

Background: eNOS function is regulated post-translationally.
Results: GIT1 phosphorylation through Src activation activates eNOS enzyme function.
Conclusion: Src and GIT1 play an important role in eNOS function.
Significance: The data highlight a novel putative eNOS post-translational modification.
Keywords: Endothelial Cell, Endothelial Dysfunction, Endothelin, G-protein, Nitric Oxide, Nitric-oxide Synthase
Abstract
Nitric oxide (NO) is a critical regulator of vascular tone and plays an especially prominent role in liver by controlling portal blood flow and pressure within liver sinusoids. Synthesis of NO in sinusoidal endothelial cells by endothelial nitric-oxide synthase (eNOS) is regulated in response to activation of endothelial cells by vasoactive signals such as endothelins. The endothelin B (ETB) receptor is a G-protein-coupled receptor, but the mechanisms by which it regulates eNOS activity in sinusoidal endothelial cells are not well understood. In this study, we built on two previous strands of work, the first showing that G-protein βγ subunits mediated activation of phosphatidylinositol 3-kinase and Akt to regulate eNOS and the second showing that eNOS directly bound to the G-protein-coupled receptor kinase-interacting protein 1 (GIT1) scaffold protein, and this association stimulated NO production. Here we investigated the mechanisms by which the GIT1-eNOS complex is formed and regulated. GIT1 was phosphorylated on tyrosine by Src, and Y293F and Y554F mutations reduced GIT1 phosphorylation as well as the ability of GIT1 to bind to and activate eNOS. Akt phosphorylation activated eNOS (at Ser1177), and Akt also regulated the ability of Src to phosphorylate GIT1 as well as GIT1-eNOS association. These pathways were activated by endothelin-1 through the ETB receptor; inhibiting receptor-activated G-protein βγ subunits blocked activation of Akt, GIT1 tyrosine phosphorylation, and ET-1-stimulated GIT1-eNOS association but did not affect Src activation. These data suggest a model in which Src and Akt cooperate to regulate association of eNOS with the GIT1 scaffold to facilitate NO production.
Introduction
Endothelial cell nitric-oxide synthase (eNOS)2 is critical in vascular homeostasis (1). eNOS function is regulated by a complex array of post-translational structural and signaling processes (1). For example, eNOS is known to bind to a number of other proteins including caveolin-1, HSP-90, G-protein-coupled receptor kinase 2 (GRK2), and G-protein-coupled receptor kinase-interacting protein 1 (GIT1) (2–5). These interactions lead to changes in eNOS function, oftentimes as a result of altered phosphorylation of critical serine residues (6).
Src, a non-receptor protein-tyrosine kinase, is involved in many of the signaling mechanisms associated with G-protein-coupled receptors (GPCRs) (7). GPCRs regulate Src family kinase activity by direct interaction with GPCRs or components of the GPCR signaling system including Gα subunits and β-arrestins and may alter their GTPase activity and affect the association of Gα and Gβγ subunits (8–10). GPCRs also activate Src family kinases indirectly through cross-talk with receptor tyrosine kinases and focal adhesion complexes, and Src plays an important role in GPCR signaling (7–8, 11).
GIT1, a GTPase-activating protein for the ADP-ribosylation factor family of small GTP-binding proteins, is a multidomain protein that links signaling proteins to distinct cellular locations (12, 13). It has been recently demonstrated that GIT1 not only functions as a scaffolding protein but also has intrinsic signaling capability (14). For example, upon adhesion of fibroblasts to fibronectin extracellular matrix, GIT1 becomes phosphorylated in fibroblasts by the tyrosine kinases Src and focal adhesion kinase (15, 16). GIT1 also undergoes tyrosine phosphorylation during cell spreading on extracellular matrix in osteoblast cells (17).
Previous work by our laboratory and others has shown that eNOS phosphorylation and activity is reduced in sinusoidal endothelial cells after liver injury (18–20), emphasizing a vascular endotheliopathy in this disease state. We have also shown that eNOS interacts with GIT1 and that this interaction leads to eNOS activation (5). These data led us to postulate that GIT1 would be regulated by phosphorylation in endothelial cells. Furthermore, we sought to identify upstream stimulators of GIT1 tyrosine phosphorylation. Thus, we examined the role of GIT1 tyrosine phosphorylation and putative signaling partners including Src and Akt in the eNOS signaling pathway in endothelial cells.
EXPERIMENTAL PROCEDURES
Cell Isolation and Culture
Sinusoidal endothelial cells were isolated from male Sprague-Dawley rats (450–500 g) (Harlan, Indianapolis, IN). In brief, after in situ perfusion of the liver with 20 mg/100 ml Pronase (Roche Applied Science) followed by collagenase (Worthington), dispersed cell suspensions were removed from a layered discontinuous density gradient of 8.2 and 15.6% Accudenz (Accurate Chemical and Scientific, Westbury, NY), further purified by centrifugal elutriation (18 ml/min flow), and grown in medium containing 20% serum (10% horse/calf). The purity of endothelial cells was documented by visual identification of cultures grown for 48 h. Only primary sinusoidal endothelial isolates of >95% purity were used for study.
siRNA
siRNA-mediated GIT1 knockdown was achieved by introducing three unique siRNA duplexes targeting GIT1 into sinusoidal endothelial cells; scrambled controls were also used. The first siRNA targeting rat GIT1 was as described previously (5); the second and third siRNA duplexes targeting rat GIT1 were 5′-A GAC CUC AGC AAG CAA CUG CAC UCG-3′ and 5′-AG UUC AAA CAU GAC AGC UU UGU GCC-3′, respectively. The scrambled control was 5′-CAT ATT GCG CGT ATA GTC GCG-3′. All were from OriGene Technologies, Inc. (Rockville, MD). We transfected siRNA into sinusoidal endothelial cells with Dharmafect (Dharmacon) according to the manufacturer's instructions.
Expression and Purification of Fusion Proteins
We generated His6-eNOS-NT and His6-eNOS-CT fusion proteins as follows. The bovine eNOS cDNA sequence (NCBI Reference Sequence NM_181037) was used to amplify the N-terminal oxidase domain plus calmodulin binding site (His6-eNOS-NT; residues 1–520) and the C-terminal reductase domain (His6-eNOS-CT; residues 521–1205) and subcloned into the vector pET30c(+) (EMD Millipore Corp., San Diego, CA) at the EcoRI and NotI sites. The full-length His6-eNOS bacterial expression plasmid was a kind gift from Paul Ortiz de Montellano (University of California, San Francisco, CA). All fusions were expressed in the Escherichia coli strain BL21-DE3 (New England Biolabs, Ipswich, MA) and purified using nickel-nitrilotriacetic acid affinity resin (Qiagen, Valencia, CA). Briefly, bound protein was rinsed five times with 50 mm sodium phosphate, 300 mm NaCl, 10% glycerol, pH 6.0 and eluted with 200 mm imidazole in PBS, pH 7.2. Glutathione S-transferase (GST) fusion proteins containing defined domain fragments of GIT1 were generated in pGEX-4T-1 vector as described previously (12), expressed in the E. coli BL21 strain (New England Biolabs), and purified on glutathione-agarose beads for use in pulldown assays.
GST Pulldown Assay
6 μg of GST or GST-GIT1 fragment fusion proteins were incubated with 4 μg of recombinant His6-eNOS (full-length or fragment fusion proteins) for 16 h at 4 °C in binding buffer (20 mm Tris, pH 8.0, 150 mm NaCl, 1% Nonidet P-40). After binding, beads were washed five times with wash buffer (50 mm Tris, 12.5 mm NaCl, 5 mm EDTA, 1 mm EGTA). Beads were eluted by boiling in 1× SDS sample buffer. eNOS (full-length or fragment fusion proteins) binding to GST-GIT1 fragment fusion proteins was detected by immunoblotting with eNOS antibody. Specifically, we used eNOS(C terminus) antibody (1:1000; BD Transduction Laboratories) to detect the C terminus of eNOS and eNOS(N terminus) antibody (1:200; Santa Cruz Biotechnology, Santa Cruz, CA) to detect the N terminus of eNOS. GST fusion proteins used in the pulldown assay were detected by immunoblotting with anti-GST antibody (1:200; Santa Cruz Biotechnology).
