

Abstract
Mutations in LRRK2 are one of the primary genetic causes of Parkinson's disease (PD). LRRK2 contains a kinase and a GTPase domain, and familial PD mutations affect both enzymatic activities. However, the signaling mechanisms regulating LRRK2 and the pathogenic effects of familial mutations remain unknown. Identifying the signaling proteins that regulate LRRK2 function and toxicity remains a critical goal for the development of effective therapeutic strategies. In this study, we apply systems biology tools to human PD brain and blood transcriptomes to reverse-engineer a LRRK2-centered gene regulatory network. This network identifies several putative master regulators of LRRK2 function. In particular, the signaling gene RGS2, which encodes for a GTPase-activating protein (GAP), is a key regulatory hub connecting the familial PD-associated genes DJ-1 and PINK1 with LRRK2 in the network. RGS2 expression levels are reduced in the striata of LRRK2 and sporadic PD patients. We identify RGS2 as a novel interacting partner of LRRK2 in vivo. RGS2 regulates both the GTPase and kinase activities of LRRK2. We show in mammalian neurons that RGS2 regulates LRRK2 function in the control of neuronal process length. RGS2 is also protective against neuronal toxicity of the most prevalent mutation in LRRK2, G2019S. We find that RGS2 regulates LRRK2 function and neuronal toxicity through its effects on kinase activity and independently of GTPase activity, which reveals a novel mode of action for GAP proteins. This work identifies RGS2 as a promising target for interfering with neurodegeneration due to LRRK2 mutations in PD patients.
INTRODUCTION
Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene have emerged as the most common genetic determinant of Parkinson's disease (PD), causing late-onset, familial autosomal dominant PD and accounting for up to 40% of PD cases in certain ethnic populations (1,2). LRRK2-associated PD is clinically and neurochemically indistinguishable from sporadic PD. The most prevalent mutation in LRRK2, G2019S, has also been found in 1–2% of sporadic PD cases (2,3). Animal models demonstrate that the G2019S mutation in LRRK2 can induce degeneration of dopaminergic (DA) neurons, which are the primary target of neurodegeneration in PD (4,5). These findings suggest that LRRK2 plays a pivotal role in the pathogenesis of human PD.
LRRK2 is a large, multi-domain protein of 2527 amino acids. It contains two catalytic domains, a kinase domain with highest sequence homology to mitogen-activated protein kinase kinase kinase (MKKK) and receptor-interacting protein (RIP) kinase families and a Ras-of-complex proteins (ROC) GTPase domain, flanked by a C-terminal of ROC (COR) domain (6). In vitro, LRRK2 can autophosphorylate and phosphorylate generic substrates (e.g. myelin basic protein or LRRKtide) as well as a range of putative substrates such as ArfGAP1, 4E-BP, moesin and β-tubulin (7–11). However few, if any, of these putative substrates have been broadly verified by multiple independent groups and validated as authentic physiological substrates of LRRK2 kinase activity in vivo (12,13). All six mutations that clearly segregate with disease occur in the kinase domain (G2019S and I2020T), Roc GTPase domain (R1441C/G/H) or COR domain (Y1699C) (6). These pathogenic mutations have been shown to affect kinase and GTPase activities to varying degrees: the R1441C/G/H and Y1699C variants impair GTP hydrolysis without consistent effects on kinase activity, while the predominant G2019S mutation enhances kinase activity with no effect on GTPase activity (6,14–18). Although the mechanisms remain unclear, these findings suggest that both activities may play a role in mediating neurodegeneration. Indeed, kinase activity is required for the pathogenic effects of the G2019S mutation in primary neurons and in rodents (5,6,17,19).
A number of studies have implicated LRRK2 in the regulation of a wide variety of biological processes such as protein translation (20), cytoskeletal processes (9,21), vesicular dynamics (22), response to mitochondrial damage (23) and autophagy (24,25). This complexity of LRRK2 biology has made it extremely difficult to understand the contributions of the kinase and GTPase domains to the function of LRRK2 (26). In particular, the upstream signaling mechanisms that control LRRK2 GTPase and kinase activities and the pathogenic effects of familial mutations remain unknown (26,27). Identifying the signaling proteins that regulate LRRK2 function and toxicity remains a critical outstanding goal for the development of effective therapeutic strategies.
In the present study, we used an in silico approach to elucidate the gene regulatory network linked to LRRK2. We applied a network-based algorithm to reverse-engineer the LRRK2 network based on human PD blood and brain transcriptomes. This work highlights in particular the role of the signaling GTPase-activating protein (GAP) RGS2 as a key regulator of LRRK2 activity, function and neuronal toxicity.
