

Abstract
Since its introduction as a public health programme in the United States in the early 1960s, newborn blood screening (NBS) has evolved from the detection of phenylalanine levels on filter paper to the application of DNA-based technologies to identify T-cell lymphopenia in infants with severe combined immunodeficiency. This latter use of NBS has required the development of an assay for T-cell lymphopenia based on the quantification of T-cell receptor excision circles (TRECs) that could be performed on dried blood spots routinely collected from newborn infants. The TREC-based NBS was developed six years ago, and there have already been 7 successful pilot studies since then. Similarly, efforts are now being made to establish a screen for B-cell defects, in particular agammaglobulinaemia, taking advantage of the introduction of the method for the quantification of K-deleting recombination excision circles (KRECs). A further achievement of NBS could be the simultaneous recognition of T- and B-cell defects using the combined quantification of TRECs and KRECs from Guthrie card blood spots. This approach may help the early identification of infants with T- and B-cell deficiencies so that they can then be referred to specialised paediatric centres, where a precise diagnosis of severe combined immunodeficiency and agammaglobulinaemia can be performed, and where then they can immediately receive specific therapy. Simultaneous TREC and KREC quantification should also allow classification of patients into subgroups and help identify children with less serious primary immunodeficiencies. This would help avoid the opportunistic infections and frequent hospitalisations that result from a late or lack of diagnosis.
Key words: primary immunodeficiency, severe combined immunodeficiency, T-cell receptor excision circles, K-deleting recombination excision circles, newborn screening.
Significance for public health
Primary immunodeficiencies include a group of more than 200 different diseases. Since the clinical severity ranges from mild to potentially life-threatening, major efforts are currently being made to develop methods to detect patients with these diseases in the neonatal period. Early identification is highly desirable. In fact, making a diagnosis before vaccines or blood products are administered, as well as before the infection develops, allows the patient to proceed to haematopoietic stem cell transplantation, and receive enzyme replacement and gene therapies. Indeed, the best outcome for the most severe form of primary immunodeficiency, as with many other conditions for which newborn screenings are now performed, is achieved when treatment is given in the first months of life, ideally before clinical presentation. The pilot studies of newborn screenings for severe combined immunodeficiency have demonstrated their cost-effectiveness and have also proved successful in terms of public health resulting from improved Quality of Life and survival of children with these diseases.
Newborn screenings
The goal of newborn blood screening (NBS) is to identify pre-symptomatic newborns with potentially serious or fatal disorders which could be successfully treated, leading to significant reductions in morbidity and mortality. NBS debuted as a public health programme in the US in the early 1960s, and has expanded to countries around the world, with different testing options in each country. NBS has progressively evolved since 1963, when Guthrie and Susi demonstrated that postnatal testing of dried blood spots on filter paper led to the identification and treatment of infants prior to the development of the cognitive deficits associated with phenylketonuria.1 The application of tandem mass spectrometry also allowed simultaneous screening for multiple disorders.2 More recently, the advent of DNA-based technologies has again shifted the paradigm of NBS, offering the potential of screening for numerous disorders at the same time. Therefore, the number of NBS-analysed disorders has progressively grown.
The American College of Medical Genetics recommends a uniform panel of 29 disorders that all infants born in every state should be screened for.3 Therefore, every year, millions of babies in the US are routinely screened for congenital deafness and, using a few drops of blood from the newborn’s heel, for blood cell disorders (sickle cell anaemia, sickle cell disease, and Hb S/beta-thalassaemia), inborn errors of amino acid metabolism (tyrosinaemia I, argininosuccinic aciduria, citrullinaemia, phenylketonuria, maple syrup urine disease, and homocystinuria), inborn errors of organic acid metabolism (glutaric acidaemia type I, hydroxymethylglutaryl lyase deficiency, isovaleric acidaemia, 3-methylcrotonyl-CoA carboxylase deficiency, methylmalonyl-CoA mutase deficiency, methylmalonic aciduria, cblA and cblB forms, beta-ketothiolase deficiency, propionic acidaemia, and multiple-CoA carboxylase deficiency), inborn errors of fatty acid metabolism (long-chain hydroxyacyl-CoA dehydrogenase deficiency, medium-chain acyl-CoA dehydrogenase deficiency, very-long-chain acyl-CoA dehydrogenase deficiency, trifunctional protein deficiency, and carnitine uptake defect) and miscellaneous multisystemic diseases (cystic fibrosis, congenital hypothyroidism, biotinidase deficiency, congenital adrenal hyperplasia, and classical galactosaemia).3 In addition, expanded screening programmes have been developed in different states in the US, with some programmes covering almost 50 conditions.4 In Europe, several countries have expanded their NBS programmes. Some programmes screen for only one or two conditions, whereas others screen for up to a few dozen. For example, the UK and France National Screening Committees recommend that all babies in their countries should be screened for phenylketonuria, congenital hypothyroidism, sickle cell disease, and cystic fibrosis, with the addition of medium-chain acyl-CoA dehydrogenase deficiency in the UK and congenital adrenal hyperplasia in France.5 In Italy, the application of the NBS programme application differs widely between regions. In some regions, only the three tests listed in the national programme are performed while other regions screen for up to 47 rare metabolic diseases. While developing countries face additional challenges related to poor economies, unstable governments, unique local cultures, geographical extremes, and different public health priorities,6-9 all other countries face challenges in implementing NBS because of the identification of the causes of genetic disorders, advances in detection technologies, and the development of better treatment regimens. One of the last NBS tests to be introduced measures the presence of T-cell deficiencies using T-cell receptor excision circle (TREC) quantification.