Adenovirus
The Ad-EV, Ad-myrAkt, Ad-dnAkt (21), Ad-Src, Ad-SrcKD (22), and Ad-GRK2ct (23) were purified from infected 293 cells by lysis in virus storage buffer followed by two sequential rounds of cesium chloride density gradient ultracentrifugation. We confirmed the efficiency of adenovirus infection of sinusoidal endothelial cells in vivo as described previously (24). Ad-ETB was a kind gift from Michael B. Fallon (The University of Texas Health Science Center, Houston, TX) (25). Sinusoidal endothelial cells were exposed to adenovirus in 2% serum for 16 h, medium was exchanged, and cells were then harvested at the specified time points.
Plasmids
pBK (Δ)-rat-GIT1/FLAG, GIT1(R39A), GIT1(Y293F), and GIT1(Y554F) have been described previously (12, 26, 27). For transient transfection, plasmid DNA encoding GIT1 (or an empty vector as control) was transfected into sinusoidal endothelial cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Transfection reagents and vectors were removed 4 h after transfection. Analysis of transfected proteins was routinely carried out 24–36 h after transfection. Transfection efficiency was confirmed using anti-FLAG antibody.
Immunoprecipitation and Immunoblotting
Immunoprecipitation assays were used to investigate the interaction of eNOS with GIT1 in cells and GIT1 tyrosine phosphorylation. Briefly, the cell lysates from sinusoidal endothelial cells (200 μg of total protein) were subjected to immunoprecipitation with antibody to GIT1 (Santa Cruz Biotechnology) overnight. A control immunoprecipitation including IgG was utilized in each experiment. Immunocomplexes were bound by incubating protein samples with protein A beads for 2 h at 4 °C. Immunoprecipitated proteins were separated by SDS-PAGE.
Immunoblotting was performed by using the specified primary antibodies including anti-eNOS antibody (1:1000; BD Transduction Laboratories), anti-phospho-eNOS-Ser1177 (1:1000; BD Transduction Laboratories or Cell Signaling Technology, Beverly, MA), anti-phosphotyrosine 4G-10 (1:1000; EMD Millipore Corp.), anti-Akt, anti-phospho-Akt-Ser473, anti-Src, anti-phospho-Src-Tyr416 (1:1000; Cell Signaling Technology), anti-GIT1 antibody H-170 (1:1000; Santa Cruz Biotechnology), and anti-FLAG M2 antibody (1:1000; Sigma-Aldrich). Anti-mouse IgG or anti-rabbit IgG horseradish peroxidase-conjugated secondary antibodies were from Promega (Madison, WI). Specific signals were visualized using enhanced chemiluminescence according to the manufacturer's instructions and scanned and quantitated with standard software. Immunoblot images shown are representative of at least three others.
Nitric Oxide Measurement
To assess NO production, we analyzed the release of nitrite, the stable breakdown product of NO, using a Sievers Chemiluminescence NO Analyzer (Sievers Instruments, Inc., Boulder, CO) according to the manufacturer's instructions. Briefly, conditioned medium was injected into a refluxing glass reaction chamber containing 0.1 m vanadium chloride and then carried in nitrogen gas to the chemiluminescence detector. Measurements of known concentrations of nitrite were used to generate a standard curve between 25 and 500 pmol of nitrite.
Statistical Analyses
All experiments were performed in replicates using cells isolated from different rats. All results were expressed as the mean ± S.E. We performed statistical analysis using the two-tailed Student's t test; p < 0.05 was considered statistically significant.
RESULTS
eNOS Directly Interacts with GIT1
We have previously demonstrated that eNOS directly interacts with GIT1 by examining in vitro binding of recombinant eNOS and GIT1 (5). To further map the GIT1 binding sites within eNOS, we created a number of constructs that were used to perform pulldown and immunoblot assays. GIT1 fragments containing amino acids 375–770 and 483–647 bound to full-length eNOS (Fig. 1A, top panel). This smaller fragment contains the synaptic localization domain (28, 29) and the majority of detected phosphorylation sites including GIT1 tyrosine phosphorylation at Tyr519, Tyr554, Tyr563, and Tyr607; GIT1 threonine phosphorylation at Thr489, Thr508, Thr546, and Thr610; and GIT1 serine phosphorylation at Ser507, Ser545, Ser555, Ser572, Ser601, Ser605, and Ser609 (29).
FIGURE 1.

Characterization of GIT1 and eNOS interaction by GST pulldown assay. In A, 6 μg of GST alone or GST fusion proteins containing fragments of individual GIT1 domains and 4 μg of His6-full-length eNOS fusion protein were subjected to GST pulldown assays as described under “Experimental Procedures.” Complexes were analyzed by immunoblotting (IB) with antibody to eNOS (upper panel). GST fusion proteins used in the pulldown assay were detected by immunoblotting with anti-GST antibody (lower panel). In B and C, 6 μg of His6-eNOS fusion protein (molecular mass, ∼146 kDa) and His6-eNOS fusion proteins containing the eNOS N-terminal oxygenase domain plus the calmodulin-binding sequence (His6-eNOS-NT; molecular mass, ∼70 kDa) and His6-eNOS fusion protein containing the C-terminal reductase domain (His6-eNOS-CT; molecular mass, ∼88 kDa) were subjected to immunoblotting with antibody that recognizes the eNOS N terminus (B, left panel) or eNOS C terminus (C, left panel). 6 μg of GST alone or a series of GST-GIT1 fragment fusion proteins and 6 μg of His6-eNOS-NT or His6-eNOS-CT were subjected to GST pulldown assays as described under “Experimental Procedures.” Complexes were analyzed by immunoblotting with antibody to the N terminus of eNOS (B, right panel) or antibody to the C terminus of eNOS (C, right panel) separately. In D, a schematic diagram of GIT1 is depicted (left panel) as well as a schematic diagram illustrating the two major domains of eNOS (right panel, top portion) and a summary of the GIT1 and eNOS interaction sites (right panel, lower portion). GAP, GTPase-activating protein; ANK, ankyrin repeats; GRK-BD, GRK binding domain; SHD, Spa2 homology domain; SLD, synaptic localization domain; PBD, paxillin-binding focal adhesion targeting domain; CaM, calmodulin binding domain; BH4, tetrahydrobiopterin binding site, H, heme binding site; ARG, arginine binding site.
Next, we generated His6-eNOS fusion proteins containing one of the two major eNOS structural domains: the eNOS N-terminal oxygenase domain plus the calmodulin-binding sequence (eNOS-NT; Fig. 1B) and the eNOS C-terminal reductase domain (eNOS-CT; Fig. 1C). We confirmed the expression of eNOS fusion proteins using N-terminal or C-terminal eNOS antibody (Fig. 1, B and C, left panels). The N-terminal reductase domain of eNOS was found to bind to GIT1(375–770) (Fig. 1B), whereas the C-terminal oxidase domain of eNOS was pulled down by both GIT1(483–647) and GIT1(647–770) but not by GIT1(375–770) (Fig. 1C). These experiments demonstrate that the direct interaction between eNOS and GIT1 involves interactions between both eNOS domains with elements of the GIT1 C terminus.
