

Background: Plant fatty acid desaturase (FAD) enzymes determine the desaturation status of plant lipids.
Results: FAD enzymes form homo- and heterodimers; FAD2-FAD3 heterodimers can channel oleate to linolenate without releasing the linoleate intermediate.
Conclusion: A physiological function for FAD2-FAD3 heterodimers has been demonstrated.
Significance: That monoenes can be channeled to trienes has implications for engineering desired plant fatty acid compositions.
Keywords: Fatty Acid, Fatty Acid Metabolism, Flavin Adenine Dinucleotide (FAD), Lipid Metabolism, Lipid Synthesis, Plant Biochemistry, Desaturation, Fatty Acid Desaturase, Metabolic Channeling, Oligomerization
Abstract
Plant desaturases comprise two independently evolved classes, a structurally well characterized soluble class responsible for the production of monoenes in the plastids of higher plants and the poorly structurally characterized integral membrane class that has members in the plastid and endoplasmic reticulum that are responsible for producing mono- and polyunsaturated fatty acids. Both require iron and oxygen for activity and are inhibited by azide and cyanide underscoring their common chemical imperatives. We previously showed that the Δ9 acyl-CoA integral membrane desaturase Ole1p from Saccharomyces cerevisiae exhibits dimeric organization, like the soluble plastidial acyl-ACP desaturases. Here we use two independent bimolecular complementation assays, i.e. yeast two-hybrid analysis and Arabidopsis leaf protoplast split luciferase assay, to demonstrate that members of the plant integral membrane fatty acid desaturase (FAD) family, FAD2, FAD3, FAD6, FAD7, and FAD8, self-associate. Further, the endoplasmic reticulum-localized desaturase FAD2 can associate with FAD3, as can the plastid-localized FAD6 desaturase with either FAD7 or FAD8. These pairings appear to be specific because pairs such as FAD3 and FAD7 (or FAD8) and FAD2 and FAD6 do not interact despite their high amino acid similarity. These results are consistent also with their known endoplasmic reticulum and plastid subcellular localizations. Chemical cross-linking experiments confirm that FAD2 and FAD3 can form dimers like the yeast Ole1p and, when coexpressed, can form FAD2-FAD3 heterodimers. Metabolic flux analysis of yeast coexpressing FAD2 and FAD3 indicates that heterodimers can form a metabolic channel in which 18:1-PC is converted to 18:3-PC without releasing a free 18:2-PC intermediate.
Introduction
Desaturase enzymes typically introduce cis double bonds into fatty acids in a process that requires a supply of reductant and molecular oxygen (1). There are two evolutionarily distinct forms of desaturase enzymes, a predominant and widespread class of integral membrane enzymes that occur in organisms as diverse as fungi, bacteria, animals, and plants and a soluble class of acyl-ACP desaturases that are primarily found in higher plant plastids (2, 3). The membrane desaturases belong to an enzyme family that includes bacterial diiron-containing alkane hydroxylase (4) and toluene monooxygenase, in addition to plant aldehyde decarbonylases and sterol methyl oxidases (4).
In plants, the saturated product of fatty acid biosynthesis, stearic acid is converted to the cis-Δ9-monounsaturated fatty acid oleate by the action of the soluble Δ9-18:0-ACP desaturase2 (5, 6). Membrane desaturases subsequently convert oleate to linoleic (cis,cis-Δ9,12-octadecadienoic) acid and α-linolenic (all cis-Δ9,12,15-octadecatrienoic) acids, which are the most abundant fatty acids in plants (7, 8). Polyunsaturated fatty acids are synthesized by one of two parallel pathways commonly referred to as the prokaryotic and eukaryotic pathways that are located in the plastids and endoplasmic reticulum, respectively (9). Fatty acids are esterified to different lipids in the two compartments, phosphatidycholine (PC)3 in the ER and glycosylglycerides in the plastid. Conversion of oleic to linoleic acid is mediated by FAD2 in the ER and by FAD6 in the plastids, and conversion of linoleic to α-linolenic acid is mediated by FAD3 in the ER and by either FAD7 or FAD8 in the plastid (10). Trigalactosyldiacylglycerol proteins have been shown to mediate the transport of lipids from the ER to the plastid, thereby providing a means of trafficking of fatty acids from the cytoplasm back to their site of synthesis (11).
Membrane-bound fatty acid desaturases share a degree of sequence similarity that includes three motifs comprised of eight conserved histidine residues thought to be important in forming a diiron center at the active site (12). They differ functionally in terms of their affinity not only for different types of glycerolipids but also for different fatty acid substrates as described above.
Soluble acyl-ACP desaturases are readily expressed in Escherichia coli in quantities sufficient for their characterization, which enabled valuable correlations to be made between their structure and function (13). For instance the crystal structure of the castor and ivy acyl-ACP desaturases confirmed their homodimeric organization mediated by deep interdigitation of loops between protomers adjacent to the four-helix bundles in which the diiron active site is located (14, 15). Based on the dimeric structure of soluble desaturase, a mechanism involving half-of-the-sites reactivity was proposed in which the desolvation energy released upon substrate binding would be transferred to the second subunit to help facilitate product release (16), the proposed limiting step in desaturation (17). Far less is known about the organization of membrane desaturases, which have generally proved recalcitrant to overexpression, purification, and characterization.
To circumvent the need for purified materials for biophysical analysis, we have undertaken a molecular genetic approach to understanding the organization of the membrane desaturases. In a previous study, we presented evidence that functional yeast Δ9-desaturase Ole1 forms a dimer in vivo (18). To determine whether the observed dimeric organization of the yeast desaturase is shared with other desaturases, we undertook the present study of a series of higher plant membrane desaturases, FAD2, FAD3, FAD6, FAD7, and FAD8, using two independent bimolecular complementation assays. The first involves two-hybrid analysis in yeast; the second employs a split luciferase assay in Arabidopsis leaf protoplasts. Data from these studies show that all the FAD desaturases investigated herein are capable of forming self-associations in vivo. In addition to self-association, we also identified specific desaturase associations between FAD2 and FAD3 of the ER and between FAD6 and FAD7 or FAD8 of the plastids. The bimolecular complementation analysis is supported by evidence from chemical cross-linking experiments visualized by Western blot analysis in which FAD2-FAD3 heterodimers were formed upon coexpression in yeast. Furthermore, metabolic flux analysis shows when FAD2 and FAD3 are coexpressed that labeled oleate can be channeled to α-linolenic acid without release of the linoleic acid intermediate.
EXPERIMENTAL PROCEDURES
Fatty Acid Analysis
Cultures were initiated from single yeast colonies. Yeast cells (YPH499) were cultured overnight in SD −Ura containing 2% glucose at 30 °C (18). Upon reaching an A600 of 0.5, cell pellets were collected by centrifugation and washed with SD −Ura lacking dextrose, after which the cells were induced at 30 °C for 4 h in SD −Ura supplemented with 2% galactose, 0.5% Tergitol Nonidet P-40, 0.1 mm oleic acid, and 0.1 mm linoleic acid, followed by incubation at 18 °C for between 1 and 4 days. The cells were collected from 0.5 ml of culture by centrifugation, washed with 1% Tergitol Nonidet P-40 followed by water, and dried under a stream of N2. Lipids were extracted with two volumes of methanol:chloroform:formic acid (2:1:1) by grinding with glass beads for 5 min before supplementing with ½ volume of 1 m KCl, 0.2 m H3PO4 and separating the organic and aqueous phases by centrifugation. Heptadecanoic acid was used as an internal standard. PC was isolated from total lipids by separation on TLC plates pretreated with 0.15 m (NH4)2SO4 and developed with acetone:toluene:H2O (91:30:7) before staining with iodine vapor. Fatty acids were esterified by incubation with 1 ml of 1 n HCl-methanol (or 1 ml of boron trichloride) at 80 °C for 1 h. Following incubation, 1 ml of 0.9% NaCl was added, and fatty acids were extracted into 2 ml of hexane. The resulting fatty acid methyl esters (FAMEs) were concentrated by evaporation under a stream of nitrogen.
