

Background: The contribution of E2F4 to hypoxic/ischemic neuronal death is understood poorly.
Results: Loss of E2F4 leads to an increase in B-Myb and contributes to hypoxic/ischemic neuronal death.
Conclusion: E2F4 is important for survival following hypoxic/ischemic neuronal death.
Significance: Targeting E2F4-repressive functions may be important in maintaining neuronal survival under hypoxic/ischemic conditions.
Keywords: Apoptosis, Cardiovascular Disease, Cell Biology, Cell Cycle, Cyclin-dependent Kinase (Cdk), E2F Transcription Factor, Hypoxia, Ischemia, Stroke, Cell Death
Abstract
Inappropriate activation of cell cycle proteins, in particular cyclin D/Cdk4, is implicated in neuronal death induced by various pathologic stresses, including DNA damage and ischemia. Key targets of Cdk4 in proliferating cells include members of the E2F transcription factors, which mediate the expression of cell cycle proteins as well as death-inducing genes. However, the presence of multiple E2F family members complicates our understanding of their role in death. We focused on whether E2F4, an E2F member believed to exhibit crucial control over the maintenance of a differentiated state of neurons, may be critical in ischemic neuronal death. We observed that, in contrast to E2F1 and E2F3, which sensitize to death, E2F4 plays a crucial protective role in neuronal death evoked by DNA damage, hypoxia, and global ischemic insult both in vitro and in vivo. E2F4 occupies promoter regions of proapoptotic factors, such as B-Myb, under basal conditions. Following stress exposure, E2F4-p130 complexes are lost rapidly along with the presence of E2F4 at E2F-containing B-Myb promoter sites. In contrast, the presence of E2F1 at B-Myb sites increases with stress. Furthermore, B-Myb and C-Myb expression increases with ischemic insult. Taken together, we propose a model by which E2F4 plays a protective role in neurons from ischemic insult by forming repressive complexes that prevent prodeath factors such as Myb from being expressed.
Introduction
Inappropriate activation of the cell cycle machinery is implicated in a number of neuronal death models induced by NGF deprivation (1), excitotoxicity (2), oxidative stress (3), DNA damage (4), and stroke (5–8). For example, an increase in cyclin D1 levels and its associated kinase activity is observed following DNA damage (an upstream mediator of stroke damage) and ischemic insult (5, 7, 9–11). In addition, treatment with pharmacologic cyclin-dependent kinase (Cdk)3 inhibitors such as flavopiridol protects neurons from both DNA damage- and ischemia-induced cell death (4, 6, 7). Expression of a dominant negative form of Cdk4, an important regulator of the G1/S phase of the cell cycle, is protective following DNA damage and global cerebral ischemia (5, 9). Together, these studies reveal a crucial role for Cdk4 in neuronal death induced by DNA damage and ischemic insult. However, the downstream effectors of cell cycle reactivation in neurons under ischemic stress remain unclear. In this regard, members of the E2F transcription factors may play a pivotal role.
The E2Fs consist of eight related family members, generally classified as activators (E2F1, E2F2, and E2F3a) or repressors (E2F3b and E2F4-E2F8) on the basis of their ability to promote or repress gene transcription (12). The activity of E2Fs (E2F1-E2F5) is regulated by their association with the pocket proteins, which include retinoblastoma protein (pRb), p107, and p130 (12, 13). E2F association with pocket proteins can promote or repress the expression of its targets, which include genes involved in cell cycle progression, DNA damage, and apoptosis (12, 13). For example, in dividing cells, pRb association with E2F prevents the expression of genes required for DNA synthesis and cell cycle progression. However, phosphorylation of pRb by Cdk4/6 disrupts its association with E2F and results in the transactivation of its target genes. In addition to transactivation, E2F complexes can also repress gene function. For example, E2F4-p130 can recruit chromatin modification factors, such as histone deacetylases (HDACs), to promoters of target genes to form an active repression complex (13–16). In this scheme, phosphorylation of p130 by Cdks disrupts its association with E2F4, resulting in the derepression of target genes. Thus, E2Fs can function as effectors of Cdk signaling via the transactivation or derepression of target genes.
The involvement of E2F and, in particular, E2F1 in the cell cycle-induced death of neurons under pathologic stress is suggested by a number of key observations. First, phosphorylation of pRb, a preferred partner for activating E2Fs such as E2F1, is observed in neuronal death models, including DNA damage and ischemia (6, 7, 17). Second, the expression of kinase-dead Cdk4 and treatment with flavopiridol attenuates pRb phosphor ylation following DNA damage and ischemia (5, 7, 17). Importantly, the expression of mutant pRB or dominant negative DP-1, a binding partner of E2Fs, prevents neuronal death following DNA damage and/or hypoxia (5, 17). Finally, E2F1 deficiency is protective following ischemic insult both in vitro and in vivo (8, 18, 19). These observations suggest that cell cycle reactivation in neurons under pathologic stress signals death through pRb inactivation and activation or derepression of E2F1 target genes.
In addition to E2F1 and pRb, other E2Fs may also be important in neuronal death. E2F4-p130 complexes are present basally in postmitotic neurons, suggesting that they may act to repress gene targets important in neuronal survival/function (16, 20). Interestingly, in the absence of insult, siRNA-mediated down-regulation of p130 or E2F4 induced apoptosis of cortical neurons (21). In addition, the expression of wild-type or phosphorylation-resistant p130 is protective following NGF deprivation of PC12 neurons (21). Whether p130-E2F4 complexes play a role in neuronal death in vivo is unclear. In this study we examined the potential involvement of E2F4 in neuronal death induced by ischemic stress and DNA damage. The results of our study demonstrate an important role for E2F4 in the survival of neurons, which is in contrast to the role of activating E2Fs such as E2F1/3.
EXPERIMENTAL PROCEDURES
Viral Construction
Plasmids containing a human E2F4 cDNA sequence were subcloned into the SpeI sites of the AM/CBA-pI-WPRE-bGH vector and used to generate a recombinant adeno-associated (AAV) virus, as described previously (22). Plasmids containing Bmyb-luciferase with a wild-type E2F site and a mutant construct harboring a mutation that abolishes E2F binding have been reported previously (23). The plasmids were subcloned into the adeno-associated vector for efficient delivery into primary neurons. The adenovirus expressing E2F1 has already been described (24).
Transgenic Mice
E2F1- (25, 26), E2F3- (25, 27), and E2F4-deficient (28) mice have been described previously. Knockout mice were generated from heterozygous breeding pairs and genotyped by PCR using primers that have been published previously (26, 28). E2F1 null mice were genotyped using the following primers: 5′-GGATATGATTCTTGGACTTCTTGG-3′, 5′-CTAAATCTGACCCCAAACGC-3′, and 5′-CAAGTGCCAGCGGGGCTGCTAAAG-3′.