Expression of GIT1 Tyrosine Phosphorylation in Sinusoidal Endothelial Cells
We have previously demonstrated that eNOS and GIT1 interact in sinusoidal endothelial cells and that GIT1 overexpression or knockdown substantially modifies eNOS activity (5). Here we hypothesized that GIT1 phosphorylation is important in the GIT1-eNOS interaction and in stimulation of eNOS phosphorylation. To better understand the relationship between GIT1 expression and GIT1 tyrosine phosphorylation, we examined GIT1 tyrosine phosphorylation before and after introduction of full-length GIT1 into sinusoidal endothelial cells. Overexpression of GIT1 was associated with increased GIT1 tyrosine phosphorylation (Fig. 2A). Additionally, knockdown of endogenous GIT1 correlated with reduction of GIT1 tyrosine phosphorylation (Fig. 2, B and C).
FIGURE 2.

GIT1 tyrosine phosphorylation in sinusoidal endothelial cells. In A, sinusoidal endothelial cells were transfected with cDNA encoding full-length GIT1 (1 μg) or an empty vector (EV; 1 μg) as described under “Experimental Procedures.” 24 h later, GIT1 tyrosine phosphorylation was assayed by immunoprecipitation (IP) with antibody to GIT1 followed by immunoblotting (IB) with 4G-10 (left panel). GIT1 levels were detected in cell lysates by immunoblotting (middle panel). In the graph shown in the right panel, bands corresponding to phospho-GIT1 were quantified (n = 4; error bars represent S.E.; *, p < 0.001 versus EV control). In B, sinusoidal endothelial cells were isolated and transduced with GIT1 siRNA at the indicated concentrations as described under “Experimental Procedures.” GIT1 tyrosine phosphorylation was assayed by immunoprecipitation with antibody to GIT1 followed by immunoblotting with antibody to 4G-10 (left, upper panel). GIT1 and β-actin protein levels were assessed by immunoblotting with antibodies to GIT1 and β-actin (left, lower panel). A scrambled siRNA (Scr) was used as a negative control, and β-actin levels were used to assess protein loading (control). In the graph shown in the bottom panel, bands corresponding to phospho-GIT1 were quantified and normalized to β-actin (n = 3; error bars represent S.E.; *, p < 0.05 versus scrambled siRNA control). In C, sinusoidal endothelial cells were isolated and transduced with two additional sets of GIT1 siRNAs (GIT1 siRNA-A and -B). GIT1 tyrosine phosphorylation was assayed by immunoprecipitation with antibody to GIT1 followed by immunoblotting with antibody to 4G-10 (left, upper panel). GIT1 and β-actin protein levels were assayed by immunoblotting with antibodies to GIT1 and β-actin (left, lower panel). In the graph shown in the left bottom panel, bands corresponding to phospho-GIT1 (P-GIT1) were quantified and normalized to β-actin (n = 3; error bars represent S.E.; *, p < 0.01; **, p < 0.005 versus scrambled siRNA control). In the right panels, GIT1 tyrosine phosphorylation was assayed by immunoprecipitation with antibody to 4G-10 followed by immunoblotting with antibody to GIT1. Total cell lysate used to detect GIT1 position and immunoprecipitation with IgG are also shown as indicated (right, upper panel). In the right bottom panel, bands corresponding to phospho-GIT1 were quantified and normalized to IgG from each immunoprecipitated sample as a loading control (n = 3; error bars represent S.E.; *, p < 0.01 versus scrambled siRNA control).
Akt Stimulates GIT1 Tyrosine Phosphorylation
We have previously demonstrated that Akt potentiates eNOS and NO signaling (5). Here we explored GIT1 tyrosine phosphorylation in the Akt signaling pathway. GIT1 tyrosine phosphorylation was significantly increased after infection of sinusoidal endothelial cells with constitutively active Akt (Ad-myrAkt), whereas the dominant negative Akt (Ad-dnAkt) decreased GIT1 tyrosine phosphorylation (Fig. 3A).
FIGURE 3.

Akt stimulates GIT1 tyrosine phosphorylation, and Src regulates GIT1/eNOS/NO signaling. In A, sinusoidal endothelial cells were infected with adenovirus (m.o.i. 250) encoding constitutively active Akt (Ad-myrAkt), dominant negative Akt (Ad-dnAkt), or an empty vector (Ad-EV) for 36 h. GIT1 tyrosine phosphorylation (upper panel) and total GIT1 (middle panel) were assayed as in Fig. 2. In the graph shown in the bottom panel, bands corresponding to phospho-GIT1 (P-GIT1) were quantified and normalized to total GIT1 (n = 3; error bars represent S.E.; *, p < 0.05; **, p < 0.01 versus Ad-EV control). In B, sinusoidal endothelial cells as in Fig. 2 were exposed to the Src inhibitor PP2 (5–10 μm) for 2 h, and then cDNA encoding full-length GIT1 (1 μg) was transduced for an additional 24 h. Cells were harvested, and cell lysates were immunoblotted (IB) to detect phospho-eNOS (P-eNOS) (Ser1177), total eNOS, and β-actin. In the bottom graph, bands corresponding to phospho-eNOS were quantified and normalized to total eNOS (n = 3; error bars represent S.E.; *, p < 0.01 versus GIT1 transfection control). In C, cells were treated as in B, conditioned medium was collected, and nitrite levels were measured as under “Experimental Procedures.” Quantitative data are shown graphically (n = 3; error bars represent S.E.; *, p < 0.05; **, p < 0.01 versus GIT1 transfection control). IP, immunoprecipitation.
Src Regulates GIT1/eNOS/NO Signaling
Because GIT1 is a known substrate for Src tyrosine phosphorylation (29–31), we hypothesized that Src may play a role in GIT1-mediated eNOS activation. The Src kinase-specific inhibitor 4-amino-5-(4-chlorophenyl)-7-t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) abrogated the effects of overexpression of GIT1, leading to a reduction of both eNOS phosphorylation (Fig. 3B) and NO production (Fig. 3C).
GIT1 Tyrosine Phosphorylation Facilitates GIT1-eNOS Interaction in a Src-dependent Manner
To understand how Src affects GIT1-mediated eNOS activation, we first examined the effect of Src manipulation on GIT1 tyrosine phosphorylation. Src overexpression led to an increase in GIT1 phosphorylation (Fig. 4A), whereas expression of the Src kinase-dead mutant (Ad-SrcKD) led to reduced GIT1 phosphorylation (Fig. 4A). Additionally, the Src kinase-specific inhibitor PP2 inhibited GIT1 and eNOS interaction in a dose-response manner (Fig. 4B), raising the possibility that Src is important in regulating GIT1-eNOS interaction. Next, we studied a series of GIT1 mutants in which known Src phosphorylation sites were mutated (GIT1(Y293F) and GIT1(Y554F)); these reduced GIT1 tyrosine phosphorylation (Fig. 4C). Wild type GIT1 and the GIT1(R39A) mutant, which lacks ADP-ribosylation factor GTPase-activating protein activity due to mutation of the arginine finger residue, appeared to increase Src and eNOS phosphorylation and NO production (Fig. 4, D and E). The two Src phosphorylation site mutants GIT1(Y293F) and GIT1(Y554F) decreased Src phosphorylation, eNOS phosphorylation, and NO production (Fig. 4, D and E). These data suggest that Src-mediated phosphorylation of GIT1 at Tyr293 and Tyr554 is important for Src docking and activation and subsequent Src-mediated eNOS phosphorylation and activation.
FIGURE 4.