Samples were analyzed on a Hewlett-Packard 6890 (Hewlett-Packard, Roseville, CA) gas chromatograph equipped with a 5973 mass selective detector (GC/MS) and a Supelco 60 m × 250 μm SP-2340 cyano capillary column. The oven temperature was raised from 100 to 240 °C at a rate of 15 °C/min and then held constant at 240 °C for 5 min, with a helium flow of 1.1 ml/min.
For the PC channeling assay, the GC/MS acquisition was performed using electron impact ionization operating in selected ion monitoring mode, which gives adequate sensitivity to quantitate the isotope ratios in the molecular ion (M+) clusters of oleic, linoleic, and linolenic acid methyl esters with m/z 296.3 (C19H36O2), m/z 264.3 (C19H34O2), and m/z 262.3 (C19H32O2), respectively (selected ion monitoring parameters: m/z are 291.20, 292.20, 293.20, 294.20, 295.20, 296.20, 297.20, 298.20, 299.20, 300.20, and 301.20). The fractions of unlabeled fatty acid molecules, as well as molecules heavier by one or two mass units (for 13C1 and d2 label, respectively), were obtained after numerical correction of the mass isotopomer distribution vector for the contribution of naturally occurring isotopes of carbon, hydrogen, and oxygen based on multiplication with the inverse of the isotope correction matrices for C19H36O2, C19H34O2, and C19H32O2, respectively. The correction matrices were derived as defined by van Winden et al. (19).
Yeast Transformation and Split Ubiquitin Analysis
Yeast transformation was performed with Yeastmaker Yeast Transformation System 2 (Clontech). All split ubiquitin analysis (20) was performed essentially according to the Frommer lab protocols.
Protoplast Preparation and Transformation
Arabidopsis protoplasts were prepared based on the “tape-Arabidopsis sandwich” method developed by Lin and co-workers (21). Leaves were harvested from 3–5-week-old plants. A tape-leaf sandwich was created by adhering 3 m tape to the upper and lower epidermis. After stripping the lower epidermis, the leaf was incubated in an enzyme solution containing 1% cellulose Onozuka R10 (Yakult, Tokyo, Japan), 0.25% macerozyme Onozuka R10 (Yakult), 0.4 m mannitol, 10 mm CaCl2, 20 mm KCl, 0.1% BSA, 20 mm MES, pH 5.7. Protoplast cells were released into the solution, collected, washed in W5 solution (154 mm NaCl, 125 mm CaCl2, 5 mm glucose, 2 mm MES, pH 5.7), and resuspended in modified MMg solution (0.4 m mannitol, 15 mm MgCl2, 4 mm MES, pH 5.7) at ∼5 × 105 cells/ml before use.
Protoplasts were transfected by established methods (18). Briefly, ∼5 × 104 protoplasts in 0.2 ml of MMg solution were mixed with 30 μg of plasmid DNA and diluted 1:1 (v:v) with a freshly prepared solution containing 40% (w/v) PEG (molecular weight, 4000; Fluka-Sigma-Aldrich), 0.1 m CaCl2, and 0.2 m mannitol. The mixture was incubated at room temperature for 5 min, before the gradual addition of 3 ml of W5 solution. Protoplasts were collected and washed with W5 solution before gentle resuspension in 1 ml of W5 solution and incubation in darkness for 16 h before analysis. The DNA vectors used for split luciferase assay were constructed according to Ref. 22.
Luminescence Detection
90 μl of protoplast cell suspensions were transferred to wells in a 96-well plate to which 10 μl of 60 μm coelenterazine (ViviRen live cell substrate; Promega, Madison, WI) was added. The plate was incubated for 40 min at 22 °C in the dark before measuring the luminescence. Luminescence from each well was quantified with 1-s integration periods using a Packard TopCount NXT microplate scintillation and luminescence counter (Packard, PerkinElmer Life Sciences).
Yeast Protein Extraction and Western Blot Analysis
Yeast cells were lysed in Y-PER yeast extraction buffer (Thermo Fisher Scientific). Following 15 min of centrifugation at 12,000 × g, the supernatant was supplemented with 0.5% protease inhibitor (Sigma-Aldrich), and the proteins were separated by 10% SDS-PAGE, transferred to a PVDF membrane, and analyzed by Western blot using monoclonal anti-FLAG M2-alkaline phosphatase antibody (Sigma-Aldrich). The reactive bands on membranes were visualized after incubation with nitroblue tetrazolium/bromochloroindolyl phosphate. Protein levels were estimated using densitometry with image analysis software (Alpha ImagerTM 2200 version 5.5; Alpha Innotech Corporation, San Leandro, CA).
Cross-linking
Yeast cells were collected by centrifugation, washed once with water, and disintegrated in PBS buffer containing 0.2% perfluorooctanoic acid (Sigma-Aldrich) by vigorous shaking with glass beads. After 15 min of centrifugation at 12,000 × g, the supernatant was supplemented with 0.5% protease inhibitor (Sigma-Aldrich). For cross-linker treatment, Lomant's reagent, dithiobis[succinimidyl propionate (Sigma-Aldrich) was added to the supernatant to a final concentration of 2 mm. After incubation for 0.5 h on ice, Tris-HCl (pH 7.5) was added to a final concentration of 20 mm to terminate the reaction. The mixture was mixed with Laemmli buffer lacking reducing reagent, and the proteins were separated on a 4–15% gradient gel (Bio-Rad), transferred to a PVDF membrane, and analyzed by Western blotting with monoclonal anti-FLAG M2-alkaline phosphatase antibodies. Immunoreactive polypeptides were visualized after incubation with nitroblue tetrazolium/bromochloroindolyl phosphate.
