

Background: PCTK3 is an uncharacterized serine/threonine kinase that belongs to the cyclin-dependent kinase family.
Results: The activity of PCTK3 is increased via interaction with cyclin A2 and phosphorylation by PKA. PCTK3 knockdown induces actin polymerization.
Conclusion: PCTK3 is activated by cyclin A2 and PKA and is involved in actin dynamics.
Significance: This study provides clues to the physiological function of PCTK3.
Keywords: Cofilin, Cyclin, Cyclin-dependent Kinase (CDK), Cytoskeleton, Protein Kinase A (PKA)
Abstract
PCTAIRE kinase 3 (PCTK3)/cyclin-dependent kinase 18 (CDK18) is an uncharacterized member of the CDK family because its activator(s) remains unidentified. Here we describe the mechanisms of catalytic activation of PCTK3 by cyclin A2 and cAMP-dependent protein kinase (PKA). Using a pulldown experiment with HEK293T cells, cyclin A2 and cyclin E1 were identified as proteins that interacted with PCTK3. An in vitro kinase assay using retinoblastoma protein as the substrate showed that PCTK3 was specifically activated by cyclin A2 but not by cyclin E1, although its activity was lower than that of CDK2. Furthermore, immunocytochemistry analysis showed that PCTK3 colocalized with cyclin A2 in the cytoplasm and regulated cyclin A2 stability. Amino acid sequence analysis revealed that PCTK3 contained four putative PKA phosphorylation sites. In vitro and in vivo kinase assays showed that PCTK3 was phosphorylated by PKA at Ser12, Ser66, and Ser109 and that PCTK3 activity significantly increased via phosphorylation at Ser12 by PKA even in the absence of cyclin A2. In the presence of cyclin A2, PCTK3 activity was comparable to CDK2 activity. We also found that PCTK3 knockdown in HEK293T cells induced polymerized actin accumulation in peripheral areas and cofilin phosphorylation. Taken together, our results provide the first evidence for the mechanisms of catalytic activation of PCTK3 by cyclin A2 and PKA and a physiological function of PCTK3.
Introduction
In mammals, the cyclin-dependent kinases (CDKs),2 a family of serine/threonine protein kinases, play an important role in regulating cellular functions such as cell proliferation, transcription, and neuron function (1). Their activities are regulated by associations with different cyclins and phosphorylation at specific sites by protein kinases (2). Because cyclins are differentially expressed and degraded by ubiquitin-mediated proteolysis at specific cell cycle phases, CDK activity oscillates during cell cycle transitions (3). Canonical CDKs contain the PSTAIRE helix, which has been implicated in binding to cyclins in the kinase domain (4). The PCTAIRE kinase (PCTK) subfamily of CDKs includes three members, PCTK1/CDK16, PCTK2/CDK17, and PCTK3/CDK18, which contain the PCTAIRE sequence instead of the PSTAIRE sequence. PCTK family members have high sequence homology in their central kinase domains, whereas the structures of their N-terminal and C-terminal regions differ. PCTK genes are conserved in higher eukaryotes, from Caenorhabditis elegans to humans. However, there are no PCTK orthologs in yeast. The PCTK subfamily is widely expressed in mammalian tissues and is relatively more abundant in post-mitotic cells, suggesting that they may function in higher eukaryotes and have different biological functions from those during the cell cycle (5). PCTK1 is the best-characterized member of this kinase family and regulates neurite outgrowth in the Neuro2A neuroblastoma cell line (6). It is also involved in membrane trafficking through the early secretory pathway via phosphorylation of N-ethylmaleimide-sensitive fusion protein (7, 8). A recent study reported that PCT-1, the C. elegans ortholog of PCTK1, is complexed with CYY-1 (ortholog of mammalian cyclin Y, a novel membrane-associated cyclin) and is also necessary for targeting presynaptic components to the axons (9). Furthermore, human PCTK1 is activated by cyclin Y and is essential for spermatogenesis (10). PCTK2 is associated with Trap (tudor repeat associator with PCTAIRE 2) and ik3–1/cables, an adaptor that functionally connects c-abl and CDK5 to support neurite growth (11, 12). The third member of this family, PCTK3, has been the least studied. Although exogenously expressed PCTK1 and PCTK2 phosphorylate myelin basic protein and histone H1 in vitro, PCTK3 kinase activity has not been detected. To characterize PCTK3 activity and elucidate its regulatory mechanism, it is necessary to identify activators for PCTK3.
Mammalian cyclin A consists of two isoforms: cyclin A1 and cyclin A2. In male mice, cyclin A1 is predominantly expressed in germ cells and is required for the entry of germ cells into the first meiotic division (13). In contrast, cyclin A2 is ubiquitously expressed in somatic cells and has a major role in S-phase progression and the G2/M phase transition by activating CDK2 and CDK1, respectively. Although cyclin A is predominantly localized in the nucleus during S phase, it also shuttles between the nucleus and cytoplasm and is degraded by ubiquitin-mediated proteolysis during the G2 phase. Cyclin A/CDK2 is transiently maintained by SCAPER (S phase cyclin A-associated protein residing in the endoplasmic reticulum), an S phase cyclin A-associated protein, in the cytoplasm (14). However, a recent study showed cyclin A2 is involved in the regulation of cell migration and invasiveness through its direct interaction with RhoA, suggesting a novel function of cyclin A in a CDK1- and CDK2-independent manner (15).
Protein kinase A (PKA) phosphorylates numerous proteins involved in various cellular phenomena and preferentially to phosphorylates a serine or threonine within the following consensus sequence: Arg-Arg-X-Ser/Thr (X represents any amino acid) (16). PKA also plays an important role in maintaining meiotic arrest (17). In Xenopus oocytes, PKA phosphorylates and inactivates Cdc25C phosphatase, whereas Wee1B kinase activity is enhanced by PKA (18, 19). The activity of the cyclin B-CDK1 complex is controlled by Wee1 kinase and Cdc25C phosphatase (20, 21). PKA regulates the activity of the cyclin B-CDK1 complex via dual regulation of the Cdc25 phosphatase and Wee1B kinase (18). On the other hand, PKA inhibits the interaction between PCTK1 and cyclin Y via phosphorylation of PCTK1 at Ser153, resulting in PCTK1 inactivation (6, 10). Thus, PKA regulates the activities of CDKs in either a direct or an indirect manner.
In this study we demonstrated that PCTK3 is activated by its association with cyclin A2 and phosphorylates retinoblastoma protein (Rb) in vitro. In addition, we found that PCTK3 contains four putative PKA phosphorylation sites (Arg-Arg-X-Ser/Thr) and that PCTK3 phosphorylation by PKA results in an increase in its catalytic activity even in the absence of cyclin A2. These findings provide some insights into the functions of PCTK3.
EXPERIMENTAL PROCEDURES
Antibodies and Materials
Antibody against cyclin A was purchased from BD Biosciences, and anti-cyclin B1, anti-cyclin D1, anti- cyclin E1, anti-cyclin E2, anti-cyclin H, anti-CDK1, anti-CDK2, anti-Rb, anti-phospho-Rb (Ser795) and (Ser807/811), anti-cofilin, anti-phospho-cofilin (Ser3), and anti-phospho-PKA substrate (RRX(S/T)) antibodies were from Cell Signaling Technology. Anti-PCTK3, anti-α-tubulin, and anti-β-actin antibodies were from Santa Cruz Biotechnology. Anti-FLAG M2 antibody was form Sigma. Anti-GST antibody was from Wako Pure Chemical Industries. Anti-Strep antibody was from Qiagen. Alexa-555 conjugated-phalloidin was from Cytoskeleton.
Plasmid Construction
cDNAs encoding mouse full-length PCTK1, PCTK2, PCTK3, CDK2, PFTAIRE kinase 1 (PFTK1), cyclin A2, cyclin E1, cyclin K, and cyclin Y and human retinoblastoma protein (779–928 amino acid; Rb C) were cloned by PCR using the respective specific primers. PCR products were cloned into TA-cloning vector pGEM-T Easy (Promega), and the inserted DNA sequences were confirmed by DNA sequencing. The inserted DNAs were subcloned in-frame into the different mammalian expression vectors including Strep-tagged expression vector pEXPR-IBA105 (IBA GmbH), glutathione S-transferase (GST) fusion expression vector pEBG, and FLAG-tagged expression vector pFLAG-CMV-2 (Sigma). A cDNA encoding for human Rb C was subcloned into the pMAL vector (New England Biolabs) of the bacterial maltose-binding protein (MBP) fusion expression system. The expression plasmid pFLAG-PKA-C encoding the catalytic subunit α isoform of mouse PKA was described previously (22). Site-directed mutagenesis was performed using PrimeSTAR Mutagenesis Basal Kit (Takara Bio) according to the manufacturer's instructions. The mutation was confirmed by DNA sequencing analysis.
