

Background: Type 1 CCK receptor function is affected by a high cholesterol environment.
Results: The CCK1R Y140A mutant has ligand binding and activation characteristics similar to wild type CCK1R in high cholesterol.
Conclusion: This mutant mimics CCK1R structure in high cholesterol.
Significance: This mutant represents a powerful and unique tool for identification of ligands to correct the abnormal conformation of CCK1R in high cholesterol.
Keywords: G Protein-coupled Receptor (GPCR), Membrane Lipid, Peptide Hormone, Radioreceptor Assays, Signal Transduction, Cholecystokinin, Receptor Binding
Abstract
Cholecystokinin (CCK) stimulates the type 1 CCK receptor (CCK1R) to elicit satiety after a meal. Agonists with this activity, although potentially useful for treatment of obesity, can also have side effects and toxicities of concern, making the development of an intrinsically inactive positive allosteric modulator quite attractive. Positive allosteric modulators also have the potential to correct the defective receptor-G protein coupling observed in the high membrane cholesterol environment described in metabolic syndrome. Current model systems to study CCK1R in such an environment are unstable and expensive to maintain. We now report that the Y140A mutation within a cholesterol-binding motif and the conserved, class A G protein-coupled receptor-specific (E/D)RY signature sequence results in ligand binding and activity characteristics similar to wild type CCK1R in a high cholesterol environment. This is true for natural CCK, as well as ligands with distinct chemistries and activity profiles. Additionally, the Y140A construct also behaved like CCK1R in high cholesterol in regard to its internalization, sensitivity to a nonhydrolyzable GTP analog, and anisotropy of a bound fluorescent CCK analog. Chimeric CCK1R/CCK2R constructs that systematically changed the residues in the allosteric ligand-binding pocket were studied in the presence of Y140A. This established increased importance of unique residues within TM3 and reduced the importance of TM2 for binding in the presence of this mutation, with the agonist trigger likely pulled away from its Leu356 target on TM7. The distinct conformation of this intramembranous pocket within Y140A CCK1R provides an opportunity to normalize this by using a small molecule allosteric ligand, thereby providing safe and effective correction of the coupling defect in metabolic syndrome.
Introduction
Cholecystokinin (CCK)2 is a gastrointestinal peptide hormone that plays an important role in nutritional homeostasis, stimulating gallbladder contraction and pancreatic exocrine secretion and affecting gastrointestinal motility and transit, as well as inducing postcibal satiety (1–4). The ability of CCK to stimulate satiety has been responsible for efforts to target this receptor as a potential treatment for obesity. Indeed, agonists of the type 1 CCK receptor (CCK1R) have been developed (5–8) and studied in clinical trials for obesity (8, 9). Although these agents did stimulate weight loss, this was not quantitatively greater than acute dieting alone. Additionally, there has been at least the theoretical concern about a long-acting CCK1R agonist having trophic effects on pancreatic cells, with possible impact to stimulate the development and/or growth of pancreatic cancer (10, 11).
A possible strategy to circumvent these concerns is the development of a nonbiologically active allosteric enhancer of the CCK1R. Such a drug could exhibit satiety effects only during the brief postcibal period when natural CCK is released into the circulation, because this hormone has a particularly brief half-life. Action at that time could limit the size of the meal, a strategy that could reinforce the motivation to maintain a normal or healthy weight in individuals.
Another set of observations is particularly relevant to this type of drug development. This receptor has been shown to be uniquely sensitive to the membrane cholesterol composition (12, 13), with the closely structurally related type 2 CCK receptor (CCK2R) not sensitive to membrane cholesterol (14). This effect has been demonstrated not only in model cell systems (15) but also in the natural cellular environment in animal models, such as the prairie dog fed a high cholesterol diet (16), and even in patients with cholesterol gallstones (17–19). Mechanistic studies have localized the cause of the reduced coupling efficiency between CCK1R and its heterotrimeric G protein (Gq) to increased cholesterol composition of the membrane (20, 21). Consistent with this, patients with pigment gallstones who are known to have normal rather than increased membrane cholesterol, such as is present in patients with cholesterol gallstones (22), have normal CCK1R function (19, 23).
We previously reported direct in vitro evidence showing that CCK1R function is sensitive to the membrane cholesterol content, which is reflected in its ligand binding characteristics and in its biological activity (12). CCK binding affinity and CCK-stimulated biological responses are reduced upon cholesterol depletion, although cholesterol augmentation also reduces CCK-stimulated biological responses, but in the setting of actually enhancing CCK binding affinity. The increased binding affinity for CCK in the setting of increased membrane cholesterol, however, is nonproductive and results in a reduction in biological responsiveness. In contrast, the closely structurally related CCK2R is not sensitive to alterations in membrane cholesterol (14).
We localized the key structural determinant within the CCK1R for cholesterol sensitivity to residues encoded by its third exon (14), including most of transmembrane segments (TM) three and four, which contains both the “cholesterol recognition/interaction amino acid consensus” (CRAC) (24) and the “cholesterol consensus motif” (CCM) (25) sequence motifs. Key residues include Tyr140 (Y3.51, based on the nomenclature of Ballesteros and Weinstein (26)). There is also an additional CRAC motif in TM5 that includes Tyr237 (Y5.66). CCK binding and signaling are negatively affected by mutation of each of these residues to alanines. It is notable that, in contrast with the mutation of the other two residues, the Y140A mutation results in elimination of the sensitivity of the CCK1R to cholesterol (14).
Tyr140 (Y3.51) is also part of the highly conserved (E/D)RY class A GPCR signature sequence. It is well established that the charged residues in this sequence, the acidic glutamic or aspartic acid and the basic arginine, form a charge-charge interaction key for the ionic lock to maintain these receptors in their inactive state (27, 28). The role of the tyrosine residue in this sequence motif is much less clear. It is less well conserved than the other residues, often replaced with a cysteine, histidine, or valine. Mutation of this residue has often had no, or minimal, functional impact (29–33).
In this study, we have focused on the Y140A mutant of the CCK1R, now establishing a stable mutant receptor-bearing cell line and expanding its functional characterization. These data support the use of this cell line as a mimic of the wild type CCK1R in the presence of elevated membrane cholesterol. As such, this provides a valuable new tool to use in the screening for positive allosteric modulators of this receptor that might correct the conformational change in the receptor induced by the high cholesterol membrane environment. Such a surrogate may offer substantial advantages in future high throughput screening strategies, because it addresses challenges of existing model systems incorporating physical enhancement in membrane cholesterol or cell lines with mutations in lipid synthetic machinery for mimicking CCK1R conformations in a high cholesterol environment, having a stable phenotype, and growing well in culture.
EXPERIMENTAL PROCEDURES
Materials
Ham's F-12 medium and Amplex Red reagent were from Invitrogen. Fetal clone 2 culture medium supplement was from Hyclone Laboratories (Logan, UT). Lipoprotein-deficient serum was obtained from Intracel (Frederick, MD). Bovine serum albumin (BSA) was from Equitech Bio, Inc. (Kerrville, TX). Quest Fluo-8-AMTM was from AAT Bioquest Inc. (Sunnyvale, CA). LDL was obtained from Sigma. All other reagents were analytical grade.
Synthetic CCK octapeptide (CCK(26–33), also known as CCK-8) was purchased from Peninsula Laboratories (Belmont, CA). The CCK-like radioactive tracer, 125I-d-Tyr-Gly-[(Nle28,31)CCK-(26–33)], was prepared by oxidative radioiodination of the parental peptide using IODO-BEADs (Pierce) with purification to homogeneity on reversed-phase HPLC, as described previously (34). The 1,5-benzodiazepine agonist ligand (GI181771X) was provided by Dr. Brad Henke at GlaxoSmithKline Research Laboratories (Research Triangle Park, NC); and BDZ-1 ligand was provided by Drs. P. S. Portoghese and E. Akgun from the University of Minnesota. A71623 was purchased from Santa Cruz Biotechnology.