GIT1 tyrosine phosphorylation is Src-dependent. In A, sinusoidal endothelial cells were infected with adenovirus (m.o.i. 250) encoding wild type c-Src (Ad-Src) or a kinase domain-deleted c-Src (Ad-SrcKD). After 36 h, cells were harvested, and GIT1 tyrosine phosphorylation and total GIT1 were detected as in Fig. 2. In the graph shown in the bottom panel, bands corresponding to tyrosine phospho-GIT1 (P-GIT1) were quantified and normalized to total GIT1 (n = 3; error bars represent S.E.; *, p < 0.001; **, p < 0.05 versus Ad-EV control). In B, sinusoidal endothelial cells were exposed to PP2 (5–20 μm) for 2 h as indicated. Cells were harvested, and cell lysates were subjected to immunoprecipitation (IP) with antibody to GIT1 followed by immunoblotting (IB) with antibodies to eNOS (upper panel), 4G-10 (middle panel), and GIT1 (lower panel). In the bottom panel, bands corresponding to eNOS-GIT1 complex were quantified and normalized to the level of immunoprecipitated GIT1 (n = 3; error bars represent S.E.; *, p < 0.05 versus control). In C, sinusoidal endothelial cells were transfected with cDNA encoding FLAG-tagged full-length GIT1, GIT1(R39A) that lacks ADP-ribosylation factor GTPase-activating protein activity, GIT1(Y293F), and GIT1(Y554F); in the latter two, Src phosphorylation sites were deleted as described under “Experimental Procedures.” After 24 h, cells were harvested, and cell lysates were subjected to immunoprecipitation with antibody to GIT1 followed by immunoblotting with antibodies to 4G-10 (upper panel) and GIT1 (middle panel). FLAG-tagged GIT1 proteins were detected by immunoblotting with FLAG tag antibody (lower panel). The blots shown are representative of three others. In D, sinusoidal endothelial cells under the same conditions as in C were harvested, and cell lysates were subjected to immunoblotting with antibodies to phospho-eNOS at Ser1177 (P-eNOS), total eNOS (eNOS), phospho-Src at Tyr416 (P-Src), and total Src (Src). Representative immunoblots are shown in the left panels, and in the graphs shown in the right panels, bands corresponding to phospho-eNOS and phospho-Src were quantified and normalized to total eNOS and Src, respectively (n = 3; error bars represent S.E.; *, p < 0.05; **, p < 0.01 versus GIT1 transfection control). In E, conditioned medium from sinusoidal endothelial cells treated as in C and D was collected, and nitrite levels were measured as described under “Experimental Procedures” (n = 3; error bars represent S.E.; *, p < 0.005 versus GIT1 alone).
Src Is Upstream of Akt in Regulating GIT1/eNOS/NO Pathways
To clarify the roles of Akt and Src signaling to GIT1 and eNOS in sinusoidal endothelial cells, we blocked Src activity using PP2 and then manipulated Akt with either active Akt (Ad-myrAkt) or dominant negative Akt (Ad-dnAkt). PP2 inhibited eNOS phosphorylation and NO production induced by Ad-myrAkt, whereas the Ad-dnAkt construct reduced eNOS phosphorylation and NO production without affecting Src phosphorylation (Fig. 5, A and B), suggesting that Akt may work downstream of Src to regulate Src-dependent GIT1-eNOS signaling. Moreover, overexpression of Ad-myrAkt or Ad-Src increased GIT1 tyrosine phosphorylation, leading to increased GIT1-eNOS association as well as increased eNOS phosphorylation within the complex (Fig. 5C, left panels). Additionally, the effect of Src overexpression was blocked by overexpression of Ad-dnAkt (Fig. 5C, right panels), suggesting that Akt is downstream of Src in this system.
FIGURE 5.

Src regulates Akt and the GIT1/eNOS/NO signaling pathway. In A, sinusoidal endothelial cells were exposed to PP2 (10 μm) for 2 h and then infected with Ad-myrAkt, Ad-dnAkt, or Ad-EV (m.o.i. 250) as indicated for 36 h. Cells were harvested, and phospho-eNOS (P-eNOS) at Ser1177, phospho-Akt (P-Akt) at Ser473, phospho-Src (P-Src) at Tyr416, total eNOS, Akt, Src, and β-actin were detected by immunoblotting as described under “Experimental Procedures.” Representative images shown in the left panels. In the graphs shown in the right panels, bands corresponding to phospho-eNOS, phospho-Akt, and phospho-Src were quantified and normalized to the level of total eNOS, Akt, and Src, respectively (n = 3; error bars represent S.E.; *, p < 0.001; **, p < 0.01 versus no PP2 control). In B, sinusoidal endothelial cells were treated as in A, nitrite levels were measured in conditioned medium as described under “Experimental Procedures”, and the data are presented graphically (n = 3; error bars represent S.E.; *, p < 0.005 versus Ad-EV control; **, p < 0.05 versus no PP2 control). In C, sinusoidal endothelial cells were infected with Ad-myrAkt, Ad-dnAkt, or Ad-EV with or without Ad-Src (m.o.i. 250) as indicated for 36 h. Cells were harvested, and cell lysates were subjected to immunoprecipitation (IP) with antibody to GIT1 followed by immunoblotting (IB) with antibody to eNOS, phospho-eNOS, 4G-10, or GIT1. Expression levels of Akt and Src (and total β-actin) after infection with specific constructs were confirmed by immunoblotting with the indicated antibodies (lower three panels). Blots shown are representative of three others.
Endothelin-1 (ET-1) Stimulates GIT1 and eNOS Signaling through Src
We demonstrated previously that ET-1 stimulates eNOS and NO release through activation of phosphatidylinositol 3-kinase and Akt (32). Therefore, we attempted to determine whether Src was a primary upstream mediator involved in ET-1-mediated NO release. First, in a control experiment, we used insulin to stimulate sinusoidal endothelial cells and found that it increased eNOS phosphorylation and NO production (Fig. 6A); the effect of insulin was blocked by PP2 (Fig. 6A). Second, to determine whether ET-1-mediated tyrosine phosphorylation of GIT1 was Src-dependent, we exposed cells to ET-1 after incubation with the Src inhibitor PP2 and found that PP2 blocked ET-1-induced GIT1 tyrosine phosphorylation (Fig. 6B). Next, we exposed sinusoidal endothelial cells to varying amounts of ET-1 and found that ET-1 stimulated Src phosphorylation in a dose-dependent manner without affecting the total Src expression (Fig. 6C). When cells were exposed to PP2, phospho-Src (Fig. 6D, left panel), phospho-Akt (Fig. 6D, middle panel), and phospho-eNOS (Fig. 6D, right panel) were reduced. Finally, as expected, PP2 blocked NO production induced by ET-1 (Fig. 6E). The data indicate that stimulation of eNOS phosphorylation by ET-1 is Src-dependent and further confirm that Src is upstream of Akt.
FIGURE 6.

