

Abstract
Histone deacetylases (HDACs) have found intense interest as drug targets for a variety of diseases, but there is disagreement about basic aspects of the inhibition and mechanism of HDACs. QM/MM calculations of HDAC8 including a large QM region provide a model that is consistent with the available crystal structures and structure–activity relationships of different HDAC inhibitors. The calculations support a spontaneous proton transfer from a hydroxamic acid to an active site histidine upon binding to the zinc. The role of the H142/D176 catalytic dyad as the general base of the reaction is elucidated. The reasons for the disagreements between previous proposals are discussed. The results provide detailed insights into the unique mechanism of HDACs, including the role of the two catalytic dyads and function of the potassium near the active site. They also have important implications for the design of novel inhibitors for a number of HDACs such as the class IIa HDACs.
Introduction
Post-translational protein modifications such as acetylation or methylation play important roles during epigenetic regulation.1−3 Among different epigenetic readers, writers, or erasers, which recognize, add, or remove these modifications, respectively, histone deacetylases (HDACs) have found particularly widespread interest as potential drug targets for novel therapeutic approaches to both cancer4−7 and noncancer8,9 disorders. Two HDAC inhibitors, suberoylanilide hydroxamic acid (SAHA) and FK228, have been approved for human use by the FDA and over 20 more are currently in various stages of clinical trials. As a result, the structure, function, and inhibition of HDACs have been the topic of intense research during the past decade.
Eukaryotic HDACs have 18 different isoforms, divided into four classes based on sequence similarity:10 class I (HDAC1–3, and 8), class II (HDAC4–7, 9 and 10), class IV (HDAC11), and class III (sirtuins 1–7). Classes I, II, and IV, commonly referred to as “classical” HDACs, are zinc-dependent hydrolases that remove an acetyl group from the ε-amino group of lysines. Class I HDACs are currently thought to be the biomedically most relevant isoforms11 and have been studied in most detail. For example, the majority of available HDAC inhibitors act most strongly on class I HDACs,12 and a large number of crystal structures have been published for class I HDACs, especially HDAC8 (see Table S2).
All known class I, II, and IV HDAC inhibitors possess a pharmacophore consisting of a zinc binding group (ZBG), a linker group mimicking the lysine side chain, and a cap group that interacts with the protein surface and contributes to isoform selectivity. The properties of the ZBG correlate strongly with the potency of the HDAC inhibitor,13 although interactions at the protein surface can also be strong.14 Among the different known ZBGs, hydroxamic acids are the most potent and best studied.15,16 We proposed a rationale for this observation based on the hypothesis of a pKa matching where the hydroxamic acid binds in the neutral form, thus circumventing the desolvation penalty, but is deprotonated by an adjacent histidine once bound to the zinc to increase Coulomb interactions.17 Similar effects have been observed in the case of TACE18 and are supported by computational studies of small models of HDAC active sites.19,20 We also studied a number of other ZBG21 to propose alternatives to hydroxamic acids.
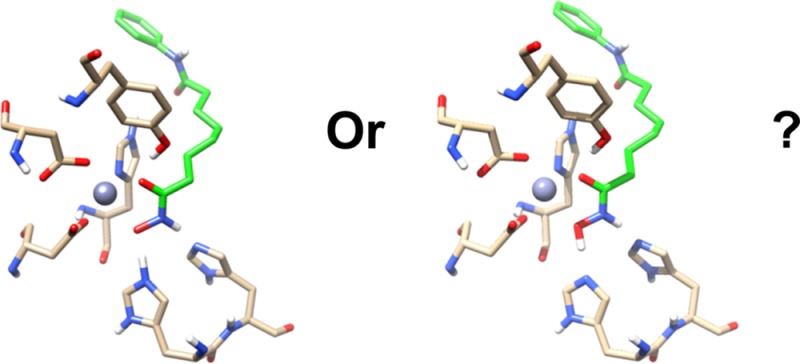
Figure 1 shows the active site of a typical class I HDAC, HDAC8 (pdb code 2V5W, resolution = 2.0 Å), bound to an acetylated lysine substrate.22,23 The active site zinc ion is buried at the bottom of a narrow hydrophobic pocket and coordinates to D178, H180, and D267 as well as to the acetyl lysine substrate and a water molecule. Other important residues include two histidine residues H142 and H143, which are in turn coordinated to two aspartates D176 and D183, respectively. These four residues are identical in the four class I human HDAC isoforms, while D183 is replaced by a Q or N in class II and IV HDACs (see Figure S1). As was pointed out previously,24 this arrangement closely resembles the catalytic triad of a serine protease, with the water taking the place of the serine. At the same time, the presence of the catalytically active zinc or other divalent or trivalent metals25 makes HDACs formally a metalloprotease, although the importance of the different overall charge state of the active site has been pointed out.17,19,26
Figure 1.
HDAC8-substrate complex crystal structure 2V5W (Y306F mutation is manually removed). The oxygen atom of water is shown as red sphere, Zn2+ and K+ are represented by gray and purple spheres, respectively. Substrate and residues considered in the QM/MM active sites are shown as sticks, other residues are shown in green cartoon.