Receptor-bearing Cells and Cell Culture
Chinese hamster ovary (CHO) cell lines engineered to stably express the wild type human type 1 CCK receptor (CHO-CCK1R) or type 2 CCK receptor (CHO-CCK2R) were utilized in this work. These cell lines have been characterized previously and have been demonstrated to express fully functional receptors that are capable of binding CCK and signaling normally, as well as undergoing agonist-induced internalization (14, 15, 35). CHO cells stably expressing the human CCK1R Y140A mutant (Fig. 1) and the analogous human CCK2R Y153A mutant were similarly engineered using previously described cDNA constructs (14). Chimeric CCK1R/CCK2R constructs involving residues lining the intramembranous inter-helical pocket (36) were also engineered to express the Y140A mutation (Fig. 1). Cell lines expressing each of these receptor constructs were established by transfecting CHO cells using Lipofectamine LTX (Invitrogen) with PlusTM reagent according to the manufacturer's directions. Receptor-expressing clones were enriched using G418 or hygromycin selection and were selected based on 125I-CCK binding after limiting dilution. CHO cell lines were grown at 37 °C in a humidified environment containing 5% CO2 in tissue culture plasticware containing Ham's F-12 medium supplemented with 5% fetal clone 2. Cells were passaged approximately two times/week for maintenance in culture.
FIGURE 1.

Topological representation and predicted helical wheel organization of residues within the TM segments of the human CCK1R. Shown is an illustration of the topology of CCK1R with amino acids predicted to form the TM segments depicted in circles containing 1-letter amino acid identifiers (A). The putative TM region is shaded, and tail and loop regions are represented by lines. Residues in black circles represent the most conserved residues within the class A GPCRs, which are denoted by TM.50 according to the Ballesteros and Weinstein numbering system (26). Residues that were mutated to the corresponding residues in CCK2R to prepare receptor chimeras are shown in gray circles within the respective TM segments. The TM segments are also shown as helical wheels with the predicted locations of the modified residues in the inter-helical pocket (B).
Modification of Membrane Cholesterol Levels
Cholesterol levels in the receptor-bearing CHO cell lines were increased either using methyl-β-cyclodextrin (MβCD)-cholesterol or by growing cells in Ham's F-12 medium containing LDL. For the first method, MβCD-cholesterol complex stock solution was prepared as described previously (37). In brief, 12 mg of cholesterol was dissolved in 80 μl of isopropyl alcohol/chloroform (2:1) solution. This was added dropwise to a solution containing 200 mg of MβCD in 2.2 ml of Krebs-Ringer/HEPES (KRH) medium (25 mm HEPES, pH 7.4, 104 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm KH2PO4, 1.2 mm MgSO4) heated to 60–80 °C with constant stirring. The receptor-bearing CHO cells were incubated with 5 mm MβCD-cholesterol in KRH medium for 35–40 min at 37 °C. In the second method, the cells were grown for 24 h in Ham's F-12 medium containing 5% lipoprotein-deficient serum with 10 μm mevastatin, followed by an additional 24-h period in which the cells were grown in Ham's F-12 medium containing 150 μg/ml LDL (12, 14, 15).
Plasma membrane cholesterol was directly determined using a morphological assay with filipin staining (12) and a quantitative biochemical assay using Amplex Red reagent (38) on lipid extracts (39). Both methods of enhancing plasma membrane cholesterol were optimized to yield levels that mimicked those found in patients with metabolic syndrome (12, 14, 15, 23). This represented an increase of 27 ± 2% above that in parental CHO cells.
Membrane Preparation
Receptor-enriched membrane fractions were prepared from receptor-bearing CHO cell lines, as described previously (40). Cells were harvested mechanically using a cell scraper and suspended in ice-cold phosphate-buffered saline, pH 7.4. Cells were homogenized by mixing the suspension with 0.3 m sucrose containing 0.01% trypsin inhibitor and 1 mm phenylmethylsulfonyl fluoride and sonicating for 10 s on ice with a Branson Sonifier 250. The sucrose concentration of the homogenate was then adjusted to 1.3 m and placed at the bottom of a centrifuge tube and overlaid with 0.3 m sucrose. Tubes were centrifuged at 225,000 × g for 1 h at 4 °C. The receptor-enriched fraction was collected at the sucrose interface, diluted with ice-cold water, and pelleted at 225,000 × g for 30 min at 4 °C. The membrane pellet was resuspended in KRH buffer and stored at −80 °C prior to use.
Radioligand Binding
CCK receptor binding experiments were performed as described previously (12, 14, 36). Two different types of CCK receptor radioligands were utilized, representing a CCK peptide-like ligand, 125I-d-Tyr-Gly-[(Nle28,31)CCK-(26–33)] (34), and an allosteric benzodiazepine radioligand 125I-BDZ-1 with selectivity for the CCK1R (41). Binding was performed with membranes or intact receptor-bearing cells in 24-well dishes at a confluence of ∼80%. The membrane suspension (7–10 μg/tube) or cells were incubated with 5 pm radioligand in the absence or presence of unlabeled competing ligand in KRH medium containing 0.2% bovine serum albumen (BSA) and 0.01% soybean trypsin inhibitor at room temperature for 60 min with gentle shaking. In certain experiments, 1 μm GppNHp was added to the reaction. Nonsaturable binding was defined as radioactivity bound in the presence of 1 μm concentration of the analogous unlabeled ligand. At the end of the incubation, the reaction was terminated by washing the cells twice with ice-cold KRH medium, lysing the cells with 0.5 m NaOH, and quantifying bound radioactivity in the lysate using a gamma counter. For the membrane binding assay, the receptor-bound fraction was separated from free radioligand by centrifugation or using vacuum filtration using GF/B filter mats (Unifilter-96-well, PerkinElmer Life Sciences) in a Filtermate Harvester. The plates were washed six times using ice-cold buffer (0.9% NaCl, 0.2% BSA) and air-dried, prior to addition of 30 μl of scintillant. Bound radioactivity was quantified using a Top Count NXT instrument (Packard Instrument Co.). Data were analyzed using the LIGAND program of Munson and Rodbard (42) and were graphed using the nonlinear least squares curve-fitting routine in GraphPad Prism 5.0 (San Diego).
Biological Activity
Increases in intracellular calcium concentrations in response to various agonist ligands were measured to assess the biological activity of the CCK receptor-bearing CHO cell lines, as described previously (15, 43). Cells were seeded in a 96-well black-walled clear bottom plate at a density of 15,000–20,000 cells/well in Ham's F-12 medium and cultured for 24 h at 37 °C in a humidified environment containing 5% CO2. Before the assay, the cells were washed once and loaded with 1.37 μm Fluo-8AM in KRH medium containing 1.2 mm MgCl2, 0.2% BSA, and 2.5 mm probenecid for 1 h at 37 °C in a humidified chamber containing 5% CO2. Following this, cells were washed once with medium, and biological activity was measured by stimulating the cells with various concentrations of the agonists at 37 °C and monitoring fluorescence over 3 min using a Flexstation 3.0 (Molecular Devices, Sunnyvale, CA) equipped with Softmax Pro 5.4 software. Emission was monitored at 520 nm after excitation at 485 nm. Data were graphed using the nonlinear least squares curve-fitting routine in GraphPad Prism 5.0.
Fluorescence Spectroscopy and Anisotropy Measurements
Fluorescence measurements in cells were performed using a Fluoromax-3 spectrophotometer. Unlabeled cells were used to correct for background fluorescence and light scattering. Receptor-bearing CHO cell lines were dislodged from the culture plates using nonenzymatic cell dissociation medium, pelleted by low speed centrifugation, and incubated with 100 nm Alexa488-Gly-[(Nle28,31)CCK-(26–33)] (Alexa488-CCK) in KRH medium containing 0.2% BSA for 90 min at 4 °C. This fluorescent ligand was fully characterized and validated previously (44). The cell suspension was then centrifuged and washed repeatedly with ice-cold KRH medium containing 0.2% BSA to remove excess unbound ligand and resuspended in KRH medium without BSA. Nonsaturable binding of the Alexa488-CCK was determined in each experiment using saturating concentrations of unconjugated CCK peptide. Fluorescence spectra for Alexa488-CCK were collected for the labeled cells by collecting emission at wavelengths ranging from 505 to 600 nm upon excitation at 485 nm, using a 4-nm bandwidth filter.
Fluorescence anisotropy was measured and calculated as described previously (12). Measurements were taken at 4 and 20 °C using an L-format-based single channel Fluoromax-3 spectrophotometer equipped with a thermostatically adjusted cuvette holder and automatic polarizer. The degrees of alignment of the excitation and emission polarizing filters were set to 55 and 0°, respectively. The excitation wavelength was set at 485 nm, and the emission wavelength was fixed at 520 nm. Data were collected with five sets of acquisitions after a delay of 10 s, each using constant wavelength profile with 10-s integration times.