Expression and Purification of Rb Recombinant Protein
The bacterial expression plasmid of Rb, pMAL-Rb C, was introduced into the bacterial strain BL21 Star DE3 (Invitrogen). An overnight culture in LB medium was diluted in fresh LB medium and incubated at 37 °C for 1 h with continuous shaking. After isopropyl-1-thio-β-d-galactopyranoside was added to the culture to a final concentration of 0.2 mm, the culture was incubated at 30 °C for an additional 6 h. The cells were resuspended in ice-cold extraction buffer A (20 mm Tris-HCl, pH 8.0, 1 mm EDTA, 10 μg/ml aprotinin, and 10 μg/ml leupeptin). After freezing and thawing, suspended cells were sonicated on ice in short bursts. The lysate was cleared by centrifugation at 10,000 × g for 10 min at 4 °C. The supernatant was then incubated with amylose resin (New England Biolabs) overnight at 4 °C. The beads were washed 5 times with ice-cold extraction buffer A and incubated with 10 mm maltose at 4 °C to elute fusion protein from the beads. After centrifugation, the supernatant was dialyzed against PBS. The purified protein was electrophoresed on SDS-PAGE and visualized by Coomassie Brilliant Blue staining, and the protein concentration was determined by Bradford assay (Bio-Rad) using BSA as a standard.
Cell Culture and Transfection
COS-7, HeLa, and HEK293T cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in 5% CO2. Cells were transfected with various expression vectors using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Subcellular Fractionations
HEK293T cells were washed twice with ice-cold PBS, harvested by scraping, and lysed in a buffer A (10 mm HEPES, pH 7.9, 10 mm KCl, 0.1 mm EDTA, 10 μg/ml leupeptin, and 10 μg/ml aprotinin). After incubation on ice for 15 min, the cells were mixed with 0.5% Nonidet P-40 and homogenized by vortex for 10 s. The homogenate was centrifuged at 2500 rpm for 3 min to sediment the nuclei. The supernatant was then centrifuged at a 10,000 rpm for 10 min, and the resulting supernatant was used as the cytoplasm fraction. The nuclear pellet was washed three times with buffer A to remove any contamination from cytoplasmic proteins. To extract nuclear proteins, the isolated nuclei were resuspended in buffer B (20 mm HEPES, pH 7.9, 400 mm NaCl, 1 mm EDTA, 10 μg/ml leupeptin, and 10 μg/ml aprotinin), and the mixture was incubated on ice for 20 min. Nuclear lysates were collected after centrifugation at 12,000 rpm for 15 min at 4 °C. The purity of nuclear and cytoplasm fractions was confirmed by immunoblotting using an anti-lamin B1 antibody (MBL) as a nuclear marker and α-tubulin antibody as a cytoplasmic marker, respectively.
Pulldown and Co-immunoprecipitation Assays
Pulldown and co-immunoprecipitation assays were performed as previously described (23). Briefly, cells were scraped in an ice-cold TNE buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.5% Nonidet P-40, and 1 mm EDTA) supplemented with protease inhibitors (10 μg/ml leupeptin and 10 μg/ml aprotinin). The cell extracts were centrifuged at 10,000 × g for 10 min at 4 °C before immunoprecipitation or immunoblotting. Equal protein amounts of the lysates were analyzed for protein expression. For pulldown assays, lysates were incubated with Strep-Tactin Sepharose (IBA GmbH) or glutathione Sepharose (GE Healthcare) overnight at 4 °C. For immunoprecipitation, lysates were incubated with anti-FLAG M2 or anti-PCTK3 antibody in the presence of protein G Sepharose (GE Healthcare) overnight at 4 °C. The beads were washed 4 times with wash buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Nonidet P-40, and 1 mm EDTA). Precipitated proteins were subjected to in vitro kinase assay or immunoblot analysis. Protein expression and phosphorylation were determined by immunoblotting of immunoprecipitates or total cell lysates.
Transfection of Small Interference RNA
The synthetic small interfering RNA (siRNA) oligonucleotides for PCTK3 (PCTK3 siRNA#1 (ID# SASI_Hs02_00334101), PCTK3 siRNA#2 (ID# SASI_Hs01_00015477), and PCTK3 siRNA#3 (ID# SASI_Hs02_00374212)) were purchased from Sigma. A MISSION siRNA Universal Negative Control #1 was used as the negative control. The transfection of siRNA was performed using Lipofectamine 2000 according to the manufacturer's instructions.
In Vitro Kinase Assay
In vitro kinase assays were carried out as previously described (23). For in vitro kinase assay of PCTK3, cell lysates were incubated with a glutathione-Sepharose or immunoprecipitated with anti-PCTK3 antibody and protein G Sepharose overnight at 4 °C by rotation. The beads were washed 3 times with wash buffer and twice with 50 mm Tris-HCl, pH 7.5. The kinase reaction was carried out by resuspending the complexes in 100 μl of kinase buffer (50 mm Tris-HCl, pH 7.5, 20 mm magnesium acetate, 20 or 50 μm ATP and phosphatase inhibitor mixture (Nacalai Tesque)) including 5 μg/ml purified MBP-Rb C in the absence or presence of 2 μCi of [γ-32P]ATP and incubating for 30 min at 30 °C. Phosphorylated MBP-Rb C was separated by SDS-PAGE and visualized with a BAS-1500 Bioimaging Analyzer (Fuji Film) or subjected to immunoblot analysis using anti-phospho-Rb (Ser795) antibody. To assay the phosphorylation of PCTK3 by PKA, cell lysate were immunoprecipitated with anti-FLAG antibody overnight at 4 °C by rotation. The kinase reaction was carried out by resuspending the complexes in 100 μl of kinase buffer with 2 μCi [γ-32P]ATP and incubating for 30 min at 30 °C. Immunocomplexes were released by heating at 95 °C in SDS sample buffer and subjected to SDS-PAGE, and phosphorylated proteins were visualized by BAS-1500 Bioimaging Analyzer. Quantitative densitometric analysis was performed using Image J software.
In Vivo Kinase Assay
HEK293T cells were treated with 10 μm forskolin for 30 min in the presence of 10% FBS and scraped in an ice-cold TNE buffer supplemented with phosphatase inhibitor mixture. The cell extracts were centrifuged at 10,000 × g for 10 min at 4 °C to remove cellular debris, and the supernatants were immunoprecipitated with anti-PCTK3 antibody with protein G-Sepharose overnight at 4 °C by rotation. The beads were washed with wash buffer, and immunocomplexes were released by heating at 95 °C in SDS sample buffer, subjected to SDS-PAGE, and immunoblotted using anti-phospho-RRX(S/T) antibody.
Immunofluorescence Analysis
Immunofluorescence analysis was performed as previously described (23). In brief, HEK293T cells grown on poly-l-lysine-coated chamber slides were cotransfected with Myc-cyclin A2 together with either FLAG-PCTK3 or FLAG-CDK2. At 24 h post-transfection, cells were washed twice with PBS and fixed for 20 min in 3.7% formaldehyde. After sequential washing with PBS, cells were permeabilized for 5 min in 0.1% Triton X-100, washed 3 times with PBS, and then treated with 5% BSA for 30 min. Cells were subsequently incubated with mouse anti-FLAG M2 IgG or rabbit anti-Myc polyclonal antibody overnight at 4 °C. After 3 washes with PBS, cells were incubated for 1 h with goat anti-mouse IgG directly conjugated to Alexa Fluor 488 or goat anti-rabbit IgG directly conjugated to Alexa Fluor 555 (Invitrogen). The slides were washed thoroughly with PBS and mounted in fluorescent mounting medium Vectashield (Vector Laboratories). A confocal laser-scanning microscope (Leica TCS-SP5) was used to obtain staining profiles.
RESULTS
Identification of Cyclin A2 as An Activator of PCTK3
PCTK1, a member of the PCTK subfamily, is activated by membrane-associated cyclin Y (10, 24). We hypothesized that PCTK3 was also activated by cyclin family members. To identify PCTK3 activators, we expressed Strep-tagged mouse PCTK3 in HEK293T cells and conducted Strep pulldown experiments. The proteins that coprecipitated with PCTK3 were analyzed by immunoblot assay using anti-cyclin A, B1, D1, E1, E2, and H antibodies. Immunoblot analysis revealed that PCTK3 interacted with cyclin A and cyclin E1, but not cyclin B1, D1, E2, or H, in HEK293T cells (Fig. 1A). To confirm the interaction between PCTK3 and cyclin A2 or E1, we performed GST pulldown experiments using lysates prepared from COS-7 cells expressing GST-fused PCTK3 and FLAG-tagged cyclins. We also examined whether cyclin K and cyclin Y bound to PCTK3, because PCTK3 was found to interact with cyclin K in a large-scale interaction study (25). As shown in Fig. 1B, GST-PCTK3 associated with FLAG-cyclin A2 and cyclin E1 but not with cyclin K or cyclin Y.