RESULTS
Elucidation of the LRRK2 regulatory network
The context likelihood of relatedness (CLR) algorithm is designed to analyze state-dependent genome-wide expression data based on the degree of synchrony of transcript levels, using mutual information as a metric for scoring the similarity between expression levels of two transcripts (28). CLR identifies the component gene regulatory networks among large numbers of subjects. We employed the CLR algorithm to analyze, in an unbiased manner, a set of 119 publicly available array data sets from the Substantia Nigra pars compacta (SNpc), frontal cortex and whole blood of human PD patients and control cases (29,30). Whole blood was included to enhance the input of data from tissues that were not in terminal stages of degeneration. Recent studies support the utility of leukocytes for study of PD by identifying putative PD biomarkers using blood transcriptional profiles from LRRK2 G2019S carriers (30,31). Gene–gene interactions were evaluated for statistical significance using the Z-score metric, which is considered an alternative for the t-test P-value metric. Interactions with a mutual information Z-score ≥2.0 (corrected for background) were considered significant. A LRRK2-centered association sub-network with first and second neighbors was derived accordingly (Fig. 1A, Supplementary Material, Fig. S1). Mutual information Z-scores between individual gene nodes in the network are listed in Supplementary Material, Table S1.
Figure 1.

Reverse-engineering of the LRRK2 gene regulatory network. (A) The CLR algorithm was applied to genomic profiling data from human PD patients and control cases to generate an in silico LRRK2 gene regulatory network. Genes among the LRRK2 centered network represent 16 functional groups. See also Supplementary Material, Figure S1 and Table S1. (B) LRRK2 centered sub-network focused on the PD genes: PINK1, DJ-1 and PARKIN. PD-linked genes are represented in pink. Genes shown in blue represent first neighbors, green represents second neighbors, yellow colored genes identify genes (or proteins) that were previously shown to be associated with LRRK2 or genes highlighted by this study; many of the yellow-colored genes are also LRRK2 first neighbors. See also Supplementary Material, Table S2.
Members of the network regulatory map were grouped to highlight the functional categories of the predicted LRRK2 interactors (Fig. 1A). The resulting LRRK2-centered network correctly identified many of the genes/proteins previously associated with LRRK2, including members of the Wnt signaling pathway, the MAPK/ERK pathway such as MKK7 (MAP2K7) and JIP1 (MAPK8IP1), beta tubulin (TUBB), FAS (which binds to FADD, a documented LRRK2-binding protein) and the PD-associated genes PARK2 (parkin), PINK1 and PARK7 (DJ-1) (Fig. 1A, Supplementary Material, Fig. S1 and Table S1) (8,32–37). Many other genes in the LRRK2 regulatory network represent intriguing novel associations such as EIF4G1, which is a first neighbor of LRRK2 and encodes for a key component of the protein translation machinery, or BCL2L1, also a LRRK2 first neighbor and member of the BCL-2 family of apoptosis regulators. Genes such as actin (ACTA1), and the Wnt signaling members FZD1 and TNKS are connected to large groups of secondary regulatory neighbors, and are highlighted in the network as regulatory ‘hubs’ (Fig. 1A, Supplementary Material, Fig. S1). The resulting network highlights the diversity of processes that coordinate with LRRK2, including processes important for synaptic transmission, cytoskeletal function, RNA processing, mitochondrial function and autophagy (Fig. 1A).
Identification of regulatory network genes that modify LRRK2 function in vivo
The MNI and CLR analyses identified 181 C. elegans and 200 human genes whose expression levels are highly coordinated with those of LRRK2. To determine the extent to which these genes modify the function of LRRK2 in vivo, we performed an RNAi high-throughput screen utilizing C. elegans.
We had previously determined that DA neurons in C. elegans are selectively sensitive to rotenone toxicity, showing neuronal death at doses 100-fold less than doses required to kill nematodes (23). This work also demonstrated that LRRK2 significantly protects C. elegans and, in particular, DA neurons, against rotenone toxicity (23). We utilized the neuroprotection phenotype as a screening platform to evaluate the contribution of the systems biology predicted interactors to LRRK2 function (Fig. 2A).
Figure 2.

Validation of LRRK2 network interactors. (A) RNAi targeted to the putative LRRK2 interactors were fed to WT LRRK2 expressing C. elegans, and DA survival in the presence of rotenone was quantified. (B) Algorithm to quantify the effects of RNAi by imaging GFP expression in the DA neurons. Scoring: 3, the cell body and dendrite were intact and similar to untreated nematodes; 2, a shrunken cell body or a loss of dendritic processes; 1, loss of or barely visible dendritic process and cell body. Hits were defined as survival outside two standard deviations from the control RNAi. (C) Representative effects of genes that modify DA neuron survival.