The unique combination of serine and metalloprotease characteristics as well as the presence of two rather than one potential proton shuttle systems poses some interesting mechanistic questions. Two mechanisms, shown in simplified form in Scheme 1, have been proposed. Based on mutation experiments and the structure of histone deacetylase-like protein (HDLP), Finin et al. proposed that the buried H142-D176 dyad acts as the general base to abstract a proton from water and facilitates water oxygen’s nucleophilic attack on the carbonyl carbon of acetyl lysine substrate.24 The role of the H143-D183 dyad is then to protonate the amine in the breakdown of the tetrahedral intermediate.
Scheme 1. HDAC Deacetylation Reaction Mechanism Proposed by Finnin et al. (Mechanism 1)24 and Zhang et al. (Mechanism 2)27,28.

An alternative mechanism for the amide hydrolysis was proposed by Zhang and co-workers based on Born–Oppenheimer ab initio QM/MM MD simulations27,28 (25 ps umbrella sampling for a total of 1.5 ns along the reaction pathway using a pseudobond approach).29 After consideration of different combination of protonation states of H142 and H143, it was suggested that HID142/HID14330 is the most stable reactant complex in HDAC8, leading to Mechanism 2 shown in Scheme 1 where the H143-D183 dyad acts as the general base in the formation of the tetrahedral intermediate and as general acid in its breakdown. The role of the H142-D176 dyad would be limited to the deprotonation of the tetrahedral intermediate concerted with the heterolysis of the C–N bond rather than a direct involvement in this step. Using the same approach, Zhang and co-workers also found that deprotonation of the hydroxamic acid upon binding to HDAC8 is energetically uphill by 3.8 kcal/mol,31 which is in contrast to the findings described earlier.17,19
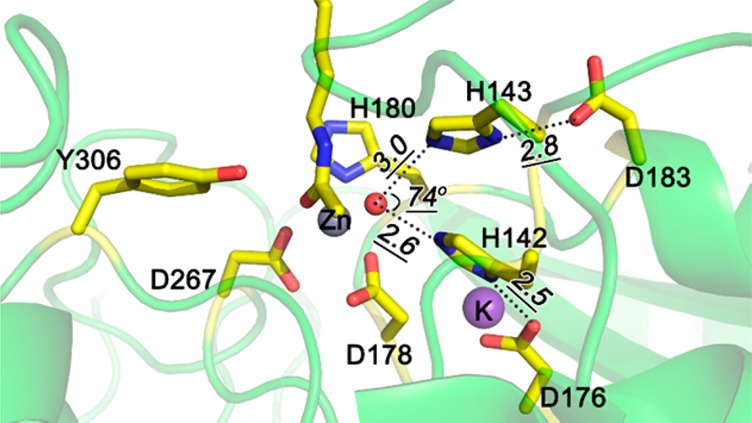
In addition to the essential metal ion Zn2+, two potassium ions have been observed in several of the HDAC8 crystal structures (Figure 2A). Site 1 for potassium binding is near the active site, and the potassium ion coordinates with the side chain oxygen of D176 and S199 and backbone oxygen atoms of D176, D178, H180, and L200 in an octahedral fashion (Figure 2B). Site 2 is positioned about 20 Å away from the catalytic metal ion, and K+ is coordinated by backbone oxygen of F189, T192, V195, and Y225, and two water molecules (Figure 2C). Experiments revealed that the structural stability of HDAC8 was increased in the presence of KCl,32 and the catalytic activity of HDAC8 was increased at lower concentrations of K+ but decreased at high concentrations.33,34 As K+ at site 2 is far away from the catalytic center, it is only possible to regulate the activity by an allosteric effect. The functional role of K+ at site 1 is unclear. Studies by Gantt et al. suggested an inhibitory role of K+ at site 1,33 while Werbeck et al. suggested an activation effect.34 Zhang’s computational work showed that occupation of site 1 by K+ could stabilize the transition state of the rate-determining step,28 and we will discuss this issue using the different theoretical model used here.
Figure 2.
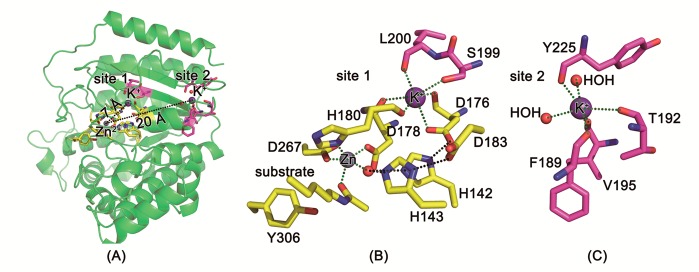
(A) crystal structure of HDAC8 (pdb code: 2V5W) with active-site Zn2+ and two K+ ions and K+ binding (B) site 1 and (C) site 2. Zn2+ and K+ are shown as gray and purple spheres, respectively.