Primary immunodeficiencies
Primary immunodeficiencies (PID) are rare inherited disorders of the immune system. They are genetically determined and may occur alone or as part of a syndrome. Several types of PID have been described by Al-Herz et al.10 and the heterogeneity within each disorder may be considerable. They typically manifest during infancy and childhood as abnormally frequent/recurrent or unusual infections, often accompanied by immunoregulatory defects.11 The vast majority of patients with PID have decreased T- and/or B-cell functions that grossly impair immunity. B-cell defects lead to antibody deficiencies. As a result of a mutation in the BTK gene, a subgroup of these patients have X-linked agammaglobulinaemia (XLA) due to a B-cell differentiation arrest in the bone marrow and the consequent absence of mature B cells and serum immunoglobulins (Ig).12 More than 30% of these patients develop irreversible organ damage in childhood or early adulthood, mainly in the lungs.12,13 Non-XLA is characterised by hypo-gammaglobulinaemia with decreased B-cell counts (less than 2% mature B cells) in the absence of the BTK gene mutation.14 In patients with XLA and non-XLA, recurrent infections appear between three and 18 months of age, whereas the mean age at diagnosis is three years.15 This delayed diagnosis results in frequent hospitalisations because of pneumonia, sepsis, meningitis, and other bacterial infections, which frequently require the intravenous administration of antibiotics and can be fatal. Thus, early diagnosis and treatment, including periodic intravenous Ig replacement therapy, are essential to improve the prognosis and Quality of Life of these patients.
T-cell defects result in combined immunodeficiencies, affecting both cellular and humoral immunity, with severe combined immunodeficiency (SCID) being the most serious and lethal form. SCID, which was reported for the first time more than 60 years ago,16 comprises a heterogeneous group of diseases, characterised by profound deficiencies of T- and B-cell functions, and, in some types, also of natural killer (NK) cells.17,18 The overall frequency of SCID is estimated to be between 1:50,000 and 1:100,000 live births.18 These infants develop failure to thrive, chronic diarrhoea, and infections in the first months of life. Infections can occur with common pathogens, but most of the time the infants also suffer from opportunistic infections, such as thrush or Pneumocystis jiroveci pneumonia. Graft-versus-host disease caused by maternal T-cell engraftment may occur in these patients, who also show skin rashes and organomegaly.
SCID is now known to be caused in humans by mutations in several different genes, such as cytokine receptor genes, antigen receptor genes and others,19 but there are many other probable causes yet to be discovered. An updated classification of SCID is based on the underlying genetics and prevalent molecular pathogenetic mechanisms and includes the following:20 impaired cytokine-mediated signalling, alterations in V(D)J recombination, impaired signalling through the pre-T cell receptor (TCR), increased lymphocyte apoptosis, absence of the thymus, alterations in thymus embryogenesis, impaired calcium flux, and other mechanisms. SCID infants, regardless of the disease genotype, show distinct lymphocyte phenotypes. They all have a profound T-cell deficiency, and those T cells that are present are usually of maternal origin, having crossed the placenta. B cells can be elevated, normal or absent, depending on the type of SCID, and the NK-cell number is variable. Therefore, according to the presence or absence of the T, B and NK lymphocytes, SCID can also be phenotypically categorised as the T−B−NK−, T−B+NK−, T−B−NK+, and T−B+NK+ (with minus meaning absence or severely reduced counts) subtypes. For example, T−B+NKSCID, accounting for up to 50% of SCID cases, is predominantly X-linked, and is caused by mutations in the IL2RG gene encoding the interleukin 2 receptor gamma chain. Although less frequent, JAK3 deficiency produces a similar T−B+NK− lymphocyte phenotype.
