

Abstract
The structural mechanism by which Hsp70-type chaperones interact with Hsp40-type co-chaperones has been of great interest, yet still remains a matter of debate. Here, we used solution NMR spectroscopy to investigate the ATP-/ADP-dependent interactions between Escherichia coli HscA and HscB, the specialized Hsp70/Hsp40 molecular chaperones that mediate iron–sulfur cluster transfer. We observed that NMR signals assigned to amino acid residues in the J-domain and its “HPD” motif of HscB broadened severely upon the addition of ATP-bound HscA, but these signals were not similarly broadened by ADP-bound HscA or the isolated nucleotide binding domain of HscA complexed with either ATP or ADP. An HscB variant with an altered HPD motif, HscB(H32A,P33A,D34A), failed to manifest WT-like NMR signal perturbations and also abolished WT-like stimulation of ATP hydrolysis by HscA. In addition, residues 153–171 in the C-terminal region of HscB exhibited NMR signal perturbations upon interaction with HscA, alone or complexed with ADP or ATP. These results demonstrate that the HPD motif in the J-domain of HscB directly interacts with ATP-bound HscA and suggest that a second, less nucleotide-dependent binding site for HscA resides in the C-terminal region of HscB.
Heat shock proteins 40 kDa (Hsp40) and 70 kDa (Hsp70) function together to maintain protein homeostasis in vivo. Hsp70 proteins are highly conserved across evolution, and many eukaryotic and prokaryotic genomes encode multiple Hsp70s and Hsp40s, a testament to their diverse cellular roles.1 Through repeated cycles of ATP binding and hydrolysis, Hsp70 allosterically binds to exposed hydrophobic residues in misfolded and unfolded proteins, thereby preventing cytotoxic protein aggregation and allowing abnormally folded proteins to refold into their native conformations.2 Hsp40 mediates this process by recognizing and escorting client proteins to the substrate binding domain (SBD) of Hsp70 while also stimulating ATP hydrolysis in Hsp70’s nucleotide binding domain (NBD). Importantly, owing to their ability to alleviate cellular stress, Hsp70s are implicated in a variety of diseases, including cancer.3
Thus, mechanistic and structural details regarding the Hsp40:Hsp70 complex are of key biological relevance, yet they have remained surprisingly elusive.4−8 Site-directed mutagenesis studies had suggested that the highly conserved “HPD” tripeptide motif located at the tip of the J-domain of Hsp40 interacts directly with Hsp70.9−12 Nevertheless, two recent investigations of the structural features of Hsp70:Hsp40 complexes have differed on this conclusion. One study reported direct interaction between the HPD motif of the isolated J-domain of the Hsp40 auxilin and the ADP-bound isolated NBD of Hsc70.4 Stabilization of the complex was achieved by introducing a non-native disulfide bridge.4 Furthermore, this study showed that the hydrophobic linker of Hsp70 is involved in the interaction with Hsp40. On the other hand, a second study, which employed NMR spectroscopy to characterize the dynamic complex between ADP-bound full-length Hsp70 and Hsp40 from Escherichia coli, provided evidence for a non-covalent complex, but the authors concluded that the HPD motif of DnaJ is not involved in the interaction with DnaK, nor is the DnaK linker implicated in binding to DnaJ.7
In addition to their roles in protein (re)folding, specialized Hsp70 and Hsp40 molecular chaperones are involved in transferring iron–sulfur (Fe–S) clusters to yield mature Fe–S proteins. Fe–S clusters comprise an ancient class of enzymatic cofactors whose biosynthesis is of critical biological importance.13 Because of the toxicities of sulfide and free iron ions, organisms have developed strictly regulated biosynthetic systems for Fe–S cluster assembly and transfer to recipient apo-Fe-S proteins.14,15 In many prokaryotes, the iron–sulfur cluster (ISC) system produces the majority Fe–S clusters to satisfy cellular needs.16 A homologous ISC system is found in eukaryotic mitochondria.17