Receptor Internalization
CCK-stimulated agonist-occupied receptor internalization was studied morphologically using the fluorescent Alexa488-CCK ligand, as described previously (12). Receptor-bearing CHO cells were seeded on glass coverslips and cultured for 48 h in Ham's F-12 medium at 37 °C in a humidified environment containing 5% CO2. Coverslips containing cells were first pre-cooled on ice for 10 min and rinsed three times with ice-cold PBS, pH 7.4 (1.5 mm NaH2PO4, 8 mm Na2HPO4, 145 mm NaCl, 0.1 mm MgCl2, and 0.08 mm CaCl2). Cells were then labeled by incubating with 100 nm Alexa488-CCK for 90 min at 4 °C, followed by washing with ice-cold PBS to eliminate unbound ligand. Coverslips were then warmed to 37 °C for various times and fixed with 2% paraformaldehyde. Morphological examination of the cells was then done using an inverted epifluorescence microscope (Axiovert 200 M, Carl Zeiss, Thornwood, NY) with a fixed YFP filter set with excitation at 500/20 nm, dichroic mirror at Q515 long pass, and emission at 535/30 nm (Chroma Technology, Brattleboro, VT). Images were collected using an ORCA-12ER charge-coupled device camera (Hamamatsu, Bridgewater, NJ) with QED-InVivo 2.03 image acquisition software (Media Cybernetics, Silver Spring, MD). Background-subtracted images were assembled using Adobe Photoshop 7.0 (Adobe Systems, Mountain View, CA).
Data Analysis
Possible differences in the binding and biological activity parameters of the constructs were determined using one-way analysis of variance and the Tukey/Dunn's post test or unpaired t test. Differences between the conditions were considered to be significant at p < 0.05 (Prism 5, GraphPad, San Diego). Fractional occupancy of receptors at the EC50 value for a particular agonist was calculated using the following formula: [ligand]/[ligand] + Kd. This can then be used to calculate number of occupied receptors by multiplication of fractional occupancy × Bmax.
RESULTS
CCK Binding and Signaling
Fig. 2 illustrates CCK radioligand binding and biological activity at wild type CCK1R and CCK2R and at the Y140A mutant of CCK1R and the analogous Y153A mutant of CCK2R. Also shown are the effects of augmentation of the membrane cholesterol in these cells. As we demonstrated previously in transient expression assays (12, 14), the type 1 CCK receptor was quite sensitive to its cholesterol environment, exhibiting higher affinity binding in the stable receptor-bearing CHO-CCK1R cell line under these conditions (3.6-fold increase in affinity in the presence of increased membrane cholesterol, p < 0.05, Fig. 2 and Table 1). Of note, this did not result in a parallel increase in natural agonist-stimulated biological activity. Instead, intracellular calcium responses to CCK in this construct were actually shifted to the right (10-fold reduction in potency, p < 0.05, see Fig. 2 and Table 2). The calculated number of wild type CCK1R receptors occupied to elicit a half-maximal biological response (EC50) was 1,138 ± 141, whereas the number of wild type receptors occupied to elicit a similar increase in biological response was 15,709 ± 4,819 when the membrane cholesterol was increased, representing an increase of 13.8-fold (p < 0.05). This is consistent with reduced G protein coupling efficiency in this environment. In contrast, there were no shifts in CCK binding affinity or biological activity in the stably expressing CHO-CCK2R cell line (p > 0.05, Fig. 2 and Tables 1 and 2).
FIGURE 2.

CCK binding and signaling responses at wild type and mutant CCK receptors in the absence or presence of elevated membrane cholesterol. Shown are CCK competition-binding (top panels, A and B) and dose-dependent calcium-response curves (bottom panels, C and D) for receptor-bearing CHO cell lines for CCK1R WT and CCK1R Y140A in the presence of excess membrane cholesterol (A and C), as well as for the corresponding mutant CCK2R Y153A (B and D). Inset shows representative images of filipin staining of cholesterol in control and cholesterol-modulated CHO-CCK1R cells, where an increase in staining is observed on cells with augmented membrane cholesterol. Radioligand binding values reflect percentages of maximum saturable CCK radioligand binding in the absence of competing CCK. Intracellular calcium responses are expressed as percentages of the maximal responses measured for each condition. The curves represent mean ± S.E. from four to nine independent experiments performed in duplicate.
TABLE 1.
Binding affinities for CCK receptor ligands at the wild type CCK1R in the absence and presence of increased membrane cholesterol and at CCK1R Y140A mutant
Values represent means ± S.E. from three to nine independent experiments performed in duplicate. chol. means cholesterol.
| Receptor | Ligand | Condition | Binding affinity Ki (nm) | Binding sites (sites/cell × 105) |
|---|---|---|---|---|
| CCK1R | CCK | WT | 12.1 ± 1.1 | 5.6 ± 1.8 |
| WT + excess chol. | 3.3 ± 0.3a | 2.9 ± 0.9a | ||
| Y140A | 1.3 ± 0.2a | 1.6 ± 0.5a | ||
| Y140A + excess chol. | 1.0 ± 3.2a | 0.4 ± 0.1a | ||
| A71623 | WT | 32.1 ± 5.4 | 8.3 ± 1.1 | |
| WT + excess chol. | 6.2 ± 0.2a | 3.3 ± 0.8a | ||
| Y140A | 5.5 ± 1.1a | 1.6 ± 1.2a | ||
| CCK2R | CCK | WT | 1.9 ± 0.6 | 4.9 ± 1.5 |
| WT + excess chol. | 1.3 ± 0.1 | 3.2 ± 0.3 | ||
| Y153A | 1.5 ± 0.4 | 5.2 ± 1.7 |
a p < 0.05 compared with the wild type receptor.
TABLE 2.
Biological activity of CCK receptor agonists at the wild type CCK1R in the absence or presence of increased membrane cholesterol and at the CCK1R Y140A mutant
Values represent means ± S.E. from three to nine independent experiments performed in duplicate. chol. means cholesterol.
| Receptor | Ligand | Condition | Calcium responses EC50 (nm) |
|---|---|---|---|
| CCK1R | CCK | WT | 0.02 ± 0.01 |
| WT + excess chol. | 0.20 ± 0.07a | ||
| Y140A | 0.37 ± 0.07a | ||
| Y140A + excess chol. | 0.44 ± 0.11a | ||
| A71623 | WT | 0.19 ± 0.06 | |
| WT + excess chol. | 1.38 ± 0.50a | ||
| Y140A | 1.24 ± 0.26a | ||
| Gl181771X | WT | 0.72 ± 0.15 | |
| WT + excess chol. | 11.2 ± 2.3a | ||
| Y140A | 6.3 ± 1.44a | ||
| CCK2R | CCK | WT | 0.30 ± 0.04 |
| WT + excess chol. | 0.28 ± 0.14 | ||
| Y153A | 0.44 ± 0.07 |
a p < 0.05 compared with the wild type receptor.
The site mutants of the analogous tyrosine residues at the bottom of TM3 in these receptors (position 3.51 based on the Ballesteros and Weinstein numbering system (26)), Y140A mutant of CCK1R and Y153A mutant of CCK2R, behaved like their parental receptors in the presence of high membrane cholesterol. The Y140A CCK1R construct exhibited higher CCK binding affinity and lower CCK-stimulated biological activity than wild type CCK1R (9.3-fold increase in binding affinity, p < 0.05, Fig. 2 and Table 1; 22-fold reduction in potency, p < 0.05, Table 2), although these characteristics of the Y153A CCK2R construct were not different from wild type CCK2R (p > 0.05, Fig. 2 and Tables 1 and 2). Of note, the cell line expressing the Y140A CCK1R construct had fewer receptors on the cell surface than the cell lines expressing wild type CCK receptors. Because this could result in a right shift in biological response curves, we also calculated the number of these receptors that are needed to be occupied to yield a half-maximal biological response. Indeed, the number of Y140A CCK1R receptors occupied to elicit an EC50 response was 33,623 ± 5,524, which is not different from the wild type CCK1R receptors necessary to elicit a similar response when present in a high cholesterol environment. This is consistent with reduced G protein coupling of this mutant receptor construct. The measured binding affinities, biological potencies of CCK, and the receptor occupancy at the EC50 at the wild type CCK1R in the presence of excess cholesterol and at the CCK1R Y140A-bearing cells were not different from each other (p > 0.05).