Metabolic Flux Analysis
Yeast YPH499 cells were cultured at 30 °C overnight in SD −Ura containing 2% glucose. Cell pellets were collected by centrifugation and washed with SD −Ura lacking dextrose. Cells were induced by the addition of SD −Ura containing 2% galactose when the A600 of the culture reached 0.5. Cultures were incubated for 4 h at 30 °C, followed by various times in the presence of 0.5% Tergitol Nonidet P-40, 0.1 mm of deuterium-labeled oleic acid 9,10-d2 (Sigma-Aldrich) and 0.1 mm of 1-13C-labeled linoleic acid (Santa Cruz Biotechnology, Inc., Dallas, TX). All experiments were performed at 18 °C. PC-bound 18:3Δ9,12,15 can be derived either via FAD3 from 18:2Δ9,12 bound to the free pool of PC, or via a FAD2-FAD3 complex from PC-bound 18:1Δ9. This relation is expressed in Equation 2,
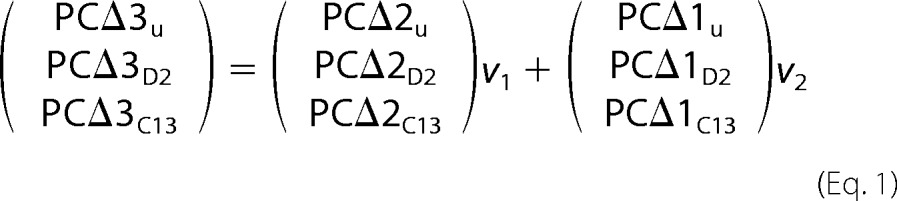 |
where v1 is the rate of conversion of PC-18:2Δ9,12 to PC-18:2Δ9,12,15 by FAD3, and v2 is the rate of conversion of PC-18:1Δ9 to PC-18:2Δ9,12,15 by combined activities of FAD2 andFAD3. The unlabeled and d2- and 13C-labeled fractions of 18:1Δ9 bound to PC are designated as PCΔ1u, PCΔ1D2, and PCΔ1C13, respectively. The fractions of the other PC-bound fatty acid species are designated accordingly, i.e. PCΔ2u, PCΔ2D2, and PCΔ2C13 (18:2Δ9,12) and PCΔ3u, PCΔ3D2, and PCΔ3C13 (18:3Δ9,12,15).
Using our experimentally measured values, the equation was solved for v1 and v2 by linear regression, using the LINEST function in Microsoft EXCEL, which solves a general linear equation of the form y = mx + b for the unknown m. To do so we set b to be 0 and substituted parts of Equation 2 into y, m, and x as follows.
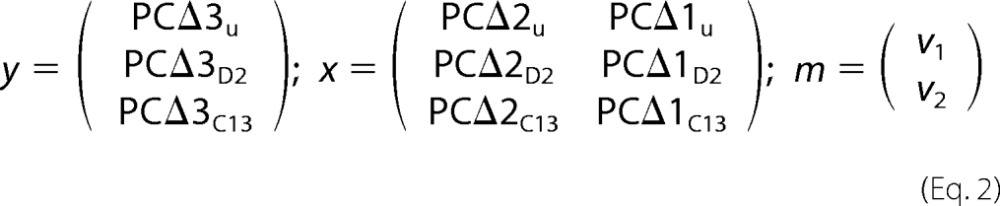 |
RESULTS
Membrane Desaturase Self-association
To test the hypothesis that plant membrane FAD-type desaturases, like their yeast counterpart Ole1, form homodimers, we subjected them to bimolecular complementation analysis using the split ubiquitin system in baker's yeast Saccharomyces cerevisiae. Membrane desaturases were individually cloned into a bait vector such that they were expressed with a C-terminal fragment of ubiquitin (Cub) followed by the artificial transcription factor protein A-LexA-VP16 (Fig. 1A). Upon interaction of bait and target proteins, a functional ubiquitin is formed that leads to release of PLV (Protein-LexA-VP16) to activate LexA, which regulates His3 and Ade2 expression to complement the prototrophy, thereby allowing growth on His−, Ade− medium.
FIGURE 1.

Membrane split ubiquitin yeast two-hybrid analysis shows FAD membrane desaturase associations. A, left panel, schematic representation of the pairs of bait and prey plasmid constructs with FAD2 as the example. Bait-Cub-artificial transcription factor PLV construct is driven by the met25 promoter; prey expression was by the alcohol dehydrogenase promoter. Right panel, growth of FAD2 test constructs on synthetic defined medium lacking L and T (SD-2). PC, positive control; NC, negative control; KanMX, kanamycin resistance module; Ole1, the yeast Ole1 Δ9-desaturase; T-H, tail to head; T-T, tail to tail. Two individual streaks are shown per treatment. B, challenge of yeast harboring the constructs illustrated in panel A on selective minimal SD-T-L-H-A-M (SD-5) medium for FAD2, FAD3, FAD6, FAD7, or FAD8 homoassociations as labeled.
To form a positive split ubiquitin association, the CubPLV and the N-terminal ubiquitin fragment (Nub) peptides must both be located on the cytosolic face of membrane. In the absence of detailed structural information such as a crystal structure, two prey vectors were constructed: a Nub G-desaturase fusion and a desaturase-Nub G fusion, the former to test for tail to head interaction and the latter to test for tail to tail interaction (by convention, head or tail is used to refer to the location of the ubiquitin fragment at the N or C terminus, respectively, as fusions to the target protein) (see Fig. 1A for a schematic diagram).
All construct-containing yeast grew on SD-L-T, but only the positive control, containing a WT Nub in the prey vector and the test prey constructs containing the tail to head construct grew on SD-L-T-H-A-M plates (Fig. 1B). Growth was not restored in yeast strains containing the plant desaturase FAD2 along with standard negative control prey vectors containing KanMX (Fig. 1B, NC1 and NC2), or when Ole1p, a related yeast desaturase, was tested as an additional negative control (Fig. 1B, NC3 and NC4). As shown in Fig. 1B, FAD2 self-associates, i.e. permits growth in a tail to head orientation, but not in a tail to tail orientation. FAD3, FAD6, FAD7, and FAD8 all conferred the same pattern of growth, indicating that like FAD2, they form specific self-associations. To confirm in vivo interactions, the FAD enzymes were subjected to a second and independent bimolecular complementation assay, employing Renilla split luciferase (RLUC) in Arabidopsis protoplast cells.
In principle, the two complementary fragments of the split luciferase can be fused to either the N or C terminus of the target protein. In practice, the functioning of combined luciferase fragments is dependent on their orientation with respect to the specific protein of interest because of steric and/or folding constraints (23). Thus, it can be useful to test several orientations. To achieve this, an N-terminal luciferase fragment was fused to the N terminus of the desaturase, and this was coexpressed with a C-terminal luciferase fragment fused to either the C or N terminus of the same desaturase creating 2H-2T and 2H-2H, respectively (Fig. 2A).
FIGURE 2.

Evaluation of the self-association of the various FADs in Arabidopsis leaves protoplasts using a RLUC bimolecular complementation assay. A, schematic representation of the orientation of FAD to the LUC (luciferase) fragment with respect to RLUC in which N- and C-terminal LUC fragments are fused to bait or prey genes via a 10-amino acid linker (-GGGGSGGGGS-). B, means and standard errors (n = 4) of relative luminescence units (RLU) of reconstituted RLUC in Arabidopsis protoplast cells.
Arabidopsis protoplasts were transformed with the DNA vectors, and luminescence was measured following an overnight incubation. Experimental signals were corrected for background signals resulting from mock-treated protoplasts.
In all cases (FAD2, FAD3, FAD6, FAD7, and FAD8), the observed signals confirmed the self-association identified by yeast two-hybrid analysis. Protoplasts transformed with head to tail orientations (2H-2T, 3H-3T, 7H-7T, and 8H-8T) showed stronger luminescence compared with those transformed with head to head (2H-2H, 3H-3H, 7H-7H, and 8H-8H) orientations (Fig. 2B).