FIGURE 1.

Identification of cyclin A2 as an activator of PCTK3. A, HEK293T cells were transfected with Strep-PCTK3. After the Strep pulldown assay, the precipitated proteins were subjected to immunoblot analysis with anti-cyclin A, anti-cyclin B1, anti-cyclin D3, anti-cyclin E1, anti-cyclin E2, and anti-cyclin H antibodies. Total cell lysates were used as the input samples and positive control. B, GST-PCTK3 was expressed in COS-7 cells with either FLAG-cyclin A2, FLAG-cyclin E1, FLAG-cyclin K, or FLAG-cyclin Y. In the control experiment (GST-PCTK3 (−)), GST alone was expressed instead of GST-PCTK3. The GST pulldown precipitates were immunoblotted with anti-FLAG antibody. Protein expression was confirmed by immunoblotting total cell lysates. C, GST and GST-PCTK3 was expressed with either FLAG-cyclin A2 or FLAG-cyclin E1 in COS-7 cells. After the GST pulldown, the precipitated samples were incubated in a kinase buffer containing [γ-32P]ATP and MBP-Rb C for 30 min. The supernatants containing MBP-Rb C were separated on SDS-PAGE, after which the gel was analyzed by bioimaging analyzer. The expression level of GST-fused and FLAG-tagged proteins was confirmed by immunoblotting with anti-FLAG and anti-GST antibody, respectively. D, COS-7 cells were transfected with Strep-PCTK3 and FLAG-cyclin A2. GST pull downed proteins were incubated in a kinase buffer containing MBP-Rb C for 30 min. The precipitates and supernatants were analyzed by SDS-PAGE and immunoblotting with anti-FLAG and anti-phospho-Rb (Ser795) and (Ser807/811) antibodies, respectively. D, COS-7 cells were transfected with Strep-PCTK3, Strep-CDK2, and FLAG-cyclin A2. Strep pull-downed proteins were incubated in a kinase buffer containing MyBP, MBP-Rb C, or histone H1 for 30 min. The supernatants were analyzed by SDS-PAGE and bioimaging analyzer. IB, immunoblot. E, Strep-PCTK3 was expressed in COS7 cells with FLAG-cyclin A2. After the Strep pulldown, bound proteins were incubated in a kinase buffer containing MBP-Rb C for 30 min. The precipitates and supernatants were analyzed by SDS-PAGE and immunoblotting with anti-FLAG and anti-phospho-Rb (Ser795) and (Ser807/811) antibodies, respectively. CBB, Coomassie Brilliant Blue.
Subsequently, to determine whether PCTK3 activity was regulated by binding to cyclin A2 or cyclin E1, we performed an in vitro kinase assay using retinoblastoma protein (Rb) as the substrate. Rb is phosphorylated by some CDKs in a cell cycle-dependent manner, and PFTK1, which is activated by cyclin D3 and cyclin Y, also phosphorylates Rb (26, 27). The C terminus of Rb contains a cluster of seven candidate CDK phosphorylation sites (Ser/Thr-Pro motifs: Ser780, Ser788, Ser795, Ser807, Ser811, Thr821, and Thr826) (28). For this in vitro kinase assay, we produced recombinant MBP-fused Rb C (amino acids 779–928) in Escherichia coli. COS-7 cells were cotransfected with GST-PCTK3 in the presence or absence of FLAG-cyclin A2 or E1, after which cell lysates were subjected to glutathione-Sepharose pulldown. In an in vitro kinase analysis with [γ-32P]ATP, MBP-Rb C was phosphorylated by PCTK3 in the presence of cyclin A2, whereas cyclin E1 did not activate PCTK3 (Fig. 1C). We also performed an in vitro kinase assay using myelin basic protein (MyBP) and histone H1 as substrates. As shown in Fig. 1D, cyclin A2-CDK2 complex strongly phosphorylated all three of the substrates tested (Rb, MyBP, and histone H1). However, the catalytic activity of PCTK3 was much lower than that of CDK2 even in the presence of cyclin A2. Among the three substrates tested, Rb was the most efficiently phosphorylated by cyclin A2-PCTK3 complex. In addition, phosphorylated MBP-Rb C was detected using antibodies against phospho-Rb (Ser795) and phospho-Rb (Ser807/811) (Fig. 1E). Both of these antibodies detected Rb phosphorylation only when PCTK3 and cyclin A2 were cotransfected, indicating that cyclin A2-PCTK3 complex phosphorylates Rb at least at Ser795 and Ser807/811 in vitro. These results showed that PCTK3 is activated through its association with cyclin A2.
Cyclin A2 Specifically Activates PCTK3
Cyclin A2 interacts with CDK1 and CDK2 during cell cycle progression (29). We performed GST pulldown experiments to evaluate whether Rb phosphorylation was dependent on PCTK3 or other kinases that coprecipitated with cyclin A2. As shown in Fig. 2A, endogenous CDK1 and CDK2 were specifically coprecipitated with GST-cyclin A (lane 4), whereas GST-PCTK3, GST-PCTK3/FLAG-cyclin A2 complex, and FLAG-cyclin A2 did not coprecipitate with endogenous CDK1 and CDK2 (lanes 1–3). In addition, we designed a PCTK3 inactive mutant on the basis of amino acids that are conserved among various members of the CDK family. An analogous K194R mutation at the putative ATP binding site of PCTK1 generates a kinase dead mutant (6). We created a kinase-dead mutant of PCTK3, in which the putative ATP binding site was replaced by arginine (PCTK3 K150R), and evaluated its catalytic activity with an in vitro kinase assay. Although both PCTK3 wild type and K150R interacted with cyclin A2, no kinase activity of PCTK3 K150R was detected (Fig. 2B). Thus, Rb phosphorylation is dependent on PCTK3 but not on other protein kinases, and it is highly likely that cyclin A2 is an activator of PCTK3.
FIGURE 2.

Cyclin A2 specifically activates PCTK3. A, COS-7 cells were transfected with GST-PCTK3, FLAG-cyclin A2, and GST-cyclin A2. In the control experiment, GST alone was transfected instead of GST-PCTK3 or GST-cyclin A2. The GST pulldown precipitates were subjected to immunoblotting with anti-FLAG, anti-CDK1, and anti-CDK2 antibodies. B, GST-PCTK3 wild type (WT) or K150R mutant was expressed in COS-7 cells with FLAG-cyclin A2. In the control experiment, GST alone was expressed instead of GST-PCTK3. After the GST pulldown, the bound proteins were incubated in a kinase buffer with MBP-Rb C. The precipitates and supernatants were analyzed by SDS-PAGE and immunoblotting with anti-FLAG and anti-phospho-Rb (Ser795) antibodies, respectively. C, FLAG-cyclin A2 was expressed in COS-7 cells with GST, GST-PCTK1, GST-PCTK2, or GST-PCTK3. The GST pulldown precipitates were subjected to in vitro kinase assay using anti-phospho-Rb (Ser795) antibody. D, GST, GST-PCTK3, GST-PFTK1, or GST-CDK2 was expressed in COS-7 cells with FLAG-cyclin A2. The GST pulled-down proteins were subjected to an in vitro kinase assay.
We also examined whether other members of the PCTK subfamily, PCTK1 and PCTK2, were activated by cyclin A2, because PCTK2 activators have not yet been identified. Although PCTK1 and PCTK2 interacted with cyclin A2, only PCTK3 was activated by cyclin A2 (Fig. 2C). In contrast, PFTK1, with high homology to PCTK3, was not associated with cyclin A2 and did not phosphorylate Rb C (Fig. 2D). However, GST pulldown and in vitro kinase assays suggested that CDK2 had a higher affinity for cyclin A2 than PCTK3 and that the catalytic activity of PCTK3 was lower than that of CDK2 in the presence of cyclin A2 (Fig. 2D). These results demonstrated that cyclin A2 specifically activates PCTK3 and suggested that there are additional mechanisms for PCTK3 activation in addition to its interaction with cyclin A2.