Cell Cultures and Treatments
Primary cerebellar granule neurons (CGNs) and cortical neuronal cultures were established as described previously (24, 29) from CD1 (Charles River Laboratories, Quebec, Canada) or E2F transgenic mice. CGNs were transfected with E2F4 siRNA, a C-Myb siRNA mixture, or control siRNA (Santa Cruz Biotechnology) using Lipofectamine 2000 (30) 5 days after plating. Alternatively, CGNs were infected with an adenovirus expressing E2F1, E2F4, or GFP at the time of plating at a multiplicity of infection of 50. Cortical neuronal cultures were cotransfected with E2F1, E2F3, or E2F4 along with GFP-containing plasmids using the calcium phosphate method 3 days after plating, as described previously (30, 31). Cortical neurons were treated with 10 μm camptothecin (Sigma) 3–4 days after plating and examined for survival at the indicated times, as described previously (30). Cortical cultures from transgenic mice and CGNs were lysed at the designated times after insult using a mildly disruptive lysis buffer and evaluated as described previously (5). Neurons cotransfected with E2F and GFP plasmids were fixed and stained with Hoechst 33342 (Sigma). Viability was assessed by nuclear integrity in GFP-positive cells in random fields, as described previously (29).
Hypoxia
CGN cultures were subjected to hypoxia at 1% O2 with 5% CO2-balanced N2 in a humidified hypoxia chamber (Coy Laboratory Products, Ann Arbor, MI) after 1 week in culture, as described previously (5). Hypoxia was induced in the presence of 10 μm MK801 (Research Biochemicals, Natick, MA), an NMDA channel blocker, for 18 h, followed by reoxygenation at normoxia for 24 h for survival studies. Alternatively, CGN cultures were subjected to varying durations of hypoxia and reoxygenation for biochemical studies. Control cultures were maintained in a humidified incubator at 37 °C and were not treated with hypoxia.
Viral Injection
Animal experiments were carried out in accordance with Canadian Council for the Use and Care of Animals in Research guidelines with approval from the University of Ottawa Animal Care Committee. Intrahippocampal, recombinant, adeno-associated viral injections in rats have been described previously (5). Briefly, male Wistar rats (80–125 g) were injected unilaterally with the recombinant adeno-associated virus carrying an enhanced green fluorescent protein (EGFP) control or E2F4 (1010 genomes/μl) 2 weeks before a four-vessel occlusion insult. 2 μl of recombinant adeno-associated virus diluted in PBS with 1 μl of mannitol (20%) was injected stereotaxically using a Harvard infusion pump (Harvard Apparatus) into the hippocampus (from the bregma, −3.6 mm anteroposterior, +2.1 mm lateral, and −2.75 mm deep).
Global Cerebral Ischemia
The four-vessel occlusion (4VO) method of global ischemia was induced as described previously for 10 min (6, 32). Four days following ischemia, rats were perfused and sacrificed, and the brains were extracted, sectioned, and stained for histological assessment. The neuronal viability of cells in the hippocampal CA1 was assessed as described previously (6, 32).
Immunohistochemistry
Antigen retrieval and deparaffinization was carried out on brain sections as described previously (32). Following permeabilization with 0.3% Triton X-100 for 10 min, rat brain sections were blocked in 10% normal goat serum (Jackson ImmunoResearch Laboratories) diluted in 2% bovine serum albumin (Fisher Scientific) in 0.01 m PBS for 1 h at room temperature. Sections were incubated with mouse monoclonal anti-GFP (Abcam) or anti-E2F4 antibodies (Abcam, 1:200) overnight at 4 °C. For visualization, sections were incubated with Alexa Fluor 488 goat anti-mouse (1:200) secondary antibody (Jackson ImmunoResearch Laboratories) for 1 h at room temperature. Neuronal nuclei were stained with Hoechst 33342 (Sigma).
Immunoprecipitation
CGN cultures were harvested and homogenized in immunoprecipitation buffer (50 mm HEPES (pH 7.5), 150 mm NaCl, 1 mm EDTA, 2.5 mm EGTA, 1 mm DTT, and 0.1% Tween 20) supplemented with protease inhibitor mixture (Roche). 500 μg of total protein lysate was incubated with 4 μg of anti-E2F4 (Santa Cruz Biotechnology) or anti-p130 antibody (Santa Cruz Biotechnology) overnight at 4 °C. As a control, samples were incubated with normal rabbit IgG (Santa Cruz Biotechnology). The antigen-antibody complex was captured with anti-rabbit Ig immunoprecipitation beads (eBiosciences) at 4 °C for 4 h. The beads were recovered, and the immunoprecipitation product was resolved on a 10% SDS-polyacrylamide gel.
Western Blotting
Cell and hippocampal tissue samples were collected at the designated times after insult and homogenized in solubilization buffer (0.0625 m Tris, 2.5 mm EDTA, 2.5 mm EGTA, 10% glycerol, 2% SDS, 0.001% bromphenol blue, and 5% β-mercaptoethanol) (24). Alternatively, E2F4 null CGNs or siRNA-transfected cultures were harvested and homogenized in solubilization buffer. Samples were run on SDS-polyacrylamide gels and transferred onto a PVDF membrane (Millipore). Membranes were probed with the following antibodies: anti-E2F4 (Abcam), anti-p130 (BD Transduction Laboratories), anti-phospho-p130 (Santa Cruz Biotechnology), anti-E2F1 (Santa Cruz Biotechnology), anti-E2F3 (Santa Cruz Biotechnology), and anti β-actin (Sigma). Densitometry was performed on Western blots using ImageJ software and normalized to the loading control. The results were expressed as the fold change over sham-operated animals in the 4VO time course and over no hypoxia control for hypoxia/reoxygenation experiments.
Semiquantitative Reverse Transcription PCR
At the indicated times following 4VO, total RNA was extracted from hippocampal tissues using QIAcube (Qiagen) following the protocol of the manufacturer. Alternatively, total RNA was extracted from siRNA-treated CGN cultures. 100 ng of total RNA was used for cDNA synthesis and targeted gene amplification using a SuperScript One-Step RT-PCR kit (Invitrogen). cDNA synthesis and amplification were conducted using the following conditions: 42 °C for 45 min, 94 °C for 2 min, followed by cycles of 94 °C for 1 min, Tm for 30 s, and 72 °C for 1 min. The rat MYB genes were amplified with the following primers: 5′-GGCTG CCGTGGCTACTACTTCTAA-3′ and 5′-CGCGC CGTTTCTTCTGTCG-3′ for B-Myb at a Tm of 59 °C for 35 cycles and 5′-ATGCCCTGGAAGTGAAC AAC-3′ and 5′-CAGCTTTTGTAAGCGGGTTC-3′ for C-Myb at a Tm of 54 °C for 35 cycles. Expression of GAPDH mRNA was used as a standard for loading control. GAPDH was amplified using the following primers: 5′-ATCCGTTGTGGATCTGACATGC-3′ and 5′-TGTCATTGAGAGCAATGCCAGC-3′ at a Tm of 52 °C for 28 cycles. The mouse C-Myb genes were amplified using the following PCR primers: 5′-GAGAGGTGGCACAACCATTT-3′ and 5′-GGGAACGTGACTGGAGATGT-3′ at a Tm of 54 °C for 31 cycles. The expression of ribosomal S 12 mRNA was used as a standard for loading control. S 12 cDNA was amplified using the following primers: 5′-GGAAGGCA TAGCTGCTGG-3′ and 5′-CCTCGATGAC ATCCTTGG-3′ at a Tm of 57 °C for 25 cycles. Densitometry was performed on results and normalized to loading control. The results were expressed as fold change over control.