Even though the biomedical importance of HDACs has overshadowed mechanistic questions, the mechanism and inhibition of HDAC8 did find significant interest.25,35−37 The ambiguities resulting from the different mechanistic proposals and possibly from limitations of the models used in earlier studies need to be resolved. Maybe more importantly, the question of the preferred protonation state and pKa’s of ZBGs for HDACs is of crucial importance for the development of novel HDAC inhibitors with new ZBGs. These are of significant interest not only because of the poor pharmacokinetic properties and potential toxicity of the currently used hydroxamic acids but also because it was shown that appropriately chosen ZBGs can contribute to isoform selectively,38 a key goal in the development of new, less toxic HDAC inhibitors.
Here, we report the results of a QM/MM study of the inhibition and mechanism of HDAC8 using a more complete representation of the enzyme active site. These studies provide a view of the protonation state of the inhibitor and the mechanism of lysine deacetylation by HDAC8. The effect of the potassium ion at site 1 and of protein dynamics are also considered here. Finally, we will discuss the importance of the findings to HDAC inhibitor design and the consequences of the presence of the zinc ion on the similarity of the mechanism to serine proteases.
Computational Methodology
Model Building
Starting structures for the simulations were built based on the available X-ray structure most similar to the relevant species. For the investigation of the protonation states of hydroxamates, the structure of the SAHA-HDAC8 complex (pdb code: 1T69,39 2.9 Å resolution), was used as the initial structure. The structure of the substrate-HDAC8 complex (pdb code: 2V5W,22 2.0 Å resolution, with F306 mutated back into Y306 manually) was used as the initial structure for the mechanism study. Water molecules near the active site that were resolved in the 2V5W crystal structure were kept in the calculation. No additional water molecules were added in the 1T69 crystal structure because previous MD simulations indicated that water is unlikely to enter the deeply buried active site if an inhibitor is bound.31,37,40 Hydrogens were added and optimized using the Protein Local Optimization Program (PLOP).41 The protonation states were manually checked to ensure that the active site aspartic and glutamic acids are negatively charged and the active site lysine and arginine are positively charged. The initial protonation state of the active site was set as both H142 and H143 singly protonated in the δ position, consistent with earlier studies42 and based on an analysis of the hydrogen bond network.
In addition to the zinc binding site, there is a second putative metal binding site 7 Å from the active site that is occupied by potassium in most of the available crystal structures of HDAC8 (see Table S2) and that was proposed to accelerate or inhibit the catalytic process.32−34 We have performed the mechanistic studies with and without the presence of potassium ion at site 1.
Before performing the QM/MM calculations, the initial structures including SAHA-HDAC8 complex structure and substrate-HDAC8 complex structures with and without the potassium ion at site 1 were minimized (500 steps steepest-descent minimization followed by 4500 conjugate gradient steps) to remove the steric repulsion with the zinc and coordinated atoms restrained using the sander module of Amber1243 and a generalized Born solvent model. The zinc parameters developed by Merz and co-workers were used.44 The Amber ff12SB forced field was used for the protein and general Amber force field (GAFF) parameters for the ligand atoms. Considering the conformational effect of the side chains on the reaction barriers, a 10 ns molecular dynamic simulation was performed for each system (with or without potassium at site 1). Three snapshots at 3, 6, and 9 ns were extracted from each simulation as initial structure for QM/MM mechanism study. Additional information on the MD simulations as well as QM/MM optimized structures from MD snapshots is provided in the Supporting Information.
Two different models were used for the study of the protonation states of hydroxamate complex: Model 1, shown in Figure 3 on the left, includes the zinc ion, the side chains of H142, H143, D178, H180, D267, and Y306, and atoms of SAHA near the catalytic zinc ion in the QM region during QM/MM calculation. This is consistent with the model of Zhang and co-workers, which lacks the aspartate portion of the catalytic dyad (D176 and D183). It was argued that in the case of serine proteases, inclusion of these residues does not lead to different results,45 but the effects for HDACs are not known. Therefore, D176 and D183 were included in Model 2, shown in Figure 3 on the right. Hydrogen atoms were added to saturate the dangling bonds at the QM/MM interface. For the mechanistic studies, the calculations were only carried out at the model 2 level, and the detailed process is shown in Figure 5.
Figure 3.

QM regions of models 1 (left) and 2 (right) shown for the optimized structures of SAHA-HDAC8 complex. Blue lines indicate the QM/MM boundary region.
Figure 5.

QM/MM deacetylation mechanism of HDAC. Energies (ONIOM(M052X/(6-31G*, SDD):AMBER), in kcal/mol) of optimized structures without or with (in parentheses) the presence of potassium at site 1 using the initial conformations from minimized structures of crystal structure without or with potassium at site 1.