Endogenous Interaction between PCTK3 and Cyclin A in the Cytoplasm
Next, we investigated the endogenous interaction between PCTK3 and cyclin A. A previous study reported that PCTK3 protein is expressed in some cell lines including HEK293 cells (7). Thus, we confirmed whether PCTK3 expression was detectable in some cell lines. As shown in Fig. 3A, an anti-PCTK3 antibody detected a specific protein band with the predicted molecular mass of PCTK3 in HEK293T cells, whereas its expression was very low or absent in human breast carcinoma MCF7cells and human cervical carcinoma HeLa cells. We confirmed PCTK3 expression in HEK293T cells by knockdown analysis using siRNA. As shown in Fig. 3B, PCTK3 expression was efficiently reduced by each of the three PCTK3 siRNAs used, whereas negative control siRNA had no effect on PCTK3 expression. We also examined the subcellular distributions of PCTK3 and cyclin A by simple cell fractionation. PCTK3 protein was found in the cytoplasm, whereas cyclin A was localized in both the cytoplasm and nucleus (Fig. 3C). Subsequently, we investigated cyclin A binding to PCTK3 in the nuclear and cytoplasmic fractions. Immunoprecipitates from each fraction with an anti-PCTK3 antibody were analyzed by immunoblotting with anti-cyclin A antibody. Cyclin A was detected in the cytoplasmic PCTK3 immunoprecipitates but not in the nuclear immunoprecipitates (Fig. 3D), indicating that PCTK3 indeed interacts with cyclin A in vivo. We also examined the intracellular localization of endogenous PCTK3 and cyclin A in HEK293T cells. Immunofluorescence analysis using confocal laser-scanning microscopy revealed that PCTK3 and cyclin A were mainly found in the cytoplasm and nucleus, respectively, and that they colocalized in the cytoplasm (Fig. 3E).
FIGURE 3.

Endogenous interaction between PCTK3 and cyclin A in the cytoplasm. A, immunoblot analysis of total proteins prepared from HeLa, MCF7, and HEK293T human cell lines. β-Actin was used as a loading control. B, siRNA knockdown of PCTK3 in HEK293T cells. Cells were transfected with three different siRNAs against PCTK3 (#1–3) or negative control siRNA (NC). Forty-eight hours after transfection, cell extracts were immunoblotted with anti-PCTK3 antibody. Expression of β-actin was used as a loading control. C, subcellular distributions of endogenous PCTK3 and cyclin A. HEK293T cells were separated into nuclear and cytoplasmic fractions as described under “Experimental Procedures.” Aliquots of each fraction were immunoblotted for PCTK3 and cyclin A. The purity of nuclear and cytoplasmic fractions was confirmed using anti-lamin B1 antibody as a nuclear marker and α-tubulin antibody as a cytoplasmic marker, respectively. D, endogenous interaction between PCTK3 and cyclin A in HEK293T cells. The fractionated lysates from HEK293T cells were immunoprecipitated (IP) using anti-PCTK3 antibody. The immunoprecipitates were analyzed by immunoblotting with anti-cyclin A antibody. Normal rabbit IgG (IgG) was used as a control. E, intracellular localization of endogenous PCTK3. HEK293T cells were fixed and incubated with rabbit anti-PCTK3 and mouse anti-cyclin A antibodies. The primary antibody was visualized with Alexa Fluor 488-conjugated anti-rabbit IgG and Alexa Fluor 555-conjugated anti-mouse IgG followed by confocal microscopy. Fluorescence for PCTK3 (green) and cyclin A (red) is shown with the merged images (merge is in yellow). Hoechst nuclear staining is represented in blue.
Additionally, we transfected HEK293T cells with FLAG-PCTK3 and Myc-cyclin A2 and performed immunofluorescence analysis. In cells that were singly transfected with either Myc-cyclin A2 or FLAG-PCTK3, cyclin A2 was strongly detected in the nucleus and moderately in the cytoplasm, whereas PCTK3 was found in only the cytoplasm (data not shown), consistent with previous data (30). In cells cotransfected with Myc-cyclin A2 and FLAG-PCTK3, cyclin A2 was translocated from the nucleus to the cytoplasm and colocalized with PCTK3 in the cytoplasm (Fig. 4, A and C). By comparison, when cyclin A2 was cotransfected with CDK2, it colocalized with CDK2 in the nucleus (Fig. 4, B and C). These findings suggested that PCTK3 and CDK2 regulate the localization of cyclin A2 in the cytoplasm and the nucleus, respectively.
FIGURE 4.
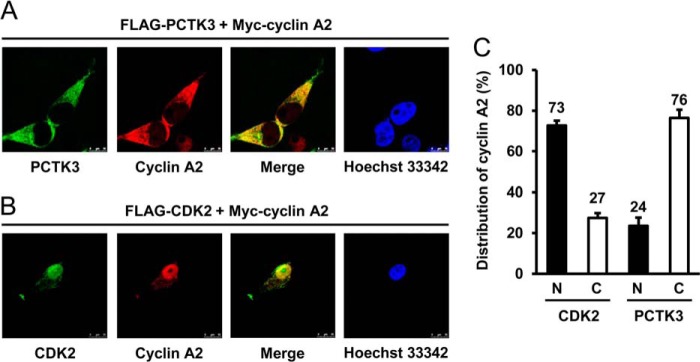
Different intracellular localization of cyclin A2 in PCTK3- and CDK2-overexpressed cells. A and B, HEK293T cells were cotransfected with Myc-cyclin A2 together with either FLAG-PCTK3 (A) or FLAG-CDK2 (B). Cells were fixed and incubated with mouse anti-FLAG and rabbit anti-Myc antibodies. The primary antibody was visualized with Alexa Fluor 488-conjugated anti-mouse IgG and Alexa Fluor 555-conjugated anti-rabbit IgG followed by confocal microscopy. Fluorescence for FLAG-PCTK3 or FLAG-CDK2 (green) and Myc-cyclin A2 (red) is shown with the merged images (merge is in yellow). Hoechst nuclear staining is represented in blue. C, the fluorescence intensities of Myc-cyclin A2 in the nuclear (N, closed bar) and cytoplasmic (C, open bar) regions were quantified using Image J software. The data shown are the means ± S.E. (n ≥ 5).
PCTK3 Protects Cyclin A2 against Degradation in the Cytoplasm
Cyclin A2 is degraded during prometaphase by anaphase-promoting complex/cyclosome (APC/C), and this degradation is necessary for mitosis progression (31). Cyclin E bound to CDK2 is also protected from degradation by the proteasome (32). We hypothesized that cyclin A2 interacted with PCTK3 is also protected from degradation in the cytoplasm. Thus, to analyze whether PCTK3 regulates cyclin A2 stability, we investigated the protein levels of cyclin A2 in PCTK3 knockdown HEK293T cells. As expected, the protein levels of cyclin A2 in PCTK3 knockdown cells was low compared with that in negative control siRNA-treated cells (Fig. 5A). To verify the protective effect of PCTK3 against cyclin A2 degradation, HEK293T cells cotransfected with FLAG-cyclin A2 and GST-PCTK3 were treated with cycloheximide, a protein synthesis inhibitor, after which they were harvested at the indicated times. Cycloheximide chase analysis showed that cyclin A2 was rapidly degraded in the absence of GST-PCTK3, whereas cyclin A2 degradation was remarkably delayed in the presence of GST-PCTK3 (Fig. 5B). Furthermore, we examined if PCTK3 sequesters cyclin A2 in the cytoplasm and negatively regulates CDK2 activity in the nucleus. Nuclear fractions of HEK293T cells transfected with FLAG-PCTK3 were immunoprecipitated using anti-CDK2 antibody, and an in vitro kinase assay was performed using Rb as the substrate. As shown in Fig. 5C, PCTK3 did not regulate CDK2 activity in the nucleus. These results suggest that PCTK3 may regulate the stability of cyclin A2, which is normally degraded in the cytoplasm by ubiquitin-proteasome pathway.
FIGURE 5.

PCTK3 protects cyclin A2 against degradation in the cytoplasm. A, HEK293T cells were transfected with PCTK3 siRNA. The cell lysates were subjected to immunoblotting (IB) with anti-PCTK3 and anti-cyclin A antibodies. Expression of α-tubulin was used as a loading control. B, HEK293T cells were cotransfected with GST or GST-PCTK3 together with FLAG-cyclin A2. After 24 h, cycloheximide (CHX) (5 μg/ml) was added to the cell culture. Cells were harvested at indicated times after cycloheximide treatment and FLAG-cyclin A2 protein levels were determined by immunoblotting (IB: anti-FLAG). β-Actin was used as a loading control (IB: anti-β-actin). FLAG-cyclin A2 protein levels were quantified by densitometric analysis and represented in a graph. C, HEK293T cells were transfected with FLAG-PCTK3 and separated into nuclear and cytoplasmic fractions. The nuclear fractions were immunoprecipitated (IP) using anti-CDK2 antibody, and the immunoprecipitates were used in an in vitro kinase assay with MBP-Rb C as a substrate. Normal rabbit IgG (IgG) was used as a control.