ChIP Assay
CGNs treated with/without hypoxia and reoxygenation were subjected to a ChIP assay as described previously (33). The mouse B-Myb promoter region was amplified using the following primers: 5′-CCTCCTCCTTCTCCTCCTTC-3′ and 5-CACTATACCCGTGCGCTTCT-3′. PCR products were resolved on an agarose gel. Densitometry was performed on results and expressed as fold change over no hypoxia control.
Luciferase Assay
One day after plating, CGNs were infected with wild-type or mutant luciferase viruses along with AAV-β-galactosidase as an internal control. After hypoxia/reoxygenation, cells were lysed in buffer provided in the Promega luciferase system (Promega). The luciferase assay was performed according to the instructions of the manufacturer. Relative luciferase activities were obtained by normalizing the luciferase activity against β-galactosidase activity. Results were presented as fold increase in reference to control values.
RESULTS
E2F Family Members Differentially Regulate Neuronal Death Mediated by Genotoxic Stress
Aberrant activation of neuronal cell cycle induced by pathologic stress such as DNA damage and ischemic insult is thought to contribute to cell death through regulation of E2F members (7, 17, 33). However, the relative contribution of different E2F members in neuronal death is not clear. To begin to examine the involvement of different E2F members, we focused initially on the effect of select activating E2Fs (E2F1 and E2F3) and the repressive E2F4 members in neuronal death. Primary cortical neurons null for these E2F members were treated with the DNA-damaging agent camptothecin and evaluated for survival. Neurons null for E2F1 (Fig. 1A) and E2F3 (Fig. 1B) were more resistant to DNA damage-induced death when compared with those from their littermate controls. E2F1−/− neurons were significantly more resistant to DNA-induced death at 8 h (59% survival, p < 0.01) and 12 h (23% survival, p < 0.05) compared with the wild-type control (43 and 13% survival at 8 and 12 h, respectively). Similarly, neuronal survival in the E2F3−/− neurons (59 and 27%) was greater than that observed for the wild type (43 and 20%, p < 0.01 and p < 0.05 at 8 and 12 h, respectively). In contrast, E2F4 deficiency resulted in sensitization to DNA damage-induced death at 12 h (21% survival) compared with the wild-type control (44%, p < 0.01, Fig. 1C).
FIGURE 1.
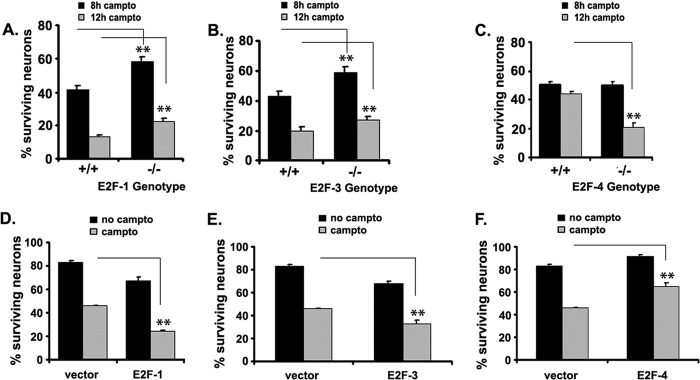
E2F family members play differential roles in neuronal death induced by genotoxic stress. E2F1 (A), E2F3 (B), and E2F4 (C) wild-type and knockout neurons were treated with 10 μm camptothecin (campto, a DNA-damaging agent) and evaluated for survival at 8 and 12 h following treatment. E2F1 (D), E2F3 (E), and E2F4 (F) containing a plasmid or vector control were transfected in cortical neuronal cultures 3 days after plating and treated with 10 μm camptothecin 24 h later. Survival was assessed 24 h following treatment. Results are expressed as percent of control ± S.E. **, p < 0.01.
We next tested the effect of expression of E2F1, E2F3, and E2F4 on neuronal death induced by DNA damage. As shown in Fig. 1, D–F, expression of E2F1 (Fig. 1D) and E2F3 (Fig. 1E) significantly (p < 0.01) reduced neuronal survival compared with neurons transfected with vector only. In contrast, overexpression of E2F4 (Fig. 1F) resulted in an increase in neuronal survival (46% in vector- and 65% in E2F4-expressing neurons, p < 0.01). Together, these results demonstrate a proapoptotic role for E2F1 and E2F3 and a prosurvival role for E2F4 in neuronal death induced by DNA damage.
E2F1 and E2F4 Have Opposing Effects on Neuronal Survival following Hypoxia/Reoxygenation
We next determined the effect of E2Fs in a more physiologically relevant model of ischemic neuronal death in vitro with a focus on E2F1 and E2F4. To this end, primary CGNs infected with an adenovirus expressing E2F1, E2F4, or GFP were subjected to 18 h of hypoxia in the presence of MK801, followed by reoxygenation for 24 h. Neurons treated in this fashion die in a delayed manner dependent upon cell cycle activation (5). Consistent with our observation with DNA damage, the expression of E2F1 (Fig. 2A) significantly reduced neuronal survival compared with the GFP control (24% survival in E2F1- versus 45% in GFP-expressing neurons, p < 0.01). In contrast, expression of E2F4 was significantly protective following hypoxia/reoxygenation compared with the GFP control (65% survival in E2F4- versus 45% in GFP-expressing neurons, p < 0.01) (Fig. 2A).
FIGURE 2.