Membrane Desaturase Heteroassociation
The Y2H experiments and split luciferase results demonstrate the propensity of FAD2, FAD3, FAD6, FAD7, and FAD8 to self-associate in yeast and plant cells, respectively. We next investigated whether members of the FAD family are capable of forming hetero-oligomers using Y2H analysis. In addition to the tested pair of FAD enzymes, Fad2-Fad2 or Fad3-Fad3 constructs were included as positive controls, and heteroassociated constructs, such as Fad2-Ole1 or Fad3-Ole1, were included as negative controls (Fig. 3A). It may be helpful to review the functions of and subcellular localizations the FAD enzymes under study here. FAD2 and FAD3 desaturate 18:1 to 18:2 and 18:2 to 18:3, respectively, within the ER, whereas FAD6 and FAD7/FAD8 perform the equivalent reactions within the plastid. Growth of yeast containing pairs of FAD enzymes is shown in Fig. 3B. Two constructs for each FAD pair were engineered, such that each was expressed as a bait and prey or vice versa. Our criterion for positive interaction is observable growth in both combinations of bait and prey (for example, FAD2-FAD3 and FAD3-FAD2). Despite their relatively low sequence similarity (55%), expression of Fad2 and Fad3 resulted in a growth phenotype in both bait and prey configurations, indicating that FAD2 interacts with FAD3 (Fig. 3B). However, the common evolutionary origins and high amino acid sequence similarity between FAD3 and FAD7 (or FAD8) (of ∼83%) raised the possibility that FAD3 might potentially interact with FAD7 or FAD8 (or both) when heterologously coexpressed in yeast. The data presented in Fig. 3B show that FAD3 did not interact with either FAD7 or FAD8. FAD2 and FAD6, which share 44% amino acid similarity, and both encode Δ-12 desaturases that localize to the ER and plastid, respectively, show no interaction by Y2H assay (Fig. 3B).
FIGURE 3.

Membrane split ubiquitin analysis of pairs of FAD enzymes. A, schematic representation of the pairs of bait and prey plasmid constructs and growth of FAD2 test constructs on synthetic defined medium lacking L and T (SD-2). The abbreviations are as defined for Fig. 1. B, Y2H assay of bait-prey pairs of desaturases as indicated on selective minimal SD-T-L-H-A-M (SD-5) medium.
Similarly, FAD6 was shown to self-associate or associate with FAD7 (42% similarity) and FAD8 (44% similarity) (Fig. 3B), but FAD6 does not interact with FAD2 or FAD3. It is notable that FAD7 and FAD8, which both localize to the plastid and share 85% similarity fail to show association by Y2H assay (Fig. 3B). Low levels of growth were observed for Fad3-Fad8, Fad3-Fad6, and Fad7-Fad2 combinations, but they were not considered significant because they did not fulfill our criteria of growth for both bait and prey relationships (see Table 1 for a summary of specific FAD-FAD amino acid similarities and interactions).
TABLE 1.
Summary of interaction/similarity for FAD enzymes used in this study
Growth (+) or lack of growth (−) and percentage of amino acid similarity are tabulated.
| Interaction/similarity | FAD2 | FAD3 | FAD6 | FAD7 | FAD8 |
|---|---|---|---|---|---|
| FAD2 | +/100% | ||||
| FAD3 | +/55% | +/100% | |||
| FAD6 | −/44% | −/47% | +/100% | ||
| FAD7 | −/54% | −/83% | +/42% | +/100% | |
| FAD8 | −/55% | −/83% | +/44% | −/85% | +/100% |
Having observed the specific FAD desaturases heteroassociations in yeast, we next evaluated them in Arabidopsis protoplast cells using the RLUC assay. The protoplasts transformed with 2H-3T, 3H-2T, 6H-7T, 7H-6T, 6H-8T, and 8H-6T showed higher signals than the protoplasts transformed with 2H-3H, 3H-2H, 6H-7H, 7H-6H, 6H-8H, and 8H-6H, respectively (Fig. 4B for significant interactions). The RLUC data are consistent with the Y2H results presented above. These results show that when two fragments of the RLUC were fused to the same terminus of two target proteins, they are not sufficiently close to facilitate productive interaction, and that their relative orientations were incompatible with productive interaction as observed previously (23). Similar observations have been reported for homo-FRET interactions (24).
FIGURE 4.

Evaluation of the associations of membrane desaturases in Arabidopsis leaf protoplasts with the split luciferase system. A, schematic representation of the plasmid constructs made for expression of genes using RLUC system in which N- and C-terminal Luc (luciferase) fragments are fused to bait or prey genes via a 10-amino acid linker (-GGGGSGGGGS-). B, assay of the activity of reconstituted RLUC by bimolecular complementation in Arabidopsis protoplast cells. Two independent experiments were performed, each consisting of three independent replicates per construct. Luminescence values are expressed as relative luminescence units (RLU). Means and standard deviations of three independent biological replicates per construct within a single representative experiment are presented. Both relative orientations of red fluorescent protein DsRed were included as a negative control.
Coexpression of FAD2 and FAD3 in Yeast
The FAD2 and FAD3 interaction was further investigated as a model to identify the potential physiological significance of a desaturase hetero-oligomer pairing. Recent studies have demonstrated that the N-terminal sequences of FAD3 proteins contain information that could mediate their rapid, proteasome-dependent protein turnover in both plant and yeast cells (25). Thus, a FAD2 fusion with a C-terminal FLAG tag was placed under the control of the GAL1 promoter, and a FAD3 was modified at its C terminus with a short flexible linker coupled to a FLAG tag and placed under the control of the GAL10 promoter in the pESC-Ura vector. There is considerable evidence for the regulation of desaturase activity primarily at the post-transcriptional level (26). For instance, PUFA content in plant cells generally increases with decreasing temperature, although changes in the corresponding FAD mRNA abundances are minor. The ER-localized FAD3 is regulated extensively by both temperature-dependent changes in translational efficiency and modulation of protein half-life (27, 28). Furthermore, expression of FAD2 in yeast cells reveals a short half-life, and its abundance is also modulated by temperature, although the details of the turnover mechanism are currently unknown (29, 30).
In an attempt to optimize the accumulation of FAD enzymes, we identified a growth regime that involves downshifting the incubation temperature after induction. Yeast cells expressing Fad3 were induced by adding 2% galactose along with the fatty acid substrate and were incubated at 30 °C for 4–6 h before being gradually cooled down to 18 °C and maintained at the lower temperature for up to 4 days. Using this approach, 18:3 accumulated to over 10% of total fatty acid upon the induction of FAD3 (Fig. 5C). Yeast cells coexpressing FAD2 and FAD3 18:3 accumulated up to 15% of the total fatty acid (Fig. 5A).
FIGURE 5.