Caenorhabditis elegans expressing wild-type human LRRK2 driven by the pan-neuronal synaptobrevin promoter (snb::LRRK2) and eGFP driven by the dopamine transporter promoter (dat-1::GFP) were used as a platform for the screen (23). The RNAi hypersensitivity mutation, rrf-3 (pk1426), was introduced to enhance the sensitivity of the neurons to RNAi. No observable phenotypic changes were observed upon introduction of the rrf-3 mutation, and the LRRK2-mediated protection phenotype was replicated (Supplementary Material, Fig. S2A). We screened a set of genes generated from the MNI and CLR analyses. A PSI blast of the 200 genes from the human CLR network was used to identify 506 putative C. elegans orthologs. This set was combined with the 181 nematode genes identified in the MNI analysis, yielding a total of 687 genes for the C. elegans study. The LRRK2 nematodes were synchronized by egg-laying on plates carrying individual RNAi vectors targeted to the regulatory network genes. Larval stage four nematodes were then treated with rotenone for 24 h, and the survival of their dopaminergic neurons evaluated by visualizing GFP expressed in the DA neurons (Fig. 2A). We devised a scoring system that took into account the number of neurons and their morphology (Fig. 2B). Previous work demonstrated a strong correlation between dat-1::GFP and DA neuron survival, and determined that the CEP and ADE DA neurons are selectively sensitive to PD-associated neurotoxins (38, 39). Replicated measurements with an empty RNAi vector, PL4440, indicated that the scoring system had a low baseline fluctuation. RNAi that significantly altered the DA neuron survival outside of two standard deviations from the empty vector control were considered hits of the screen. Selected RNAi constructs were sequenced and found to be specific for the predicted target gene and did not exhibit significant off-target effects (Supplementary Material, Fig. S2B and C).
The cumulative results indicated that 280 (40%) of the systems biology predicted genes altered LRRK2-mediated DA neuron survival. The validated systems biology candidates were distributed across the LRRK2-centered PD network (Supplementary Material, Table S3). Seventy-one percent of the validated candidates decreased DA neuron survival, while 29% increased survival. The hits were annotated using Wormbase and GO analysis (Supplementary Material, Table S3). The ‘autophagy/endosomal/lysosomal’ and the ‘Parkinson's disease related’ groups consistently exhibited strong negative effect sizes with −51 and −43%, respectively (Fig. 2C, Supplementary Material, Table S3). The autophagy-related genes, including unc-51, vps-34 (VPS34, PIK3C3), vps-33.1, hda-6 (HDAC6), ctns-1, num-1 and unc-57, showed a consistent inhibitory effect on the protection phenotype, with the largest effect evident for knockdown of vps-34 (PIK3C3). The 81% reduction of DA neuron survival observed upon knockdown of vps-34(PIK3C3) was one of the strongest decreases of DA neuron survival in the C. elegans LRRK2 line (Fig. 2C, Supplementary Material, Table S3). These results suggest that protection by WT LRRK2 requires normal autophagic function.
In contrast, the ‘NFkB/TGFbeta/WNT’ and ‘Axon Guidance/Cytoskeleton’ groups had the largest positive effect sizes with 22 and 18%, respectively (Supplementary Material, Table S3); the smaller average effect sizes reflect the interaction of positive and negative effectors in these groups. It is interesting to note that knockdown of transcripts in the mitochondrial gene category improved survival, while knockdown of the PD-associated genes djr-1.1(DJ-1), pdr-1(PARKIN) and pink-1(PINK1) resulted in −43, −49 and −38% decreases in DA neuron survival, respectively (Fig. 2C, Supplementary Material, Table S3). Previous work has identified functional interactions between all four of PD-linked genes, which supports the current results and suggests that these genes function together in an integrated PD gene network (37) (Fig. 2C).
Genes associated with MAPK signaling have been highly implicated in LRRK2 function. Knockdown of MAPK signaling genes, including jkk-1 (MAP2K7), mek-2 (MAP2K2) and mpk-1 (MAPK1) reduced DA neuronal survival by −25, −47 and −69%, respectively (Supplementary Material, Table S3). This replicates findings from our group and others showing a strong interaction between MKK6 (MAP2K6), MKK7 (MAP2K7), ERK (MAPK) and JNK (MAP2K8) in LRRK2 function (32, 34, 35, 40, 41). Another kinase implicated in neurodegenerative processes, cdk-5 (CDK5), also exhibited strong effects. Knockdown of cdk-5 enhanced neuron survival by 99% (Supplementary Material, Table S3).
We identified novel putative LRRK2 interactions as well. The gene, mom-5 (FZD1), shows a striking role as a hub in the LRRK2 regulatory network, and knockdown of FZD1 increased DA neuron survival by 49% (Fig. 1A, Supplementary Material, Table S3). This result mirrors a recent study identifying FZD2 as a central regulator of progranulin function (42). Other WNT genes, such as cwn-1 (WNT4) and cfz-2 (FZD9), strongly reduced DA neuron survival upon knockdown (−75 and −52%, respectively, Supplementary Material, Table S3). There were 12 G protein signaling genes, including the regulators of G protein signaling, rgs-2, rgs-6, rgs-8.1 and eat-16. These genes showed differential regulation; inhibition of rgs-2 (RGS1,RGS2) and rgs-6 (RGS1,RGS2) enhanced DA neuron survival by +20–30%, while inhibition of rgs-8.1 and eat-16 decreased DA neuron survival by −40 to −50%. (Fig. 2C, Supplementary Material, Table S3).