Computational Details
All QM/MM optimizations were carried out using the ONIOM method with electronic embedding in Gaussian 09.46 The TAO package47 was used for the preparation and analysis of the ONIOM calculations. The QM region (indicated by the blue lines in Figure 3) was treated using the M05-2X48 functional together with the 6-31G* basis set for all main group elements and the Stuttgart ECP basis set (SDD)49 for Zn2+. This level of theory was found to give the best results in earlier benchmark studies.21,48 The results from single point calculations at a larger basis set or other functionals are given in Supporting Information. The Amber force field was used to treat the MM region. Atoms within a distance of 6 Å from the model 2 QM boundary were fully optimized, while the other atoms in the system were fixed to decrease the energy fluctuation and to reduce CPU requirements. A small step size (set by Gaussian iop: 1/8 = 1) was used during optimization, and structures were gradually changed to avoid abnormal conformational change in the classically treated region. Transition states were confirmed by the eigenvectors with negative eigenvalues. The lowest 10 frequencies were calculated with Gaussian 09 keywords freq(Nfreq = 10), and only one imaginary frequency was found for transition state. Partial charges were derived using the ESP fitting scheme.
Results and Discussion
Protonation States of Hydroxamates
The simplest, yet biomedically most important model to study the effect of the expanded model 2 is the investigation of the protonation state of hydroxamic acids bound to HDAC8. The pKa of aliphatic hydroxamic acids is 9.4.50 Computational studies of the zinc complexation of acids in TACE suggest that their acidity increases by ∼3.3 pKa units upon binding to the metal.18 Experimentally, the position of the proton is difficult to observe directly, but computational studies of small active site models by us17 and others19,26 indicated a negative hydroxamate in the active site, while QM/MM studies31 suggest a neutral hydroxamic acid.
To investigate the model dependence of the protonation state, we started the QM/MM optimization of models 1 and 2 from the same initial structure where SAHA is protonated and both H142 and H143 are singly protonated at the δ position. As shown in Figure 3 left, optimization of model 1 leads to a structure very similar to the one obtained by Zhang and co-workers31 where the hydroxamic acid remains uncharged. However, optimization of model 2 leads to a spontaneous proton transfer from SAHA to the nearby histidine H142 with very little reorganization of the remaining QM region. This suggests that D176 stabilizes the doubly protonated H142 by sharing of the proton between the two residues as is to be expected in a charge-relay system. Indeed, calculation of the Wiberg bond index between the δ-H and δ-N of H142 gave a value of 0.54, indicating a weak N–H bond. This is also indicated by the elongated N–H bond length of 1.07 Å. For comparison, the Wiberg bond index and the distance between the δ-H and δ-N of H143 were 0.65 and 1.03 Å, respectively.
To further study whether these differences are a function of the different models or rather result from the differences in the QM/MM methodology used here and in the earlier studies,31 we scanned the O–H distance in the hydroxamic acid as the deprotonation coordinate to obtain relative energies of the protonated and deprotonated states in the two models. As shown in Figure 4, the neutral hydroxamic acid in model 1 is calculated to be 4.4 kcal/mol lower in energy than the deprotonated form, similar to the 3.8 kcal/mol found in Zhang’s work.31 In contrast, the deprotonated hydroxamate is calculated to be 3.5 kcal/mol more stable for the case of model 2. The TSA-HDAC8 complex (pdb code: 1T64) was also investigated, and the results are consistent with those of the SAHA-HDAC8 complex (see Supporting Information). These results strongly support the hypothesis that the hydroxamic acid is deprotonated after binding with the zinc ion in the active site of HDAC8.
Figure 4.

Energy change during O–H distance scan for two active site models.
The finding that inclusion of the complete catalytic dyad in the quantum region shown in Figure 4 has a profound effect in HDAC8, but not in Born–Oppenheimer ab initio QM/MM MD simulations of a serine protease45 points to some interesting consequences of the dual character of HDACs as both serine- and metalloproteases. While in the serine protease, the stabilization of the protonated histidine by the aspartate is not sufficient to deprotonate the serine side chain, the combination of the lower pKa of hydroxamic acid in combination with zinc binding leads to the formation of the bidentate hydroxamate binder. Although a method dependence of these results (i.e., optimization vs MD on a subnanosecond time scale) cannot be excluded, our results suggest that for a proper description of this process, the aspartate residue needs to be included in the quantum region of the QM/MM optimizations.
The results presented also provide a rationalization of a number of experimental observations. Analysis of the available high-resolution (<2.3 Å) X-ray structures of HDAC8 bound to hydroxamic acids (pdb codes 1T64, 2V5X, and 1VKG), shown in Table 1, indicates that the distance between the zinc and the carbonyl oxygen is longer than that between the zinc and the hydroxyl oxygen. The same trend is observed in the three HDAC8 structures where the zinc is replaced by a different divalent metal (pdb codes 3MZ4, 3MZ6, and 3MZ7, see Table S1 in the Supporting Information). These observations are in good agreement with the charge distribution derived from model 2, where the hydroxyl oxygen has a substantially more negative partial charge after deprotonation but are more difficult to explain for a protonated hydroxamic acid as obtained for model 1.