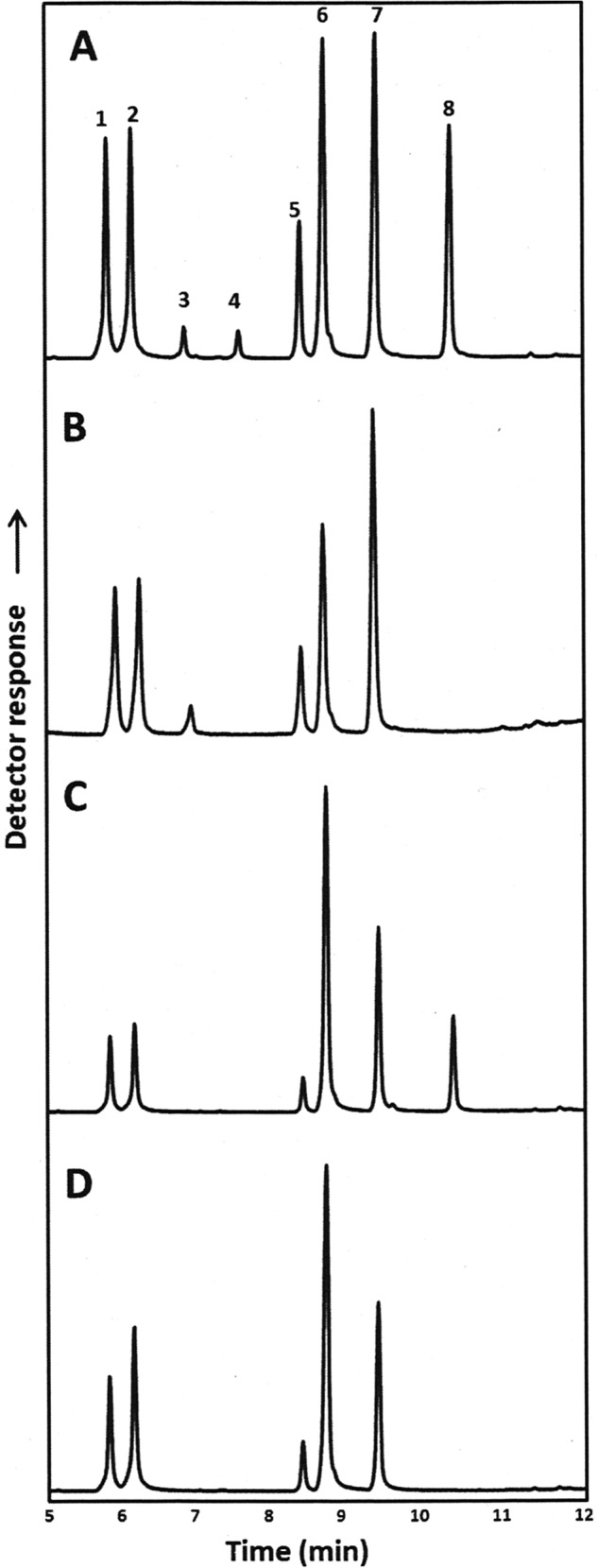
GC analysis of fatty acid methyl esters from yeast cells. YPH499 yeast cells transformed with pESC-Ura and expressing Fad2 and Fad3 (A), Fad2 (B), Fad3 (C), or the negative control empty vector (D) were cultured with medium containing 0.1 mm oleic and 0.1 mm linoleic acid at 30 °C for 4 h followed by 4 days at 18 °C. FAMEs correspond to 16:0 (peak 1), 16:1 (peak 2), 16:2 (peak 3), 16:3 (peak 4), 18:0 (peak 5), 18:1 (peak 6), 18:2 (peak 7), and 18:3 (peak 8).
Cross-linking Experiment Provides Evidence for Dimerization of FAD2 and FAD3
We recently established that the yeast Δ9 integral membrane desaturase Ole1p forms dimers in vivo (18), in a similar fashion to those reported for soluble desaturases (14). In the present work, we show that plant FAD enzymes form self- and specific heteroassociations when expressed both in yeast and in Arabidopsis protoplasts. We further investigated yeast lysates from cells coexpressing FAD2 and FAD3 by Western blotting following cross-linking treatment. Immunoreactive species were detected at ∼45 and 90 kDa, consistent with mobility of FAD monomers and dimers, respectively (Fig. 6). After cross-linking, the dimer represents the dominant signal, and no higher order oligomers were detected. From this, we conclude that FAD2 and FAD3, like the related yeast Ole1P, forms dimers.
FIGURE 6.
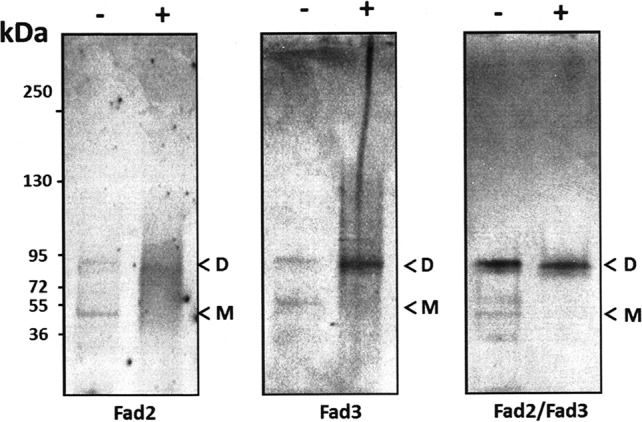
FAD2 and FAD3 can form dimers in yeast cells. Shown are Western blots of extracts of yeast expressing FLAG-tagged constructs of Fad2, Fad3, or Fad2 and Fad3 probed with anti-FLAG antibodies with (+) or without (−) chemical cross-linking (as described under “Experimental Procedures”). Yeast lysates were from cells expressing Fad2 (left panel), Fad3 (middle panel), or Fad2/Fad3 (right panel). The numbers correspond to the molecular masses of protein standards; molecular masses of monomer and dimer are indicated by M and D, respectively. Each panel is representative of at least three independent experiments.
Metabolic Channeling Occurs upon Coexpression of Fad2 and Fad3
Yeast does not naturally synthesize linoleic and linolenic acids and therefore represents an ideal host for testing the functional expression of FAD2, FAD3, and FAD2-FAD3 combinations by accumulation of their products. Because FAD2 and FAD3 can form both homodimers and heterodimers in yeast, we hypothesized that the FAD2-FAD3 heterodimers could form the basis for a metabolic channel capable of converting PC-18:1 to PC-18:3 without free exchange of a PC-18:2 intermediate. We tested this hypothesis by coexpressing FAD2 and FAD3 in yeast and culturing the cells in a medium containing mass labeled oleic acid (in which two hydrogens were replaced by deuterons) or linoleic acid in which the C1 carbon was replaced by 13C.
GAL1 and GAL10 are distinct galactose-inducible promoters that drive luciferase expression at approximately equal levels (18). However, because FAD3 is regulated extensively at the translational and post-transcriptional levels (27, 28), we first characterized the relative accumulation of FAD2 and FAD3 polypeptides. To achieve this, a C-terminal epitope FLAG tag was added to FAD2, and because FAD2 and FAD3 are similar in molecular mass, a short flexible linker (LK) was inserted between the C terminus of FAD3 and its FLAG tag to create FAD3-LK-FLAG (Fig. 7A). This strategy resulted in a mass difference detectable by Western analysis using anti-FLAG antibody (Fig. 7B). Quantitation of the Western signal showed that the protein amount of FAD3-LK-FLAG is ∼26% of FAD2-FLAG (Fig. 7C).
FIGURE 7.

Protein quantification of FAD2 and FAD3 in yeast cells. A, schematic representation of the plasmid constructs made for expression of genes in the yeast cells. A 36-amino acid flexible peptide linker (GGSAGGSGSGSSGGSSGASGTGTAGGTGSGSGTGSG) was inserted between the C terminus of FAD3 and the FLAG epitope tag. B, yeast cells from strain YPH499 expressing Fad2 or Fad3 or coexpressing Fad2 and Fad3 (Fad2/Fad3) in pESC-Ura vectors were grown in medium containing 0.1 mm oleic acids and 0.1 mm linoleic acids at 30 °C for 4 h followed by incubation at 18 °C for 24 h. Extracts were separated by SDS-PAGE, immunoblotted, and visualized with the use of anti-FLAG antibodies. C, quantitation of Western blotting signal for FAD2-FLAG and FAD3-LK-FLAG.