We investigated whether the effects of knockdown would be the same with a different assay. We examined the effects of selected genes using a thrashing assay, which is a phenotype strongly influenced by synaptic function. We examined five genes linked to autophagy: parkin, pink-1, hda-6, atp6a2 as well as lrk-1. Knockdown of four of the genes did not alter thrashing to extents that were significantly different (% change in thrashing versus vector: parkin, 2.9 ± 2.7; pink-1, 8.3 ± 2.5; atp6a2, 14.5 ± 3.1; lrk-1, 3.0 ± 2.7). Knockdown of hda-6 decreased thrashing by 40.1 ± 8.1%. This indicates that the effects on DA neuron survival can be quite specific for neuroprotection.
Endogenous lrk-1 is required for effects on DA neuron survival in C. elegans
The strong effects of genes linked to autophagy on DA neuron survival in the LRRK2 C. elegans line prompted us to examine whether these genes required LRRK2/lrk-1 for their actions. We focused on vps-34, unc-51 and hda-6 because these are particularly important regulators of the autophagic-lysosomal degradation pathway (5,43,44). Repeat experiments validated that the knockdown of vps-34 leads to a significant reduction in survival score compared with empty vector treated LRRK2-expressing C. elegans (P < 0.0001) (Fig. 3A). Knockdown of vps-34 in the lrk-1(km17) exhibited an inverse effect on DA neuron survival, significantly enhancing survival compared with empty vector control (P < 0.05) (Fig. 3A). Knockdown of unc-51 also produced a significant reduction in survival score compared with empty vector treated LRRK2-expressing C. elegans (P < 0.05) (Fig. 3B), consistent with the results of our initial screen. Knockdown of unc-51 in the lrk-1(km17) strain did not modify DA neuron survival beyond that observed with lrk-1(km17) alone (Fig. 3B).
Figure 3.

LRK-1 deletion studies. (A) Knockdown of vps-34 (RNAi) significantly reduced DA neuron survival in WT LRRK2/dat-1::GFP/rrf3(pk1426) nematodes, but increased survival in the lrk-1 [km-17] deletion line. The data for (A)–(C) are normalized to the results for pDAT::GFP nematodes grown under basal conditions. (B) Knockdown of unc-51(RNAi) significantly reduced DA neuron survival in the WT LRRK2/dat-1::GFP/rrf3(pk1426) line, but had no effect on the km-17. (C) Quantification showing a significant increase in DA survival score upon knockdown of rgs-2 (RNAi) in LRRK2 expressing C. elegans (P < 0.05). Inhibition of rgs-2 in the km17 strain does not cause a significant change in survival, indicating an effect specific to LRRK2 function.
We also examined RGS2, which is abundant in dopaminergic neurons, and is decreased in the striatum of D1 receptor knockout mice (45, 46). Yeast two hybrid studies also show an interaction between RGS2 and parkin (47). The enhanced DA neuron protection produced by knockdown of rgs-2 was replicated in the LRRK2-expressing C. elegans line following treatment with rgs-2 RNAi (P < 0.05) (Fig. 3C). Protection mediated by rgs-2 knockdown required the presence of LRRK2/lrk-1; knockdown of rgs-2 did not cause a significant change in the lrk-1(km17) strain, which has a deletion in the endogenous ortholog of LRRK2 (Fig. 3C). These data suggest that rgs-2 modulates LRRK2/lrk-1 function, and that the two genes act through the same pathway.