PCTK3 Is Phosphorylated by PKA in Vitro and in Vivo
PKA is an essential factor that has been implicated in various physiological functions, including cytoskeletal organization, cell motility, and signal transduction (16). PKA preferentially phosphorylates serine/threonine residues within the consensus sequence Arg-Arg-X-Ser/Thr. A previous report demonstrated that PKA inactivates PCTK1 via phosphorylation at Ser153 (6). Amino acid sequence analysis revealed that mouse and human PCTK3 each contained four putative PKA phosphorylation sites, RRLS12, RRFS66, RRAS109, and RRQS449 in mouse and RRFS14, RRFS89, RRAS162, and RRQS502 in human. This suggested that PCTK3 is a substrate for PKA. To determine whether endogenous PCTK3 was phosphorylated by PKA, HEK293T cells were treated with the adenylate cyclase activator forskolin. We immunoprecipitated cell lysates with an anti-PCTK3 antibody, and followed it by immunoblot analysis using an anti-phospho-RRX(S/T) antibody. As shown in Fig. 6A, PCTK3 proteins were specifically precipitated by the anti-PCTK3 antibody and were efficiently phosphorylated after treatment with forskolin. Next, we examined whether PKA directly phosphorylates PCTK3. COS-7 cells were cotransfected with FLAG-PCTK3 and FLAG-PKA catalytic subunit, after which an in vitro kinase assay was performed. As shown in Fig. 6B, PKA directly phosphorylated PCTK3. Furthermore, to identify the PKA phosphorylation sites of PCTK3, we constructed four non-phosphorylatable mutants of mouse PCTK3 in which each of the putative PKA phosphorylation sites was replaced by alanine: S12A, S66A, S109A, and S449A. We tested these using in vitro and in vivo kinase assays. Phosphorylation of the S12A, S66A, and S109A mutants was reduced as compared with that of wild type PCTK3, whereas the phosphorylation intensity of the S449A mutant was almost equal to that of wild-type PCTK3 (Fig. 6B). Although PKA phosphorylated the S12A/S66A, S66A/S109A, and S12A/S109A mutants of PCTK3 (Fig. 6C), it failed to phosphorylate the S12A/S66A/S109A mutant (Fig. 6, B and C). These results identified three serine residues, Ser12, Ser66, and Ser109, as possible PKA phosphorylation sites for mouse PCTK3. Furthermore, the PKA phosphorylation sites of PCTK3 were confirmed by an in vivo kinase assay using an anti-phospho-RRX(S/T) antibody. HeLa cells transiently transfected with FLAG-tagged PCTK3 were treated with 10 μm forskolin. Cell lysates were immunoprecipitated with an anti-FLAG antibody and then immunoblotted with an anti-phospho-RRX(S/T) antibody. As expected, forskolin treatment resulted in a 1.7-fold increase in phosphorylation of PCTK3 wild type, whereas the phosphorylation of the S12A, S66A, and S109A mutants was reduced as compared with that of wild-type PCTK3 (Fig. 6D). These findings provided evidence that PCTK3 is phosphorylated by PKA at three sites (RRLS12, RRFS66, and RRAS109 in mouse) both in vitro and in vivo.
FIGURE 6.

PCTK3 is phosphorylated by PKA in vitro and in vivo. A, endogenous PCTK3 is phosphorylated by PKA. HEK293T cells were treated with 10 μm forskolin for 30 min in the presence of 10% FBS. Cell lysates were immunoprecipitated (IP) with anti-PCTK3 antibody. Immunoprecipitates were analyzed by immunoblot (IB) analysis using anti-phospho-RRX(S/T) or anti-PCTK3 antibodies. The bands of phosphorylated and total PCTK3 were indicated by arrows. B, FLAG-PKA C, FLAG-PCTK3 wild type (WT), and mutants (S12A, S66A, S109, S449, S12A/S66A/S109A/S449A (4SA), and S12A/S66A/S109A (3SA)) were expressed in COS-7 cells. Cell lysates were immunoprecipitated with anti-FLAG antibody. Immunoprecipitates were incubated with kinase buffer containing [γ-32P]ATP. Equal amounts of cell lysates were analyzed by immunoblotting with anti-FLAG antibody. The graph indicates the phosphorylation rate of PCTK3. The phosphorylation rate of wild-type PCTK3 was taken as 100%. C, the double or triple mutants of PCTK3 (S12A/S66A, S12A/S109A, S66A/S109A, and S12A/S66A/S109A (3SA)) were subjected to in vitro kinase assay using PKA. D, HeLa cells were transfected with FLAG-PCTK3 wild type (WT) and mutants (S12A, S66A, S109A, and S12A/S66A/S109A (3SA)). After 24 h, transfected cells were treated with 10 μm forskolin at 30 min. Cell extracts were subjected to immunoprecipitated with anti-FLAG antibody (IP: anti-FLAG). Immunoprecipitates were subsequently analyzed by immunoblotting with anti-phospho-RRX(S/T) or anti-FLAG antibody.
PCTK3 Activity Is Regulated via Phosphorylation at Ser12 by PKA
We then investigated the effects of PKA phosphorylation of PCTK3 on its kinase activity. PCTK3 activity was determined by GST pulldown and in vitro kinase assays using Rb C as the substrate. Three phosphomimic PCTK3 mutants (PCTK3 S12D, S66D, and S109D), in which the PKA phosphorylation sites were replaced by aspartic acid, were generated, and the kinase activities of these phospho-mimic mutants were examined. As shown in Fig. 7A, the PCTK3 S12D mutant had relatively high activity for Rb phosphorylation even in the absence of cyclin A2. Furthermore, the catalytic activity of the cyclin A2-S12D mutant complex was significantly increased (>2-fold) as compared with that of cyclin A2-wild-type PCTK3, although the ability of S12D mutant to bind to cyclin A was not changed. By comparison, the PCTK3 S66D and S109D mutants had no effect on kinase activity. On the other hand, the activity of the phospho-null mutant S12A was obviously decreased as compared with that of wild-type PCTK3 (Fig. 7B), strongly supporting that the phosphorylation at Ser12 influences the catalytic activation. In addition, the kinase activity of the PCTK3 S12D mutant was compared with that of the cyclin A2-CDK2 complex. Although cyclin A2-wild-type PCTK3 activity was six times less than that of cyclin A2-CDK2, the cyclin A2-S12D mutant had strong activity and reached a level comparable with the kinase activity of cyclin A2-CDK2 (Fig. 7C).
FIGURE 7.

PCTK3 activity is regulated via phosphorylation at Ser12 by PKA. A, COS-7 cell lysates expressing GST, GST-PCTK3 wild-type (WT), or GST-PCTK3 mutants (S12D, S66D, S109D, and S12D/S66D/S109D) and FLAG-cyclin A2 were used in a kinase assay using Rb as the substrate. The relative kinase activity of PCTK3 was quantified by densitometric analysis. The activity of wild-type PCTK3 complexed with FLAG-cyclin A2 was taken as 1. Results are expressed as the means ± S.E. from three independent experiments. Statistical significance was determined by analysis of variance. ***, p < 0.001. B, FLAG-PCTK3 WT and S12D and S12A mutants were expressed with Myc-cyclin A2 in COS-7 cells. The cell lysates were immunoprecipitated (IP) with anti-FLAG antibody (IP: anti-FLAG), and an in vitro kinase assay was performed using MBP-Rb C as the substrate. C, GST-PCTK3 WT, S12D mutant, and GST-CDK2 was expressed in COS-7 cells with or without FLAG-cyclin A2. The protein complexes were pulled down by glutathione-Sepharose beads, and an in vitro kinase assay was performed using MBP-Rb C protein as the substrate. The graph shows the average of three independent experiments. CBB, Coomassie Brilliant Blue. D, expression of PCTK3 in HEK293T cells was knocked down by siRNA. PCTK3 knockdown cells were treated with 10 μm forskolin for 30 min in medium containing 10% FBS. Cell lysates were immunoprecipitated with anti-PCTK3 antibody. Immunoprecipitates were subjected to in vitro kinase assay using Rb as the substrate. The activity of endogenous PCTK3 in negative control (NC) knockdown cells was taken as 100%. Experiments were performed three times independently. Results are expressed as the means ± S.E. Statistical significance was determined by analysis of variance. *, p < 0.05. E, HEK293T cells were transfected with FLAG-PCTK3 wild type and S12D mutant, FLAG-CDK2, and FLAG-cyclin A2. After 24 h, cell extracts were analyzed by immunoblotting with anti-phospho-Rb (Ser795), anti-Rb, and anti-FLAG antibodies.
We also determined whether endogenous PCTK3 was activated by PKA. First, to determine endogenous PCTK3 activity, we immunoprecipitated PCTK3 from HEK293Tcells transfected with negative control siRNA or PCTK3 siRNA and determined in vitro kinase activity using Rb as the substrate. As shown in Fig. 7D, significantly high Rb phosphorylation activity was detected in immunoprecipitates from cells transfected with negative control siRNA as compared with those from cells transfected with PCTK3 siRNA, suggesting that this activity was dependent on endogenous PCTK3 activity. Therefore, we assessed the effects of forskolin treatment on the kinase activity of PCTK3. Rb phosphorylation was significantly increased after forskolin treatment. These data demonstrated that PCTK3 is activated via phosphorylation at Ser12 by PKA.