Fluorescence Anisotropy of Receptor-bound Alexa488-CCK
The fluorescence characteristics of a receptor-bound ligand can provide insights into the microenvironment in which the fluorophore resides when bound, thereby providing indirect insights into the conformation of the receptor. We previously reported that the anisotropy of Alexa488-CCK bound to the CCK1R changed in the presence of increased membrane cholesterol, increasing to reflect decreased rotational motion of the fluorophore (12). This observation was repeated here (Fig. 3), demonstrating a 1.2-fold increase in anisotropy at 4 °C (p < 0.05). The trend was similar at 20 °C, although it did not reach statistical significance. The anisotropy of this fluorescent probe when bound to the CCK1R Y140A construct was in the same range as when bound to the wild type CCK1R in the presence of high membrane cholesterol, also exhibiting a significantly higher value than that observed for wild type CCK1R at 4 °C (Fig. 3). Of note, this anisotropy value did not change when the Y140A CCK1R construct was expressed in a high membrane cholesterol environment.
FIGURE 3.

Fluorescence anisotropy of Alexa488-CCK bound to the CCK1 receptor-bearing CHO cells. Shown are the values of fluorescence anisotropy of receptor-bound Alexa488-CCK at two different temperatures, 20 °C (left) and 4 °C (right). For each temperature, fluorescence anisotropy values were measured for the CCK1R WT or CCK1R Y140A-bound Alexa488-CCK in the absence or presence of excess cholesterol (chol.). Data represent means ± S.E. of data from four to eight independent experiments. *, p < 0.05, compared with control.
Internalization of CCK in Receptor-bearing CHO Cells
We previously demonstrated that agonist-stimulated internalization of CCK1R is not affected by the presence of high membrane cholesterol (12). Here, we studied Alexa488-CCK internalization in wild type CCK1R and CCK1R Y140A-bearing cell lines. The CCK1R Y140A construct behaved like wild type CCK1R, with the fluorescent agonist ligand probe internalizing normally, with the same time course as that observed for the wild type CCK1R (Fig. 4). Indeed, internalization of wild type CCK1R was not affected by increasing membrane cholesterol.
FIGURE 4.
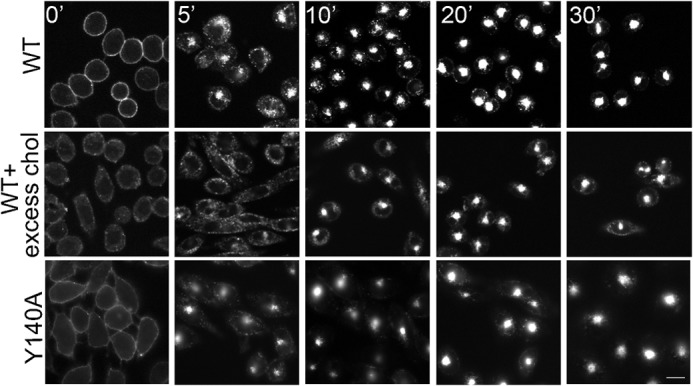
Internalization kinetics of CCK1 receptors on CHO cell lines. Shown are representative fluorescence images for internalization of Alexa488-CCK bound to CCK1R WT on CHO cells under normal or excess cholesterol conditions, as well as on the CCK1R Y140A CHO cells at various time points. Cells internalized the ligand normally at each condition. Images are representative of three independent experiments. Scale bar, 10 μm.
Non-natural Ligand Binding and Activity at Wild Type and Mutant CCK Receptors
Presumably, the changes in CCK ligand binding and CCK-stimulated biological activity at the CCK1R in the presence of increased membrane cholesterol reflect the impact of this membrane environment on receptor conformation. If the conformation of the Y140A CCK1R mutant in a normal membrane environment mimics the abnormal conformation of wild type CCK1R in a high cholesterol environment, it might also be expected to bind and respond to a variety of ligands having distinct structures in a manner similar to the wild type receptor in a high cholesterol environment.
In this series of studies, we compared the binding and agonist-stimulated biological activity of a series of CCK1R ligands. The tetrapeptide agonist ligand, A71623 (45), exhibited increased binding affinity for wild type CCK1R in the presence of increased membrane cholesterol (5.1-fold increase in affinity, p < 0.05, Fig. 5 and Table 1). Its binding to CCK1R Y140A was similar to that in the high cholesterol environment (5.8-fold higher than wild type CCK1R, p < 0.05, Fig. 5 and Table 1). Also, as shown in Fig. 5, the high binding affinity was nonproductive, as measured by a significant decrease in its potency to stimulate a biological response (7.2-fold reduction in potency at the wild type CCK1R in the presence of increased membrane cholesterol, p < 0.05, Fig. 5 and Table 2). The potency of A71623 at the CCK1R Y140A mutant was similar to that at the wild type CCK1R in the high cholesterol environment (6.5-fold decrease in potency relative to the wild type CCK1R in a normal environment, p < 0.05, Fig. 5 and Table 2). For this compound, as for CCK, the calculated number of ligand-occupied receptors necessary to elicit an EC50 response was similar to the Y140A CCK1R construct and the wild type CCK1R in a high membrane cholesterol environment (28,063 ± 4,908 and 54,678 ± 15,781, respectively, representing 5.6- and 10.9-fold higher numbers of receptors (p < 0.05) than for wild type CCK1R in a normal membrane cholesterol environment (5,007 ± 1,548)). For A71623, the binding affinities, biological potencies, and fractional receptor occupation at EC50 at the wild type CCK1R in the presence of excess cholesterol and at the CCK1R Y140A-bearing cells were not different (p > 0.05).
FIGURE 5.

A71623 binding and biological activity in cholesterol-modulated CCK1R WT CHO cells, as well as CCK1R Y140A cells. Shown are the heterologous competition binding curves with increasing concentrations of A71623 competing for CCK-radioligand binding to the CCK1R WT CHO cells in the absence or presence of excess cholesterol (chol.), as well as on the CCK1R Y140A CHO cells (left). Values reflect saturable binding as a percentage of that occurring in the absence of competitor. Also shown are the concentration-dependent A71623-stimulated intracellular calcium responses on the same cells (right). Intracellular calcium responses are expressed as percentages of the maximal responses measured for each condition. Values represent mean ± S.E. from three to six independent experiments performed in duplicate.
The molecular basis of CCK1R binding of benzodiazepine ligands has been most extensively studied. These ligands bind to an allosteric site within the intramembranous helical bundle that is distinct from the orthosteric CCK peptide-binding site of CCK1R (46–48). We used the benzodiazepine antagonist, BDZ-1, which has been shown to bind within the intramembranous allosteric pocket, with major determinants in transmembrane segments six and seven (36), as well as the benzodiazepine agonist, GI181771X, which binds within the same pocket, but with distinct determinants (43). Fig. 6 illustrates data from competition binding studies using the BDZ-1 radioligand, 125I-BDZ-1. BDZ-1 exhibited higher binding affinity to the wild type CCK1R in the presence of elevated membrane cholesterol (Ki, 0.55 ± 0.1 versus 2.1 ± 0.4 nm, increased by 3.8-fold, p < 0.05, Fig. 6A), and its binding affinity to the Y140A CCK1R mutant was similar to that of wild type CCK1R in the presence of high cholesterol (Ki, 0.3 ± 0.1 nm). The affinity of BDZ-1 for the CCK1R Y140A construct was observed to be significantly higher than its binding affinity for wild type CCK1R (7-fold increase, p < 0.05, Fig. 6A).
FIGURE 6.

Binding and signaling behavior of benzodiazepine ligands in CCK1R-bearing CHO cells. Shown are the curves for the homologous competition binding of the benzodiazepine antagonist, BDZ-1 (A), and competition binding for the CCK-like radioligand (B) and calcium responses (C) of the agonist, GI181771X, on CCK1R WT bearing CHO cells in the absence or presence of excess cholesterol, as well as on the CCK1R Y140A-bearing CHO cells. Nonsaturable binding of BDZ-1 was measured using a 1 μm concentration of the same unlabeled ligand, whereas in the case of GI181771X, 1 μm unlabeled CCK was used. Values represent means ± S.E. from three to five independent experiments performed in duplicate.