Interaction of LRRK2 with RGS2 in mammalian cells and in vivo
Having validated the role of RGS2 in LRRK2 function in C. elegans, we proceeded to explore its interactions with LRRK2 in mammalian systems. We first examined the capacity of RGS2 to bind LRRK2 by co-immunoprecipitation (IP) of overexpressed proteins in HEK293T cells. Following IP of FLAG-LRRK2, we found a robust interaction with full-length HA-RGS2 (Fig. 4A). PD-associated mutations in LRRK2 (G2019S, R1441C and Y1699C) had no effect on the strength of the interaction (Fig. 4A). In the reverse IP, Myc-LRRK2 interacted with immunoprecipitated HA-RGS2 (Fig. 4B). We next validated the interaction of endogenous LRRK2 and RGS2 in vivo, by showing that RGS2 is co-immunoprecipitated with endogenous LRRK2 from mouse striatal tissue (Fig. 4C). We then examined the domain requirements for association of LRRK2 and RGS2. RGS2 was overexpressed with LRRK2 constructs coding for individual functional domains (Fig. 4D). Following IP of LRRK2 deletion constructs, we found that RGS2 interacts most strongly with residues 480–895 of LRRK2, a region containing LRRK2-specific repeats (residues 1–660), armadillo repeats (residues 180–660) and putative ankyrin repeats (residues 690–860) known to mediate protein–protein interactions (Fig. 4D). Conversely, we wanted to establish the domain of RGS2 with which LRRK2 interacts. RGS2 contains a core domain of ∼130 amino acids, which is responsible for its GAP activity and a 78-amino-acid N-terminal domain, which mediates its selective binding to specific G protein-coupled receptors (GPCRs) to modulate linked G protein signaling (48) (Fig. 4E). We overexpressed FLAG-LRRK2 with full-length RGS2 or a truncated version of the protein lacking the first 78 amino acids (ΔN-RGS2) (Fig. 4E). Expression levels of ΔN-RGS2 were lower compared with the full-length protein, presumably because of the importance of the N-terminal domain in stabilizing the protein (49). Following IP of FLAG-LRRK2, we found that truncated ΔN-RGS2 was still able to interact with LRRK2 (Fig. 4E). This suggests that RGS2 most likely binds to LRRK2 via its GAP domain. Finally, we assessed the co-localization of both proteins in mammalian cells by confocal microscopy. Consistent with previous reports (48,50), we found that RGS2 exhibits a ubiquitous and diffuse distribution in the nucleus and cytoplasm, whereas LRRK2 is also diffusely expressed in the cytoplasm but excluded from the nucleus (Supplementary Material, Fig. S4). Upon co-expression, although both proteins do not uniformly co-localize in the cytoplasm, we identified multiple areas of co-localization in all visualized HEK cells and primary neurons, which likely account for the biochemical and functional interactions identified between LRRK2 and RGS2 (from three independent experiments, n = 10 HEK cells and neurons visualized/experiment) (Supplementary Material, Fig. S4). No obvious changes in the cellular localization of either protein were observed upon co-expression compared with expression of either protein alone (data not shown). It was not possible to reliably assess co-localization of endogenous RGS2 and LRRK2 with currently available antibodies because of insufficient specificity when used for immunocytochemistry.
Figure 4.

Interaction of LRRK2 with RGS2 in mammalian cells and in vivo. (A) Overexpressed HA-RGS2 interacts with WT and mutant (R1441C and G2019S) FLAG-LRRK2, following IP with anti-FLAG antibody from HEK293T cells. (B) In the reverse IP, wild-type Myc-LRRK2 interacts with HA-RGS2 following IP with anti-HA antibody from HEK293T cells. (C) Endogenous RGS2 interacts with LRRK2 in mouse cerebral cortex following IP with anti-LRRK2 antibody but not with a rabbit IgG control. IgG heavy chain (HC) indicates the equivalent amounts of IgG used for IP. (D) HA-RGS2 was overexpressed with individual WT LRRK2 deletion constructs in HEK293T cells. Immunobloting for HA-RGS2 following IP with anti-FLAG antibody revealed that RGS2 interacts most strongly with the F2 fragment containing residues 480–895. Domain organization of LRRK2 deletion constructs is indicated. (E) Full-length WT FLAG-LRRK2 was overexpressed in HEK293T cells with either full-length WT RGS2-HA (∼25 kDa) or N-terminally truncated RGS2-HA (ΔN-RGS2-HA, ∼16 kDa). Following IP of FLAG-LRRK2, we found an interaction with both full-length and truncated RGS2, suggesting that LRRK2 interacts with the GAP domain of RGS2. Domain organization of RGS2 constructs is indicated. Data are representative of at least two independent experiments. See also Supplementary Material, Figure S4.
RGS2 regulates the GTPase and kinase activities of LRRK2
RGS2 is known to function as a GAP protein, acting as negative modulator of GPCR signaling by stimulating the GTPase activity of the α subunits of G proteins (48). We therefore examined whether RGS2 may also regulate the GTPase activity of LRRK2. Recombinant RGS2 increased the GTPase activity of immunopurified full-length LRRK2 in a dose-dependent manner in vitro (Fig. 5A, left panel). Recombinant RGS2 itself lacks intrinsic GTPase activity (Fig. 5A, right panel). We next monitored the effects of RGS2 on steady-state levels of GTP-bound LRRK2 in vivo. We conducted GTP-sepharose pull-down assays on HEK293T cell extracts expressing Myc-LRRK2 in the presence or absence of HA-RGS2. We found that RGS2 did not affect the steady-state levels of GTP-bound WT or mutant (R1441C and G2019S) LRRK2 (Fig. 5B). The specificity of the assay was demonstrated by showing that the K1347A mutant, which abolishes binding to guanine nucleotides (51), is not pulled down by the GTP-sepharose, and that excess amounts of free GTP compete for binding and eliminate precipitation of LRRK2 (Fig. 5C).
Figure 5.