E2F1 and E2F4 have opposing roles in neuronal death induced by hypoxia. A, primary CGN cultures were infected with adenovirus expressing E2F1, E2F4, or GFP control at the time of plating and treated with hypoxia after a week in culture for 18 h, followed by reoxygenation for 24 h. B, Western blot analysis showing E2F4 knockdown in CGN cultures treated with E2F4 siRNA mixture. Ctrl, control. C, CGNs were transfected with E2F4 or control siRNA 5 days after plating. Cultures were treated with 18 h of hypoxia followed by 24 h of reoxygenation 2 days after transfection. D, CGNs from E2F4 KO and wild-type mice were similarly treated with 18 h of hypoxia and 24 h of reoxygenation. Neuronal survival was evaluated 24 h after hypoxia. Results are expressed as percent of control ± S.E. E, E2F1 protein levels are unaffected by E2F4 deficiency. CGNs from E2F4+/+ and E2F4−/− neurons were subjected to Western blot analysis and probed with anti-E2F1 antibody and anti-β-actin as a control. F, quantitation of E2F1 levels as in E. G, E2F3 protein levels are diminished in E2F4 knockout neurons. A Western blot analysis was conducted on E2F4+/+ and E2F4−/− CGNs. E2F3 was detected using anti-E2F3 antibody. β-actin was used as a loading control. H, quantitation of E2F3 protein levels as in G by densitometry. The protein levels of E2F1 (E) and E2F3 (G) were normalized to β-actin. Error bars represents the mean ± S.E. (n ≥ 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Loss of function studies were consistent with the observations above (Fig. 2, C and D). In this regard, we focused on the role of E2F4 because loss of function studies with E2F1 following ischemic insult have been shown previously to promote survival (8, 18, 19, 34). CGNs were transfected with siRNA to E2F4 or control siRNA and subjected to hypoxia and reoxygenation. E2F4 knockdown in transfected cultures was verified by Western blot analysis (Fig. 2B). Evaluation of other E2Fs, including E2F1 and E2F3, showed that the levels of these proteins were not affected significantly by E2F4 siRNA-mediated knockdown.4 We observed sensitization of neurons to death induced by hypoxia/reoxygenation when E2F4 was transiently knocked down (3% survival) compared with the siRNA control (29% survival, p < 0.001). Interestingly, neurons transfected with E2F4 siRNA were remarkably more vulnerable to neuronal death even in the absence of insult (19% survival in E2F4 knockdown versus 84% in siRNA control cultures, p < 0.001) (Fig. 2C). CGNs from E2F4−/− and E2F4+/+ mice were also similarly subjected to hypoxia/reoxygenation. E2F4−/− neurons also showed an increased sensitivity to hypoxia-induced neuronal death (23% survival in E2F4 null versus 58% in wild-type neurons, p < 0.01) (Fig. 2D). These results suggest that, unlike E2F1, E2F4 plays a prosurvival role in neuronal death induced by hypoxia. Interestingly, the increased sensitization of E2F4 siRNA-treated neurons to death in the absence of insult suggests a potential compensatory effect in E2F4 knockout neurons. To examine this possibility, we analyzed E2F1 and E2F3 protein levels in E2F4 wild-type and knockout neurons by Western blot analysis. Our results showed that the E2F1 protein level is unaffected by the loss of E2F4 (Fig. 2, E and F). However, the E2F3 protein level was reduced significantly (p < 0.05) with germ line E2F4 loss (Fig. 2, G and H). This is in contrast with our earlier observation that E2F3 levels are unaffected by the transient knockdown of E2F4.4
E2F4 and p130 Proteins Are Reduced following Hypoxia/Reoxygenation in Vitro
E2F4 is known to form repressive complexes with p130 in neurons, which may be important in neuronal survival (21). Coimmunoprecipitation experiments showed that E2F4 and p130 normally form complexes in CGN (Fig. 3A). This is in accordance with previous reports (23, 35). We then asked whether there were perturbations in total E2F4 and p130 protein levels following hypoxia/reoxygenation insult. CGN cultures were subjected to varying durations of hypoxia or 18 h of hypoxia, followed by varying reoxygenation times. Total protein was harvested at the indicated times and analyzed by Western blot analysis (Fig. 3, B and D). The blots were probed with antibody against E2F4 (Fig. 3B) or p130 (Fig. 3D). E2F4 (Fig. 3, B and C) protein levels were diminished significantly (p < 0.05) following 18 h of hypoxia (R0) and remained reduced for up to 8 h following reoxygenation when compared with untreated cultures. Although there was a reinduction of E2F4 levels at 2 and 4 h of reoxygenation, it did not reach the levels of untreated controls. Similar to E2F4, p130 (Fig. 3, D and E) protein levels were also reduced drastically (p < 0.05) immediately following hypoxia, with a brief reinduction at 2 and 4 h of reoxygenation. The reason for this induction is unclear at the moment. However, levels appeared to diminish again thereafter. The loss of p130 could be related to the loss of E2F4 following hypoxia and reoxygenation. Indeed, a Western blot analysis performed on E2F4−/− CGN cultures showed that p130 protein levels were diminished significantly (p < 0.05) in the absence of E2F4 (Fig. 3, F and G).
FIGURE 3.

p130 and E2F4 protein levels are down-regulated following hypoxia/reoxygenation of CGNs in culture. A, E2F4 forms a complex with p130 in CGNs. Total cell lysates from CGN cultures were subjected to reciprocal immunoprecipitation (IP) with anti-E2F4 and anti-p130 antibodies. Immune complexes were resolved by Western blot (WB) analysis and probed with anti-E2F4 and anti-p130 antibodies. B, Western blot analysis showing a loss of E2F4 protein in CGNs subjected to varying durations of hypoxia or 18 h of hypoxia with varying durations of reoxygenation. NH, no hypoxia. C, densitometry was performed on E2F4 blot as in B. E2F4 protein levels were normalized to the actin control. H, hypoxia; R, reoxygenation; R0, 18 h of hypoxia and 0 h of reoxygenation. D, Western blot analysis showing the loss of p130 following different durations of hypoxia or 18 h of hypoxia with varying reoxygenation times. E, densitometry was performed on p130 protein levels as in D and normalized to the β-actin control. F, basal p130 protein levels were diminished in E2F4−/− CGNs. A Western blot analysis was conducted on total protein lysate obtained from E2F4+/+ and E2F4−/− CGN cultures. The blots were probed with anti-p130 antibody and anti-β-actin as a control. G, densitometry was conducted on p130 blots as in F. p130 levels were normalized to β-actin. Error bars represent the mean ± S.E. (n ≥ 3). *, p < 0.05.