In competition-binding studies using the CCK-like radioligand, the GI181771X competition curves were not statistically different for the Y140A CCK1R mutant (Ki, 380 ± 76 nm) and the wild type CCK1R in the absence or presence of increased membrane cholesterol (Ki, 1,100 ± 340 versus 660 ± 190 nm, respectively, p > 0.05, Fig. 6B). Also shown in this figure, GI181771X stimulated intracellular calcium responses in a concentration-dependent manner, with its potency reduced in the presence of increased membrane cholesterol (15-fold reduction, p < 0.05, Fig. 6C and Table 2). Its ability to stimulate intracellular calcium in the Y140A CCK1R construct was similar to that for the wild type CCK1R in the presence of increased cholesterol, and it reflected a significant reduction relative to wild type CCK1R in its natural environment (8.8-fold reduction, p < 0.05, Fig. 6C and Table 2). When compared with CCK1R WT, the fraction of ligand-occupied receptors at the EC50 was significantly higher for CCK1R Y140A (∼25-fold) and CCK1R WT in the presence of excess membrane cholesterol (∼25.5-fold). The measured binding affinities, biological potencies of GI181771X, and fractional receptor occupation at EC50 at the wild type CCK1R in the presence of excess cholesterol and at the CCK1R Y140A-bearing cells were not different (p > 0.05).
Effect of the Nonhydrolyzable GTP Analog GppNHp
Receptor binding in the presence of GppNHp is known to move this receptor toward its G protein-uncoupled state, often resulting in a lower binding affinity (49). Indeed, this effect was observed for wild type CCK1R, where treatment with 1 μm GppNHp resulted in a 2.8-fold reduction in the binding affinity of CCK for this receptor when compared with control (Ki, 40.3 ± 4.3 versus 14.2 ± 1.7 nm, respectively, p < 0.05, Fig. 7). To determine the sensitivity of the wild type CCK1R in high membrane cholesterol environment, we used cell membranes from CCK1R-bearing SRD15 cells that have increased levels of cholesterol in their plasma membrane (2.0-fold higher than control) (15). We previously reported the functional characteristics of this cell line as similar to CCK1R behavior in a high membrane cholesterol environment (15). We observed that the wild type CCK1R in these membranes was no longer sensitive to 1 μm GppNHp when compared with control (Ki, 4.6 ± 2.6 versus 2.7 ± 0.9 nm, respectively, Fig. 7), indicating less efficient G protein-coupling. Similar to this, the CCK1R Y140A mutant also did not exhibit a shift to a lower binding affinity in the presence of this concentration of GppNHp when compared with the control (Ki, 1.2 ± 0.2 versus 1.1 ± 0.3 nm respectively, Fig. 7).
FIGURE 7.

Effect of GppNHp on receptor binding of 125I-CCK. Shown are homologous competition binding curves demonstrating the effect of 1 μm GppNHp on CCK binding at the wild type receptor expressed on membranes from CHO cells with normal cholesterol (chol.) (left), on membranes from CCK1R-bearing SRD15 cells with high membrane cholesterol (middle), and on membranes from the CCK1R Y140A mutant expressed on CHO cells (right). Saturable binding of the 125I-CCK was determined by competing with 1 μm nonradioactive CCK. Data represent means ± S.E. of values from three to four independent experiments performed in duplicate.
Nature of the Allosteric Ligand Binding Pocket in Y140A CCK1R
Similar to our previous approach to map the allosteric small molecule ligand-binding site within the CCK1R using chimeric CCK1R/CCK2R constructs (36, 43), we introduced the Y140A mutation into each of these constructs to test for significant differences in this pocket that this mutation might introduce. Fig. 8A shows that the benzodiazepine radioligand, 125I-BDZ-1, exhibited saturable binding to the wild type CCK1 receptor, with concentration-dependent competition by GI181771X. In contrast, this radioligand did not bind saturably to CCK2R. Because of this clear differential in binding, chimeric CCK1R/CCK2R constructs in which all of the differences in the small molecule binding pocket between these two receptors were exchanged were particularly useful to define molecular determinants. The CCK1R Y140A constructs in which the distinct residues from CCK2R in TM2 (N2.61T), TM6 (I6.51V,F6.52Y), and TM7 (L7.39H) were introduced, continued to bind this ligand saturably (Fig. 8A). Whereas the affinities of the TM2 and TM6 chimeric constructs were not different from that of the CCK1R Y140A parental construct, that of the TM7 chimeric construct was actually higher. This establishes that these residues are not the determinants responsible for the poor binding to CCK2R. In contrast, introducing the distinct residues from CCK2R in TM3 (T3.28V,T3.29S) into CCK1R Y140A eliminated saturable binding, indicating that this region in CCK1R is clearly important for binding of GI181771X.
FIGURE 8.

Receptor binding and biological activity of chimeric CCK receptor constructs. Shown are competition binding curves demonstrating the abilities of GI181771X to compete for binding of the CCK1 receptor-selective benzodiazepine radioligand, 125I-BDZ-1 (A), and curves representing the abilities of GI181771X to stimulate intracellular calcium responses in wild type and chimeric Y140A CCK receptor-bearing cells (B). Saturable binding of the 125I-BDZ-1 was determined by competing with 1 μm nonradioactive benzodiazepine ligand. Calcium data are plotted relative to the maximal responses of each cell line to CCK. Data represent means ± S.E. of values from four to six independent experiments performed in duplicate.
The biological activity studies with the CCK1R/CCK2R chimeric constructs were even more informative. Here, too, there were clear functional differences between the two CCK receptor subtypes, with GI181771X acting as a full agonist at CCK1R and having no biological activity at CCK2R (43). When the analogous CCK2R residues were used to replace the CCK1R residues in each TM segment of the CCK1R Y140A construct, significant calcium responses were still observed for the replacements in TM6 and TM2, but there were no observed calcium responses when these residues in TM7 or TM3 were introduced (Fig. 8B). Because the TM3 chimeric construct did not bind saturably, the absence in biological activity was expected. However, the TM7 chimeric construct bound with even higher affinity than CCK1R Y140A, establishing L7.39 as a critical residue for biological activity. GI181771X was a more potent stimulant of calcium responses in the TM2 chimeric construct than in the CCK1R Y140A construct, establishing that N2.61 is not an important residue for biological activity.
Comparing the data for these chimeric constructs in the presence of Y140A with those previously reported for the same constructs in the absence of this mutation (43), there were some key differences (Table 3). For the binding determinants, there was a major difference for the impact of the TM3 chimeric construct (T3.28V,T3.29S), with this actually increasing affinity for the benzodiazepine at the wild type CCK1R, although it eliminated binding at the CCK1R Y140A construct. We know that this construct was expressed on the cell surface because the natural peptide agonist, CCK, bound to this receptor in intact cells (Ki, 0.22 ± 0.12 nm) and stimulated a full biological response (EC50, 0.74 ± 0.16 nm). The other noteworthy change in binding involved the TM7 chimeric construct in which there was no effect on binding of the benzodiazepine to the wild type CCK1R, although there was higher affinity binding for the CCK1R Y140A mutant. Thus, T3.28 and T3.29 appear to be more important for binding in the Y140A mutant than at the wild type CCK1R receptor, and L7.39 appears to be less important for binding in this mutant. Looking at the biological activity data, the major difference observed was for the TM2 chimeric construct in which potency for stimulating intracellular calcium responses was reduced relative to the wild type CCK1R in the absence of Y140A and increased in the presence of this mutation. This suggests that N2.61 is less important for biological activity in the presence of this mutation. In contrast, the negative impact of the TM3 construct was much more significant in the presence of Y140A, suggesting that T3.28 and T3.29 are more important determinants of activity in the presence of this mutation.
TABLE 3.