ET-1 regulates GIT1 and eNOS signaling. In A, sinusoidal endothelial cells were exposed to PP2 (10 μm) for 2 h and then stimulated with insulin (10 nm) for 30 min. Phospho-eNOS (P-eNOS) and phospho-Src (P-Src) were detected by immunoblotting as described under “Experimental Procedures.” Conditioned medium was collected, and nitrite levels were measured and are shown in the bottom panel (n = 4; error bars represent S.E.; *, p < 0.005 versus control; **, p < 0.001 versus insulin stimulation control). In B, sinusoidal endothelial cells were exposed to PP2 (20 nm) for 2 h and then stimulated with ET-1 for 30 min. GIT1 tyrosine phosphorylation was detected as in Fig. 2. In the graphs shown in the bottom panel, bands corresponding to phospho-GIT1 were quantified and normalized to the level of immunoprecipitated (IP) GIT1 (n = 5; error bars represent S.E.; *, p < 0.005 versus ET-1 control). In C, sinusoidal endothelial cells were exposed to ET-1 from 10 to 20 nm for 30 min, and cell lysates were subjected to immunoblotting (IB) to detect phospho-Src or total Src. In the bottom graph, bands corresponding to Src phosphorylation were quantified (n = 3; error bars represent S.E.; *, p < 0.05; **, p < 0.01 versus control). In D, sinusoidal endothelial cells were exposed to PP2 (10 μm) for 2 h and then stimulated with ET-1 (10 nm) for 30 min. Cells were collected, and phospho-Src at Tyr416, phospho-Akt (P-Akt) at Ser473, phospho-eNOS at Ser1177, total Src, Akt, and eNOS were detected by immunoblotting as described under “Experimental Procedures.” In the graphs shown in the bottom panels, bands corresponding to phospho-Src, phospho-Akt, and phospho-eNOS were quantified and normalized to the level of total eNOS, Akt, and Src, respectively (n = 3; error bars represent S.E.; *, p < 0.001; **, p < 0.05 versus ET-1 control). In E, cells were exposed to PP2 (10 μm) for 2 h and then stimulated with ET-1 (10 nm) for 30 min. Conditioned medium was collected, and nitrite levels were measured (n = 4; error bars represent S.E.; *, p < 0.001 versus control; **, p < 0.005 versus ET-1 control).
Role of Gβγ in ET-1-mediated GIT1 and eNOS Signaling
To determine whether Gβγ may play a role in GIT1-mediated eNOS signaling, we examined the role of the G-protein-coupled ETB receptor, which is prominently expressed in primary sinusoidal endothelial cells unlike the ETA receptor, which is absent (19, 33–35). GIT1 tyrosine phosphorylation was more prominent after overexpression of the ETB receptor (Fig. 7A). Next, we investigated expression of the C terminus of GRK2 (GRK2ct), which binds and inhibits G-protein βγ subunit function (23). We found that overexpression of GRK2ct blocked Akt phosphorylation (Fig. 7B) as demonstrated previously (32). Importantly, ET-1 was found to stimulate the interaction of GIT1 and eNOS (Fig. 7C), and it also stimulated GIT1 tyrosine phosphorylation after ET-1 stimulation (Fig. 7D). Ad-GRK2ct abrogated ET-1-stimulated GIT1-eNOS interaction, GIT1 tyrosine phosphorylation, and eNOS phosphorylation (Fig. 7, C and D); however, it had no affect on Src phosphorylation (Fig. 7E). These data suggest that ET-1 stimulates Src independently of Gβγ and that Gβγ promotes tyrosine phosphorylation of GIT1. Because pathways downstream of GPCRs have been classified as either pertussis toxin (PTX)-sensitive (i.e. coupled to Gαi/o) or PTX-insensitive (i.e. coupled to Gαq/11, Gαs, or Gα12/13) (36–38), we studied whether ET-1 utilizes the PTX-sensitive G-protein to regulate GIT1 and Src activation. We found that PTX pretreatment did not block either ET-1-stimulated GIT1 tyrosine phosphorylation (Fig. 7F) or Src phosphorylation (Fig. 7G), suggesting that GIT1 phosphorylation is not linked to Gαi/o.
FIGURE 7.

The ETB receptor signals through Gβγ to GIT1 and eNOS. In A, sinusoidal endothelial cells were infected with adenovirus encoding ETB receptor (m.o.i. 250) or empty virus (Ad-EV) for 36 h before exposure to ET-1 (10 nm) for 30 min. Cells were harvested, and GIT1 tyrosine phosphorylation and total GIT1 were detected as in Fig. 2. In the right graph, bands corresponding to tyrosine phospho-GIT1 (P-GIT1) were quantified and normalized to total GIT1 (n = 3; error bars represent S.E.; *, p < 0.05 versus Ad-EV control; **, p < 0.01 versus ET-1 control). In B, sinusoidal endothelial cells were infected with adenovirus (m.o.i. 250) encoding the GRK2ct (Ad-GRK2ct; the C terminus of G-protein-coupled receptor kinase) or Ad-EV for 36 h before exposure to ET-1 (10 nm) for 30 min as indicated. Cells were harvested, and Akt phosphorylation (P-Akt) at Ser473 and total Akt were detected by immunoblotting (IB) as described under “Experimental Procedures.” In C, sinusoidal endothelial cells from the same treatment as in B were harvested, and GIT1-eNOS interaction was measured by immunoprecipitation (IP) with antibody to GIT1 and immunoblotting with antibody to eNOS. The level of eNOS from 10% of total cell lysate used for immunoprecipitation is also shown as indicated. In D, sinusoidal endothelial cells treated as in B were harvested, and GIT1 tyrosine phosphorylation (P-GIT1) and total GIT1 were detected as in Fig. 2. GIT1-dependent eNOS phosphorylation was measured by immunoprecipitation with antibody to GIT1 and immunoblotting with antibody to phospho-eNOS (P-eNOS) at Ser1177. Immunoprecipitation with IgG is shown. The level of GIT1 from cell lysate used for immunoprecipitation is also shown as indicated. In the bottom graph, bands corresponding to tyrosine phospho-GIT1 were quantified and normalized to the level of immunoprecipitated GIT1 (n = 3; error bars represent S.E.; *, p < 0.05; **, p < 0.01 versus Ad-EV control; #, p < 0.01 versus ET-1 control). In E, sinusoidal endothelial cells from the same treatment as in B were harvested, and cell lysates were subjected to immunoblotted with antibodies to phospho-Src (P-Src) at Tyr416. In the bottom graph, bands corresponding to Src phosphorylation were quantified and normalized (n = 3; error bars represent S.E.; *, p < 0.05 versus Ad-EV control; **, p < 0.05 versus Ad-GRK2ct control). In F, sinusoidal endothelial cells were exposed to PTX (100 ng/ml) for 6 h and then stimulated with ET-1 (10 nm) for 30 min as indicated. Cells were harvested, and GIT1 tyrosine phosphorylation was detected as in Fig. 2. Immunoprecipitation with IgG is also shown. In the bottom graph, bands corresponding to tyrosine phospho-GIT1 were quantified (n = 3; error bars represent S.E.; *, p < 0.05 versus no treatment control). In G, cells from the same treatment as in D were harvested, and cell lysates were subjected to immunoblotting to detect phospho-Src as described under “Experimental Procedures.” In the bottom graph, bands corresponding to Src phosphorylation were quantified and normalized (n = 3; error bars represent S.E.; *, p < 0.005 versus no treatment control).
DISCUSSION
We have previously reported that the signaling scaffold protein GIT1 binds directly to eNOS and increases its activity in sinusoidal endothelial cells (5). Here we extend those findings by exploring the localization of GIT1 and eNOS binding domains and how GIT1 helps integrate signals carried by Src and Akt downstream of endothelin receptor stimulation to regulate eNOS activity.
eNOS is a complex tripartite protein harboring oxygenase, calmodulin binding, and reductase domains. The N-terminal oxygenase domain of eNOS represents the catalytic center and contains binding sites for heme, arginine, and tetrahydrobiopterin, which the reductase domain supports with binding sites for FMN, FAD, and NADPH. These two domains are linked by a connecting region where calmodulin binds (Fig. 1D) (39, 40). GIT1 contains an N-terminal ADP-ribosylation factor-GTPase-activating protein domain, ankyrin repeats, and Spa2 homology domain 1, and a C-terminal paxillin-binding focal adhesion targeting domain (amino acids 647–770) with less well defined C-terminal GRK binding domain (amino acids 375–770), and synaptic localization domain (amino acids 375–596) (Fig. 1D) (29). In our study, using a GST pulldown assay, we found that the N terminus of eNOS was pulled down by GIT1(375–770) but not GIT1(483–647), whereas the C terminus of eNOS was pulled down by GIT1(483–647) and GIT1(647–770). This suggests that direct GIT1-eNOS interactions are complex. The complex GIT1 C-terminal interactions with eNOS help to explain our previous finding that two non-overlapping fragments within the GIT1 C terminus were able to pull down eNOS activity (5). Note also that bacterially expressed GIT1 and eNOS fragments are not subject to tyrosine phosphorylation, providing strong evidence that GIT1-eNOS interaction can occur in the absence of any tyrosine phosphorylation (although this basal binding may be modulated by phosphorylation events). Also, these two distinct interactions are indeed direct protein-protein binding events.