Hypoxia/Reoxygenation Induces a Concomitant Reduction of E2F4 and Induction of E2F1 Binding at the B-Myb Promoter
The previous evidence indicates that p130 is lost following ischemia. We next determined how E2F members such as E2F1 or E2F4 might differ in binding to known E2F sites. In this regard, we focused on B-Myb, a prodeath factor described previously for neurons and regulated by E2Fs (21, 35). We examined the relative promoter occupancy of both E2F1 and E2F4 following hypoxia/reoxygenation in vitro. ChIP was carried out on lysates from CGN cultures treated with 18 h of hypoxia and 16 h of reoxygenation using E2F1- and E2F4-specific antibodies. E2F1- and E2F4-associated chromatin was subjected to PCR using B-Myb promoter-specific primers. As shown in Fig. 4A, endogenous E2F4 occupancy at the B-Myb promoter was present in the untreated, no hypoxia samples under basal conditions. This occupancy decreased with hypoxia/reoxygenation (Fig. 4, A and B). This contrasts with E2F1, where occupancy is low under basal conditions and increases with hypoxic stress (Fig. 4, A and C). Consistent with the loss of E2F4, a ChIP analysis performed using p130-specific antibody also showed that it is lost from the B-Myb promoter following hypoxia/reoxygenation (Fig. 4, D and E). Interestingly, we found that, in the absence of E2F4, p130 is lost from the B-Myb promoter under basal conditions.4 This result indicates that E2F1 and E2F4 may mutually regulate B-Myb in an opposing manner to increase B-Myb expression and promote death. The ChIP experiments were performed at later time points where we could observe robust biochemical changes. To query the effects of E2F regulation at earlier time points, we next examined changes in E2F-mediated activity at the B-Myb promoter utilizing more sensitive, luciferase-based promoter assays. To this end, CGNs were infected with AAV expressing E2F reporter constructs, B-Myb-promoter-luciferase containing wild-type or mutated E2F sites as a control, and β-galactosidase 1 day after plating. The cells were treated with hypoxia and varying duration of reoxygenation after a week in culture. Luciferase and β-galactosidase analyses showed that E2F activity at the B-Myb promoter was induced significantly (2.5-fold, p < 0.05) immediately following hypoxia and remained elevated during reoxygenation (Fig. 4F). Because E2F4 is known to form repressive complexes and our results indicated that it is lost following hypoxic stress, we next investigated the effects of its deficiency on overall E2F activity at the B-Myb promoter. E2F4 wild-type and knockout CGNs were similarly infected with AAV expressing the same E2F reporter constructs as above, and luciferase activity was measured following 18 h of hypoxia and 4 h of reoxygenation (Fig. 4G). Consistent with our earlier results, luciferase activity increased 2-fold (p < 0.01) following hypoxia/reoxygenation in the E2F4 wild-type CGNs (Fig. 4G). In the E2F4 null CGNs, the basal luciferase activity was elevated (2-fold, p < 0.05) compared with E2F4 wild-type cultures and was not increased further with hypoxia/reoxygenation (Fig. 4G). This result indicates that E2F4 is important in the basal suppression of B-myb. It is important to note here that, because no death is observed basally (at least with non-stressed E2F4 knockout neurons), this suggests that multiple signals, including Myb, act in concert to promote death following ischemic insult.
FIGURE 4.

Reduction of E2F4 and induction of E2F1 binding at the B-Myb promoter following hypoxia and reoxygenation. A, ChIP was conducted with E2F4 and E2F1 antibody on CGNs treated with hypoxia (hyp) for 18 h, followed by reoxygenation (reoxy) for 16 h. B, densitometry of the B-Myb signal following ChIP performed with anti-E2F4 antibody as shown in A. Loss of E2F4 was observed at the B-Myb promoter following hypoxia and reoxygenation. C, quantitation of the B-Myb signal following ChIP with anti-E2F1 antibody. D, ChIP was performed with anti-p130 antibody. p130 binding at the B-Myb promoter was lost following hypoxia and reoxygenation. E, densitometry of the B-Myb signal following ChIP as shown in D. F, E2F activity increased after hypoxia and reoxygenation in CGNs. Cells were infected with AAV expressing B-Myb-promoter-luciferase with wild-type E2F or a mutated E2F site and β-galactosidase. Cells were treated with 18 h of hypoxia followed by reoxygenation for up to 8 h. Luciferase activity and β-galactosidase were measured at the indicated times. Data represent values of luciferase/β-galactosidase activity. G, a luciferase assay was conducted in E2F4 wild-type and knockout CGNs treated with 18 h of hypoxia followed by 4 h of reoxygenation. Error bars represent mean ± S.E. (n ≥ 3). *, p < 0.05; **, p < 0.01; ***, p < 0.005.
E2F4 Expression Is Protective following Transient Cerebral Ischemia in Vivo
To further investigate the role of E2F4 under ischemic conditions, we next examined its effects in an in vivo model of stroke induced by global ischemia. AAV expressing E2F4 or GFP control were injected unilaterally into the hippocampal CA1 region 2 weeks prior to insult, as described previously (5). Global ischemia was induced for 10 min using the four-vessel occlusion method, as described previously (6, 32). E2F4 overexpression and efficiency of viral delivery were verified by immunohistochemistry staining of CA1 neurons (Fig. 5A) and confirmed by Western blot analysis performed on hippocampal protein lysate from AAV-injected rats (Fig. 5D). Analysis of CA1 neurons 4 days following global ischemia showed significantly more live CA1 neurons (55% survival) in E2F4-expressing rats compared with those expressing GFP (3%, p < 0.05) (Fig. 5, B and C). GFP or E2F4 overexpression alone had no effect on neuronal viability in the absence of insult (Fig. 5C).
FIGURE 5.

E2F4 expression protects CA1 neurons from global cerebral ischemia. A, immunofluorescence staining showing E2F4 overexpression in hippocampal CA1 neurons of rats injected with AAV expressing E2F4. B, hematoxylin and eosin-stained sections of CA1 neurons in sham- and 4VO-operated rats injected with GFP or E2F4 expressing AAV. C, quantitation of live CA1 neurons following 4VO in GFP and E2F4 injected rats. Rats were injected with AAV expressing E2F4 or a GFP control and subjected to sham surgery of 10 min of 4VO. Brains were extracted 4 days following reperfusion, sectioned, and stained with hematoxylin and eosin. H&E-stained coronal sections were evaluated for cell survival in the hippocampal CA1 (n ≥ 4/group, data are mean ± S.E). *, p < 0.05 compared with GFP control. D, Western blot analysis of E2F4 expression in the hippocampus of GFP and E2F4 injected rats.
E2F4 and p130 Proteins Are Diminished following Global Ischemia
We also asked whether perturbations in E2F4 and p130 levels occurred in vivo following cerebral ischemia. Similar to the results obtained in CGNs, coimmunoprecipitation results showed that E2F4 and p130 also normally form complexes in rat hippocampal lysates.4 To examine potential changes in E2F4 and p130 protein levels in vivo following ischemia, rats were subjected to 10 min of 4VO and sacrificed at various times following reperfusion, as described under “Experimental Procedures.” Hippocampal lysates were extracted and subjected to Western blot analysis (Fig. 6A). The blots were probed with antibody against p130, E2F4, and actin for a loading control. p130 levels were increased (although not statistically significant) at early time points following ischemia but declined precipitously 24 h following reperfusion (Fig. 6, A and B). The early increase in p130 could be a compensatory response. Similarly to p130, the E2F4 protein level was reduced dramatically reduced 24 h following ischemia (Fig. 6, A and C). The biological function of p130 (14, 36) as well as its stability and down-regulation (37) are known to be regulated by Cdk-mediated phosphorylation. p130 has been shown to contain multiple Cdk phosphorylation sites, including Ser-952 (36). Therefore, we examined p130 Ser-952 phosphorylation following ischemia in vivo. Cdk-mediated phosphorylation of Ser-952 was increased following cerebral ischemia (Fig. 6, D and E). These results indicate that p130 is phosphorylated at early time points following ischemic insult and that both p130 and E2F4 proteins are subsequently lost in the death process. These results are similar to those observed with hypoxia/reoxygenation in vitro.