The early recognition of SCID should be considered a paediatric emergency because a diagnosis before live vaccines or prior to the development of infections permits lifesaving unfractionated HLA-identical or T-cell depleted haploidentical non-ablative haematopoietic stem cell transplantation, enzyme replacement therapy, or gene therapy.21-29 However, these infants often appear normal at birth and have no family history of immunodeficiency,30 and consequently, many of them are not identified until a life-threatening infection occurs. This is important because the long-term prognosis of infants with SCID and other serious immunodeficiencies can be markedly improved if the diagnosis is made early, before the onset of severe infections.31 Indeed, if SCID is not detected until the infant is older, there is a much higher likelihood of death from live vaccines, graft-versus-host disease from non-irradiated blood products, or infections, before a successful definitive therapy can be adopted.21,23,24
Pilot studies for newborn blood screening for severe combined immunodeficiency using T-cell receptor excision circles quantification
Because SCID is not apparent at birth and early recognition is essential for life-saving treatment, it has, for many years now, been recognised as a candidate for NBS.32-35 Indeed, the disease satisfies the criteria that the Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children recommends for routine inclusion in NBS. These criteria are that: i) the frequency of the disease in the population is high enough to warrant screening; ii) the untreated natural history of the disease is well-defined; iii) the untreated disease clearly confers significant morbidity and/or mortality; iv) the disease is treatable, and treatment significantly improves outcome; v) disease testing is safe, simple, and sufficiently sensitive to detect all cases; vi) specific confirmatory testing is available for the disease; and vii) the test, treatment, and treatment outcomes are cost-effective with respect to the non-treatment of the disease.36
An initial screening test for SCID was based on the detection of peripheral blood lymphopenia. However, a complete blood count cannot be performed using filter paper-dried blood spots, making this test unsuitable for NBS. In addition, the quantification of peripheral blood lymphocytes may fail to identify some infants with SCID due to the presence of normal B-cell or NK-cell numbers, the maternal engraftment of T cells, and/or the expansion of a few T-cell clonotypes.37,38 Therefore, no laboratory test on newborn dried blood spots was available to detect T-cell lymphopenia in infants with SCID until 2005 when real-time polymerase chain reaction (PCR) for the quantification of TRECs was developed and validated for population-based screening.39 TRECs are created during the process of lymphocyte differentiation (Figure 1A) when TCR rearrangements occur within the thymus. The rearrangement of the TCR alpha (TCRA) chain has the peculiarity of involving the excision of delta-coding segments, which encode the delta chain of gamma/delta T cells that, being nestled in the TCRA locus between the variable (V) and junctional (J) gene segments, must be excised to generate the TCRA chain. The excised circularised DNA elements are called TRECs (Figure 1B).
Figure 1.
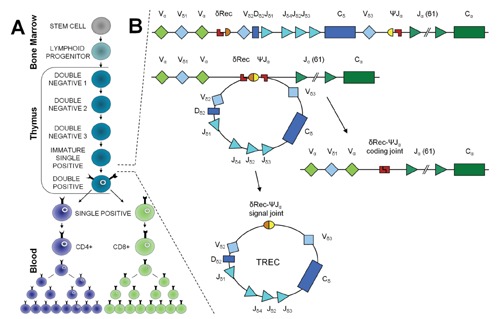
T-cell differentiation and T-cell receptor excision circle formation. (A) Lymphoid progenitors, which have developed from haematopoietic stem cells in the bone marrow, migrate to the thymus to complete their antigen-independent maturation into functional T cells. In the thymus, the T cells develop their specific T-cell markers and undergo thymic education through positive and negative selection. (B) Because TCRD gene segments are interspersed within TCRA gene segments along chromosome 14q11 (TCRAD locus), following V(D)J recombination of the TCRD locus, the entire locus is preferentially deleted by δREC–ψJα rearrangements. This process gives rise to a δREC–ψJα signal joint on an excision circle (TREC) and a δREC–ψJα coding joint in the genome.
Approximately 70% of T cells generate a TREC.40 Several properties were established for TRECs that make them useful markers of thymic output: they are stable, do not degrade easily over time, are not replicated when a cell divides, and are (almost) exclusively of thymic origin, without extrathymic sources of TCR rearrangements.41,42
After the demonstration that the TREC assay detects SCID patients regardless of the genetic cause,43 the test was modified and refined so it could be performed using blood from dried spots, was highly sensitive and specific for SCID, and was cost-effective and reproducible, thereby becoming amenable to high-throughput population-based testing.44 At that point, pilot studies of NBS for SCID, integrated with plans for its definitive diagnosis and management, were established in some US states.30 In 2008, Wisconsin became the first state to implement mandatory NBS for T-cell deficiency,45 followed by Massachusetts in February 2009,46,47 and later by New York, the Navajo Nation, California, Puerto Rico and Louisiana.29 With the exception of California, where a TREC assay kit under development by a medical technology company is being used, all the other states are using in-house modifications of the TREC assay.39,44,46,47 By April 30th 2011, these pilot studies had been performed on 961,925 newborns, and had identified 14 cases of SCID, 6 cases of SCID variants and 40 cases of T-cell lymphopenia that were not related to SCID.29 All infants diagnosed with SCID in Wisconsin and Massachusetts have undergone transplantation or enzyme replacement therapy and no deaths have been reported.45,48
Identification of patients with agammaglobulinaemia using K-deleting recombination excision circle quantification
Taking advantage of the introduction of the K-deleting recombination excision circle (KREC) method to study the replication history of B cells,49 efforts have recently been made to establish a NBS for B-cell maturation defects.14 During the process of B-cell maturation, KRECs are produced by recombination events that determine the allelic and isotypic exclusion of the Kappa chain.50 Indeed, during B-cell development, KRECs are produced in those B lymphocytes that, after the Ig heavy chain rearrangement, have failed to productively rearrange the Ig light Kappa chain genes (IGK) on one or both alleles.51 In these cells, the IGK locus becomes non-functional through the deletion of the constant K-gene segment (IGKC) by recombination of the K-deleting element (Kde), which is a sequence located approximately 24 kb downstream of the constant K-gene segment, with one of the two upstream recombination signal sequences.51 When the Kde recombines with the recombination signal sequence located in the intron between the junction and constant K-gene segments, portions of the DNA are deleted, and the ligation of the excised recombination-signal ends generates the circularised DNA elements defined as KRECs (Figure 2A). These products persist in the cell, are unable to replicate, and are diluted as a result of cell division.