In E. coli, protein components necessary for Fe–S cluster biosynthesis are encoded by the isc operon. This operon contains genes for the specialized Hsp70-type chaperone (HscA) and Hsp40-type co-chaperone (HscB) pair known to facilitate Fe–S cluster transfer.18 The isc operon also encodes an Fe–S cluster scaffold protein (IscU), which interconverts between two metamorphic conformations, one more structured (S-state) and one more disordered (D-state).19−21 We previously demonstrated that HscB binds preferentially to the S-state,22 whereas HscA binds preferentially to the D-state of IscU.23
Despite their specialized roles in mediating Fe–S cluster transfer, many structural and functional features of HscA and HscB are similar to those of DnaK and DnaJ (in E. coli, the sequence identities between HscA/DnaK and HscB/DnaJ are 42.4% and 17.2%, respectively). Like DnaK, HscA has two domains: an NBD that binds and hydrolyzes ATP24 and an SBD that binds IscU as the substrate.25 IscU binding affinity in the SBD is allosterically regulated by the nucleotide bound in the NBD. The SBD of ATP-bound HscA (HscA:ATP) has an open conformation with weak affinity for IscU, whereas the SBD of ADP-bound HscA (HscA:ADP) has a closed conformation with strong affinity for IscU.18,26,27 Several studies of Hsp70s have reported that the hydrophobic linker, which connects the NBD and SBD and is highly conserved across Hsp70-type chaperones, mediates Hsp70 allosteric conformational changes and Hsp70-Hsp40 interactions.4,28,29 HscB serves as a bridge between HscA:ATP and IscU:26 the C-terminal domain (C-domain) of HscB binds to IscU in its S-state (i.e., cluster-bound IscU) while its N-terminal domain (J-domain) interacts with HscA:ATP.30,31 Together, IscU and HscB synergistically stimulate the ATPase activity of HscA.26
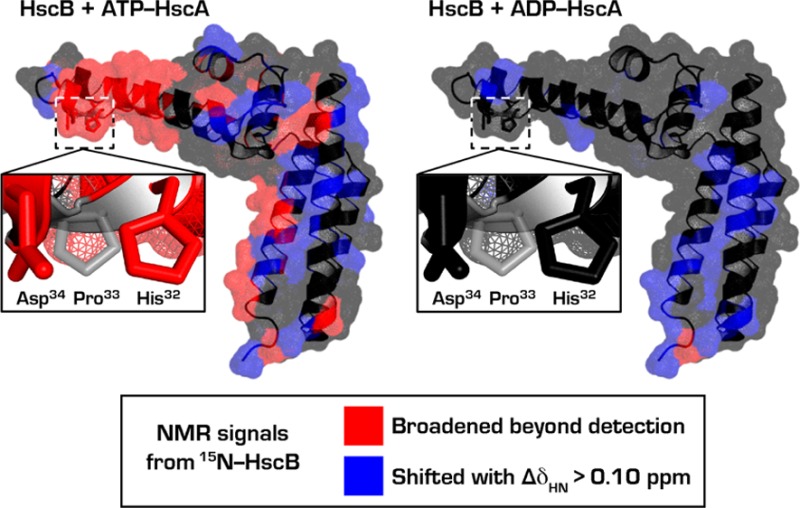
Here we employed solution NMR spectroscopy to investigate the interaction between HscB and HscA. We monitored the NMR spectrum of [U-15N]-HscB, which has been fully assigned [Biological Magnetic Resonance Data Bank (BMRB) entry 15541],31,32 upon the addition of HscA(T212V), a variant that lacks ATPase activity but still exhibits wild-type-like conformational changes in response to binding ADP or ATP.23,33 This variant enabled us to distinguish among the effects of HscA alone, ATP-bound HscA (HscA:ATP), and ADP-bound HscA (HscA:ADP). Titration of [U-15N]-HscB with 3-fold excess of unlabeled HscA(T212V) in the absence (Figure 1, panels A–C) or the presence of excess ADP (Figure 1, panels D–F) led to similar changes in 2D 1H–15N heteronuclear single-quantum correlation (HSQC) spectra. Under both conditions, the major changes localized to signals from residues (153–171) in C-terminal region of HscB. The addition of HscA(T212V) alone also led to broadening of signals from residues L6, Y13, H63, and L169 (Figures 2 and S1, upper panel; colored red), and a signal of the residue L169 in the presence of ADP (Figures 2 and S1, middle panel; colored red). In contrast, in the presence of ATP, much greater signal perturbations were observed (Figure 1, panels G–I). Signals from residues Y3, F4, F7, Y13, A19, F24–H32, D34–F36, S40, A46–A52, I54, A57, T60, L61, M66, L74, A112, V133, L136, R152, D155, K156, R158, A161, Q163, E166, and L169 broadened beyond detection upon addition of 3-fold HscA(T212V) (Figure 2 and S1, lower panel; colored red). Notably, the signals corresponding to the 32HPD34 motif of [U-15N]-HscB were perturbed significantly by HscA(T212V) solely in the presence of ATP. Yet, the addition of HscA affected signals from residues in the C-terminal region of [U-15N]-HscB regardless of nucleotide, although more were broadened in the presence of ATP.