FIGURE 6.

p130 and E2F4 protein levels are decreased following global cerebral ischemia in rats. A, Western blot analyses of p130 and E2F4 expression following 4VO in rats. Animals were subjected to 10 min of 4VO and sacrificed at the indicated times following reperfusion. Hippocampal tissues were extracted and subjected to Western blot analysis. Densitometry was performed on p130 protein levels (B) as well as E2F4 protein levels (C) and normalized to the actin control. D, Western blot analysis of p130 phosphorylation following 4VO. Rats were treated as in A and subjected to analysis using antibody directed at p130 phosphorylated at Ser-952. E, densitometry of Ser-952-phosphorylated p130. Expression levels were normalized to the actin control (n ≥ 3). *, p < 0.05 compared with sham.
B- and C-myb Transcripts Are Induced following Global Ischemia in Vivo
Because our results showed that E2F activity is increased at the B-myb promoter in vitro, we examined potential changes in its transcript levels as well as that of the related C-Myb following ischemic insult in vivo. We observed that both B-Myb (Fig. 7, A and B) and C-Myb (Fig. 7, C and D) mRNA transcript levels were increased 3-fold (p < 0.05) 24 h following global ischemia. Overall, our results suggest that E2F4-p130 complexes provide neuronal protection under ischemic stress by regulating the expression of proapoptotic factors. Consistent with this hypothesis and previous reports (23, 35), we found that siRNA-mediated knockdown of the C-Myb gene provided protection for neurons subjected hypoxia/reoxygenation insult (Fig. 7F). CGN cultures were transfected with siRNA to C-Myb or control siRNA 5 days after plating and subjected to 18 h of hypoxia followed by 16 h of reoxygenation. Analysis of neuronal survival showed increased neuronal survival in neurons transfected with C-Myb siRNA (56%) compared with control siRNA (37%) (Fig. 7F). Knockdown of the C-Myb message was verified by semiquantitative RT-PCR in siRNA-treated cultures 24 h after transfection (Fig. 7E).
FIGURE 7.

B- and C-Myb mRNA transcript levels are increased following global cerebral ischemia. A, semiquantitative RT-PCR of the B-Myb message following global ischemia. Rats were subjected to 10 min of 4VO followed by 12 and 24 h of reperfusion. Hippocampal tissues were extracted and analyzed by semiquantitative RT-PCR. GAPDH is shown as a control. B, densitometry of B-Myb levels. Data are expressed as fold over sham-operated control. C, semiquantitative RT-PCR of the C-Myb message following 4VO as described in A. E, semiquantitative RT-PCR showing siRNA mediated knockdown of the C-Myb message 24 h following transfection. S 12 is shown as a loading control. F, C-Myb knockdown protects CGNs from hypoxia/reoxygenation insult. Neurons were transfected with C-Myb siRNA and subjected to 18 h of hypoxia followed by 16 h of reoxygenation. Error bars represent the mean ± S.E. (n ≥ 3). *, p < 0.05.
DISCUSSION
Cell cycle induced death of neurons has been described in numerous cell death paradigms (1, 20, 38–40). However, the downstream effectors of this pathway in the context of pathologic neuronal death are not fully understood, particularly in the context of ischemic damage. In this regard, a growing body of evidence suggests that death following ischemic insult is mediated via pRb-E2F1.
In line with this, we found that E2F1 deficiency was protective, whereas its overexpression exacerbated death induced by DNA damage and hypoxia/reoxygenation. Our data are consistent with a proapoptotic role demonstrated previously for E2F1 in other contexts, including death induced by staurosporine (41), β-amyloid (42), potassium deprivation (24, 43, 44), kainic acid (34), oxygen-glucose deprivation (OGD) (8), and ischemia (8, 24, 34, 41, 42). Similar to E2F1, E2f3, another member of the activating E2F family, is proapoptotic. Overexpression of E2F3 promoted cell death in response to camptothecin treatment, whereas its down-regulation was protective. This is in line with a previous report demonstrating a protective role for E2F3 deficiency in the developing CNS in response to DNA damage (45).
The role of the repressive E2F4 contrasts that of activating E2F members by promoting survival under conditions of ischemic stress. This is supported by several findings in this study. Acute down-regulation of E2F4 in CGNs using siRNA caused death in the absence of insult. Hypoxia/reoxygenation resulted in death that was exacerbated with E2F4 deficiency and knockdown in culture. In contrast, we found that overexpression of E2F4 protected neurons from death induced by hypoxia/reoxygenation in vitro and global cerebral ischemia in vivo. The data presented here as well as by others (8, 18, 19) demonstrate that, in the context of cerebral ischemic damage, the activities of both the activating and repressive E2Fs are an important determinant of neuronal survival and death.
The differential roles of activating and repressive E2Fs in neuronal survival is interesting. Although the exact reasoning behind this role is not clear, there are some intriguing hypotheses generated by our data. First, we showed that p130-E2F4 levels are present basally but reduced following ischemic insult. Second, we show that E2F4 binding to the E2F site on the B-Myb promoter is reduced following hypoxia. This suggests a model where p130-E2F4 exists basally to form active repressive complexes but is lost after death-inducing insult. These repressive complexes may be critical for neuronal survival. In support of this, p130 predominantly occupies E2F sites in cultured neurons (21) and has been shown, along with E2F4, to participate in the mammalian DP, Rb-like, E2F and MuvB-like (DREAM) protein complex (46). Additionally, p130 levels are high in neurons (47, 48), and p130 loss has been suggested to promote death (49). In particular, p130 deficiency leads to strain-dependent lethality in null mice (49), and its down-regulation in cultured neurons promotes apoptosis (21).
In this scenario, we suggest that E2F4 is protective because it would facilitate the formation of repressive complexes, which are lost following ischemic insult. In contrast to E2F4, we propose that E2F1 acts to promote death by directly activating prodeath genes. This is supported in this study by the observation that E2F1 is prodeath and that its occupancy on death-inducing genes is increased following hypoxic insult. Therefore, we propose a model whereby hypoxia/ischemia leads to the loss of E2F4-repressive complexes on target promoters and allows for increase in E2F1 transactivating activity on those genes. Both would then contribute to an increase in expression of targets genes such as Myb, leading to death. Indeed, we show that targets such as B-Myb and C-Myb are increased following ischemia in vivo. It will be important to fully test this hypothesis in the future.