Binding and biological activity parameters of GI181771X in CCK1R mutants in CHO cells
Values represent means ± S.E. from four to six independent experiments performed in duplicate. The following abbreviations are used: NDB, no detectable binding; NR, no response.
| Receptors | Binding affinity |
Biological activity |
|||
|---|---|---|---|---|---|
| WTa Ki (nm) | Y140A Ki (nm) | WTa EC50 (nm) | Y140A EC50 (nm) | ||
| CCK1R | WT | 97 ± 17 | 42 ± 4 | 0.8 ± 0.1 | 5.1 ± 0.02b |
| CCK1R TM2 | N2.61T | 89 ± 37 | 84 ± 35 | 4.5 ± 1.0 | 1.2 ± 0.2b |
| CCK1R TM3 | T3.28V,T3.29S | 6 ± 1 | NDBb | 2.5 ± 1.3 | NRb |
| CCK1R TM6 | I6.51V,F6.52Y | 74 ± 8 | 39 ± 8 | 19.6 ± 5.5 | 28 ± 8 |
| CCK1R TM7 | L7.39H | 109 ± 44 | 15 ± 3b | NR | NR |
| CCK2R | WT | NDB | NR | NR | |
a Data for chimeric wild type CCK1R/CCK2R constructs previously reported (43) are shown for comparison with Y140A mutant data.
b p < 0.05 compared with WT receptor.
DISCUSSION
A prominent feature of GPCRs is their shape-changing character (50). This is responsible for their ability to bind an extracellular agonist ligand and change the conformation of their intracellular face that couples with its G protein to initiate signaling. It is also responsible for the possibility of allosteric modulation of these receptors to change other characteristics, such as agonist specificity and signaling characteristics. Lateral allosteric regulation of the CCK1R, induced by a membrane environment with elevated cholesterol content, is responsible for the defective coupling of this receptor with its G protein (20, 21), resulting in a defective biological response observed in the setting of cholesterol gallstone disease (19). This is also likely present to reduce the effectiveness of the negative feedback mechanism regulating postprandial satiety through CCK action on vagal afferent nerves in obese subjects (3).
We are particularly interested in a strategy to utilize an allosteric drug to correct the abnormal conformation of the CCK1R when it is in a high cholesterol membrane environment. If such a drug has no endogenous agonist activity, it could simply correct the biological response to CCK released after a meal, thereby stimulating satiety and reducing food intake. This effect would be active only when CCK is in the circulation. Because this hormone has a half-life of only a few minutes, the duration of this effect would be limited, thereby reducing the risk of excess activation of this receptor.
A strategy to identify such an allosteric modulator is challenging, due to the absence of stable, easily handled model systems to reflect the CCK receptor in a high cholesterol membrane environment. Most studies have utilized animal models or limited precious human biospecimens that are obviously finite and not readily available (16, 18, 19). Cholesterol has been increased in the membrane using acute cholesterol loading strategies (12, 14), but this does not yield reproducible levels of cholesterol enrichment and is not stable over time. Similarly, cell lines have been engineered to include mutations in the lipid metabolic machinery to yield elevated cholesterol, but these require highly specialized and often expensive media and grow quite slowly, as well as carrying the risk of reversion of phenotype (15, 51, 52).
For these reasons, we have been encouraged by the possibility of developing a mutant CCK1R cell line that could act as a surrogate, mimicking the structure and function of the CCK1R in a high cholesterol environment. Such a cell line could be readily propagated with a stable phenotype and could be used in a high throughput screening strategy. We are hopeful that the Y140A CCK1R-bearing cell line developed for this work could be such a cell line. In this work, we demonstrate that this line has the CCK binding and biological activity properties observed for the wild type receptor in a high cholesterol environment. Of interest, augmentation of cholesterol in the membrane of the Y140A CCK1R-bearing cells did not further modify these functions. Additionally, other ligands of the CCK1R behaved similarly at the Y140A CCK1R-bearing cells as they did at the wild type CCK1R in a high cholesterol environment. We also show that the resultant decrease in the potencies of the agonists at these two conditions is likely due to reduced coupling efficiency of the receptor to its G protein, as indicated by an increased fractional occupation of receptors at these two conditions that is necessary to elicit an EC50 response, than at the wild type receptor. Although this was a limited series of ligands, it spanned distinct chemotypes and included both agonists and antagonists. Behavior like the wild type receptor in a high cholesterol membrane environment was further confirmed by the fluorescence studies probing the microenvironment of CCK bound to this receptor. Even the internalization behavior of this mutant was similar to that of wild type CCK1R in a high cholesterol environment. It is believed that increased membrane cholesterol is responsible for the defective G protein coupling of the CCK1R in cholesterol gallstone disease (19–21). Consistent with this, we now show that the CCK1R Y140A mutant also exhibits defective G protein coupling, contributing to the defective signaling observed with this mutant.
It was particularly interesting that the mutation that resulted in this phenotype affected a residue in the (E/D)RY motif, which is known to play a very important role for stabilizing the inactive state of class A GPCRs (27, 28). However, it involved the tyrosine residue in this motif that has been the least conserved residue in the motif, which has been mutated in a variety of receptors, with minimal or no functional impact (29–33). Perhaps the dual role of this tyrosine as also being a part of a cholesterol-binding motif in CCK1R (14) makes it different from other analogous residues in related class A GPCRs. When this tyrosine is mutated to an alanine, it can no longer bind cholesterol, but it seems to mimic the effect on receptor structure and function of the cholesterol-bound tyrosine in that position.
The molecular basis for the binding of the natural orthosteric agonist ligand of this receptor, the CCK peptide, has been carefully determined (53–55). We have direct spatial approximation data for five of the six residues within the hormone pharmacophore, establishing the receptor residue adjacent to it when docked at this receptor (49, 53, 55). These studies demonstrate that the orthosteric site is at the external surface of the receptor, utilizing molecular determinants within the receptor amino-terminal tail and external loop regions. In contrast, benzodiazepine ligands have had their intramembranous site of action mapped using mutagenesis (36, 56) and even direct photoaffinity labeling (47). This has been demonstrated using pharmacological approaches to represent an allosteric site that is distinct from that of docking the natural peptide agonist (36, 48).
The molecular determinants for this allosteric small molecule ligand pocket within the CCK1R have been carefully mapped using site-directed mutagenesis and chimeric CCK1R/CCK2R approaches (36). This was recently extended to provide insights into the differences between the active conformation and inactive conformations of this pocket (43). With all these insights, however, we still have little understanding of the conformational changes in this pocket that are influenced by a high cholesterol membrane environment. This work provides the first insights into what these changes might include. This was achieved by studying the chimeric constructs involving each TM segment lining this pocket that are distinct in CCK1R and CCK2R. With the addition of Y140A into these chimeric CCK1R/CCK2R constructs, differences were observed for both binding and biological activity. Key observations included profound reduction in benzodiazepine binding and biological activity by changing the residues in TM3 (T3.28V,T3.29S) of the Y140A mutant, although the binding affinity was actually increased at the wild type CCK1R, with its biological activity retained. Another important difference was the effect of the TM2 change (N2.61T) that improved biological activity at the CCK1R Y140A construct, while reducing it at the wild type CCK1R. These observations suggest a more important role of TM3 in binding and biological activity and a less important role of TM2 for binding in the presence of the Y140A than in the wild type receptor. With normal docking of the benzodiazepine to the wild type CCK1R, the benzo ring points toward TM3 and the N1 isopropyl group believed to represent the agonist trigger points toward L7.39. Presumably, the conformational change in this allosteric pocket may move the ligand toward TM3, moving the trigger further away from its target on TM7. Unfortunately, these experimental observations are qualitative and not adequate to provide specific constraints to build a meaningful molecular model reflecting the changes implied by these data. However, we can be certain that the cholesterol-enhanced membrane exerts substantial lateral allosteric impact on the CCK1R that results in a conformational change that affects receptor-G protein coupling. Once there are adequate data for ligands that preferentially recognize this conformation over that of the wild type CCK1R, a ligand-directed approach to modeling should provide insights into the details of this conformation.
Acknowledgments
We thank Dr. Brad Henke at GlaxoSmithKline Research Laboratories for providing GI181771X, Drs. P. S. Portoghese and E. Akgun from the University of Minnesota for providing BDZ ligands, and M. L. Augustine and A. M. Ball for their excellent technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant DK032878. This work was also supported by the Mayo Clinic.