Figure 1.

2D 1H–15N TROSY-HSQC NMR spectra reveal differential interactions between [U-15N]-HscB and HscA(T212V) in the absence of nucleotide (A–C), in the presence of 10 mM ADP (D–F), or in the presence of 10 mM ATP (G–I). The spectra shown in red (A, D, and G) are of 0.2 mM [U-15N]-HscB in the absence of HscA(T212V). The spectra shown in green (B, E, and H; overlaid on those in red) were collected following the addition of HscA(T212V) to a concentration of 0.6 mM. The buffer contained 50 mM HEPES pH 7, 2 mM MgCl2, and 1 mM DTT. The graphs shown in C, F, and I plot the chemical shift changes of the cross peaks (ΔδNH = [(ΔδN/5)2+(ΔδH)2]1/2) as a function of the HscB residue number. Residues whose signals broadened beyond detection are designated by red triangles.
Figure 2.
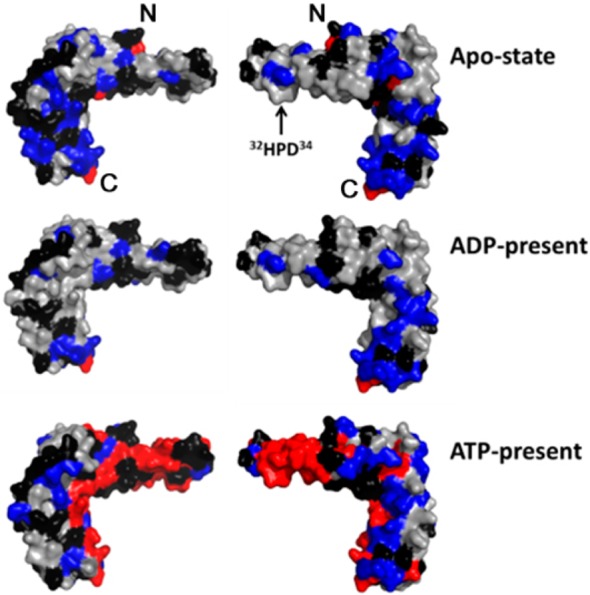
Results from the NMR signal perturbation profiles (panels C, F, and I of Figure 1) of [U-15N]-HscB with HscA(T212V) mapped onto the structure of HscB (PDB 1FPO).30 Color code: black, residues with no signal (Pro), unassigned residues, or residues whose signals could not be followed upon addition of HscA(T212V); gray, residues whose signals were minimally affected (ΔδNH ≤ 0.01 ppm) by addition of HscA(T212V); blue, residues with ΔδNH > 0.01 ppm; and red, residues whose signals broadened beyond detection. Only the surface of the structure is shown to better represent the putative HscA binding interface. The N- and C-terminals of HscB are denoted by “N” and “C”, respectively.
In order to further study the interaction between HscB and HscA:ATP, we collected 2D 1H–15N TROSY-HSQC NMR data for 0.2 mM [U-15N]-HscB with excess ATP in the presence of increasing amounts of HscA(T212V). The addition of equimolar HscA(T212V) in the presence of ATP led to selective decreases in the amplitudes (<40% of the original) of 1H–15N peaks from several residues: F7, R29, H32, D34, K35, S40, A46, V48, S51, A52, and A57 (data not shown). Increasing the molar ratio of HscA(T212V)/[U-15N]-HscB from 1 to 3, led to progressive shifts and broadening of signals from both the J- and C-terminal domains (Figure S2). Significant shifts and broadening of signals from 32HPD34 only appeared at the higher molar ratios (2 and 3). Broadening of signals from the C-terminal residues occurred at a 2-fold excess of HscA(T212V), but most signals broadened beyond detection only at a 3-fold excess of HscA(T212V).
We conclude from the above observations that, in addition to the HPD motif of its J-domain, HscB has a secondary binding site for HscA at its C-terminal domain. From its relatively weaker signal responses, it appears that this additional interaction would not confer a meaningful effect with the nucleotide-free or ADP-bound state of HscA. On the other hand, the fortified interaction of the C-terminal domain with HscA:ATP may have physiological importance: HscB bridges the interaction between HscA:ATP and IscU, and IscU binds to the C-terminal domain of HscB. Previous studies have shown that the residues R87, L92, L96, R99, E100, and F153 of HscB constitute the binding site for IscU,32 and some of the residues of HscB, namely R152, D155, K156, R158, and Q163, that broadened upon binding to HscA(T212V):ATP reside near this IscU binding site (Figure S3). In addition, the signal from F153 of HscB, the residue that is essential for interacting with IscU,31,32 was noticeably perturbed upon addition of HscA(T212V):ATP (Figure 1I). These results suggest that the structure of the HscB-IscU binary complex may be influenced by interaction with HscA:ATP.