Several members of the CDK family preferentially phosphorylate the canonical motif Ser/Thr-Pro-X-Lys/Arg/His (where X represents any amino acid). Because PCTK3 phosphorylated Rb in vitro, we investigated whether Rb was phosphorylated by PCTK3 in vivo. HEK293T cells were transfected with FLAG-tagged PCTK3 wild type or S12D mutant, after which cell lysates were subjected to immunoblotting using anti-phospho-Rb (Ser795). Both PCTK3 wild type and S12D failed to phosphorylate endogenous Rb, although Rb phosphorylation was detected in cyclin A2-CDK2-transfected cells (Fig. 7E). This suggested that Rb is a poor substrate for PCTK3 in vivo, possibly due to its different subcellular localization.
Suppression of PCTK3 Induces Actin Cytoskeletal Changes through Cofilin Phosphorylation
In a final set of experiments, we explored possible physiological functions of PCTK3 using a RNA interference approach. We noted that PCTK3 knockdown in HEK293T cells induced morphological changes, cell spreading, and extensions (Fig. 8, A and B). To investigate whether this is responsible for alteration of the actin cytoskeleton, we visualized F-actin by staining cells with phalloidin. Control HEK293T cells exhibited a relatively weak cortical distribution of F-actin (Fig. 8B, upper panel). In PCTK3 knockdown cells, lamellipodia extensions were induced, and F-actin was concentrated at these lamellipodia. PCTK3 knockdown also stimulated the appearance of stress fibers (Fig. 8B, lower panel). Furthermore, the effects of PCTK3 overexpression on actin filament dynamics were examined. Constitutively active mutant of PCTK3 (PCTK3 S12D) suppressed formation of actin stress fibers, whereas wild-type PCTK3 and S12A mutant did not (Fig. 8C).
FIGURE 8.

Suppression of PCTK3 induces actin cytoskeletal changes through cofilin phosphorylation. A and B, HEK293T cells were transfected with negative control siRNA or PCTK3 siRNA. After 48 h, cells were fixed and incubated with Alexa 555-conjugated phalloidin (B, red). Hoechst nuclear staining is represented in blue. C, HEK293T cells were transfected with PCTK3 siRNA for 48 h and then transfected with FLAG-tagged mouse PCTK3 wild type (WT) or S12D or S12A mutants. After 24 h, cells were fixed and incubated with mouse anti-FLAG antibody. The primary antibody was visualized with Alexa Fluor 488-conjugated anti-mouse IgG followed by confocal microscopy. Fluorescence for FLAG-PCTK3 (green) and F-actin (Alexa 555-conjugated phalloidin staining, red) are shown with merged images (merge is in yellow). Hoechst nuclear staining is represented in blue. D, cell lysates of PCTK3 knock-down HEK293T cells were subjected to immunoblotting with anti-PCTK3, anti-cofilin, and anti-phospho-cofilin (Ser3) antibodies. Expression of β-actin was used as a loading control. E, HEK293T cells were transfected with FLAG-PCTK3 (WT or S12D or S12A mutant). After 24 h, cell lysates were subjected to immunoblotting using anti-phospho-cofilin (Ser3), anti-cofilin, and anti-FLAG antibodies.
Actin filament dynamics are regulated by actin-depolymerizing factor/cofilin proteins, and cofilin is inactivated through the phosphorylation at Ser3 (33, 34). Therefore, we investigated the phosphorylation states of cofilin in PCTK3 knockdown HEK293T cells by immunoblotting using anti-phospho-cofilin (Ser3). As shown in Fig. 8D, cofilin phosphorylation in PCTK3 knockdown HEK293T cells was markedly increased as compared with that in control cells. In contrast, overexpression of PCTK3 S12D effectively suppressed cofilin phosphorylation without affecting total cofilin protein levels, although wild-type PCTK3 did not affect (Fig. 8E). The phospho-null S12A mutant rather slightly increased. These data suggested that PCTK3 was involved in the actin cytoskeleton organization.
DISCUSSION
In this study we provided evidence for the activation mechanisms of PCTK3, the activity of which was regulated by two pathways: binding to cyclin A2 and phosphorylation by PKA. Cyclin A2 is widely expressed in various tissues and has been proposed to regulate the transition from the S phase to the M phase during the mitotic cell cycle. The canonical partners for cyclin A2 are CDK1 and CDK2, and cyclin A2-CDK2 complex has been generally used as a structural model of cyclin-CDK. Crystal structure analysis revealed that cyclin A2 enhanced the catalytic activity of CDK2 through rearrangement of its catalytic core element, including the PSTAIRE helix (35). The kinase domain of PCTK3 is highly conserved among isoforms (>80% identity) and has high sequence homology with that of CDK2 (52% identity in humans) (5). Although the crystal structure of PCTK3 remains to be determined, that of PCTK1 is available from the Protein Data Bank (PDB code 3MTL). By comparison with the crystal structure of the cyclin A2-CDK2 complex, two amino acid residues of cyclin Y, Lys225 and Glu253, have been identified as important residues for binding to PCTK1 (24). This Lys-Glu pair is also found in other cyclins and is likely to be a conserved structural motif in cyclin-CDK complexes. This suggests that the Lys-Glu pair of cyclin A2 is also likely to mediate the interaction with PCTK3. In addition, we demonstrated that all three PCTK isoforms bound to cyclin A2 at nearly equal affinity. These data suggested that PCTK3 may be associated with cyclin A2 through a common region of PCTK isoforms, such as the PCTAIRE sequence. On the other hand, the catalytic activities of PCTK1 and PCTK2 were not affected by binding to cyclin A2. Furthermore, we found that cyclin E1 did not affect the catalytic activity of PCTK3, although it had a greater affinity for PCTK3 than cyclin A2. Several studies have showed that substrate recognition by CDKs is modulated by their associations with different cyclins (36–39). For example, although both cyclin A-CDK2 and cyclin E/CDK2 phosphorylate histone H1, only the cyclin A-CDK2 complex phosphorylates lamin B in vitro (39). Furthermore, although both cyclin A and cyclin E associate with CDK1, only cyclin A activates CDK1 kinase activity, indicating that cyclin A and E could modulate the substrate specificity of CDKs. These facts raise the possibility that cyclin E1-PCTK3, cyclin A2-PCTK1, and cyclin A2-PCTK2 complexes may have kinase activity against other substrates but not Rb. On the other hand, cyclin E inhibits the activity of CDK5 by competing with p35 for CDK5 binding (40). This suggests that cyclin E1 may inactivate PCTK3 by dissociating cyclin A2 from PCTK3. To obtain more detailed information regarding the regulatory mechanism of PCTK3 by cyclin A2 and cyclin E1, it will be necessary to perform crystal structure analysis. In an in vitro kinase assay, we also found that cyclin A2-PCTK3 efficiently phosphorylates Rb among Rb, MyBP, and histone H1. However, PCTK3, but not CDK2, failed to phosphorylate endogenous Rb. Previous studies indicated that Rb regulates cell proliferation by controlling the activity of various transcription factors in the nucleus (41, 42), suggesting that PCTK3 may not be able to make contact with Rb because of the differences in their subcellular localizations. Taken together, PCTK3 may be involved in other cellular functions independent of the cell cycle via the phosphorylation of its true substrates in the cytoplasm.
Cyclin A2 lacks a nuclear localization signal and is dependent on binding partners for its translocation between the nucleus and the cytoplasm (30, 43). However, the precise translocation mechanism for cyclin A2 remains obscure because CDK2 also has no nuclear localization signal motif. Recently, SCAPER was identified and characterized as a substrate for cyclin A2-CDK2, which is exclusively localized in the cytoplasm (14). Although the function of SCAPER remains unknown, it was suggested that SCAPER may act as a cytoplasmic pool for cyclin A2 and regulate cell cycle progression. On the other hand, we found that PCTK3 bound to cyclin A2 in the cytoplasm and promoted the translocation of cyclin A2 from the nucleus to the cytoplasm. These results demonstrated that PCTK3 is a cytoplasmic binding partner of cyclin A2 and that cyclin A2 alters its subcellular localization by binding to different partners.
The expression level of cyclin A2 is tightly controlled by transcription and ubiquitin-mediated degradation. Cyclin A2 degradation begins in early prometaphase and is completed by metaphase. The major ubiquitin ligase required for mitosis is APC/C (anaphase-promoting complex/cyclosome) (31). Because the level of cyclin A2 protein was greatly reduced in PCTK3 knockdown cells as compared with that in control cells, we hypothesized that PCTK3 protects cyclin A2 from proteolytic degradation. As expected, cycloheximide chase analysis showed that cyclin A2 degradation is suppressed in the presence of PCTK3, whereas free cyclin A2 is readily degraded. A previous study showed that cyclin E bound to CDK2 is also protected from degradation by the proteasome (32). These observations strongly support the physiological significance of the association of between PCTK3 and cyclin A2.