Figure 2.
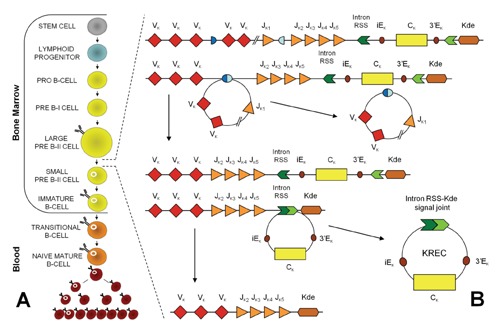
B-cell differentiation and K-deleting recombination excision circle formation. (A) B-cell development begins as lymphoid stem cells differentiate into progenitor B cells (pro-B cells), which are the earliest distinctive B-lineage cells. During the antigen-independent maturation phase, immune competent B cells expressing membrane IgM and IgD are generated in the bone marrow. Only approximately 10% of the potential B cells reach maturity and leave the bone marrow. Naïve B cells in the periphery die within a few days unless they encounter soluble protein antigen and activated Th cells. Once activated, B cells proliferate within secondary lymphoid organs. (B) B-cell differentiation from the stem-cell to the plasma-cell stages is characterised by Ig V(D)J rearrangement, somatic mutation and Ig class switching. The V(D)J recombination on the IGK locus results in a Vκ-Jκ coding joint. Subsequent rearrangement between the intronRSS and the Kde element can render the IGK allele non-functional by deleting the IGKC exon and the enhancers. Consequently, the coding joint precludes any further rearrangements in the IGK locus and, therefore, remains present in the genome, whereas an intronRSS-Kde signal joint is formed on the KREC.
The frequency of these recombination events in human B-cell malignancies ranged from 31% to 69%; approximately 50% of transformed B cells should contain KRECs.52 Therefore, the quantification of KRECs was initially used to determine the number of developing B lymphocytes in the bone marrow of children with B-precursor acute lymphoblastic leukaemia treated with allogeneic human stem cell transplantation.53 However, as IGK gene deletion occurs physiologically in all B lymphocytes that fail to productively rearrange the IGK genes on one or both alleles, the number of KRECs has been be proposed to be a quantitative marker of bone marrow output in all individuals.54 Furthermore, because KRECs are not supposed to be produced in XLA and non-XLA patients because their B-cell maturation defects occur before K-deleting recombination occurs (Figure 2B), the KREC measurement can potentially be applied to identify these types of B-cell deficiencies, which account for approximately 20% of all B-cell defects. In addition, some other types of combined immunodeficiencies show an arrest at the B-cell maturation stage and can also be identified using KREC detection.
Indeed, more than 200 KREC copies/μg DNA were present in the dried blood spots of healthy children, whereas no KRECs were detected in 30 XLA and 5 non-XLA patients.12,14 This suggests that the measurement of KRECs in neonatal dried blood spots could be used in an NBS that identifies neonates with early B-cell maturation defects. This early identification of children at risk should improve their future Quality of Life and help reduce health care costs, considering that the clinical phenotype of agammaglobulinaemia is milder than that of SCID. Therefore, the disease would be underdiagnosed in the first years of life in the absence of a screening programme, with patients developing serious organ damage before an accurate diagnosis could be made.
Simultaneous quantification of T- and B-cell defects using the T-cell receptor excision circles/K-deleting recombination excision circles assay
In 2010, we improved the methods for TREC and KREC quantification by developing a duplex quantitative real-time PCR protocol that allows an accurate and simultaneous assessment of both targets.54 For their absolute quantification, a standard curve is created using serial dilutions of TREC and KREC signal joint constructs cloned in a bacterial plasmid, together with a control gene, the TCRA constant gene (TCRAC). The primers and probes used for the duplex real-time PCR assay are listed in Table 1.
Table 1.
Primer and probe sequences for TREC and KREC quantification.