Subsequently, we tested whether the interaction with HscB solely involves the nucleotide binding domain of HscA (HscA(NBD)) as previously hypothesized.18,30 We replaced a codon for M386 of HscA(T212V) with a translation termination codon. The resulting construct codes for HscA(NBD;T212V), a protein that includes the entire NBD domain but neither the linker nor the SBD. The 2D 1H–15N TROSY-HSQC NMR spectrum of [U-15N]-HscB in the presence of 10 mM ATP (Figure 3A) showed minor perturbations following the addition of a 3-fold molar excess of HscA(NBD;T212V) (Figure 3B). Residues in the C-terminal region showed larger chemical shift changes than residues in the J-domain (Figure 3C). We conclude that the isolated NBD of HscA does not bind to the J-domain region of HscB, but that it may interact with HscB residues in the C-terminal domain. We also found that, whereas the ATPase activity of full-length HscA was stimulated up to 10-fold by HscB, the ATPase activity of HscA(NBD) was unaffected by an equivalent amount of HscB (Figure S4).
Figure 3.
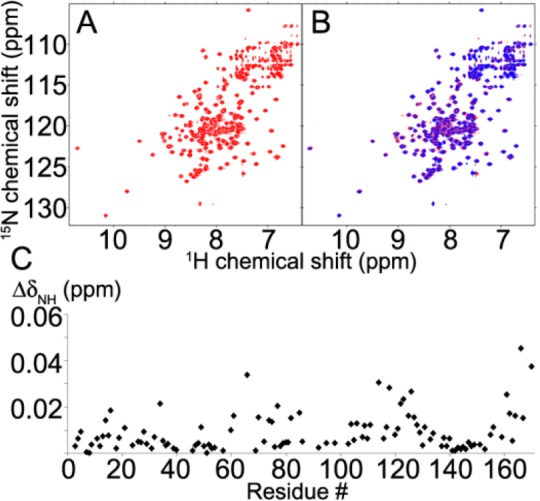
Comparison of 2D 1H–15N TROSY-HSQC NMR spectra of 0.2 mM HscB with 10 mM ATP in the absence (A, red) and presence (B, blue) of 0.6 mM HscA(NBD;T212V). (C) Chemical shift changes (ΔδNH) plotted as a function of HscB residue number.
Previous studies of yeast and human orthologues of HscB, Jac1p and HSC20, respectively, showed that mutation of their HPD motif to AAA decreased the binding affinity to their respective Hsp70 proteins.11,34,35 We prepared [U-15N]-HscB(H32A,P33A,D34A) in order to investigate the effect of this change on its interaction with HscA(T212V). 2D 1H–15N TROSY-HSQC spectra were acquired for [U-15N]-HscB(H32A,P33A,D34A) in the presence of 10 mM ADP without (Figure S5A) and with 3-fold excess HscA(T212V) (Figure S5B) and in the presence of 10 mM ATP without (Figure S5C) and with 3-fold excess HscA(T212V) (Figure S5D). The spectral changes were similar in the presence of ADP and ATP with the largest ΔδNH values for the C-terminal residues (Figure S6). Moreover, HscB(H32A,P33A,D34A) was unable to stimulate the ATPase activity of HscA (Figure S4). These results indicate that the 32HPD34 motif of HscB is essential for promoting the interaction with HscA:ATP and subsequently activating ATP hydrolysis. Importantly, spectral similarity between wild-type HscB and HscB(H32A,P33A,D34A) excludes the possibility that the triple alanine substitution affects the overall structural fold of HscB (Figure S7).