Yeast cultures coexpressing FAD2 and FAD3 were supplemented with 0.1 mm 9,10-d2 oleic acid (18:1Δ9) and 0.1 mm 13C-C1 linoleic acid (18:2Δ9,12). The concentration of exogenous free fatty acids in the medium decreased to a steady state after incubation for 4 h at 30 °C followed by 1 day at 18 °C.
Because FAD2 and FAD3 both act on fatty acids esterified to PC, we isolated PC from the total lipids by TLC visualized by iodine staining. The PC fraction was isolated and converted to FAMES, which were subjected to GC-MS for separation and quantitation of the individual fatty acids. Labeled FAMES were identified with the use of single ion monitoring MS in which d2 oleic acid was identified by an increase of 2 atomic mass units, and 13C-labeled linoleic acid was identified by an increase of 1 atomic mass unit. The proportions of endogenous unlabeled, d2, or 13C-labeled fatty acid in PC are presented in Table 2. Although both labeled precursors were efficiently incorporated into PC-18:3Δ9,12,15, the label distribution in PC-bound 18:2 and 18:3 differs considerably. However, if all PC-bound 18:3 acyl chains had been derived from 18:2 acyl chains bound to the same PC pool, the label distribution should have been equivalent. As described above, the conversion of PC-bound 18:1Δ9 to 18:3Δ9,12,15 can take place by two distinct routes, either via desaturation by discrete FAD2 and FAD3 homodimers or via sequential coupled desaturation by FAD2-FAD3 heterodimers (Fig. 8). The latter path predicts that d2-labeled 18:1 fed to the cells are converted into d2–18:3 without the intermediary d2–18:2 being released into the general PC pool. Similarly, exogenously supplied 1-13C 18:2 should be excluded from conversion to 1-13C-18:3. Consistent with these two predictions of the metabolic channel model, we observed that the fraction of d2 label in PC-18:3 is higher than that of PC-18:2, whereas 1-13C label is higher in PC-18:2 than in PC-18:3 (Table 2). Based on the scheme outlined in Fig. 8, the relative contribution of the metabolic channel to 18:3 formation was quantitatively assessed by applying Equation 2 to the Table 2 data. The result indicates that ∼80% of the linolenate was derived from discrete FAD2 and FAD3 enzyme steps, whereas ∼20% was channeled via the FAD2-FAD3 heterodimer. These data show that a significant portion of the conversion of 18:1 to 18:3 is channeled through a FAD2-FAD3 complex. The proportion of channeling might be improved if expression of the desaturases could be better balanced.
TABLE 2.
C18 fatty acid analysis of PC from yeast cells transformed with Fad2/Fad3
Cells were grown in medium containing 0.1 mm deuterated oleic acid and 0.1 mm 13C-labeled linoleic acids at 30 °C for 4 h followed by 20 h of growth at 18 °C. The values are the means ± S.E. (n = 4).
| 18:1Δ9 | 18:2Δ9,12 | 18:3Δ9,12,15 | |
|---|---|---|---|
| Unlabeled FA | 49.9 ± 0.7 | 26.3 ± 0.1 | 24.7 ± 0.6 |
| 9,10-d2 FA | 50.1 ± 0.7 | 29.1 ± 0.3 | 39.7 ± 0.4 |
| 13C-C1 FA | 0 | 44.6 ± 0.4 | 35.6 ± 0.3 |
FIGURE 8.
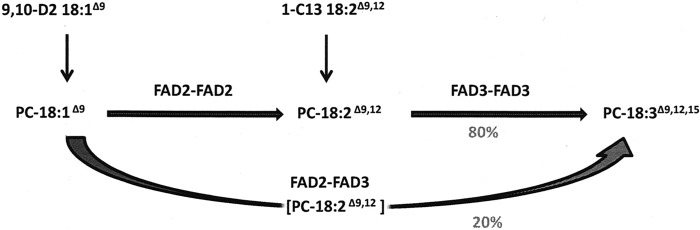
Schematic view of the metabolic conversions between PC-bound 18:1, 18:2, and 18:3 in yeast cells expressing FAD2 and FAD3. Different fates of exogenously supplied isotope-labeled fatty acids according to sequential desaturation via two discrete homodimers of FAFD2 and FAD3 or via a FAD2-FAD3 heterodimer. Via the heterodimer, d2-labeled oleic acid (18:1) is converted to d2-linolenic acid (18:3) via metabolic channeling, without the intermediary d2–18:2 being released into the general PC pool. Similarly, the metabolic channel excludes exogenously supplied 1-13C 18:2 from conversion into 1-13C-18:3 via the heterodimer.
DISCUSSION
The quaternary structure of enzymes has broad implications for many aspects of enzyme function including catalytic rate, specificity, cooperativity, allostery, and metabolic channeling. Evidence from ubiquitin and luciferase bimolecular complementation analyses performed in yeast and Arabidopsis cells, respectively, provides independent biochemical evidence that FAD2, 3, 6, 7, and 8 can form homoassociations in vivo. Immunological cross-linking experiments show that FAD enzymes form dimers, like their distant integral membrane desaturase homolog, yeast Ole1p. The existence of FAD dimers is also consistent with published genetic studies in which the heterologous expression of a nonfunctional Brassica oleate desaturase FAD2 mutant (Bnfad2) resulted in a dominant negative reduction in linoleic acid and a corresponding increase in oleic acid in transgenic cotton seeds (31, 32). Interpretation of these results in the context of the present work suggests that the defective Bnfad2 subunit may associate with the functional WT cotton subunit, resulting in its inactivation. This phenomenon is sometimes referred to as subunit poisoning (33).
Similar high oleate phenotypes have been reported upon the expression of FAD2 homologs that catalyze the production of unusual fatty acids. For instance, expression of the Momordica conjugase or castor hydroxylase in Arabidopsis resulted in the accumulation of the unusual fatty acids accompanied by an increase in the level of oleate (34–36). In this case, the increase in oleate could result either from subunit poisoning or from inhibition of the native FAD2 upon the buildup of unusual fatty acids in the PC pool, thereby displacing the 18:1-PC substrate available to FAD2.
The FAD enzyme data presented here raise the question as to why the two independently evolved classes of soluble and membrane desaturases share a dimeric organization. Details of the catalytic cycle of the membrane desaturases are poorly defined relative to those of the soluble desaturases for which the occurrence of dimers has been linked to a proposed mechanism involving half of the sites. Under this proposal, the desolvation energy gained upon substrate binding to one subunit is directed toward the energetically unfavorable resolvation of the fatty acid upon product release from the second subunit, which is the rate-limiting step (16, 17). It is unlikely that mechanism is applicable to the membrane desaturases because the hydrophobic products are released into a hydrophobic membrane environment, a process that has a lower energy requirement than that of the soluble desaturases. Indeed, for membrane desaturases, abstraction of the first hydrogen, rather than product release, is rate-limiting (37). However, both soluble and membrane desaturases require two electrons for catalysis that are supplied by ferredoxin or cytochrome b5, respectively (2). Both of these cofactors are capable only of transferring one electron at a time; thus for desaturation to proceed, two discrete electron transfers must occur (38). It is possible that each subunit of the dimer is reduced, and then one subunit becomes two-electron-reduced, whereas the other becomes oxidized by disproportionation. It remains to be tested whether the need for two discrete one-electron transfers underlies the observed dimeric organization.