Table 1. Zinc–Oxygen Distances and Partial Charges.
| Ocarbonyl | Ohydroxyl | |
|---|---|---|
| Zn–O Distance [Å] | ||
| 1T64 | 2.22 | 2.00 |
| 2V5X | 2.47 | 2.07 |
| 1VKG | 1.91 | 1.97 |
| Partial Charge | ||
| model 1 | –0.31 | –0.36 |
| model 2 | –0.54 | –0.82 |
The hypothesis that the acidity of hydroxamic acid (pKa = 9.4) is increased by ∼3.3 pKa units upon binding to the active site of HDAC8 to allow deprotonation by a histine (pKa ∼ 6)17 also provides insights into the experimental findings for class IIa HDACs (HDAC4, 5, 7, and 9), where Y306 is mutated to a conserved histidine. It can be hypothesized that because the hydrogen bond of Y306 to the carbonyl oxygen is missing, the negative charge is not sufficiently stabilized to allow the deprotonation of hydroxamic acids in class IIa HDACs. This is consistent with the observation that hydroxamic acids are relatively weak inhibitors of class IIa HDACs12 and bind in a monodentate fashion in the crystal structure of HDAC7.51 It also suggests the intriguing possibility that the inhibition of class IIa HDACs by hydroxamic acids could be greatly improved by a subtle shift in acidity by appropriate substitution. This would provide new approaches to the inhibition of class IIa HDACs, for which relatively few potent inhibitors are available at this time.12
The Reaction Mechanism of HDAC8
The results discussed above emphasize the importance of a suitable model system in the calculations and question the mechanistic conclusions drawn from model 1. We therefore revisited the two mechanisms shown in Scheme 1 to define the role of each of the two potential proton shuttle systems H142/D176 and H143/D183, which will crucially depend on the protonation states in the active site: one where H143 acts as the general base, which is in analogy to the mechanism proposed previously by Zhang and co-workers,27,28 and the other derived from the mechanism proposed by Finnin et al.,24 where H142 serves as the general base. The results of these studies are summarized in Figure 5.
We first discuss the mechanism without the presence of the potassium at site 1. As the initial structure, we chose the structure where both H142 and H143 are protonated in the δ-position. Both Zhang’s study28 and our own work (Figure S4) indicate that the optimized structure, 1, (Figure 6) has better structural agreement with the available X-ray data than other possible protonation states, e.g. with H142 or H143 doubly protonated. The agreement between the previous28 and the present studies also shows that the effect of the expanded QM region is small for the reactant state. The coordinated water forms two hydrogen bonds with H142 and H143, respectively. One of the His-Asp dyads may function as a base to abstract one proton from the coordinated water and then facilitate the nucleophilic attack of water oxygen on carbonyl carbon.
Figure 6.
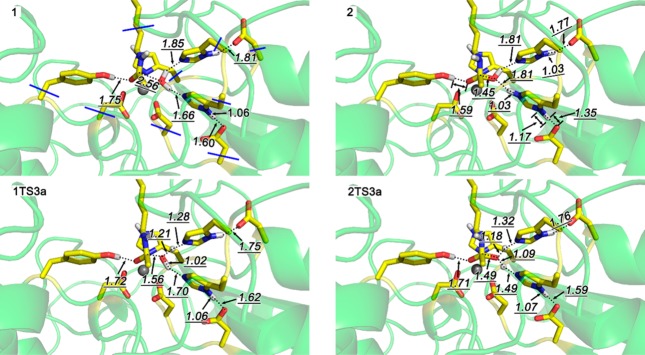
Optimized structures of selected stationary points without the presence of potassium at site 1.
In agreement with the previously described mechanism, we calculated 1TS3a for the activation of the water nucleophile by H143 as the general base, with an activation energy of 12.4 kcal/mol. However, we also identified an alternative pathway where H142 acts as the general base and that is, with an activation energy of 8.5 kcal/mol, significantly more favorable. More importantly, the product 2 with the protonated H142/D176 dyad formed in this pathway is predicted to be more stable than the corresponding product 3a by 8.0 kcal/mol. This pathway requires the transfer of the proton from H142 to H143 for all the latter to act as a general acid for the cleavage of the amide bond. Although the difference to 1TS3a is small, this step, with an activation energy of 11.1/kcal/mol, is the lowest-energy pathway.
The preference for H142 as the general base can be understood in terms of the environment of the two catalytic dyads. The H142/D176 is more deeply buried and positioned in a hydrophobic environment. The results of our calculations indicate, in agreement with earlier qualitative arguments24 and the results of small quantum models,19 that neutralization of this dyad will be thermodynamically and kinetically more favorable compared to the more solvent exposed H143/D183 dyad.
The next steps in the reaction are a reorientation of H143 toward the amide nitrogen, followed by an essentially barrierless proton transfer to form the N-protonated amide 4. This undergoes rapid cleavage of the C–N bond, leading to the product complex 5. During this process, a proton is transferred from the tetrahedral intermediate to H143 in a concerted fashion.