The results presented here demonstrate that the highly conserved HPD motif of HscB directly mediates the interaction with HscA:ATP; this interaction does not occur with apo-HscA, HscA:ADP, or HscA(NBD):ATP. Our data suggest that a global rather than local domain-specific, allosteric conformational transition of HscA in response to ATP-binding is required to generate the binding site for HscB.4,28,29
Our evidence that HscB contains a second site in its C-terminal region that appears to interact with the NBD of HscA is intriguing. All studies of the activation of the ATPase activity of HscA by the HscB:IscU complex have utilized apo-IscU. We have presented a hypothesis that the complex of HscB:IscU[2Fe-2S] with HscA:ATP does not stimulate ATPase activity, but instead that activation is triggered by the attack of cysteine side chains of an acceptor protein on the iron atoms of the cluster.21 Such an attack would lead to release of IscU side chains (likely from histidine and cysteine) that ligated the cluster. The proximity of the binding sites on HscB for IscU and HscA (Figure S3) suggests that a conformational change in IscU could affect the structure of HscB that would alter its secondary interaction, via its C-terminal region, with the NBD of HscA so as to trigger ATP hydrolysis. Conversion of bound ATP to ADP leads to a change in the conformation of HscA to a state that no longer interacts with the J-domain of HscB and has a strong binding interaction with the D-state of IscU, which no longer binds the Fe–S cluster. These changes would lead to the complete release of the cluster to the acceptor protein as well as the dissociation of HscB from the complex. It will be of interest to investigate in greater detail the possible role of this secondary interaction in modifying the ATPase activity of HscA.
Furthermore, results presented here with HscA and HscB are likely relevant to interactions between general classes of Hsp70 and Hsp40. We provide direct evidence for the proposed interaction of the Hsp40 HPD motif with ATP-bound Hsp70.9−12 In addition, the present data are consistent with the proposition that ATP-induced allosteric changes in the hydrophobic linker of Hsp70 are necessary to accommodate the interaction with Hsp40.4,28,29 On the other hand, our observations suggest that Hsp40s can interact with Hsp70s in diverse ways. Thus, the HPD-mediated interaction between ADP-bound mammalian Hsc70 and auxilin4 and the non-HPD-mediated dynamic complex formation between ADP-bound E. coli DnaK and DnaJ7 may simply represent different aspects of the Hsp70–Hsp40 interactome.
Acknowledgments
We thank Dr. Anna K. Füzéry (Department of Biochemistry, University of Wisconsin-Madison, Madison, WI) for preparing the expression plasmid of HscB(H32A,P33A,D34A). This work was supported by NIH grant U01 GM94622. This study made use of the National Magnetic Resonance Facility at Madison, which is supported by NIH grant P41GM66326 (NIGMS). Equipment was purchased with funds from the University of Wisconsin-Madison, the NIH (P41RR02301, P41GM66326, S10RR02781, S10RR08438, S10RR023438, S10RR025062, S10RR029220), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA.
Supporting Information Available
Additional experimental procedures; figure documenting changes in 2D 1H–15N NMR peaks from [U-15N]-HscB upon adding 1-, 2-, and 3-fold HscA(T212V) and where the affected residues are located in the structure of HscB; structural model of HscB showing the binding sites for IscU and HscA:ATP; ATPase activity assays of HscA and HscA(NBD) in the absence/presence of HscB; NMR signal perturbation profile of [U-15N]-HscB(H32A,P33A,D34A) upon adding HscA(T212V) in the presence of ADP or ATP; comparison of 2D 1H–15N TROSY-HSQC spectra of wild-type [U-15N]-HscB and [U-15N]-HscB(H32A,P33A,D34A). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ J.H.K.: Max Planck Institute for Biophysical Chemistry, Göttingen, Germany
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kampinga H. H.; Craig E. A. Nat. Rev. Cell Biol. 2010, 11, 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattoo R. U.; Sharma S. K.; Priya S.; Finka A.; Goloubinoff P. J. Biol. Chem. 2013, 288, 21399–21411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M. E. Carcinogenesis 2013, 34, 1181–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; Maes E. G.; Taylor A. B.; Wang L.; Hinck A. P.; Lafer E. M.; Sousa R. Mol. Cell 2007, 28, 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene M. K.; Maskos K.; Landry S. J. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6108–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garimella R.; Liu X.; Qiao W.; Liang X.; Zuiderweg E. R.; Riley M. I.; Van Doren S. R. Biochemistry 2006, 45, 6917–6929. [DOI] [PubMed] [Google Scholar]