Acknowledgment
We thank Yasmilde Rodriguez Gonzalez for critical reading of the manuscript.
This work was supported by grants from the Heart and Stroke Foundation of Canada, the Heart and Stroke Foundation of Ontario, the Canadian Institutes of Health Research, the Center for Stroke Recovery, and the Neuroscience Canada/Krembil Foundation (to D. S. P.).
G. O. Iyirhiaro, Y. Zhang, F. Safarpour, S. M. Callaghan, R. S. Slack, and D. S. Park, unpublished data.
- Cdk
- cyclin-dependent kinase
- AAV
- adeno-associated virus
- CGN
- cerebellar granule neuron
- 4VO
- four-vessel occlusion
- CA1
- cornu ammonis 1.
REFERENCES
- 1. Park D. S., Levine B., Ferrari G., Greene L. A. (1997) Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J. Neurosci. 17, 8975–8983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Park D. S., Obeidat A., Giovanni A., Greene L. A. (2000) Cell cycle regulators in neuronal death evoked by excitotoxic stress: implications for neurodegeneration and its treatment. Neurobiol. Aging 21, 771–781 [DOI] [PubMed] [Google Scholar]
- 3. Schwartz E. I., Smilenov L. B., Price M. A., Osredkar T., Baker R. A., Ghosh S., Shi F. D., Vollmer T. L., Lencinas A., Stearns D. M., Gorospe M., Kruman I. I. (2007) Cell cycle activation in postmitotic neurons is essential for DNA repair. Cell Cycle 6, 318–329 [DOI] [PubMed] [Google Scholar]
- 4. Park D. S., Morris E. J., Greene L. A., Geller H. M. (1997) G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J. Neurosci. 17, 1256–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rashidian J., Iyirhiaro G., Aleyasin H., Rios M., Vincent I., Callaghan S., Bland R. J., Slack R. S., During M. J., Park D. S. (2005) Multiple cyclin-dependent kinases signals are critical mediators of ischemia/hypoxic neuronal death in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 14080–14085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang F., Corbett D., Osuga H., Osuga S., Ikeda J. E., Slack R. S., Hogan M. J., Hakim A. M., Park D. S. (2002) Inhibition of cyclin-dependent kinases improves CA1 neuronal survival and behavioral performance after global ischemia in the rat. J. Cereb. Blood Flow Metab. 22, 171–182 [DOI] [PubMed] [Google Scholar]
- 7. Osuga H., Osuga S., Wang F., Fetni R., Hogan M. J., Slack R. S., Hakim A. M., Ikeda J. E., Park D. S. (2000) Cyclin-dependent kinases as a therapeutic target for stroke. Proc. Natl. Acad. Sci. U.S.A. 97, 10254–10259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gendron T. F., Mealing G. A., Paris J., Lou A., Edwards A., Hou S. T., MacManus J. P., Hakim A. M., Morley P. (2001) Attenuation of neurotoxicity in cortical cultures and hippocampal slices from E2F1 knockout mice. J. Neurochem. 78, 316–324 [DOI] [PubMed] [Google Scholar]
- 9. Ferguson K. L., Callaghan S. M., O'Hare M. J., Park D. S., Slack R. S. (2000) The Rb-CDK4/6 signaling pathway is critical in neural precursor cell cycle regulation. J. Biol. Chem. 275, 33593–33600 [DOI] [PubMed] [Google Scholar]
- 10. Katchanov J., Harms C., Gertz K., Hauck L., Waeber C., Hirt L., Priller J., von Harsdorf R., Bruck W., Hortnagl H., Dirnagl U., Bhide P. G., Endres M. (2001) Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J. Neurosci. 21, 5045–5053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wen Y., Yang S., Liu R., Simpkins J. W. (2005) Cell-cycle regulators are involved in transient cerebral ischemia induced neuronal apoptosis in female rats. FEBS Lett. 579, 4591–4599 [DOI] [PubMed] [Google Scholar]
- 12. Iaquinta P. J., Lees J. A. (2007) Life and death decisions by the E2F transcription factors. Curr. Opin. Cell Biol. 19, 649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dimova D. K., Dyson N. J. (2005) The E2F transcriptional network: old acquaintances with new faces. Oncogene 24, 2810–2826 [DOI] [PubMed] [Google Scholar]
- 14. Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262 [DOI] [PubMed] [Google Scholar]
- 15. Frolov M. V., Dyson N. J. (2004) Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J. Cell Sci. 117, 2173–2181 [DOI] [PubMed] [Google Scholar]
- 16. Greene L. A., Biswas S. C., Liu D. X. (2004) Cell cycle molecules and vertebrate neuron death: E2F at the hub. Cell Death Differ. 11, 49–60 [DOI] [PubMed] [Google Scholar]
- 17. Park D. S., Morris E. J., Bremner R., Keramaris E., Padmanabhan J., Rosenbaum M., Shelanski M. L., Geller H. M., Greene L. A. (2000) Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J. Neurosci. 20, 3104–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. MacManus J. P., Jian M., Preston E., Rasquinha I., Webster J., Zurakowski B. (2003) Absence of the transcription factor E2F1 attenuates brain injury and improves behavior after focal ischemia in mice. J. Cereb Blood Flow Metab. 23, 1020–1028 [DOI] [PubMed] [Google Scholar]
- 19. MacManus J. P., Koch C. J., Jian M., Walker T., Zurakowski B. (1999) Decreased brain infarct following focal ischemia in mice lacking the transcription factor E2F1. Neuroreport 10, 2711–2714 [DOI] [PubMed] [Google Scholar]
- 20. Greene L. A., Liu D. X., Troy C. M., Biswas S. C. (2007) Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim. Biophys. Acta 1772, 392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu D. X., Nath N., Chellappan S. P., Greene L. A. (2005) Regulation of neuron survival and death by p130 and associated chromatin modifiers. Genes Dev. 19, 719–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zolotukhin S., Potter M., Zolotukhin I., Sakai Y., Loiler S., Fraites T. J., Jr., Chiodo V. A., Phillipsberg T., Muzyczka N., Hauswirth W. W., Flotte T. R., Byrne B. J., Snyder R. O. (2002) Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods 28, 158–167 [DOI] [PubMed] [Google Scholar]
- 23. Liu D. X., Greene L. A. (2001) Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron 32, 425–438 [DOI] [PubMed] [Google Scholar]
- 24. O'Hare M. J., Hou S. T., Morris E. J., Cregan S. P., Xu Q., Slack R. S., Park D. S. (2000) Induction and modulation of cerebellar granule neuron death by E2F-1. J. Biol. Chem. 275, 25358–25364 [DOI] [PubMed] [Google Scholar]