Figure 6 shows the geometries of selected structures along the reaction pathway (for other structures, see Figure S7 in the Supporting Information). In 1 (top left), the common starting material of both mechanisms, the hydrogen bond from the water nucleophile to H142 is with 1.66 Å substantially shorter than the one to H143 with 1.85 Å, in agreement with the energetic argument discussed above. As a result, the proton is transferred from the active site water to H142 to yield the tetrahedral intermediate 2 (top right), where the negative charge is stabilized by a hydrogen bond of 1.59 Å lengths to Y306. It is noteworthy that in the H142/D176 proton shuttle system, the proton is not fully transferred. Instead, the N–H and O–H bonds are with 1.17 and 1.35 Å, respectively, quite similar. This is consistent with a partial sharing of the proton and rationalizes the stabilization of 2. The rate-determining step for the mechanism proposed by Zhang is the transition structure for the proton transfer to H143, 1TS3a (bottom left), leading directly to 3a. The structure is very similar to the one originally reported28 and does not exhibit the proton sharing, presumably due to the solvation stabilization of the charges in the H143/D183 dyad on the surface of the protein. The rate-determining step in the second mechanism is the concerted transfer of two protons from the H142/D176 to the tetrahedral intermediate and from the tetrahedral intermediate to the H143/D183 dyad. At this point, the two mechanisms converge in 3a, as shown in Figure 5.
The presence of a potassium at site 1 (denoted here by apostrophe) leads to some interesting differences in the reaction pathway. As shown in Figure 5, 2′ and 5′ significantly destabilized to the point where the energy difference between 1TS3a′ and 2TS3a′ is very small. This is noteworthy because in previous simulations, the model with a potassium at site 1 was found to have a lower energy of activation than the one without.28 Interestingly, both acceleration and inhibition of the reaction by potassium have been reported in experimental studies.33,34 The instability of 5′ creates a driving force for further proton transfers between the neutral lysine, acetic acid, and the protonated H142, to get a to more stable state. However, these steps occur beyond the actual enzymatic reaction and are therefore not discussed here.
Analysis of the structures of 2′ and the relevant transition structures, shown in Figure 7, provides a structural rationale for the increase in energy. Coordination of K+ with the Oδ of D176 reduces the hydrogen-bond interaction between D176 and H142, which increases the energies of species where H142 is protonated by over 10 kcal/mol. This analysis is further supported by the direct comparison of the two sets of structures shown in Figures 6 and 7, which are derived from the same initial structure, substrate-HDAC8 complex crystal structure. Comparison of the structure of 2 (without potassium at site 1) and 2′ (with potassium at site 1) shows that the proton sharing between D176 and H142 is decreased, presumably due to electrostatic effect of the nearby potassium. Similarly, in pairs of structures (1 and 1′, 2 and 2′, 1TS3a and 1TS3a′, 2TS3a and 2TS3a′), the distance between tyrosine hydroxyl hydrogen and substrate carbonyl oxygen as well as the distance between substrate carbonyl carbon and water oxygen atom is increased. This is consistent with a decreased relative stability of these structures due to the presence of potassium at site 1.
Figure 7.

Optimized structures of selected stationary points with the presence of potassium at site 1; apostrophe symbol indicates the presence of potassium at site 1.
Finally, we investigated the question whether the results are dependent on the initial conformation. Snapshots at 3, 6, and 9 ns were extracted from 10 ns MD simulations with and without potassium ion at site 1, and structures of the process from 1 to 3a were optimized by QM/MM computations using the MD snapshots as initial structures. The structures and the energies along the pathways are shown in the Supporting Information. These results show that the basic mechanistic findings described do not substantially change as a function of the side chain orientations. Specifically, the stepwise pathway via intermediate 2 is predicted to be preferred in the absence of the potassium at site 1, while the pathways are too close to distinguish in the presence of the potassium for all three starting structures. However, it should be noted that larger conformational changes relative to the X-ray structures will not be uncovered by the 10 ns MD simulations.
Conclusion
The results presented here show that the inclusion of the aspartic acid residues of the two HDAC charge-relay systems into the QM region of the QM/MM optimization significantly affects the results of computational studies of inhibition and mechanism of class I HDACs. Several of the findings are in disagreement with previous Born–Oppenheimer ab initio QM/MM MD simulations,27,28,31 but in good agreement with QM studies of model systems as well as the available experimental results. Specifically, the present study strongly suggest that the charge state of the widely used hydroxamic acid HDAC inhibitors in the active site is indeed negative. This confirms the computational results from small model studies17,19 and explains a number of experimental observations discussed earlier. It is also consistent with the proposal that the active species in the HDAC inhibition by largazole is the thiolate,14 which would be hard to rationalize with a protonated thiol. Although HDAC8, a class I HDAC, was studied here, the findings have also experimentally verifiable implications for the design of more potent class IIa HDAC inhibitors by emphasizing the importance of the pKa of the zinc binding group.