The identification of hetero-oligomers by yeast two-hybrid analysis was unexpected based on their low sequence similarity; indeed this was discovered when FAD2 and FAD3 were coexpressed in what was envisioned as a negative control experiment for the expression of FAD2 and FAD3 separately. However, this result is consistent with published data showing that FAD2 and FAD3 colocalize to the same subdomain of the endoplasmic reticulum (39). That FAD2 interacts with FAD3 prompted us to perform all combinations of the FADs, and from this we discovered that FAD6 can interact with both FAD7 and FAD8. The fact that enzymes that mediate oleate and linoleate desaturations, i.e. mediating sequential desaturation steps within both the ER and the plastid, prompted us to ask whether there was a physiological significance to the observed pattern of interactions. Support for the significance of these specific interactions came from the observation that FAD2/FAD3, FAD6/FAD7, and FAD6/FAD8 interact despite their low homologies (55, 42, and 44% similarity, respectively), whereas FAD7 and FAD8, which colocalize to the plastid and are 85% similar, do not associate.
Labeling patterns of fatty acid products resulting from metabolic flux analysis provided evidence that heterodimers of FAD2 and FAD3 are able to channel oleate to linolenate without releasing a linoleoyl-PC intermediate. The identification of a metabolic channel for the production of triunsaturated fatty acids provides physiological context for the observation that FAD2-FAD3 heterodimers form in vivo. It remains to be determined whether FAD6-FAD7 or FAD6-FAD8 heterodimers form a similar metabolic channel in plastids. The foundation for specific hetero-oligomer formation likely arises from the ability of ancestral membrane desaturases to form homodimers in organisms such as yeast that contain only one desaturase. Subsequent gene duplication and functional differentiation could then occur with maintenance of shared dimer interfaces. As desaturase genes duplicated and isoforms became targeted to distinct compartments, selection for dimerization, even between close homologs, would be lost. The impact of FAD heterodimer formation on temperature regulation of membrane desaturation remains to be investigated.
There is currently considerable interest in engineering plants to accumulate high levels of polyunsaturated fatty acids and unusual fatty acids in production plants (40, 41). Coexpressing desaturases or desaturase-like enzymes that can interact to achieve the accumulation a desired product may provide another opportunity for optimizing their accumulation via metabolite channeling. Further work to understand details of desaturase-desaturase interactions may enable rational engineering of specific interactions between pairs of desaturase enzymes that do not naturally associate.
Acknowledgment
We thank Xiao-Hong Yu for helpful discussion.
This work was supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy (DOE) under Grant DOE KC0304000.
Fatty acid nomenclature: X:Y indicates that the fatty acid contains X numbers of carbon atoms and Y numbers of double bonds; Δz indicates that a double bond is positioned at the zth carbon atom from the C terminus.
- PC
- phosphatidylcholine
- ACP
- acyl carrier protein
- ER
- endoplasmic reticulum
- FAD
- fatty acid desaturase
- FAME
- fatty acid methyl ester
- Cub
- C-terminal fragment of ubiquitin
- Nub
- N-terminal ubiquitin fragment
- RLUC
- Renilla split luciferase
- Y2H
- yeast two hybrid.
REFERENCES
- 1. Bloch K. (1969) Enzymatic synthesis of monounsaturated fatty acids. Acc. Chem. Res. 2, 193–202 [Google Scholar]
- 2. Shanklin J., Cahoon E. B. (1998) Desaturation and related modifications of fatty acids. Annu. Rev. Plant Physiol. Plant. Mol. Biol. 49, 611–641 [DOI] [PubMed] [Google Scholar]
- 3. Sperling P., Ternes P., Zank T. K., Heinz E. (2003) The evolution of desaturases. Prostaglandins Leukot. Essent. Fatty Acids 68, 73–95 [DOI] [PubMed] [Google Scholar]
- 4. Shanklin J., Achim C., Schmidt H., Fox B. G., Münck E. (1997) Mössbauer studies of alkane omega-hydroxylase: evidence for a diiron cluster in an integral-membrane enzyme. Proc. Natl. Acad. Sci. U.S.A. 94, 2981–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thompson G. A., Scherer D. E., Foxall-Van Aken S., Kenny J. W., Young H. L., Shintani D. K., Kridl J. C., Knauf V. C. (1991) Primary structures of the precursor and mature forms of stearoyl-acyl carrier protein desaturase from safflower embryos and requirement of ferredoxin for enzyme activity. Proc. Natl. Acad. Sci. U.S.A. 88, 2578–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shanklin J., Somerville C. (1991) Stearoyl-acyl-carrier-protein desaturase from higher plants is structurally unrelated to the animal and fungal homologs. Proc. Natl. Acad. Sci. U.S.A. 88, 2510–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Okuley J., Lightner J., Feldmann K., Yadav N., Lark E., Browse J. (1994) Arabidopsis FAD2 gene encodes the enzyme that is essential for polyunsaturated lipid synthesis. Plant Cell 6, 147–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arondel V., Lemieux B., Hwang I., Gibson S., Goodman H. M., Somerville C. R. (1992) Map-based cloning of a gene controlling omega-3 fatty acid desaturation in Arabidopsis. Science 258, 1353–1355 [DOI] [PubMed] [Google Scholar]
- 9. Somerville C., Browse J. (1991) Plant lipids: metabolism, mutants, and membranes. Science 252, 80–87 [DOI] [PubMed] [Google Scholar]
- 10. Ohlrogge J. B., Browse J., Somerville C. R. (1991) The genetics of plant lipids. Biochim. Biophys. Acta 1082, 1–26 [DOI] [PubMed] [Google Scholar]
- 11. Xu C., Moellering E. R., Muthan B., Fan J., Benning C. (2010) Lipid transport mediated by Arabidopsis TGD proteins is unidirectional from the endoplasmic reticulum to the plastid. Plant Cell Physiol. 51, 1019–1028 [DOI] [PubMed] [Google Scholar]
- 12. Shanklin J., Whittle E., Fox B. G. (1994) Eight histidine residues are catalytically essential in a membrane-associated iron enzyme, stearoyl-CoA desaturase, and are conserved in alkane hydroxylase and xylene monooxygenase. Biochemistry 33, 12787–12794 [DOI] [PubMed] [Google Scholar]
- 13. Lindqvist Y. (2001) Delta Nine Stearoyl-Acyl Carrier Protein Desaturase, John Wiley & Sons, Chichester UK [Google Scholar]
- 14. Lindqvist Y., Huang W., Schneider G., Shanklin J. (1996) Crystal structure of a delta nine stearoyl-acyl carrier protein desaturase from castor seed and its relationship to other diiron proteins. EMBO J. 15, 4081–4092 [PMC free article] [PubMed] [Google Scholar]
