y., Hesson T., Zhao J., Vilenchik L., Liu Y. H., Mayhood T. W., Le H. V. (2011) Fully activated MEK1 exhibits compromised affinity for binding of allosteric inhibitors U0126 and PD0325901. Biochemistry 50, 7964–7976 [DOI] [PubMed] [Google Scholar]
- 35. Coales S. J., Tuske S. J., Tomasso J. C., Hamuro Y. (2009) Epitope mapping by amide hydrogen/deuterium exchange coupled with immobilization of antibody, on-line proteolysis, liquid chromatography and mass spectrometry. Rapid Commun. Mass Spectrom. 23, 639–647 [DOI] [PubMed] [Google Scholar]
- 36. Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., Bricogne G. (2011) Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67, 293–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 38. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 39. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luzzati V. (1952) Traitement statistique des erreurs dans la determination des structures cristallines. Acta Cryst. 5, 802–810 [Google Scholar]
- 41. Hernaez B., Alonso C. (2010) Dynamin- and clathrin-dependent endocytosis in African swine fever virus entry. J. Virol. 84, 2100–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saphire E. O., Parren P. W., Pantophlet R., Zwick M. B., Morris G. M., Rudd P. M., Dwek R. A., Stanfield R. L., Burton D. R., Wilson I. A. (2001) Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science 293, 1155–1159 [DOI] [PubMed] [Google Scholar]
- 43. Hussack G., Arbabi-Ghahroudi M., van Faassen H., Songer J. G., Ng K. K., MacKenzie R., Tanha J. (2011) Neutralization of Clostridium difficile toxin A with single-domain antibodies targeting the cell receptor binding domain. J. Biol. Chem. 286, 8961–8976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. United States Centers for Disease Control and Prevention (2013) Antibiotic Resistance Threats in the United States, 2013, United States Department of Health and Human Services, Atlanta, GA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.