RGS2 regulates the GTPase and kinase activities of LRRK2. (A) Recombinant RGS2 increases the GTPase activity of immunopurified full-length LRRK2 in a dose-dependent manner, as measured by the production of α-32P-GDP in the α-32P-GTP hydrolysis reaction (Vi μm/min). Recombinant RGS2 was pre-incubated at various concentrations with 32 nm of immunopurified full-length WT LRRK2 to give increasing ratios of [RGS2]/[LRRK2]. Background reaction was conducted in the absence of LRRK2. Recombinant RGS2 itself lacks intrinsic GTPase activity. Reactions were conducted in triplicate. Error bars represent SEM. (B) RGS2 does not affect the steady-state levels of GTP-bound WT or mutant (R1441C and G2019S) LRRK2, as indicated by GTP-sepharose pull-down assays on HEK293T cell extracts expressing Myc-LRRK2 in the presence or absence of HA-RGS2. (C) The GDP/GTP binding-deficient K1347A mutant is not pulled down by the GTP-sepharose and excess amounts of free GTP (2 mm) compete for binding and eliminate precipitation of LRRK2 in the presence or absence of RGS2. (D) In vitro kinase activity assay with γ-32P-ATP showing that recombinant RGS2 inhibits kinase activity of full-length LRRK2 in a dose-dependent manner, as measured by scintillation counting of LRRKtide peptide phosphorylation. Background reaction was conducted in the absence of LRRK2. Recombinant RGS2 was pre-incubated at various concentrations with 100 nm of immunopurified full-length WT LRRK2 to give increasing ratios of [RGS2]/[LRRK2]. Reactions were conducted in duplicate. Error bars represent SEM. (E) HEK cells were transfected with V5-LRRK2 (WT) ± RGS2. Lysates were collected and probed for total LRRK2 (anti-V5, left panel), RGS2, actin, phospho-Ser910-LRRK2 (middle panel) and phospho-Ser935-LRRK2 (right panel). Only phospho-Ser910-LRRK2 was reduced by co-expression with RGS2. (F) 32P autoradiograph shows the robust phosphorylation of recombinant GST-RGS2 by full-length WT LRRK2. Phosphorylation is further enhanced by the kinase-hyperactive G2019S mutation, whereas a kinase-dead LRRK2 variant (D1994A) has negligible effects on RGS2 phosphorylation. Immunoblot of input levels of recombinant GST-RGS2 and full-length LRRK2 variants immunoprecipitated from HEK 293T cells shows equal loading for each condition. LRRK2 autophosphorylation was also detected in this assay. * denotes a non-specific band. Data are representative of at least two independent experiments. See also Supplementary Material, Figure S5.
We then assessed the effects of RGS2 on the kinase activity of LRRK2. We found that recombinant RGS2 inhibited kinase activity of full-length LRRK2 in vitro in a dose-dependent manner (Fig. 5D). Analysis of the dose–response curves for RGS2 modulation of LRRK2 enzymatic activities shows that RGS2 achieves maximal inhibition of LRRK2 kinase activity (∼50% inhibition) at a dose of RGS2 that is at least one-tenth the dose required to maximally stimulate GTPase activity (i.e. RGS2:LRRK2 ratios of 1 versus >10, Fig. 5D versus Fig. 5A). We also found that RGS2 was unable to modulate the activity of truncated LRRK2 (970–2527 amino acids) (data not shown), indicating a requirement for full-length LRRK2, which is consistent with RGS2 binding most strongly with residues 480–895 of LRRK2 (Fig. 4D). Inhibition of LRRK2 kinase activity was verified by examining LRRK2 autophosphorylation sites. Lysates of HEK 293 cells expressing V5-LRRK2±RGS2 were probed with antibodies against LRRK2 (anti-V5), or phosphospecific LRRK2 antibodies (Fig. 5E). Phosphorylation of LRRK2 at Ser910 was reduced by co-expression with RGS2 (Fig. 5E), consistent with the in vitro results from Figure 5D. In contrast, phosphorylation at Ser935 was not affected (Fig. 5E). Immunopurified full-length LRRK2 was therefore used in an in vitro assay to establish whether RGS2 is also a substrate for the kinase activity of LRRK2. We found that full-length WT LRRK2 could robustly phosphorylate recombinant RGS2 (Fig. 5F). The specificity of the assay was demonstrated by showing that phosphorylation of RGS2 was enhanced by the kinase-hyperactive G2019S mutation, whereas a kinase-dead LRRK2 variant (D1994A) had negligible effects on RGS2 phosphorylation (Fig. 5F). Finally, the oligomerization/dimerization state of LRRK2 has been shown to be a critical determinant of the kinase activity of the protein (52). To investigate whether RGS2 affects the oligomerization of LRRK2, we ran native PAGE gels of HEK293T cell extracts expressing Myc-LRRK2 in the presence or absence of RGS2 (Supplementary Material, Fig. S5). We found that the high-molecular weight distribution and dimerization of wild-type or mutant (R1441C and G2019S) LRRK2 was similar in the presence or absence of RGS2 (Supplementary Material, Fig. S5). Taken together, these results show that RGS2 regulates both the GTPase and kinase activities of LRRK2, without affecting the oligomeric structure of the protein.