In contrast to a simple model where activated Akt simply phosphorylates eNOS to stimulate NO production, we found evidence for a tightly linked system of regulatory events centered on the GIT1 scaffold that coordinate activation of eNOS (Fig. 8). Upon endothelin stimulation, sinusoidal endothelial cell GIT1 becomes phosphorylated on tyrosine residues, predominantly Tyr293 and Tyr554. These sites have been described previously as sites for phosphorylation by the Src kinase family (29, 31). Accordingly, treatment of cells with the Src family inhibitor PP2 reduced GIT1 tyrosine phosphorylation as well as GIT1-eNOS association, eNOS activity, and eNOS activity-related phosphorylation at Ser1177. Similarly, overexpression of GIT1(Y293F) and GIT1(Y554F) mutants also blocked eNOS activity and eNOS phosphorylation at Ser1177. Thus, Src phosphorylation of GIT1 at these sites is critical to formation of the GIT1-eNOS complex and subsequent activation of eNOS.
FIGURE 8.
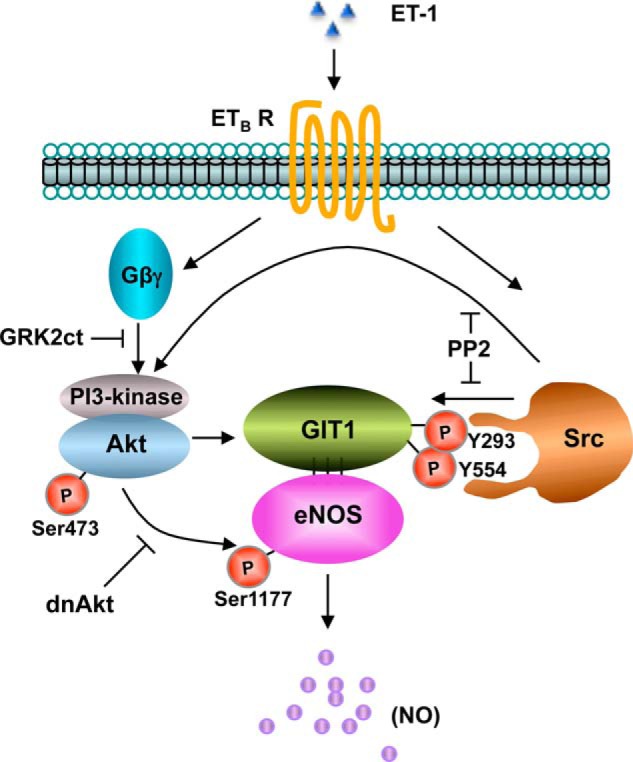
A proposed model for GIT1/eNOS signaling in sinusoidal endothelial cells. In sinusoidal endothelial cells, ET-1 activates its cognate G-protein-coupled receptor (ETB receptor (ETB R) and causes Gα and Gβγ activation and dissociation (see also Ref. 32). The mechanism of Src activation is currently unknown but may occur through ETB receptor-mediated β-arrestin recruitment of Src (54). Activated Src phosphorylates GIT1 at Tyr293 and Tyr554 and perhaps also eNOS at Tyr83 to partially activate it (41, 42). Phosphorylated GIT1 then associates with eNOS to facilitate its activating phosphorylation at Ser1177 by Akt. Receptor-activated Gβγ simultaneously stimulates Akt (at Ser473) activation through phosphatidylinositol (PI) 3-kinase (32), and this activation may be amplified by other signaling pathways (55). Thus, both Src and Akt kinases are crucial in the resultant phosphorylation-enhanced association of GIT1 and eNOS and in stimulating eNOS activity and NO production. dnAkt, dominant negative Akt.
We had previously described a pathway linking endothelin receptors to activation of Akt through G-protein βγ subunits (32). In this pathway, Gβγ released by activated receptor then stimulates phosphatidylinositol 3-kinase to increase Akt activity, leading to eNOS phosphorylation and NO production. We now show that Gβγ also contributes to the GIT1-eNOS pathway because blockade of Gβγ signaling also blocked the tyrosine phosphorylation of GIT1. However, Gβγ does not appear to have a role in activation of Src activity as measured by phospho-Src accumulation, leaving a question as to how Gβγ acts on GIT1. One possibility is that Gβγ acts through activation of Akt as GIT1 has a predicted Akt phosphorylation site at Ser419 (29) that may be important for subsequent Src action on GIT1.
Overall, it is clear from our inhibition studies that Src and Akt both act upstream and downstream of GIT1 in the GIT1-eNOS pathway, consistent with its role as a tightly regulated signaling scaffold. For Akt, it is well known that Akt directly phosphorylates eNOS at Ser1177. Although phosphorylated eNOS can be found in complex with GIT1, there is no enrichment in phosphorylated eNOS in GIT1-bound versus unbound fractions (5), suggesting that Ser1177 phosphorylation does not regulate GIT1-eNOS association. However, Akt activity clearly does regulate GIT1-eNOS association with elevated Akt activity favoring complex formation (5). Similarly, we now found that Src phosphorylation of GIT1 at Tyr293 and Tyr554 was required for GIT1-eNOS complex formation. Although inhibition of Akt had no effect on Src activity, preventing Src phosphorylation of (and presumably subsequent Src homology 2 domain-mediated binding to) GIT1 also reduced total Src activity. Src is known to directly phosphorylate eNOS at Tyr83 (41–43), and Src also has been reported to regulate eNOS through Akt (44–47). These studies are consistent with the deep entwining of the Src and Akt pathways leading to eNOS activation that we saw mediated through the GIT1 scaffold.
GIT1 has been reported to have multiple scaffolding roles that regulate numerous cellular processes in diverse cell types (48). These include roles in regulating endothelial barrier function in the vasculature (49, 50) and in vascular development in the lung (51). Endothelin-1 initiates multiple signaling pathways in sinusoidal endothelial cells through G-protein pathways (both Gαi- and Gβγ-mediated) including Akt and Src activation (32, 52, 53). Our working model for GIT1 action in the eNOS pathway (Fig. 8) proposes that GIT1 functions as a scaffold for these two distinct signaling mediators, facilitating both their actions to stimulate eNOS as well as critical regulatory interactions between the two kinase pathways. Future work will be needed to more clearly delineate the extent of these regulatory interactions and how they are facilitated by GIT1 and to determine how this signal cross-talk regulates eNOS recruitment to the GIT1 complex for activation.
Acknowledgments
We thank Yingyu Ren and Xianjie Zeng for valuable assistance with primary cell isolation and cell culture and Serhan Karvar and Pankaj Srivastava for valuable assistance with fusion protein work.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DK 57830 (to D. C. R.). This work was also supported by a Burroughs Wellcome Fund Translational Scientist Award (to D. C. R.) and an American Liver Fund Liver Scholar Award (to S. L.).
- eNOS
- endothelial nitric-oxide synthase
- GRK
- G-protein-coupled receptor kinase
- GIT1
- G-protein-coupled receptor kinase-interacting protein 1
- ET
- endothelin
- GPCR
- G-protein-coupled receptor
- PP2
- 4-amino-5-(4-chlorophenyl)-7-t-butyl)pyrazolo[3,4-d]pyrimidine
- PTX
- pertussis toxin
- m.o.i.