Consistent with the relative basicity of the two catalytic dyads in HDAC8, but in contrast to the mechanism suggested by the earlier Born–Oppenheimer ab initio QM/MM MD simulations, the computational results support the original mechanistic proposal by Finnin et al.24 The atomistic details provided by the calculations not only point to the role of the H142/D176 dyad as the general base, but also provide structural support for the previously postulated role51 of Y306 in stabilizing the tetrahedral intermediate. This has again important consequences for the design of inhibitors of class IIa HDACs, where Y306 is mutated to a histidine. Our results show that the catalytic activity is inhibited by the presence of a potassium ion at site 1. The calculated energies for this system are too close in energy to unambiguously distinguish between the classic mechanism by Finnin24 and the “unexpected” mechanism proposed by Zhang and co-workers,27 but they provide a structural rational for the experimentally observed inhibition of the reaction by potassium.33,34
Finally, the results provide new insights into the unique position of HDACs as a mechanistic cross between serine and metalloproteases containing two rather than one charge relay system. A shift of pKa of substrates and inhibitors upon binding to the zinc modulates the protonation state of inhibitors as well as the mechanism of the reaction to a point where both dyads play an active role in the mechanism of the amide hydrolysis by HDACs. As a result, neither serine proteases, where inclusion of the aspartate into the QM region of Born–Oppenheimer ab initio QM/MM MD simulations did not change the results,45 nor metalloproteases alone are good models for the mechanism of class I HDACs. Rather, they are interesting examples of how different active site motifs can be combined to generate new mechanistic variations.
Acknowledgments
We gratefully acknowledge the support of this work by the National Institutes of Health (RO1 CA152314), the National Science Foundation of China (21133002, 21232001), the MOST of China (2013CB911501), and the Shenzhen Peacock Program (KQTD201103). We also thank Dr. Lung-Wa Chung(SUSTC) for useful suggestions with the ONIOM calculations in G09 and Dr. Xinhao Zhang(PKUSZ) for helpful discussions.
Supporting Information Available
Results of the TSA-HDAC8 ONIOM calculations, single point energy calculations at other basis sets or functionals, conformational effects on reaction barrier, and pdb files of all ONIOM optimized structures and full refs (43) and (46). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. Nat. Rev. Drug Discovery 2012, 11, 384. [DOI] [PubMed] [Google Scholar]
- Dawson M. A.; Kouzarides T. Cell 2012, 150, 12. [DOI] [PubMed] [Google Scholar]
- Suvà M. L.; Riggi N.; Bernstein B. E. Science 2013, 339, 1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane A. A.; Chabner B. A. J. Clin. Oncol. 2009, 27, 5459. [DOI] [PubMed] [Google Scholar]
- Carew J. S.; Giles F. J.; Nawrocki S. T. Cancer Lett. 2008, 269, 7. [DOI] [PubMed] [Google Scholar]
- Johnstone R. W.; Licht J. D. Cancer Cell 2003, 4, 13. [DOI] [PubMed] [Google Scholar]
- Marks P. A.; Rifkind R. A.; Richon V. M.; Breslow R.; Miller T.; Kelly W. K. Nat. Rev. Cancer 2001, 1, 194. [DOI] [PubMed] [Google Scholar]
- Wiech N. L.; Fisher J. F.; Helquist P.; Wiest O. Curr. Top. Med. Chem. 2009, 9, 257. [DOI] [PubMed] [Google Scholar]
- Pipalia N. H.; Cosner C. C.; Huang A.; Chatterjee A.; Bourbon P.; Farley N.; Helquist P.; Wiest O.; Maxfield F. R. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoretti L.; Lee Y.; Goodson H. V. J. Mol. Biol. 2004, 17. [DOI] [PubMed] [Google Scholar]
- Witt O.; Deubzer H. E.; Milde T.; Oehme I. Cancer Lett. 2009, 277, 8. [DOI] [PubMed] [Google Scholar]
- Bradner J. E.; West N.; Grachan M. L.; Greenberg E. F.; Haggarty S. J.; Warnow T.; Mazitschek R. Nat. Chem. Biol. 2010, 6, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D. F.; Wiest O.; Helquist P.; Lan-Hargest H. Y.; Wiech N. L. Bioorg. Med. Chem. Lett. 2004, 14, 707. [DOI] [PubMed] [Google Scholar]
- Bowers A.; Greshock T.; West N.; Estiu G.; Schreiber S.; Wiest O.; Williams R.; Bradner J. J. Am. Chem. Soc. 2009, 131, 2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minucci S.; Pelicci P. G. Nat. Rev. Cancer 2006, 6, 38. [DOI] [PubMed] [Google Scholar]
- Bertrand P. Eur. J. Med. Chem. 2010, 45, 2095. [DOI] [PubMed] [Google Scholar]
- Wang D.; Helquist P.; Wiest O. J. Org. Chem. 2007, 72, 5446. [DOI] [PubMed] [Google Scholar]
- Cross J. B.; Duca J. S.; Kaminski J. J.; Madison V. S. J. Am. Chem. Soc. 2002, 124, 11004. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Proft F. D.; Loverix S.; Tourwé D.; Geerlings P. Bioorg. Med. Chem. 2005, 13, 3987. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Loverix S.; Geerlings P.; Tourwé D. Bioorg. Med. Chem. 2005, 13, 6070. [DOI] [PubMed] [Google Scholar]
- Chen K.; Xu L.; Wiest O. J. Org. Chem. 2013, 78, 5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A.; Volpari C.; Gallinari P.; Jones P.; Mattu M.; Carfi A.; De Francesco R.; Steinkuhler C.; Di Marco S. EMBO Rep. 2007, 8, 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 306F mutation used to render the enzyme catalytically inactive was manually reverted.