- Ahmad A.; Bhattacharya A.; McDonald R. A.; Cordes M.; Ellington B.; Bertelsen E. B.; Zuiderweg E. R. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 18966–18971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa R.; Jiang J.; Lafer E. M.; Hinck A. P.; Wang L.; Taylor A. B.; Maes E. G. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, E734.(author reply E735). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall D.; Zylicz M.; Georgopoulos C. J. Biol. Chem. 1994, 269, 5446–5451. [PubMed] [Google Scholar]
- Suh W. C.; Burkholder W. F.; Lu C. Z.; Zhao X.; Gottesman M. E.; Gross C. A. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 15223–15228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisine C.; Cheng Y. C.; Ohlson M.; Schilke B.; Hoff K.; Beinert H.; Marszalek J.; Craig E. A. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 1483–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genevaux P.; Schwager F.; Georgopoulos C.; Kelley W. L. Genetics 2002, 162, 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinert H.; Holm R. H.; Münck E. Science 1997, 277, 653–659. [DOI] [PubMed] [Google Scholar]
- Johnson D. C.; Dean D. R.; Smith A. D.; Johnson M. K. Annu. Rev. Biochem. 2005, 74, 247–281. [DOI] [PubMed] [Google Scholar]
- Lill R.; Muhlenhoff U. Annu. Rev. Biochem. 2008, 77, 669–700. [DOI] [PubMed] [Google Scholar]
- Roche B.; Aussel L.; Ezraty B.; Mandin P.; Py B.; Barras F. Biochim. Biophys. Acta 2013, 1827, 455–469. [DOI] [PubMed] [Google Scholar]
- Lill R.; Hoffmann B.; Molik S.; Pierik A. J.; Rietzschel N.; Stehling O.; Uzarska M. A.; Webert H.; Wilbrecht C.; Muhlenhoff U. Biochim. Biophys. Acta 2012, 1823, 1491–1508. [DOI] [PubMed] [Google Scholar]
- Vickery L. E.; Cupp-Vickery J. R. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 95–111. [DOI] [PubMed] [Google Scholar]
- Bonomi F.; Iametti S.; Ta D.; Vickery L. E. J. Biol. Chem. 2005, 280, 29513–29518. [DOI] [PubMed] [Google Scholar]
- Kim J. H.; Tonelli M.; Markley J. L. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley J. L.; Kim J. H.; Dai Z.; Bothe J. R.; Cai K.; Frederick R. O.; Tonelli M. FEBS Lett. 2013, 587, 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H.; Füzéry A. K.; Tonelli M.; Ta D. T.; Westler W. M.; Vickery L. E.; Markley J. L. Biochemistry 2009, 48, 6062–6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H.; Tonelli M.; Frederick R. O.; Chow D. C.; Markley J. L. J. Biol. Chem. 2012, 287, 31406–31413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickery L. E.; Silberg J. J.; Ta D. T. Protein Sci. 1997, 6, 1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberg J. J.; Hoff K. G.; Tapley T. L.; Vickery L. E. J. Biol. Chem. 2001, 276, 1696–1700. [DOI] [PubMed] [Google Scholar]
- Silberg J. J.; Tapley T. L.; Hoff K. G.; Vickery L. E. J. Biol. Chem. 2004, 279, 53924–53931. [DOI] [PubMed] [Google Scholar]
- Cupp-Vickery J. R.; Peterson J. C.; Ta D. T.; Vickery L. E. J. Mol. Biol. 2004, 342, 1265–1278. [DOI] [PubMed] [Google Scholar]
- Kumar D. P.; Vorvis C.; Sarbeng E. B.; Cabra Ledesma V. C.; Willis J. E.; Liu Q. J. Mol. Biol. 2011, 411, 1099–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuravleva A.; Gierasch L. M. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6987–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupp-Vickery J. R.; Vickery L. E. J. Mol. Biol. 2000, 304, 835–845. [DOI] [PubMed] [Google Scholar]
- Füzéry A. K.; Tonelli M.; Ta D. T.; Cornilescu G.; Vickery L. E.; Markley J. L. Biochemistry 2008, 47, 9394–9404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füzéry A. K.; Oh J. J.; Ta D. T.; Vickery L. E.; Markley J. L. BMC Biochem. 2011, 12, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomi F.; Iametti S.; Morleo A.; Ta D.; Vickery L. E. Biochemistry 2008, 47, 12795–12801. [DOI] [PubMed] [Google Scholar]
- Dutkiewicz R.; Schilke B.; Cheng S.; Knieszner H.; Craig E. A.; Marszalek J. J. Biol. Chem. 2004, 279, 29167–29174. [DOI] [PubMed] [Google Scholar]
- Uhrigshardt H.; Singh A.; Kovtunovych G.; Ghosh M.; Rouault T. A. Hum. Mol. Genet. 2010, 19, 3816–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.