- multiplicity of infection
- EV
- empty vector
- GRK2ct
- C terminus of GRK2.
REFERENCES
- 1. Rafikov R., Fonseca F. V., Kumar S., Pardo D., Darragh C., Elms S., Fulton D., Black S. M. (2011) eNOS activation and NO function: structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J. Endocrinol. 210, 271–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. García-Cardeña G., Martasek P., Masters B. S., Skidd P. M., Couet J., Li S., Lisanti M. P., Sessa W. C. (1997) Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J. Biol. Chem. 272, 25437–25440 [DOI] [PubMed] [Google Scholar]
- 3. García-Cardeña G., Fan R., Shah V., Sorrentino R., Cirino G., Papapetropoulos A., Sessa W. C. (1998) Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 392, 821–824 [DOI] [PubMed] [Google Scholar]
- 4. Whalen E. J., Foster M. W., Matsumoto A., Ozawa K., Violin J. D., Que L. G., Nelson C. D., Benhar M., Keys J. R., Rockman H. A., Koch W. J., Daaka Y., Lefkowitz R. J., Stamler J. S. (2007) Regulation of β-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell 129, 511–522 [DOI] [PubMed] [Google Scholar]
- 5. Liu S., Premont R. T., Rockey D. C. (2012) G-protein-coupled receptor kinase interactor-1 (GIT1) is a new endothelial nitric-oxide synthase (eNOS) interactor with functional effects on vascular homeostasis. J. Biol. Chem. 287, 12309–12320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bauer P. M., Fulton D., Boo Y. C., Sorescu G. P., Kemp B. E., Jo H., Sessa W. C. (2003) Compensatory phosphorylation and protein-protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J. Biol. Chem. 278, 14841–14849 [DOI] [PubMed] [Google Scholar]
- 7. McGarrigle D., Huang X. Y. (2007) GPCRs signaling directly through Src-family kinases. Sci. STKE 2007, pe35. [DOI] [PubMed] [Google Scholar]
- 8. Luttrell D. K., Luttrell L. M. (2004) Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene 23, 7969–7978 [DOI] [PubMed] [Google Scholar]
- 9. Ma Y. C., Huang J., Ali S., Lowry W., Huang X. Y. (2000) Src tyrosine kinase is a novel direct effector of G proteins. Cell 102, 635–646 [DOI] [PubMed] [Google Scholar]
- 10. Bence K., Ma W., Kozasa T., Huang X. Y. (1997) Direct stimulation of Bruton's tyrosine kinase by Gq-protein α-subunit. Nature 389, 296–299 [DOI] [PubMed] [Google Scholar]
- 11. Natarajan K., Berk B. C. (2006) Crosstalk coregulation mechanisms of G protein-coupled receptors and receptor tyrosine kinases. Methods Mol. Biol. 332, 51–77 [DOI] [PubMed] [Google Scholar]
- 12. Premont R. T., Perry S. J., Schmalzigaug R., Roseman J. T., Xing Y., Claing A. (2004) The GIT/PIX complex: an oligomeric assembly of GIT family ARF GTPase-activating proteins and PIX family Rac1/Cdc42 guanine nucleotide exchange factors. Cell. Signal. 16, 1001–1011 [DOI] [PubMed] [Google Scholar]
- 13. Schmalzigaug R., Phee H., Davidson C. E., Weiss A., Premont R. T. (2007) Differential expression of the ARF GAP genes GIT1 and GIT2 in mouse tissues. J. Histochem. Cytochem. 55, 1039–1048 [DOI] [PubMed] [Google Scholar]
- 14. Segura I., Essmann C. L., Weinges S., Acker-Palmer A. (2007) Grb4 and GIT1 transduce ephrinB reverse signals modulating spine morphogenesis and synapse formation. Nat. Neurosci. 10, 301–310 [DOI] [PubMed] [Google Scholar]
- 15. Manabe R., Kovalenko M., Webb D. J., Horwitz A. R. (2002) GIT1 functions in a motile, multi-molecular signaling complex that regulates protrusive activity and cell migration. J. Cell Sci. 115, 1497–1510 [DOI] [PubMed] [Google Scholar]
- 16. Wang J., Yin G., Menon P., Pang J., Smolock E. M., Yan C., Berk B. C. (2010) Phosphorylation of G protein-coupled receptor kinase 2-interacting protein 1 tyrosine 392 is required for phospholipase C-γ activation and podosome formation in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 30, 1976–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Menon P., Yin G., Smolock E. M., Zuscik M. J., Yan C., Berk B. C. (2010) GPCR kinase 2 interacting protein 1 (GIT1) regulates osteoclast function and bone mass. J. Cell. Physiol. 225, 777–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shah V., Toruner M., Haddad F., Cadelina G., Papapetropoulos A., Choo K., Sessa W. C., Groszmann R. J. (1999) Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology 117, 1222–1228 [DOI] [PubMed] [Google Scholar]
- 19. Rockey D. C., Chung J. J. (1998) Reduced nitric oxide production by endothelial cells in cirrhotic rat liver: endothelial dysfunction in portal hypertension. Gastroenterology 114, 344–351 [DOI] [PubMed] [Google Scholar]
- 20. Liu S., Premont R. T., Kontos C. D., Zhu S., Rockey D. C. (2005) A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat. Med. 11, 952–958 [DOI] [PubMed] [Google Scholar]
- 21. Fujio Y., Guo K., Mano T., Mitsuuchi Y., Testa J. R., Walsh K. (1999) Cell cycle withdrawal promotes myogenic induction of Akt, a positive modulator of myocyte survival. Mol. Cell. Biol. 19, 5073–5082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhan S., Chan C. C., Serdar B., Rockey D. C. (2009) Fibronectin stimulates endothelin-1 synthesis in rat hepatic myofibroblasts via a Src/ERK-regulated signaling pathway. Gastroenterology 136, 2345–55.e1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koch W. J., Inglese J., Stone W. C., Lefkowitz R. J. (1993) The binding site for the βγ subunits of heterotrimeric G proteins on the β-adrenergic receptor kinase. J. Biol. Chem. 268, 8256–8260 [PubMed] [Google Scholar]
- 24. Yu Q., Que L. G., Rockey D. C. (2002) Adenovirus-mediated gene transfer to nonparenchymal cells in normal and injured liver. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G565–G572 [DOI] [PubMed] [Google Scholar]
- 25. Tang L., Luo B., Patel R. P., Ling Y., Zhang J., Fallon M. B. (2007) Modulation of pulmonary endothelial endothelin B receptor expression and signaling: implications for experimental hepatopulmonary syndrome. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L1467–L1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Premont R. T., Claing A., Vitale N., Freeman J. L., Pitcher J. A., Patton W. A., Moss J., Vaughan M., Lefkowitz R. J. (1998) β2-Adrenergic receptor regulation by GIT1, a G protein-coupled receptor kinase-associated ADP ribosylation factor GTPase-activating protein. Proc. Natl. Acad. Sci. U.S.A. 95, 14082–14087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmalzigaug R., Garron M. L., Roseman J. T., Xing Y., Davidson C. E., Arold S. T., Premont R. T. (2007) GIT1 utilizes a focal adhesion targeting-homology domain to bind paxillin. Cell. Signal. 19, 1733–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang H., Webb D. J., Asmussen H., Horwitz A. F. (2003) Synapse formation is regulated by the signaling adaptor GIT1. J. Cell Biol. 161, 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Webb D. J., Mayhew M. W., Kovalenko M., Schroeder M. J., Jeffery E. D., Whitmore L., Shabanowitz J., Hunt D. F., Horwitz A. F. (2006) Identification of phosphorylation sites in GIT1. J. Cell Sci. 119, 2847–2850 [DOI] [PubMed] [Google Scholar]