- Finnin M. S.; Donigian J. R.; Cohen A.; Richon V. M.; Rifkind R. A.; Marks P. A.; Breslow R.; Pavletich N. P. Nature 1999, 401, 188. [DOI] [PubMed] [Google Scholar]
- Gantt S. L.; Gattis S. G.; Fierke C. A. Biochemistry 2006, 45, 6170. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Van Alsenoy C.; De Proft F.; Martins J. C.; Tourwe D.; Geerlings P. Org. Biomol. Chem. 2003, 1, 2951. [DOI] [PubMed] [Google Scholar]
- Corminboeuf C.; Hu P.; Tuckerman M. E.; Zhang Y. J. Am. Chem. Soc. 2006, 128, 4530. [DOI] [PubMed] [Google Scholar]
- Wu R.; Wang S.; Zhou N.; Cao Z.; Zhang Y. J. Am. Chem. Soc. 2010, 132, 9471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Theor. Chem. Acta 2006, 116, 43. [Google Scholar]
- HID means that histidine is singly protonated at δ nitrogen position.
- Wu R.; Lu Z.; Cao Z.; Zhang Y. J. Am. Chem. Soc. 2011, 133, 6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A.; Volpari C.; Filocamo G.; Casavola E. C.; Brunetti M.; Renzoni D.; Chakravarty P.; Paolini C.; De Francesco R.; Gallinari P.; Steinkühler C.; Di Marco S. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 15064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantt S. L.; Joseph C. G.; Fierke C. A. J. Biol. Chem. 2010, 285, 6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werbeck N. D.; Kirkpatrick J.; Reinstein J.; Hansen D. F. ChemBioChem 2014, 15, 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunze M. B. A.; Wright D. W.; Werbeck N. D.; Kirkpatrick J.; Coveney P. V.; F H. D. J. Am. Chem. Soc. 2013, 135, 17862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.; Po H.; Wang S.; Cao Z.; Zhang Y. J. Chem. Theory Comput. 2010, 6, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estiu G.; West N.; Mazitschek R.; Greenberg E.; Bradner J.; Wiest O. Bioorg. Med. Chem. 2010, 18, 4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot J.; Chakravarty P.; Chenard M.; Close J.; Cruz J.; Dahlberg W.; Fleming J.; Hamblett C.; Hamill J.; Harrington P.; Harsch A.; Heidebrecht R.; Hughes B.; Jung J.; Kenific C.; Kral A.; Meinke P.; Middleton R.; Ozerova N.; Sloman D.; Stanton M.; Szewczak A.; Tyagarajan S.; Witter D.; Secrist P.; Miller T. A. Bioorg. Med. Chem. Lett. 2008, 18, 973. [DOI] [PubMed] [Google Scholar]
- Somoza J. R.; Skene R. J.; Katz B. A.; Mol C.; Ho J. D.; Jennings A. J.; Luong C.; Arvai A.; Buggy J. J.; Chi E.; Tang J.; Sang B.-C.; Verner E.; Wynands R.; Leahy E. M.; Dougan D. R.; Snell G.; Navre M.; Knuth M. W.; Swanson R. V.; McRee D. E.; Tari L. W. Structure 2004, 12, 1325. [DOI] [PubMed] [Google Scholar]
- Weerasinghe S. V. W.; Estiu G.; Wiest O.; Pflum M. K. H. J. Med. Chem. 2008, 51, 5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M. P.; Pincus D. L.; Rapp C. S.; Day T. J. F.; Honig B.; Shaw D. E.; Friesner R. A. Proteins: Struct., Funct., Bioinform. 2004, 55, 351. [DOI] [PubMed] [Google Scholar]
- Estiu G.; West N.; Mazitschek R.; Greenberg E.; Bradner J.; Wiest O. Bioorg. Med. Chem. 2010, 18, 4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; . et al. Amber12; University of California: San Francisco, CA, 2012.
- Hoops S. C.; Anderson K. W.; Merz K. M. J. Am. Chem. Soc. 1991, 113, 8262. [Google Scholar]
- Zhang Y.; Ku J.; McCammon J. A. J. Am. Chem. Soc. 2002, 124, 10572. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; . et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2009.
- Tao P.; Schlegel H. B. J. Comput. Chem. 2010, 31, 2363. [DOI] [PubMed] [Google Scholar]
- Sorkin A.; Truhlar D. G.; Amin E. A. J. Chem. Theory Comput. 2009, 5, 1254. [DOI] [PubMed] [Google Scholar]
- Dolg M.; Wedig U.; Stoll H.; Preuss H. J. Chem. Phys. 1987, 86, 866. [Google Scholar]
- Wise W. M.; Brandt W. W. J. Am. Chem. Soc. 1955, 77, 1058. [Google Scholar]
- Schuetz A.; Min J.; Allali-Hassani A.; Schapira M.; Shuen M.; Loppnau P.; Mazitschek R.; Kwiatkowski N. P.; Lewis T. A.; Maglathin R. L.; McLean T. H.; Bochkarev A.; Plotnikov A. N.; Vedadi M.; Arrowsmith C. H. J. Biol. Chem. 2008, 283, 11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.