| Primer* | Sequence |
|---|---|
| SJ TRECs forward | 5’-CAC ATC CCT TTC AAC CAT GCT-3’ |
| SJ TRECs reverse | 5’-TGC AGG TGC CTA TGC ATC A-3’ |
| SJ KRECs forward | 5’-TCC CTT AGT GGC ATT ATT TGT ATC ACT-3’ |
| SJ KRECs reverse | 5’-AGG AGC CAG CTC TTA CCC TAG AGT-3’ |
| TCRAC forward | 5’-TGG CCT AAC CCT GAT CCT CTT-3’ |
| TCRAC reverse | 5’-GGA TTT AGA GTC TCT CAG CTG GTA CAC-3’ |
| Probe* | Sequence |
| TRECs probe | 5’-FAM-ACA CCT CTG GTT TTT GTA AAG GTG CCC ACT-TAMRA-3’ |
| KRECs probe | 5’-HEX-TCT GCA CGG GCA GCA GGT TGG-TAMRA-3’ |
| TCRAC probe | 5’-FAM-TCC CAC AGA TAT CCA GAA CCC TGA CCC-TAMRA-3’ |
*Sottini et al.54
The main advantage of the combined TREC/KREC assay is that the variability related to DNA quantification is eliminated by the use of the triple-insert plasmid in which the TREC, KREC and TCRAC gene fragments are present in a 1:1:1 ratio. This allows a more accurate assessment of the TREC and KREC copy number. Furthermore, the simultaneous quantification of the two targets in the same reaction contains the reagent costs. The number of TRECs or KRECs per 106 peripheral blood mononuclear cells (PBMCs) is calculated by dividing the mean quantity of TRECs or KRECs by the mean number of TCRAC gene copies, which has to be divided by 2 because there are two TCRAC gene copies in each cell (i.e. one for each chromosome) and then multiplied by 106. This value, together with the lymphocyte plus monocyte count (which are the cells obtained in the PBMC preparation) in 1 mL of blood, was used to calculate the number of TRECs or KRECs per mL of blood as follows:
| TRECs or KRECs/mL = (TRECs or KRECs per 106 PBMCs) x (lymphocyte + monocyte count in 1 mL of blood)/106.54 |
Using this strategy, we found that in healthy subjects, the number of TRECs and KRECs is correlated to age (Figure 3) and that, in particular, it significantly decreases over time, with a steeper slope in the first three years of life.55 With this assay, we demonstrated that the T- and B-cell reconstitution in children with different PIDs who underwent human stem cell transplantation or were cured with enzyme replacement therapy using pegylated bovine adenosine deaminase involves the mobilisation of both T and B cells from the respective production and maturation sites, and that the increase in TRECs and KRECs can be either strictly associated with or independent from one another.54,56 These results demonstrated that knowledge of the numbers of TRECs and KRECs, obtained with an assay that can be easily introduced into routine laboratory practice, is highly informative and can be used for a more precise identification of patients with PID or for their therapeutic monitoring. Accordingly, this method has been recently validated in a cohort of 2560 anonymised NBS cards and in 49 originally stored Guthrie cards from patients diagnosed with different PIDs, allowing the identification of patients with SCID, XLA, ataxia-telangiectasia (AT) and Nijmegen-breakage syndrome.57 Therefore, the set up of a single assay for the simultaneous quantification of TRECs and KRECs will permit NBS for both SCID and agammaglobulinaemia, which together have a combined estimated incidence of 1:30,000-1:50,000 births. Furthermore, this test allows the separation of T-cell deficient cases into B-cell positive and B-cell negative subgroups, guiding additional diagnostics toward either T−B+ or T−B− SCID.
Figure 3.

Age-dependence of T-cell receptor excision circles and K-deleting recombination excision circles. The numbers of T-cell receptor excision circles (TRECs) and K-deleting recombination excision circles (KRECs) in healthy individuals. The graphics were prepared using data from Sottini et al.54 and Serana et al.55,56
Limitations of the introduction of newborn blood screening for primary immunodeficiencies
The use of the TREC assay has identified a large number of subjects that do not meet the criteria for SCID. Some of these other conditions, such as DiGeorge syndrome and Down syndrome, are known to result in T-cell lymphopenia,45,58 whereas others were unexpected and many remain undefined. In particular, premature babies have been shown to have a lower number of TRECs than full-term babies, despite not being immunodeficient.59 Moreover, the TREC and KREC assay may identify other forms of immunodeficiency, some of them severe, for which a definitive treatment is not available, e.g. AT.57,60 Therefore, it is important to emphasise that the identification of SCID and agammaglobulinaemia at birth must be based on a two-tier assay, with TREC/KREC determination only being the first tier. Appropriate second-tier assays must be used to define the disease (if any) that results in the low TREC/KREC values. For some disorders, such as AT, defining the disease at birth may indeed prove challenging because the values of alphafetoprotein in newborns are elevated.61 This requirement of a two-tiered system may represent an important limitation of the use of the TREC/KREC assay for the population-based screening of genetic conditions in newborns. Other principal obstacles to the full implementation of NBS for PID could be the costs of setting up the screening test and the possibility that the combined measurement of TRECs and KRECs may generate higher call rates for newborns to be on clinical follow up, which in turn has an impact on the total screening costs. However, the introduction of the SCID NBS in the pilot studies had an approximate cost of less than 1 million dollars,29 whereas in the US, the cost of treating one baby with SCID that is not diagnosed until he or she has a serious infection can easily exceed 2 million dollars. There is also a striking difference with regard to the costs for transplantation in SCID infants under 3.5 months of age (approx. 1 million dollars) and the costs for those aged over 3.5 months (more than 4 million dollars).29,62 An initial study, performed before the introduction of the TREC assay on dried blood spots, demonstrated that a SCID screening test that cost approximately 5 US Dollars would be considered cost-effective.63 However, the study incorporated limited information about the care costs or outcome differences between infants identified before and after becoming symptomatic. This limitation was bypassed by a recent study that evaluated the cost-effectiveness of universal screening using a model incorporating the impact of early detection on the natural history of SCID.62 The authors demonstrated that assuming a SCID incidence of 1:75,000 births and test specificity and sensitivity of 0.99 each, the screen remained cost-effective up to a maximum cost of 15 dollars per infant screened.62 Therefore, considering that the TREC assay is relatively inexpensive, with a cost of approximately 5.50 dollars per assay,45 that the incidence of T-cell lymphopenia is relatively high, and that better health outcomes and lower costs are associated with earlier stem cell transplantation, NBS for SCID meets the reasonable standards of willingness-to-pay and could be a worthwhile addition to NBS panels. Despite the favourable evaluation of SCID based upon the NBS guidelines, national implementation is pending in most countries which have long-lasting traditions of their own NBS programmes. The inclusion of KREC detection in the test only slightly increases the cost because it only involves the addition of a single reagent to the assay. Thus, a single assay capable of screening for both SCID and agammaglobulinaemia, which together have a combined estimated incidence of 1:30,000-1:50,000 births, should further improve the cost-effectiveness of NBS. Additional preventive medical research for other severe PIDs, including, but not limited to, inherited agammaglobulinaemias and haemophagocytic syndromes,64 would be advisable.