RGS2 protects against G2019S LRRK2-induced neurite shortening and neuronal toxicity
LRRK2 regulates neuronal process length and complexity in primary neurons with silencing or knockout of LRRK2 increasing neurite length and branching, whereas overexpression of the PD-associated G2019S LRRK2 mutant induces neurite shortening and concomitant neuronal toxicity (9,10,53). We therefore investigated the effects of RGS2 expression on G2019S LRRK2-induced neurite shortening and toxicity. Mouse primary cortical neurons were transfected at days-in-vitro (DIV) 5 with empty vector control, wild-type LRRK2 or G2019S LRRK2 in the presence or absence of overexpressed RGS2. We observed no changes in myc-LRRK2 (WT or G2019S) or HA-RGS2 expression levels upon co-expression of both proteins, compared with levels of either protein expressed alone (Fig. 6A). A GFP-expressing plasmid was co-transfected in each condition to morphologically label transfected neurons (LRRK2/RGS2/GFP; 10:10:1 molar ratio). Double- or triple-immunolabeling with overexpressed myc-LRRK2 and HA-RGS2 was confirmed in >95% of GFP-positive neurons (Supplementary Material, Fig. S6A). Neurons were subsequently fixed at DIV20 and immunolabeled for GFP (Fig. 6B). For neurite length measurements, we determined the length of the longest neurite process of individual GFP-labeled neurons, corresponding to the axon. Neurite length analysis was restricted to MAP2-positive neurons, in which axons could be distinguished from dendrites by the absence of MAP2 staining (Supplementary Material, Fig. S6B). Consistent with previous observations, we found that G2019S LRRK2 induced a robust shortening of axonal processes relative to empty vector control, with a smaller but still significant effect of WT LRRK2 (Fig. 6B and C). We found that RGS2 robustly protected against the effects of the G2019S mutant and could also rescue WT LRRK2-induced axonal shortening (Fig. 6B and C). RGS2 expression alone had no effect on axonal length (Fig. 6C). Next, to determine whether neuronal toxicity occurred concomitant to neurite shortening in our model, we assessed the viability of transfected GFP-positive neurons at DIV20. Neurons with no obvious processes or processes shorter than twice the size of the cell body were counted as non-viable (Fig. 6D). Expression of G201S LRRK2 resulted in significant neuronal toxicity, which was rescued by co-expression of RGS2 (Fig. 6D and E). RGS2 expression alone had no effect on neuronal viability relative to control (Fig. 6D and E). Taken together, these results show that RGS2 protects against G2019S LRRK2-induced neurite shortening and neuronal toxicity.
Figure 6.

RGS2 protects against G2019S LRRK2-induced neurite shortening and neuronal toxicity. (A) Western blot analysis with anti-myc (LRRK2), anti-HA (RGS2) and anti-actin antibodies of cell extracts of mouse cortical neurons harvested at DIV20 following transfection with indicated constructs at DIV5. No changes in myc-LRRK2 (WT or G2019S) or HA-RGS2 expression levels were observed upon co-expression of both proteins, compared with levels of either protein expressed alone. (B) Mouse primary cortical neurons were transfected at DIV5 with empty vector control or Myc-LRRK2 (WT or G2019S), HA-RGS2 and GFP at a 10:10:1 molar ratio and fixed at DIV20. Fluorescence microscopy with anti-GFP antibody allows labeling of cell body and neuritic processes of transfected neurons. Arrows indicate the neuronal soma and arrowheads mark the axonal processes. Photomicrographs are representative of three independent experiments/cultures. Scale bar: 200 μm. (C) Measurement of the length of GFP-labeled longest neuritic processes, i.e. axons, reveals that G2019S LRRK2 induces a significant shortening of axonal process relative to empty vector control, with a smaller but significant effect of WT LRRK2. RGS2 rescues WT LRRK2-induced neurite shortening and robustly protects against the effects of the G2019S mutant. RGS2 expression alone has negligible effects on axonal length relative to control, ***P < 0.001, **P < 0.01, *P < 0.05, by one-way ANOVA with the Newman–Keuls post hoc analysis. ns, non-significant. Bars represent the mean ± SEM, from three independent experiments, with n = 30–45 neurons measured/experiment. (D) Representative photomicrographs of the viability of GFP-positive neurons at DIV20 following transfection with indicated constructs at DIV5. Representative photomicrographs of the viability of GFP-positive neurons at DIV20 following transfection with indicated constructs at DIV5. Neurons with no obvious processes or processes shorter than twice the size of the cell body were counted as non-viable. Arrows indicate neurons counted as non-viable. Scale bar: 100 μm. (E) Quantification of the number of viable transfected neurons as a % of total transfected neurons. Bars represent the mean ± SEM, from three independent experiments, with n = 150–200 transfected neurons counted/experiment. RGS2 overexpression protects against neuronal toxicity induced by G2019S LRRK2. *P < 0.05, by one-way ANOVA with the Newman–Keuls post-hoc analysis. See also Supplementary Material, Figure S6.