- 30. Kawachi H., Fujikawa A., Maeda N., Noda M. (2001) Identification of GIT1/Cat-1 as a substrate molecule of protein tyrosine phosphatase ζ/β by the yeast substrate-trapping system. Proc. Natl. Acad. Sci. U.S.A. 98, 6593–6598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haendeler J., Yin G., Hojo Y., Saito Y., Melaragno M., Yan C., Sharma V. K., Heller M., Aebersold R., Berk B. C. (2003) GIT1 mediates Src-dependent activation of phospholipase Cγ by angiotensin II and epidermal growth factor. J. Biol. Chem. 278, 49936–49944 [DOI] [PubMed] [Google Scholar]
- 32. Liu S., Premont R. T., Kontos C. D., Huang J., Rockey D. C. (2003) Endothelin-1 activates endothelial cell nitric-oxide synthase via heterotrimeric G-protein βγ subunit signaling to protein kinase B/Akt. J. Biol. Chem. 278, 49929–49935 [DOI] [PubMed] [Google Scholar]
- 33. Housset C., Rockey D. C., Bissell D. M. (1993) Endothelin receptors in rat liver: lipocytes as a contractile target for endothelin 1. Proc. Natl. Acad. Sci. U.S.A. 90, 9266–9270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sakurai T., Yanagisawa M., Takuwa Y., Miyazaki H., Kimura S., Goto K., Masaki T. (1990) Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature 348, 732–735 [DOI] [PubMed] [Google Scholar]
- 35. Lamas S., Marsden P. A., Li G. K., Tempst P., Michel T. (1992) Endothelial nitric oxide synthase: molecular cloning and characterization of a distinct constitutive enzyme isoform. Proc. Natl. Acad. Sci. U.S.A. 89, 6348–6352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 37. Albert P. R., Robillard L. (2002) G protein specificity: traffic direction required. Cell. Signal. 14, 407–418 [DOI] [PubMed] [Google Scholar]
- 38. Ribas C., Takesono A., Sato M., Hildebrandt J. D., Lanier S. M. (2002) Pertussis toxin-insensitive activation of the heterotrimeric G-proteins Gi/Go by the NG108-15 G-protein activator. J. Biol. Chem. 277, 50223–50225 [DOI] [PubMed] [Google Scholar]
- 39. Daff S. (2010) NO synthase: structures and mechanisms. Nitric Oxide 23, 1–11 [DOI] [PubMed] [Google Scholar]
- 40. Stuehr D. J., Tejero J., Haque M. M. (2009) Structural and mechanistic aspects of flavoproteins: electron transfer through the nitric oxide synthase flavoprotein domain. FEBS J. 276, 3959–3974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fulton D., Church J. E., Ruan L., Li C., Sood S. G., Kemp B. E., Jennings I. G., Venema R. C. (2005) Src kinase activates endothelial nitric-oxide synthase by phosphorylating Tyr-83. J. Biol. Chem. 280, 35943–35952 [DOI] [PubMed] [Google Scholar]
- 42. Fulton D., Ruan L., Sood S. G., Li C., Zhang Q., Venema R. C. (2008) Agonist-stimulated endothelial nitric oxide synthase activation and vascular relaxation. Role of eNOS phosphorylation at Tyr83. Circ. Res. 102, 497–504 [DOI] [PubMed] [Google Scholar]
- 43. Ruan L., Torres C. M., Buffett R. J., Kennard S., Fulton D., Venema R. C. (2013) Calcineurin-mediated dephosphorylation of eNOS at serine 116 affects eNOS enzymatic activity indirectly by facilitating c-Src binding and tyrosine 83 phosphorylation. Vascul. Pharmacol. 59, 27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haynes M. P., Li L., Sinha D., Russell K. S., Hisamoto K., Baron R., Collinge M., Sessa W. C., Bender J. R. (2003) Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J. Biol. Chem. 278, 2118–2123 [DOI] [PubMed] [Google Scholar]
- 45. Ciccarelli M., Cipolletta E., Santulli G., Campanile A., Pumiglia K., Cervero P., Pastore L., Astone D., Trimarco B., Iaccarino G. (2007) Endothelial β2 adrenergic signaling to AKT: role of Gi and SRC. Cell. Signal. 19, 1949–1955 [DOI] [PubMed] [Google Scholar]
- 46. Su K. H., Tsai J. Y., Kou Y. R., Chiang A. N., Hsiao S. H., Wu Y. L., Hou H. H., Pan C. C., Shyue S. K., Lee T. S. (2009) Valsartan regulates the interaction of angiotensin II type 1 receptor and endothelial nitric oxide synthase via Src/PI3K/Akt signalling. Cardiovasc. Res. 82, 468–475 [DOI] [PubMed] [Google Scholar]
- 47. Banquet S., Delannoy E., Agouni A., Dessy C., Lacomme S., Hubert F., Richard V., Muller B., Leblais V. (2011) Role of Gi/o-Src kinase-PI3K/Akt pathway and caveolin-1 in β2-adrenoceptor coupling to endothelial NO synthase in mouse pulmonary artery. Cell. Signal. 23, 1136–1143 [DOI] [PubMed] [Google Scholar]
- 48. Frank S. R., Hansen S. H. (2008) The PIX-GIT complex: a G protein signaling cassette in control of cell shape. Semin. Cell Dev. Biol. 19, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van Nieuw Amerongen G. P., Natarajan K., Yin G., Hoefen R. J., Osawa M., Haendeler J., Ridley A. J., Fujiwara K., van Hinsbergh V. W., Berk B. C. (2004) GIT1 mediates thrombin signaling in endothelial cells: role in turnover of RhoA-type focal adhesions. Circ. Res. 94, 1041–1049 [DOI] [PubMed] [Google Scholar]
- 50. Shikata Y., Birukov K. G., Birukova A. A., Verin A., Garcia J. G. (2003) Involvement of site-specific FAK phosphorylation in sphingosine-1 phosphate- and thrombin-induced focal adhesion remodeling: role of Src and GIT. FASEB J. 17, 2240–2249 [DOI] [PubMed] [Google Scholar]
- 51. Pang J., Hoefen R., Pryhuber G. S., Wang J., Yin G., White R. J., Xu X., O'Dell M. R., Mohan A., Michaloski H., Massett M. P., Yan C., Berk B. C. (2009) G-protein-coupled receptor kinase interacting protein-1 is required for pulmonary vascular development. Circulation 119, 1524–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hsieh H. L., Lin C. C., Chan H. J., Yang C. M. (2012) c-Src-dependent EGF receptor transactivation contributes to ET-1-induced COX-2 expression in brain microvascular endothelial cells. J. Neuroinflammation 9, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spinella F., Caprara V., Di Castro V., Rosanò L., Cianfrocca R., Natali P. G., Bagnato A. (2013) Endothelin-1 induces the transactivation of vascular endothelial growth factor receptor-3 and modulates cell migration and vasculogenic mimicry in melanoma cells. J. Mol. Med. 91, 395–405 [DOI] [PubMed] [Google Scholar]
- 54. Imamura T., Huang J., Dalle S., Ugi S., Usui I., Luttrell L. M., Miller W. E., Lefkowitz R. J., Olefsky J. M. (2001) β-Arrestin-mediated recruitment of the Src family kinase Yes mediates endothelin-1-stimulated glucose transport. J. Biol. Chem. 276, 43663–43667 [DOI] [PubMed] [Google Scholar]
- 55. Lu Y., Yu Q., Liu J. H., Zhang J., Wang H., Koul D., McMurray J. S., Fang X., Yung W. K., Siminovitch K. A., Mills G. B. (2003) Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J. Biol. Chem. 278, 40057–40066 [DOI] [PubMed] [Google Scholar]