The implications of NBS for PID in countries other than the US cannot be easily established as the attributed value for a quality life-year considerably differs among countries, particularly among the US, Europe and other regions. Indeed, although PIDs are mentioned within those disorders that might be considered for the gradual expansion of NBS in the member states of the European Union,65 developing countries will probably face additional challenges related to poor economies, unstable governments, unique local cultures, geographical extremes, and different public health priorities.
Conclusions
Newborn blood screening for SCID and other T-cell deficiencies, based on TREC quantification, was the first screening introduced for PID, but with the advent of modern molecular technologies, screening for other immune system defects will probably be available soon.
One of the most promising candidates for NBS protocols is the combined quantification of TRECs and KRECs that could support early diagnosis, classification of patient subgroups, and evaluation of stem cell replacement therapy for both T- and B-cell defects.
In order to achieve this, clinicians have to play an active role in promoting NBS for PID and other T- and B-lymphocyte abnormalities, and work closely with public health officials and NBS committees. They also have to offer their expertise to advise laboratories that are developing the new techniques. They will have an even more important role in establishing an accurate diagnosis for those infants who have NBS-positive results and in ensuring that these children are given the best possible and most appropriate treatment.
References
- 1.Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963;32:318-43 [PubMed] [Google Scholar]
- 2.Wilcken B. Recent advances in newborn screening. J Inherit Metab Dis 2007;30:129-33 [DOI] [PubMed] [Google Scholar]
- 3.American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 481: Newborn screening. Obstet Gynecol 2011;117:762-5 [DOI] [PubMed] [Google Scholar]
- 4.Newborn Screening Status Report (updated 09/06/12). National Newborn Screening and Genetics Resource Center. Available from: http://genes-r-us.uthscsa.edu/sites/genes-r-us/files/nbsdisorders.pdf
- 5.Burton H, Moorthie S. Expanded newborn screening: a review of the evidence. PHG Foundation. 2010. Available from: http://www.phgfoundation.org/file/5502/ [Google Scholar]
- 6.Padilla CD, Therrell BL. Newborn screening in the Asia Pacific region. J Inherit Metab Dis 2007;30:490-506 [DOI] [PubMed] [Google Scholar]
- 7.Padilla CD. Towards universal newborn screening in developing countries: obstacles and the way forward. Ann Acad Med Singapore 2008;37 Suppl 12:6-9 [PubMed] [Google Scholar]
- 8.Krotoski D, Namaste S, Raouf RK, et al. Conference report: second conference of the Middle East and North Africa newborn screening initiative: partnerships for sustainable newborn screening infrastructure and research opportunities. Genet Med 2009;11:663-8 [DOI] [PubMed] [Google Scholar]
- 9.Saadallah AA, Rashed MS. Newborn screening: experiences in the Middle East and North Africa. J Inherit Metab Dis 2007;30:482-9 [DOI] [PubMed] [Google Scholar]
- 10.Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol 2011;2:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol 2010;125:S182-94 [DOI] [PubMed] [Google Scholar]
- 12.van Zelm MC, van der Burg M, Langerak AW, van Dongen JJ. PID comes full circle: applications of V(D)J recombination excision circles in research, diagnostics and newborn screening of primary immunodeficiency disorders. Front Immunol 2011;2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quinti I, Soresina A, Spadaro G, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol 2007;27:308-16 [DOI] [PubMed] [Google Scholar]
- 14.Nakagawa N, Imai K, Kanegane H, et al. Quantification of κ-deleting recombination excision circles in Guthrie cards for the identification of early B-cell maturation defects. J Allergy Clin Immunol 2011;128:223-5 [DOI] [PubMed] [Google Scholar]
- 15.Kanegane H, Futatani T, Wang Y, et al. Clinical and mutational characteristics of X-linked agammaglobulinemia and its carrier identified by flow cytometric assessment combined with genetic analysis. J Allergy Clin Immunol 2001;108:1012-20 [DOI] [PubMed] [Google Scholar]
- 16.Glanzmann E, Riniker P. [Essential lymphocytophthisis; new clinical aspect of infant pathology]. Ann Paediatr 1950;175:1-32 [Article in German]. [PubMed] [Google Scholar]
- 17.Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol 2004;22:625-55 [DOI] [PubMed] [Google Scholar]