Furthermore, PCTK3 was also activated by PKA via phosphorylation at Ser12, independently of its interaction with cyclin A2. Endogenous PCTK3 in HEK293T cells was phosphorylated and activated by the PKA activator forskolin. In addition, site-directed mutagenesis analysis revealed that PKA phosphorylates Ser12, Ser66, and Ser109 of PCTK3. These results indicate that PCTK3 is a bona fide substrate of PKA. Furthermore, the Rb phosphorylation activity of a phosphomimic mutant of Ser12 (S12D) was comparable with the activity of CDK2 in the presence of cyclin A2, suggesting that the complete activation of PCTK3 is required for not only binding to cyclin A2 but also for its phosphorylation at Ser12. Although the S12D mutant caused an increase in PCTK3 activity, phosphomimic mutations of Ser66 and Ser109 (homologous to Ser119 and Ser153 of PCTK1, respectively) had no effects on the Rb phosphorylation activity of PCTK3. Recently, Mikolcevic et al. (10) reported that PKA plays an inhibitory role in the interaction between PCTK1 and cyclin Y by phosphorylation at Ser153 of PCTK1. On the other hand, phosphorylation at Ser153 of PCTK1 creates the binding motif of 14-3-3 proteins and transfers PCTK1 from the cell membrane to the cytoplasm. The 14-3-3 protein family includes seven members (β, γ, ϵ, σ, ζ, τ, and η) in mammals, which are highly conserved and ubiquitously expressed proteins (44). These 14-3-3 proteins can interact with various proteins, usually by recognizing phosphoserine or phosphothreonine motifs, and alter the activity, subcellular localization, and interacting partners of their target proteins, which in turn modulate diverse biological processes. A previous report showed that PCTK3 interacts with 14-3-3ζ (45). Consistent with this, we found that PCTK3 binds to several 14-3-3 isoforms.3 Therefore, PKA phosphorylation at Ser66 and Ser109 of PCTK3 may mediate the interactions with 14-3-3 proteins and alter the subcellular localization of PCTK3.
Furthermore, PKA is involved in regulating of cell morphology through phosphorylation of cytoskeletal proteins including microtubules, intermediate filaments, and actin (46, 47). PKA also regulates actin filament turnover by modulating the activity of LIM domain kinase 1 (LIMK1) (48). A recent study reported that cyclin A2 plays a role in cytoskeletal reorganization via its association with RhoA, a member of the small GTPase family. Suppression of cyclin A2 by shRNA resulted in an increase in cell size and cortical localization of F-actin independent of the cell cycle, which led to increased cell migration and invasion (15). Similar to the cyclin A2-deficient phenotype, PCTK3 knockdown HEK293T cells also exhibited an increase in cell size and F-actin accumulation at the edges of cell membranes and increased cofilin phosphorylation. These data suggest that PCTK3 activated through binding to cyclin A2, and phosphorylation by PKA is involved in regulating cell motility via reorganization of actin filaments. Although CDKs were originally identified as enzymes that controlled cell cycle events, some members of the CDK family are involved in other cellular processes (49). For example, CDK5 is not considered to play a significant role in cell cycle regulation; however, it has an important function in the control of neurogenesis, including neurite outgrowth, axon guidance, and cell migration. CDK5 activated by neuron-specific regulator p35 inhibits p21-activated kinase 1 (PAK1) activity through phosphorylation at Thr212 (PVTPTRD, where T is Thr212) (50). PAK1, an effector of Rac, has a pivotal role in cell morphology and motility via the LIM domain kinase/cofilin pathway (51). Phosphorylation of PAK1 by p35/CDK5 is likely to be implicated in the dynamics of the actin cytoskeleton reorganization. PCTK3 may also phosphorylate substrate proteins, such as PAK1, and control actin dynamics.
In summary, we demonstrated that cyclin A2 is a specific activator of PCTK3 and that phosphorylation at Ser12 of PCTK3 significantly enhances its kinase activity in the absence of cyclin A2. These findings shed light on the activation mechanism of not only PCTK3 but also other uncharacterized members of the CDK family, such as PCTK2. Further investigations will be needed to clarify the physiological functions and pathological roles of PCTK3.
Acknowledgments
We thank Shiori Sato for continuous encouragement and Taito Matsuda and Towa Sasakura for technical assistance. We are also grateful to Taishi Hirase for critical reading of the manuscript.
This work was supported in part by a grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to K. Y.).
S. Matsuda, K. Kominato, and K. Yuasa, unpublished data.
- CDK
- cyclin-dependent kinase
- PCTK
- PCTAIRE kinase
- PFTK
- PFTAIRE kinase
- PKA
- protein kinase A
- Rb
- retinoblastoma protein
- MBP
- maltose-binding protein
- MyBP
- myelin basic protein
- PAK1
- p21-activated kinase 1
- SCAPER
- S phase cyclin A-associated protein residing in the endoplasmic reticulum.
REFERENCES
- 1. Malumbres M., Barbacid M. (2005) Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 30, 630–641 [DOI] [PubMed] [Google Scholar]
- 2. Kaldis P. (1999) The cdk-activating kinase (CAK): from yeast to mammals. Cell. Mol. Life Sci. 55, 284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nakayama K. I., Nakayama K. (2006) Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer 6, 369–381 [DOI] [PubMed] [Google Scholar]
- 4. Morgan D. O. (1997) Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 13, 261–291 [DOI] [PubMed] [Google Scholar]
- 5. Cole A. R. (2009) PCTK proteins: the forgotten brain kinases? Neurosignals 17, 288–297 [DOI] [PubMed] [Google Scholar]
- 6. Graeser R., Gannon J., Poon R. Y., Dubois T., Aitken A., Hunt T. (2002) Regulation of the CDK-related protein kinase PCTAIRE-1 and its possible role in neurite outgrowth in Neuro-2A cells. J. Cell Sci. 115, 3479–3490 [DOI] [PubMed] [Google Scholar]
- 7. Palmer K. J., Konkel J. E., Stephens D. J. (2005) PCTAIRE protein kinases interact directly with the COPII complex and modulate secretory cargo transport. J. Cell Sci. 118, 3839–3847 [DOI] [PubMed] [Google Scholar]
- 8. Liu Y., Cheng K., Gong K., Fu A. K., Ip N. Y. (2006) Pctaire1 phosphorylates N-ethylmaleimide-sensitive fusion protein: implications in the regulation of its hexamerization and exocytosis. J. Biol. Chem. 281, 9852–9858 [DOI] [PubMed] [Google Scholar]
- 9. Ou C. Y., Poon V. Y., Maeder C. I., Watanabe S., Lehrman E. K., Fu A. K., Park M., Fu W. Y., Jorgensen E. M., Ip N. Y., Shen K. (2010) Two cyclin-dependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell 141, 846–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mikolcevic P., Sigl R., Rauch V., Hess M. W., Pfaller K., Barisic M., Pelliniemi L. J., Boesl M., Geley S. (2012) Cyclin-dependent kinase 16/PCTAIRE kinase 1 is activated by cyclin Y and is essential for spermatogenesis. Mol. Cell Biol. 32, 868–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hirose T., Kawabuchi M., Tamaru T., Okumura N., Nagai K., Okada M. (2000) Identification of tudor repeat associator with PCTAIRE 2 (Trap). A novel protein that interacts with the N-terminal domain of PCTAIRE 2 in rat brain. Eur. J. Biochem. 267, 2113–2121 [DOI] [PubMed] [Google Scholar]
- 12. Yamochi T., Nishimoto I., Okuda T., Matsuoka M. (2001) ik3–1/Cables is associated with Trap and Pctaire2. Biochem. Biophys. Res. Commun. 286, 1045–1050 [DOI] [PubMed] [Google Scholar]
- 13. Liu D., Matzuk M. M., Sung W. K., Guo Q., Wang P., Wolgemuth D. J. (1998) Cyclin A1 is required for meiosis in the male mouse. Nat. Genet. 20, 377–380 [DOI] [PubMed] [Google Scholar]