- CCK
- cholecystokinin
- BDZ
- benzodiazepine
- CCK1R
- type 1 cholecystokinin receptor
- CCK2R
- type 2 cholecystokinin receptor
- CRAC
- cholesterol recognition/interaction amino acid consensus
- GPCR
- G protein-coupled receptor
- MβCD
- methyl-β-cyclodextrin
- TM
- transmembrane
- GppNHp
- guanosine 5′-[β,γ-imido]triphosphate.
REFERENCES
- 1. Rehfeld J. F. (1978) Immunochemical studies on cholecystokinin. II. Distribution and molecular heterogeneity in the central nervous system and small intestine of man and hog. J. Biol. Chem. 253, 4022–4030 [PubMed] [Google Scholar]
- 2. Kissileff H. R., Pi-Sunyer F. X., Thornton J., Smith G. P. (1981) C-terminal octapeptide of cholecystokinin decreases food intake in man. Am. J. Clin. Nutr. 34, 154–160 [DOI] [PubMed] [Google Scholar]
- 3. Smith G. P., Gibbs J. (1985) The satiety effect of cholecystokinin. Recent progress and current problems. Ann. N.Y. Acad. Sci. 448, 417–423 [DOI] [PubMed] [Google Scholar]
- 4. Beglinger C., Degen L., Matzinger D., D'Amato M., Drewe J. (2001) Loxiglumide, a CCK-A receptor antagonist, stimulates calorie intake and hunger feelings in humans. Am. J. Physiol. 280, R1149–R1154 [DOI] [PubMed] [Google Scholar]
- 5. Aquino C. J., Armour D. R., Berman J. M., Birkemo L. S., Carr R. A., Croom D. K., Dezube M., Dougherty R. W., Jr., Ervin G. N., Grizzle M. K., Head J. E., Hirst G. C., James M. K., Johnson M. F., Miller L. J., Queen K. L., Rimele T. J., Smith D. N., Sugg E. E. (1996) Discovery of 1,5-benzodiazepines with peripheral cholecystokinin (CCK-A) receptor agonist activity. 1. Optimization of the agonist “trigger”. J. Med. Chem. 39, 562–569 [DOI] [PubMed] [Google Scholar]
- 6. Berger R., Zhu C., Hansen A. R., Harper B., Chen Z., Holt T. G., Hubert J., Lee S. J., Pan J., Qian S., Reitman M. L., Strack A. M., Weingarth D. T., Wolff M., Macneil D. J., Weber A. E., Edmondson S. D. (2008) 2-Substituted piperazine-derived imidazole carboxamides as potent and selective CCK1R agonists for the treatment of obesity. Bioorg. Med. Chem. Lett. 18, 4833–4837 [DOI] [PubMed] [Google Scholar]
- 7. Sherrill R. G., Berman J. M., Birkemo L., Croom D. K., Dezube M., Ervin G. N., Grizzle M. K., James M. K., Johnson M. F., Queen K. L., Rimele T. J., Vanmiddlesworth F., Sugg E. E. (2001) 1,4-Benzodiazepine peripheral cholecystokinin (CCK-A) receptor agonists. Bioorg. Med. Chem. Lett. 11, 1145–1148 [DOI] [PubMed] [Google Scholar]
- 8. Zhu C., Hansen A. R., Bateman T., Chen Z., Holt T. G., Hubert J. A., Karanam B. V., Lee S. J., Pan J., Qian S., Reddy V. B., Reitman M. L., Strack A. M., Tong V., Weingarth D. T., Wolff M. S., MacNeil D. J., Weber A. E., Duffy J. L., Edmondson S. D. (2008) Discovery of imidazole carboxamides as potent and selective CCK1R agonists. Bioorg. Med. Chem. Lett. 18, 4393–4396 [DOI] [PubMed] [Google Scholar]
- 9. Jordan J., Greenway F. L., Leiter L. A., Li Z., Jacobson P., Murphy K., Hill J., Kler L., Aftring R. P. (2008) Stimulation of cholecystokinin-A receptors with GI181771X does not cause weight loss in overweight or obese patients. Clin. Pharmacol. Ther. 83, 281–287 [DOI] [PubMed] [Google Scholar]
- 10. Hoshi H., Logsdon C. D. (1993) Both low- and high-affinity CCK receptor states mediate trophic effects on rat pancreatic acinar cells. Am. J. Physiol. 265, G1177–G1181 [DOI] [PubMed] [Google Scholar]
- 11. Smith J. P., Solomon T. E. (2014) Cholecystokinin and pancreatic cancer: the chicken or the egg? Am. J. Physiol. 306, G91–G101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harikumar K. G., Puri V., Singh R. D., Hanada K., Pagano R. E., Miller L. J. (2005) Differential effects of modification of membrane cholesterol and sphingolipids on the conformation, function, and trafficking of the G protein-coupled cholecystokinin receptor. J. Biol. Chem. 280, 2176–2185 [DOI] [PubMed] [Google Scholar]
- 13. Desai A. J., Miller L. J. (2012) Sensitivity of cholecystokinin receptors to membrane cholesterol content. Front Endocrinol. 3, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Potter R. M., Harikumar K. G., Wu S. V., Miller L. J. (2012) Differential sensitivity of types 1 and 2 cholecystokinin receptors to membrane cholesterol. J. Lipid Res. 53, 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harikumar K. G., Potter R. M., Patil A., Echeveste V., Miller L. J. (2013) Membrane cholesterol affects stimulus-activity coupling in type 1, but not type 2, CCK receptors: use of cell lines with elevated cholesterol. Lipids 48, 231–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu P., Chen Q., Biancani P., Behar J. (1996) Membrane cholesterol alters gallbladder muscle contractility in prairie dogs. Am. J. Physiol. 271, G56–G61 [DOI] [PubMed] [Google Scholar]
- 17. Behar J., Lee K. Y., Thompson W. R., Biancani P. (1989) Gallbladder contraction in patients with pigment and cholesterol stones. Gastroenterology 97, 1479–1484 [DOI] [PubMed] [Google Scholar]
- 18. Chen Q., Amaral J., Oh S., Biancani P., Behar J. (1997) Gallbladder relaxation in patients with pigment and cholesterol stones. Gastroenterology 113, 930–937 [DOI] [PubMed] [Google Scholar]
- 19. Xiao Z. L., Chen Q., Amaral J., Biancani P., Jensen R. T., Behar J. (1999) CCK receptor dysfunction in muscle membranes from human gallbladders with cholesterol stones. Am. J. Physiol. 276, G1401–G1407 [DOI] [PubMed] [Google Scholar]
- 20. Yu P., Chen Q., Harnett K. M., Amaral J., Biancani P., Behar J. (1995) Direct G protein activation reverses impaired CCK signaling in human gallbladders with cholesterol stones. Am. J. Physiol. 269, G659–G665 [DOI] [PubMed] [Google Scholar]
- 21. Xiao Z. L., Chen Q., Amaral J., Biancani P., Behar J. (2000) Defect of receptor-G protein coupling in human gallbladder with cholesterol stones. Am. J. Physiol. 278, G251–G258 [DOI] [PubMed] [Google Scholar]
- 22. Seres I., Fóris G., Varga Z., Kosztáczky B., Kassai A., Balogh Z., Fülöp P., Paragh G. (2006) The association between angiotensin II-induced free radical generation and membrane fluidity in neutrophils of patients with metabolic syndrome. J. Membr. Biol. 214, 91–98 [DOI] [PubMed] [Google Scholar]
- 23. Chen Q., Amaral J., Biancani P., Behar J. (1999) Excess membrane cholesterol alters human gallbladder muscle contractility and membrane fluidity. Gastroenterology 116, 678–685 [DOI] [PubMed] [Google Scholar]
- 24. Li H., Papadopoulos V. (1998) Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 139, 4991–4997 [DOI] [PubMed] [Google Scholar]
- 25. Hanson M. A., Cherezov V., Griffith M. T., Roth C. B., Jaakola V. P., Chien E. Y., Velasquez J., Kuhn P., Stevens R. C. (2008) A specific cholesterol binding site is established by the 2.8 A structure of the human β2-adrenergic receptor. Structure 16, 897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ballesteros J. A., Weinstein H. (1992) Analysis and refinement of criteria for predicting the structure and relative orientations of transmembranal helical domains. Biophys. J. 62, 107–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rovati G. E., Capra V., Neubig R. R. (2007) The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol. Pharmacol. 71, 959–964 [DOI] [PubMed] [Google Scholar]