- 18.Fischer A, Le Deist F, Hacein-Bey-Abina S, et al. Severe combined immunodeficiency. A model disease for molecular immunology and therapy. Immunol Rev 2005;203:98-109 [DOI] [PubMed] [Google Scholar]
- 19.International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies, Notarangelo LD, Fischer A, Geha RS, Casanova JL, Chapel H, et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol 2009;124:1161-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cossu F. Genetics of SCID. Ital J Pediatr 2010;36:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buckley RH, Schiff SE, Schiff RI, et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med 1999;340:508-16 [DOI] [PubMed] [Google Scholar]
- 22.Kane L, Gennery AR, Crooks BN, et al. Neonatal bone marrow transplantation for severe combined immunodeficiency. Arch Dis Child Fetal Neonatal Ed 2001;85:F110-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood 2002;99:872-8 [DOI] [PubMed] [Google Scholar]
- 24.Buckley RH. Transplantation of hematopoietic stem cells in human severe combined immunodeficiency: longterm outcomes. Immunol Res 2011;49:25-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hershfield MS, Buckley RH, Greenberg ML, et al. Treatment of adenosine deaminase deficiency with polyethylene glycol-modified adenosine deaminase. N Engl J Med 1987;316:589-96 [DOI] [PubMed] [Google Scholar]
- 26.Aiuti A, Cattaneo F, Galimberti S, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med 2009;360:447-58 [DOI] [PubMed] [Google Scholar]
- 27.Hacein-Bey-Abina S, Hauer J, Lim A, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 2010;363:355-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaspar HB, Cooray S, Gilmour KC, et al. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med 2011;3:97ra79. [DOI] [PubMed] [Google Scholar]
- 29.Buckley RH. The long quest for neonatal screening for severe combined immunodeficiency. J Allergy Clin Immunol 2012;129:597-604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puck JM; SCID Newborn Screening Working Group. Population-based newborn screening for severe combined immunodeficiency: steps toward implementation. J Allergy Clin Immunol 2007;120: 760-8 [DOI] [PubMed] [Google Scholar]
- 31.Brown L, Xu-Bayford J, Allwood Z, et al. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood 2011;117:3243-6 [DOI] [PubMed] [Google Scholar]
- 32.Buckley RH, Schiff RI, Schiff SE, et al. Human severe combined immunodeficiency: genetic, phenotypic, and functional diversity in one hundred eight infants. J Pediatr 1997;130:378-87 [DOI] [PubMed] [Google Scholar]
- 33.Lindegren ML, Kobrynski L, Rasmussen SA, et al. Applying public health strategies to primary immunodeficiency diseases: a potential approach to genetic disorders. MMWR Recomm Rep 2004;53:1-29 [PubMed] [Google Scholar]
- 34.McGhee SA, Stiehm ER, McCabe ER. Potential costs and benefits of newborn screening for severe combined immunodeficiency. J Pediatr 2005;147:603-8 [DOI] [PubMed] [Google Scholar]
- 35.McGhee SA, Stiehm ER, Cowan M, et al. Two-tiered universal newborn screening strategy for severe combined immunodeficiency. Mol Genet Metab 2005;86:427-30 [DOI] [PubMed] [Google Scholar]
- 36.Wilson JM, Jungner YG. [Principles and practice of mass screening for disease]. Bol Oficina Sanit Panam 1968;65:281-393 [Article in Spanish]. [PubMed] [Google Scholar]
- 37.Müller SM, Ege M, Pottharst A, et al. Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: a study of 121 patients. Blood 2001;98:1847-51 [DOI] [PubMed] [Google Scholar]
- 38.Palmer K, Green TD, Roberts JL, et al. Unusual clinical and immunologic manifestations of transplacentally acquired maternal T cells in severe combined immunodeficiency. J Allergy Clin Immunol 2007;120:423-8 [DOI] [PubMed] [Google Scholar]
- 39.Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol 2005;115:391-8 [DOI] [PubMed] [Google Scholar]
- 40.Verschuren MC, Wolvers-Tettero IL, Breit TM, et al. Preferential rearrangements of the T cell receptor-delta-deleting elements in human T cells. J Immunol 1997;158:1208-16 [PubMed] [Google Scholar]
- 41.Douek DC, McFarland RD, Keiser PH, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature 1998;396:690-5 [DOI] [PubMed] [Google Scholar]
- 42.Zanotti C, Caimi L, Imberti L. Severe combined immunodeficiency patients: immune recovery after stem cell transplantation. Stem Cells Cancer Stem Cells 2012;4:287-96 [Google Scholar]
- 43.Morinishi Y, Imai K, Nakagawa N, et al. Identification of severe combined immunodeficiency by T-cell receptor excision circles quantification using neonatal guthrie cards. J Pediatr 2009;155:829-33 [DOI] [PubMed] [Google Scholar]