- 15. Guy J. E., Whittle E., Kumaran D., Lindqvist Y., Shanklin J. (2007) The crystal structure of the ivy delta4-16:0-ACP desaturase reveals structural details of the oxidized active site and potential determinants of regioselectivity. J. Biol. Chem. 282, 19863–19871 [DOI] [PubMed] [Google Scholar]
- 16. Broadwater J. A., Ai J., Loehr T. M., Sanders-Loehr J., Fox B. G. (1998) Peroxodiferric intermediate of stearoyl-acyl carrier protein delta 9 desaturase: oxidase reactivity during single turnover and implications for the mechanism of desaturation. Biochemistry 37, 14664–14671 [DOI] [PubMed] [Google Scholar]
- 17. Fox B. G., Lyle K. S., Rogge C. E. (2004) Reactions of the diiron enzyme stearoyl-acyl carrier protein desaturase. Acc Chem Res 37, 421–429 [DOI] [PubMed] [Google Scholar]
- 18. Lou Y., Shanklin J. (2010) Evidence that the yeast desaturase Ole1p exists as a dimer in vivo. J. Biol. Chem. 285, 19384–19390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Winden W. A., Wittmann C., Heinzle E., Heijnen J. J. (2002) Correcting mass isotopomer distributions for naturally occurring isotopes. Biotechnol. Bioeng. 80, 477–479 [DOI] [PubMed] [Google Scholar]
- 20. Obrdlik P., El-Bakkoury M., Hamacher T., Cappellaro C., Vilarino C., Fleischer C., Ellerbrok H., Kamuzinzi R., Ledent V., Blaudez D., Sanders D., Revuelta J. L., Boles E., André B., Frommer W. B. (2004) K+ channel interactions detected by a genetic system optimized for systematic studies of membrane protein interactions. Proc. Natl. Acad. Sci. U.S.A. 101, 12242–12247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu F. H., Shen S. C., Lee L. Y., Lee S. H., Chan M. T., Lin C. S. (2009) Tape-Arabidopsis sandwich: a simpler Arabidopsis protoplast isolation method. Plant Methods 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fujikawa Y., Kato N. (2007) Split luciferase complementation assay to study protein-protein interactions in Arabidopsis protoplasts. Plant J 52, 185–195 [DOI] [PubMed] [Google Scholar]
- 23. Massoud T. F., Paulmurugan R., Gambhir S. S. (2004) Molecular imaging of homodimeric protein-protein interactions in living subjects. FASEB J. 18, 1105–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sorkin A., McClure M., Huang F., Carter R. (2000) Interaction of EGF receptor and grb2 in living cells visualized by fluorescence resonance energy transfer (FRET) microscopy. Curr. Biol. 10, 1395–1398 [DOI] [PubMed] [Google Scholar]
- 25. Khuu N., Gidda S., Shockey J. M., Dyer J. M., Mullen R. T. (2011) The N termini of Brassica and tung omega-3 fatty acid desaturases mediate proteasome-dependent protein degradation in plant cells. Plant Signal Behav. 6, 422–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Somerville C. (1995) Direct tests of the role of membrane lipid composition in low-temperature-induced photoinhibition and chilling sensitivity in plants and cyanobacteria [comment]. Proc. Natl. Acad. Sci. U.S.A. 92, 6215–6218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Horiguchi G., Fuse T., Kawakami N., Kodama H., Iba K. (2000) Temperature-dependent translational regulation of the ER omega-3 fatty acid desaturase gene in wheat root tips. Plant J. 24, 805–813 [DOI] [PubMed] [Google Scholar]
- 28. O'Quin J. B., Bourassa L., Zhang D., Shockey J. M., Gidda S. K., Fosnot S., Chapman K. D., Mullen R. T., Dyer J. M. (2010) Temperature-sensitive post-translational regulation of plant omega-3 fatty-acid desaturases is mediated by the endoplasmic reticulum-associated degradation pathway. J. Biol. Chem. 285, 21781–21796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Covello P. S., Reed D. W. (1996) Functional expression of the extraplastidial Arabidopsis thaliana oleate desaturase gene (FAD2) in Saccharomyces cerevisiae. Plant Physiol. 111, 223–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tang G. Q., Novitzky W. P., Carol Griffin H., Huber S. C., Dewey R. E. (2005) Oleate desaturase enzymes of soybean: evidence of regulation through differential stability and phosphorylation. Plant J. 44, 433–446 [DOI] [PubMed] [Google Scholar]
- 31. Chapman K. D., Austin-Brown S., Sparace S. A., Kinney A., Ripp K. G., Pirtle I. L., Rirtle R. M. (2001) Transgenic cotton plants with increased seed oleic acid content. J. Am. Oil Chem. Soc. 78, 941–947 [Google Scholar]
- 32. Chapman K. D., Neogi P. B., Hake K. D., Stawska A. A., Speed T. R., Cotter M. Q., Garrett D. C., Kerby T., Richardson C. D., Ayre B. G., Ghosh S., Kinney A. (2008) Reduced oil accumulation in cottonseeds transformed with a Brassica nonfunctional allele of a delta-12 fatty acid desaturase (FAD2). Crop Sci. 48, 1470–1481 [Google Scholar]
- 33. Sturino J. M., Klaenhammer T. R. (2006) Engineered bacteriophage-defence systems in bioprocessing. Nat. Rev. Microbiol. 4, 395–404 [DOI] [PubMed] [Google Scholar]
- 34. Cahoon E. B., Carlson T. J., Ripp K. G., Schweiger B. J., Cook G. A., Hall S. E., Kinney A. J. (1999) Biosynthetic origin of conjugated double bonds: production of fatty acid components of high-value drying oils in transgenic soybean embryos. Proc. Natl. Acad. Sci. U.S.A. 96, 12935–12940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Broun P., Boddupalli S., Somerville C. (1998) A bifunctional oleate 12-hydroxylase:desaturase from Lesquerella fendleri. Plant J. 13, 201–210 [DOI] [PubMed] [Google Scholar]
- 36. Broun P., Somerville C. (1997) Accumulation of ricinoleic, lesquerolic, and densipolic acids in seeds of transgenic Arabidopsis plants that express a fatty acyl hydroxylase cDNA from castor bean. Plant Physiol. 113, 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Buist P. H. (2004) Fatty acid desaturases: selecting the dehydrogenation channel. Nat. Prod. Rep. 21, 249–262 [DOI] [PubMed] [Google Scholar]
- 38. Sobrado P., Lyle K. S., Kaul S. P., Turco M. M., Arabshahi I., Marwah A., Fox B. G. (2006) Identification of the binding region of the [2Fe-2S] ferredoxin in stearoyl-acyl carrier protein desaturase: insight into the catalytic complex and mechanism of action. Biochemistry 45, 4848–4858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dyer J. M., Mullen R. T. (2001) Immunocytological localization of two plant fatty acid desaturases in the endoplasmic reticulum. FEBS Lett. 494, 44–47 [DOI] [PubMed] [Google Scholar]
- 40. Napier J. A., Sayanova O. (2005) The production of very-long-chain PUFA biosynthesis in transgenic plants: towards a sustainable source of fish oils. Proc. Nutr. Soc. 64, 387–393 [DOI] [PubMed] [Google Scholar]
- 41. Napier J. A. (2007) The production of unusual fatty acids in transgenic plants. Annu. Rev. Plant Biol. 58, 295–319 [DOI] [PubMed] [Google Scholar]