- 28. Palczewski K., Kumasaka T., Hori T., Behnke C. A., Motoshima H., Fox B. A., Le Trong I., Teller D. C., Okada T., Stenkamp R. E., Yamamoto M., Miyano M. (2000) Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739–745 [DOI] [PubMed] [Google Scholar]
- 29. Gáborik Z., Jagadeesh G., Zhang M., Spát A., Catt K. J., Hunyady L. (2003) The role of a conserved region of the second intracellular loop in AT1 angiotensin receptor activation and signaling. Endocrinology 144, 2220–2228 [DOI] [PubMed] [Google Scholar]
- 30. Ohyama K., Yamano Y., Sano T., Nakagomi Y., Wada M., Inagami T. (2002) Role of the conserved DRY motif on G protein activation of rat angiotensin II receptor type 1A. Biochem. Biophys. Res. Commun. 292, 362–367 [DOI] [PubMed] [Google Scholar]
- 31. Zhu S. Z., Wang S. Z., Hu J., el-Fakahany E. E. (1994) An arginine residue conserved in most G protein-coupled receptors is essential for the function of the m1 muscarinic receptor. Mol. Pharmacol. 45, 517–523 [PubMed] [Google Scholar]
- 32. Hawtin S. R. (2005) Charged residues of the conserved DRY triplet of the vasopressin V1a receptor provide molecular determinants for cell surface delivery and internalization. Mol. Pharmacol. 68, 1172–1182 [DOI] [PubMed] [Google Scholar]
- 33. Proulx C. D., Holleran B. J., Boucard A. A., Escher E., Guillemette G., Leduc R. (2008) Mutational analysis of the conserved Asp2.50 and ERY motif reveals signaling bias of the urotensin II receptor. Mol. Pharmacol. 74, 552–561 [DOI] [PubMed] [Google Scholar]
- 34. Powers S. P., Pinon D. I., Miller L. J. (1988) Use of N,O-bis-Fmoc-d-Tyr-ONSu for introduction of an oxidative iodination site into cholecystokinin family peptides. Int. J. Pept. Protein Res. 31, 429–434 [DOI] [PubMed] [Google Scholar]
- 35. Cheng Z. J., Harikumar K. G., Holicky E. L., Miller L. J. (2003) Heterodimerization of type A and B cholecystokinin receptors enhance signaling and promote cell growth. J. Biol. Chem. 278, 52972–52979 [DOI] [PubMed] [Google Scholar]
- 36. Cawston E. E., Lam P. C., Harikumar K. G., Dong M., Ball A. M., Augustine M. L., Akgün E., Portoghese P. S., Orry A., Abagyan R., Sexton P. M., Miller L. J. (2012) Molecular basis for binding and subtype selectivity of 1,4-benzodiazepine antagonist ligands of the cholecystokinin receptor. J. Biol. Chem. 287, 18618–18635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pang L., Graziano M., Wang S. (1999) Membrane cholesterol modulates galanin-GalR2 interaction. Biochemistry 38, 12003–12011 [DOI] [PubMed] [Google Scholar]
- 38. Amundson D. M., Zhou M. (1999) Fluorometric method for the enzymatic determination of cholesterol. J. Biochem. Biophys. Methods 38, 43–52 [DOI] [PubMed] [Google Scholar]
- 39. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 40. Hadac E. M., Ghanekar D. V., Holicky E. L., Pinon D. I., Dougherty R. W., Miller L. J. (1996) Relationship between native and recombinant cholecystokinin receptors: role of differential glycosylation. Pancreas 13, 130–139 [DOI] [PubMed] [Google Scholar]
- 41. Akgün E., Körner M., Gao F., Harikumar K. G., Waser B., Reubi J. C., Portoghese P. S., Miller L. J. (2009) Synthesis and in vitro characterization of radioiodinatable benzodiazepines selective for type 1 and type 2 cholecystokinin receptors. J. Med. Chem. 52, 2138–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Munson P. J., Rodbard D. (1980) Ligand: a versatile computerized approach for characterization of ligand-binding systems. Anal. Biochem. 107, 220–239 [DOI] [PubMed] [Google Scholar]
- 43. Harikumar K. G., Cawston E. E., Lam P. C., Patil A., Orry A., Henke B. R., Abagyan R., Christopoulos A., Sexton P. M., Miller L. J. (2013) Molecular basis for benzodiazepine agonist action at the type 1 cholecystokinin receptor. J. Biol. Chem. 288, 21082–21095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roettger B. F., Rentsch R. U., Pinon D., Holicky E., Hadac E., Larkin J. M., Miller L. J. (1995) Dual pathways of internalization of the cholecystokinin receptor. J. Cell Biol. 128, 1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin C. W., Shiosaki K., Miller T. R., Witte D. G., Bianchi B. R., Wolfram C. A., Kopecka H., Craig R., Wagenaar F., Nadzan A. M. (1991) Characterization of two novel cholecystokinin tetrapeptide (30–33) analogues, A-71623 and A-70874, that exhibit high potency and selectivity for cholecystokinin-A receptors. Mol. Pharmacol. 39, 346–351 [PubMed] [Google Scholar]
- 46. Kopin A. S., Beinborn M., Lee Y. M., McBride E. W., Quinn S. M. (1994) The CCK-B/gastrin receptor. Identification of amino acids that determine nonpeptide antagonist affinity. Ann. N.Y. Acad. Sci. 713, 67–78 [DOI] [PubMed] [Google Scholar]
- 47. Hadac E. M., Dawson E. S., Darrow J. W., Sugg E. E., Lybrand T. P., Miller L. J. (2006) Novel benzodiazepine photoaffinity probe stereoselectively labels a site deep within the membrane-spanning domain of the cholecystokinin receptor. J. Med. Chem. 49, 850–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gao F., Sexton P. M., Christopoulos A., Miller L. J. (2008) Benzodiazepine ligands can act as allosteric modulators of the Type 1 cholecystokinin receptor. Bioorg. Med. Chem. Lett. 18, 4401–4404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Harikumar K. G., Clain J., Pinon D. I., Dong M., Miller L. J. (2005) Distinct molecular mechanisms for agonist peptide binding to types A and B cholecystokinin receptors demonstrated using fluorescence spectroscopy. J. Biol. Chem. 280, 1044–1050 [DOI] [PubMed] [Google Scholar]
- 50. Kenakin T., Miller L. J. (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 62, 265–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee P. C., Sever N., Debose-Boyd R. A. (2005) Isolation of sterol-resistant Chinese hamster ovary cells with genetic deficiencies in both Insig-1 and Insig-2. J. Biol. Chem. 280, 25242–25249 [DOI] [PubMed] [Google Scholar]
- 52. Chang T. Y., Limanek J. S. (1980) Regulation of cytosolic acetoacetyl coenzyme A thiolase, 3-hydroxy-3-methylglutaryl coenzyme A synthase, 3-hydroxy-3-methylglutaryl coenzyme A reductase, and mevalonate kinase by low density lipoprotein and by 25-hydroxycholesterol in Chinese hamster ovary cells. J. Biol. Chem. 255, 7787–7795 [PubMed] [Google Scholar]
- 53. Miller L. J., Lybrand T. P. (2002) Molecular basis of agonist binding to the type A cholecystokinin receptor. Pharmacol. Toxicol. 91, 282–285 [DOI] [PubMed] [Google Scholar]
- 54. Dong M., Liu G., Pinon D. I., Miller L. J. (2005) Differential docking of high-affinity peptide ligands to type A and B cholecystokinin receptors demonstrated by photoaffinity labeling. Biochemistry. 44, 6693–6700 [DOI] [PubMed] [Google Scholar]
- 55. Harikumar K. G., Pinon D. I., Miller L. J. (2006) Fluorescent indicators distributed throughout the pharmacophore of cholecystokinin provide insights into distinct modes of binding and activation of type A and B cholecystokinin receptors. J. Biol. Chem. 281, 27072–27080 [DOI] [PubMed] [Google Scholar]
- 56. Kopin A. S., McBride E. W., Quinn S. M., Kolakowski L. F., Jr., Beinborn M. (1995) The role of the cholecystokinin-B/gastrin receptor transmembrane domains in determining affinity for subtype-selective ligands. J. Biol. Chem. 270, 5019–5023 [DOI] [PubMed] [Google Scholar]