- 44.Baker MW, Grossman WJ, Laessig RH, et al. Development of a routine newborn screening protocol for severe combined immunodeficiency. J Allergy Clin Immunol 2009;124:522-7 [DOI] [PubMed] [Google Scholar]
- 45.Routes JM, Grossman WJ, Verbsky J, et al. Statewide newborn screening for severe T-cell lymphopenia. JAMA 2009;302:2465-70 [DOI] [PubMed] [Google Scholar]
- 46.Comeau AM, Hale JE, Pai SY, et al. Guidelines for implementation of population-based newborn screening for severe combined immunodeficiency. J Inherit Metab Dis 2010;33 Suppl 2:S273-81 [DOI] [PubMed] [Google Scholar]
- 47.Gerstel-Thompson JL, Wilkey JF, Baptiste JC, et al. High-throughput multiplexed T-cell-receptor excision circle quantitative PCR assay with internal controls for detection of severe combined immunodeficiency in population-based newborn screening. Clin Chem 2010;56:1466-74 [DOI] [PubMed] [Google Scholar]
- 48.Hale JE, Bonilla FA, Pai SY, et al. Identification of an infant with severe combined immunodeficiency by newborn screening. J Allergy Clin Immunol 2010;126:1073-4 [DOI] [PubMed] [Google Scholar]
- 49.van Zelm MC, Szczepanski T, van der Burg M, van Dongen JJ. Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion. J Exp Med 2007;204:645-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Siminovitch KA, Bakhshi A, Goldman P, Korsmeyer SJ. A uniform deleting element mediates the loss of kappa genes in human B cells. Nature 1985;316:260-2 [DOI] [PubMed] [Google Scholar]
- 51.Beishuizen A, de Bruijn MA, Pongers-Willemse MJ, et al. Heterogeneity in junctional regions of immunoglobulin kappa deleting element rearrangements in B cell leukemias: a new molecular target for detection of minimal residual disease. Leukemia 1997;11:2200-7 [DOI] [PubMed] [Google Scholar]
- 52.van Dongen JJ, Langerak AW, Brüggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003;17:2257-317 [DOI] [PubMed] [Google Scholar]
- 53.Fronkova E, Muzikova K, Mejstrikova E, et al. B-cell reconstitution after allogeneic SCT impairs minimal residual disease monitoring in children with ALL. Bone Marrow Transplant 2008;42:187-96 [DOI] [PubMed] [Google Scholar]
- 54.Sottini A, Ghidini C, Zanotti C, et al. Simultaneous quantification of recent thymic T-cell and bone marrow B-cell emigrants in patients with primary immunodeficiency undergone to stem cell transplantation. Clin Immunol 2010;136:217-27 [DOI] [PubMed] [Google Scholar]
- 55.Serana F, Airò P, Chiarini M, et al. Thymic and bone marrow output in patients with common variable immunodeficiency. J Clin Immunol 2011;31:540-9 [DOI] [PubMed] [Google Scholar]
- 56.Serana F, Sottini A, Chiarini M, et al. The different extent of B and T cell immune reconstitution after hematopoietic stem cell transplantation and enzyme replacement therapies in SCID patients with adenosine deaminase deficiency. J Immunol 2010;185:7713-22 [DOI] [PubMed] [Google Scholar]
- 57.Borte S, von Döbeln U, Fasth A, et al. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood 2012;119:2552-5 [DOI] [PubMed] [Google Scholar]
- 58.Ram G, Chinen J. Infections and immunodeficiency in Down syndrome. Clin Exp Immunol 2011;164:9-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baker MW, Laessig RH, Katcher ML, et al. Implementing routine testing for severe combined immunodeficiency within Wisconsin’s newborn screening program. Public Health Rep 2010;125Suppl 2:88-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Borte S, Wang N, Oskarsdóttir S, et al. Newborn screening for primary immunodeficiencies: beyond SCID and XLA. Ann N Y Acad Sci. 2011;1246:118-30 [DOI] [PubMed] [Google Scholar]
- 61.Bader D, Riskin A, Vafsi O, et al. Alpha-fetoprotein in the early neonatal period--a large study and review of the literature. Clin Chim Acta 2004;349:15-23 [DOI] [PubMed] [Google Scholar]
- 62.Chan K, Davis J, Pai SY, et al. A Markov model to analyze cost-effectiveness of screening for severe combined immunodeficiency (SCID). Mol Genet Metab 2011;104:383-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McGhee SA, Stiehm ER, McCabe ER. Potential costs and benefits of newborn screening for severe combined immunodeficiency. J Pediatr 2005;147:603-8 [DOI] [PubMed] [Google Scholar]
- 64.Borte S, von Döbeln U, Hammarström L. Guidelines for newborn screening of primary immunodeficiency diseases. Curr Opin Hematol 2013;20:48-54 [DOI] [PubMed] [Google Scholar]
- 65.Burgard P, Rupp K, Lindner M, et al. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 2. From screening laboratory results to treatment, follow-up and quality assurance. J Inherit Metab Dis 2012;35:613-25 [DOI] [PubMed] [Google Scholar]