- 25. McClellan K. A., Ruzhynsky V. A., Douda D. N., Vanderluit J. L., Ferguson K. L., Chen D., Bremner R., Park D. S., Leone G., Slack R. S. (2007) Unique requirement for Rb/E2F3 in neuronal migration: evidence for cell cycle-independent functions. Mol. Cell. Biol. 27, 4825–4843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Field S. J., Tsai F. Y., Kuo F., Zubiaga A. M., Kaelin W. G., Jr., Livingston D. M., Orkin S. H., Greenberg M. E. (1996) E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell 85, 549–561 [DOI] [PubMed] [Google Scholar]
- 27. Leone G., Sears R., Huang E., Rempel R., Nuckolls F., Park C. H., Giangrande P., Wu L., Saavedra H. I., Field S. J., Thompson M. A., Yang H., Fujiwara Y., Greenberg M. E., Orkin S., Smith C., Nevins J. R. (2001) Myc requires distinct E2F activities to induce S phase and apoptosis. Mol. Cell 8, 105–113 [DOI] [PubMed] [Google Scholar]
- 28. Humbert P. O., Rogers C., Ganiatsas S., Landsberg R. L., Trimarchi J. M., Dandapani S., Brugnara C., Erdman S., Schrenzel M., Bronson R. T., Lees J. A. (2000) E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol. Cell 6, 281–291 [DOI] [PubMed] [Google Scholar]
- 29. O'Hare M. J., Kushwaha N., Zhang Y., Aleyasin H., Callaghan S. M., Slack R. S., Albert P. R., Vincent I., Park D. S. (2005) Differential roles of nuclear and cytoplasmic cyclin-dependent kinase 5 in apoptotic and excitotoxic neuronal death. J. Neurosci. 25, 8954–8966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gonzalez Y. R., Zhang Y., Behzadpoor D., Cregan S., Bamforth S., Slack R. S., Park D. S. (2008) CITED2 signals through peroxisome proliferator-activated receptor-γ to regulate death of cortical neurons after DNA damage. J. Neurosci. 28, 5559–5569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xia Z., Dudek H., Miranti C. K., Greenberg M. E. (1996) Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J. Neurosci. 16, 5425–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iyirhiaro G. O., Brust T. B., Rashidian J., Galehdar Z., Osman A., Phillips M., Slack R. S., Macvicar B. A., Park D. S. (2008) Delayed combinatorial treatment with flavopiridol and minocycline provides longer term protection for neuronal soma but not dendrites following global ischemia. J. Neurochem. 105, 703–713 [DOI] [PubMed] [Google Scholar]
- 33. Zhang Y., Parsanejad M., Huang E., Qu D., Aleyasin H., Rousseaux M. W., Gonzalez Y. R., Cregan S. P., Slack R. S., Park D. S. (2010) Pim-1 kinase as activator of the cell cycle pathway in neuronal death induced by DNA damage. J. Neurochem. 112, 497–510 [DOI] [PubMed] [Google Scholar]
- 34. Smith R. A., Walker T., Xie X., Hou S. T. (2003) Involvement of the transcription factor E2F1/Rb in kainic acid-induced death of murine cerebellar granule cells. Brain Res. Mol. Brain Res. 116, 70–79 [DOI] [PubMed] [Google Scholar]
- 35. Liu D. X., Biswas S. C., Greene L. A. (2004) B-myb and C-myb play required roles in neuronal apoptosis evoked by nerve growth factor deprivation and DNA damage. J. Neurosci. 24, 8720–8725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hansen K., Farkas T., Lukas J., Holm K., Rönnstrand L., Bartek J. (2001) Phosphorylation-dependent and -independent functions of p130 cooperate to evoke a sustained G1 block. EMBO J. 20, 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tedesco D., Lukas J., Reed S. I. (2002) The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCF(Skp2). Genes Dev. 16, 2946–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hernández-Ortega K., Quiroz-Baez R., Arias C. (2011) Cell cycle reactivation in mature neurons: a link with brain plasticity, neuronal injury and neurodegenerative diseases? Neurosci. Bull. 27, 185–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moh C., Kubiak J. Z., Bajic V. P., Zhu X., Smith M. A., Lee H. G. (2011) Cell cycle deregulation in the neurons of Alzheimer's disease. Results Probl. Cell Differ. 53, 565–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rashidian J., Iyirhiaro G. O., Park D. S. (2007) Cell cycle machinery and stroke. Biochim. Biophys. Acta 1772, 484–493 [DOI] [PubMed] [Google Scholar]
- 41. Hou S. T., Callaghan D., Fournier M. C., Hill I., Kang L., Massie B., Morley P., Murray C., Rasquinha I., Slack R., MacManus J. P. (2000) The transcription factor E2F1 modulates apoptosis of neurons. J. Neurochem. 75, 91–100 [DOI] [PubMed] [Google Scholar]
- 42. Giovanni A., Keramaris E., Morris E. J., Hou S. T., O'Hare M., Dyson N., Robertson G. S., Slack R. S., Park D. S. (2000) E2F1 mediates death of B-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J. Biol. Chem. 275, 11553–11560 [DOI] [PubMed] [Google Scholar]
- 43. Konishi Y., Bonni A. (2003) The E2F-Cdc2 cell-cycle pathway specifically mediates activity deprivation-induced apoptosis of postmitotic neurons. J. Neurosci. 23, 1649–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yuan Z., Yao L., Li M., Liu S., He W., Lu Y. (2011) Opposing roles for E2F1 in survival and death of cerebellar granule neurons. Neurosci. Lett. 499, 164–169 [DOI] [PubMed] [Google Scholar]
- 45. Martinez L. A., Goluszko E., Chen H. Z., Leone G., Post S., Lozano G., Chen Z., Chauchereau A. (2010) E2F3 is a mediator of DNA damage-induced apoptosis. Mol. Cell. Biol. 30, 524–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Litovchick L., Sadasivam S., Florens L., Zhu X., Swanson S. K., Velmurugan S., Chen R., Washburn M. P., Liu X. S., DeCaprio J. A. (2007) Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell 26, 539–551 [DOI] [PubMed] [Google Scholar]
- 47. Kusek J. C., Greene R. M., Pisano M. M. (2001) Expression of the E2F and retinoblastoma families of proteins during neural differentiation. Brain Res. Bull. 54, 187–198 [DOI] [PubMed] [Google Scholar]
- 48. Baldi A., Esposito V., De Luca A., Fu Y., Meoli I., Giordano G. G., Caputi M., Baldi F., Giordano A. (1997) Differential expression of Rb2/p130 and p107 in normal human tissues and in primary lung cancer. Clin. Cancer Res. 3, 1691–1697 [PubMed] [Google Scholar]
- 49. LeCouter J. E., Kablar B., Whyte P. F., Ying C., Rudnicki M. A. (1998) Strain-dependent embryonic lethality in mice lacking the retinoblastoma-related p130 gene. Development 125, 4669–4679 [DOI] [PubMed] [Google Scholar]