- 14. Tsang W. Y., Wang L., Chen Z., Sánchez I., Dynlacht B. D. (2007) SCAPER, a novel cyclin A: interacting protein that regulates cell cycle progression. J. Cell Biol. 178, 621–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arsic N., Bendris N., Peter M., Begon-Pescia C., Rebouissou C., Gadéa G., Bouquier N., Bibeau F., Lemmers B., Blanchard J. M. (2012) A novel function for cyclin A2: control of cell invasion via RhoA signaling. J. Cell Biol. 196, 147–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pearce L. R., Komander D., Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22 [DOI] [PubMed] [Google Scholar]
- 17. Eyers P. A., Liu J., Hayashi N. R., Lewellyn A. L., Gautier J., Maller J. L. (2005) Regulation of the G2/M transition in Xenopus oocytes by the cAMP-dependent protein kinase. J. Biol. Chem. 280, 24339–24346 [DOI] [PubMed] [Google Scholar]
- 18. Shibuya E. K. (2003) G2 cell cycle arrest: a direct link between PKA and Cdc25C. Cell Cycle 2, 39–41 [DOI] [PubMed] [Google Scholar]
- 19. Han S. J., Conti M. (2006) New pathways from PKA to the Cdc2/cyclin B complex in oocytes: Wee1B as a potential PKA substrate. Cell Cycle 5, 227–231 [DOI] [PubMed] [Google Scholar]
- 20. Parker L. L., Piwnica-Worms H. (1992) Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 257, 1955–1957 [DOI] [PubMed] [Google Scholar]
- 21. Nilsson I., Hoffmann I. (2000) Cell cycle regulation by the Cdc25 phosphatase family. Prog. Cell Cycle Res. 4, 107–114 [DOI] [PubMed] [Google Scholar]
- 22. Yuasa K., Michibata H., Omori K., Yanaka N. (1999) A novel interaction of cGMP-dependent protein kinase I with troponin T. J. Biol. Chem. 274, 37429–37434 [DOI] [PubMed] [Google Scholar]
- 23. Yuasa K., Matsuda T., Tsuji A. (2011) Functional regulation of transient receptor potential canonical 7 by cGMP-dependent protein kinase Iα. Cell Signal. 23, 1179–1187 [DOI] [PubMed] [Google Scholar]
- 24. Shehata S. N., Hunter R. W., Ohta E., Peggie M. W., Lou H. J., Sicheri F., Zeqiraj E., Turk B. E., Sakamoto K. (2012) Analysis of substrate specificity and cyclin Y binding of PCTAIRE-1 kinase. Cell. Signal. 24, 2085–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rual J. F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G. F., Gibbons F. D., Dreze M., Ayivi-Guedehoussou N., Klitgord N., Simon C., Boxem M., Milstein S., Rosenberg J., Goldberg D. S., Zhang L. V., Wong S. L., Franklin G., Li S., Albala J. S., Lim J., Fraughton C., Llamosas E., Cevik S., Bex C., Lamesch P., Sikorski R. S., Vandenhaute J., Zoghbi H. Y., Smolyar A., Bosak S., Sequerra R., Doucette-Stamm L., Cusick M. E., Hill D. E., Roth F. P., Vidal M. (2005) Towards a proteome-scale map of the human protein-protein interaction network. Nature 437, 1173–1178 [DOI] [PubMed] [Google Scholar]
- 26. Shu F., Lv S., Qin Y., Ma X., Wang X., Peng X., Luo Y., Xu B. E., Sun X., Wu J. (2007) Functional characterization of human PFTK1 as a cyclin-dependent kinase. Proc. Natl. Acad. Sci. U.S.A. 104, 9248–9253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang M., Gao Y., Yang T., Zhu X., Chen J. (2009) Cyclin Y, a novel membrane-associated cyclin, interacts with PFTK1. FEBS Lett. 583, 2171–2178 [DOI] [PubMed] [Google Scholar]
- 28. Adams P. D., Li X., Sellers W. R., Baker K. B., Leng X., Harper J. W., Taya Y., Kaelin W. G., Jr. (1999) Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complexes. Mol. Cell Biol. 19, 1068–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meyerson M., Enders G. H., Wu C. L., Su L. K., Gorka C., Nelson C., Harlow E., Tsai L. H. (1992) A family of human cdc2-related protein kinases. EMBO J. 11, 2909–2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jackman M., Kubota Y., den Elzen N., Hagting A., Pines J. (2002) Cyclin A- and cyclin E-Cdk complexes shuttle between the nucleus and the cytoplasm. Mol. Biol. Cell 13, 1030–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. den Elzen N., Pines J. (2001) Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J. Cell Biol. 153, 121–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clurman B. E., Sheaff R. J., Thress K., Groudine M., Roberts J. M. (1996) Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 10, 1979–1990 [DOI] [PubMed] [Google Scholar]
- 33. Moon A., Drubin D. G. (1995) The ADF/cofilin proteins: stimulus-responsive modulators of actin dynamics. Mol. Biol. Cell 6, 1423–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moriyama K., Iida K., Yahara I. (1996) Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells 1, 73–86 [DOI] [PubMed] [Google Scholar]
- 35. Jeffrey P. D., Russo A. A., Polyak K., Gibbs E., Hurwitz J., Massagué J., Pavletich N. P. (1995) Mechanism of CDK activation revealed by the structure of a cyclin A-CDK2 complex. Nature 376, 313–320 [DOI] [PubMed] [Google Scholar]
- 36. Brown N. R., Noble M. E., Endicott J. A., Johnson L. N. (1999) The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol. 1, 438–443 [DOI] [PubMed] [Google Scholar]
- 37. Peeper D. S., Parker L. L., Ewen M. E., Toebes M., Hall F. L., Xu M., Zantema A., van der Eb A. J., Piwnica-Worms H. (1993) A- and B-type cyclins differentially modulate substrate specificity of cyclin-cdk complexes. EMBO J. 12, 1947–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sarcevic B., Lilischkis R., Sutherland R. L. (1997) Differential phosphorylation of T-47D human breast cancer cell substrates by D1-, D3-, E-, and A-type cyclin-CDK complexes. J. Biol. Chem. 272, 33327–33337 [DOI] [PubMed] [Google Scholar]
- 39. Horton L. E., Templeton D. J. (1997) The cyclin box and C terminus of cyclins A and E specify CDK activation and substrate specificity. Oncogene 14, 491–498 [DOI] [PubMed] [Google Scholar]
- 40. Odajima J., Wills Z. P., Ndassa Y. M., Terunuma M., Kretschmannova K., Deeb T. Z., Geng Y., Gawrzak S., Quadros I. M., Newman J., Das M., Jecrois M. E., Yu Q., Li N., Bienvenu F., Moss S. J., Greenberg M. E., Marto J. A., Sicinski P. (2011) Cyclin E constrains Cdk5 activity to regulate synaptic plasticity and memory formation. Dev. Cell 21, 655–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Classon M., Harlow E. (2002) The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2, 910–917 [DOI] [PubMed] [Google Scholar]
- 42. Zacksenhaus E., Jiang Z., Hei Y. J., Phillips R. A., Gallie B. L. (1999) Nuclear localization conferred by the pocket domain of the retinoblastoma gene product. Biochim. Biophys. Acta 1451, 288–296 [DOI] [PubMed] [Google Scholar]
- 43. Maridor G., Gallant P., Golsteyn R., Nigg E. A. (1993) Nuclear localization of vertebrate cyclin A correlates with its ability to form complexes with cdk catalytic subunits. J. Cell Sci. 106, 535–544 [DOI] [PubMed] [Google Scholar]
- 44. Fu H., Subramanian R. R., Masters S. C. (2000) 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40, 617–647 [DOI] [PubMed] [Google Scholar]
- 45. Meek S. E., Lane W. S., Piwnica-Worms H. (2004) Comprehensive proteomic analysis of interphase and mitotic 14-3-3-binding proteins. J. Biol. Chem. 279, 32046–32054 [DOI] [PubMed] [Google Scholar]
- 46. Shabb J. B. (2001) Physiological substrates of cAMP-dependent protein kinase. Chem. Rev. 101, 2381–2411 [DOI] [PubMed] [Google Scholar]
- 47. Liu F., Verin A. D., Borbiev T., Garcia J. G. (2001) Role of cAMP-dependent protein kinase A activity in endothelial cell cytoskeleton rearrangement. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L1309–L1317 [DOI] [PubMed] [Google Scholar]
- 48. Nadella K. S., Saji M., Jacob N. K., Pavel E., Ringel M. D., Kirschner L. S. (2009) Regulation of actin function by protein kinase A-mediated phosphorylation of Limk1. EMBO Rep. 10, 599–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dhavan R., Tsai L. H. (2001) A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2, 749–759 [DOI] [PubMed] [Google Scholar]
- 50. Rashid T., Banerjee M., Nikolic M. (2001) Phosphorylation of Pak1 by the p35/Cdk5 kinase affects neuronal morphology. J. Biol. Chem. 276, 49043–49052 [DOI] [PubMed] [Google Scholar]
- 51. Hanna S., El-Sibai M. (2013) Signaling networks of Rho GTPases in cell motility. Cell. Signal. 25, 1955–1961 [DOI] [PubMed] [Google Scholar]