Silencing of RGS2 induces neurite shortening in a LRRK2-dependent manner
Having shown that RGS2 overexpression could protect against mutant LRRK2-induced neurite shortening, we then investigated whether endogenous RGS2 played a role in the regulation of neurite process length. We first tested plasmids expressing short hairpin RNAs (shRNAs) targeted against mouse RGS2 for their ability to silence mouse HA-RGS2 overexpressed in HEK293T cells, relative to a non-silencing control shRNA plasmid carrying a ‘scramble’ sequence (sh-scramble) (Supplementary Material, Fig. S7A). The sh-RGS2 sequence #3 was found to highly efficiently silence mouse RGS2 (Supplementary Material, Fig. S7A) and was chosen for all subsequent experiments. We also chose a sh-LRRK2 construct, which was previously shown by western blot to efficiently silence mouse LRRK2 (54). Mouse primary cortical neurons were then transfected at DIV5 with combinations of sh-scramble, sh-RGS2 and sh-LRRK2 plasmids (Fig. 7A). As above, a GFP plasmid was co-transfected in each condition to morphologically label transfected neurons. Neurons were fixed at DIV20 and immunolabeled for GFP. We found that silencing of endogenous RGS2 led to a significant reduction in axonal length relative to the control group transfected with sh-scramble only (Fig. 7A and B). This effect was no longer visible upon co-silencing of LRRK2 (Fig. 7A and B), which, by itself, promoted a significant increase in axonal length (Fig. 7A and B), as previously reported (9,53). These data therefore identify a novel cellular function for RGS2 in the control of neurite length, which is dependent on LRRK2.
Figure 7.

Silencing of RGS2 induces neurite shortening in a LRRK2-dependent manner and has a kinase-dependent synergistic effect on LRRK2-induced neurite retraction. (A) Mouse primary cortical neurons were transfected at DIV5 with either sh-scramble alone (control group), sh-scramble + sh-RGS2, sh-scramble + sh-LRRK2 or sh-RGS2 + sh-LRRK2 plasmids combined with GFP at a 10:10:1 molar ratio. Neurons were fixed at DIV20. Note that sh-scramble was co-transfected with sh-RGS2 or sh-LRRK2 to keep the levels of transfected DNA constant. Photomicrographs are representative of three independent experiments/cultures. Scale bar: 200 μm. (B) Silencing of endogenous RGS2 (sh-scramble + sh-RGS2) leads to a significant reduction in axonal length relative to the control group transfected with sh-scramble only. In contrast, RGS2 silencing no longer had a significant effect on axonal length upon co-silencing of LRRK2 (sh-RGS2 + sh-LRRK2 compared with sh-scramble + sh-LRRK2). LRRK2 silencing alone promoted a significant increase in axonal length relative to the sh-scramble control group. *P < 0.05, by one-way ANOVA with the Newman–Keuls post-hoc analysis. ns, non-significant. Bars represent the mean ± SEM, from three independent experiments, with n = 30–45 neurons measured/experiment. (C) Mouse primary cortical neurons were infected with LV-sh-scramble or LV-sh-RGS2 at DIV3, followed by transfection at DIV5 with empty vector control or WT LRRK2 combined with GFP at a 10:1 molar ratio. Neurons were fixed at DIV20. Overexpression of WT-LRRK2 alone (LV-sh-scramble + WT-LRRK2 group) and silencing of RGS2 alone (LV-sh-RGS2) led to a significant retraction of axons compared with the LV-sh-scramble control group. Expression of WT LRRK2 in the presence of RGS2 silencing (LV-sh-RGS2 + WT LRRK2) induced a significant synergistic shortening of axons as determined by the two-way factorial ANOVA analysis (WT LRRK2 overexpression × RGS2 silencing, ***P < 0.001). (D) The kinase-inactive K1906M LRRK2 variant by itself has negligible effects on axonal length. Silencing of RGS2 alone reduced axonal length by ∼15%. No synergistic effects were produced by overexpressing K1906M LRRK2 in the presence of RGS2 silencing. ***P < 0.001, **P < 0.01 *P < 0.05, by two-way ANOVA with the Newman–Keuls post hoc analysis. Bars represent the mean ± SEM, from three independent experiments, with n = 30–45 neurons measured/experiment. See also Supplementary Material, Figure S7.
Synergistic effect of RGS2 silencing on LRRK2-induced neurite shortening is dependent on LRRK2 kinase activity
Since silencing of endogenous RGS2 promotes neurite shortening in a LRRK2-dependent manner, we now sought to directly investigate the effect of RGS2 silencing on LRRK2-induced neurite shortening. Lentiviral vectors (LV) were produced based on the sh-scramble and sh-RGS2 silencing constructs described above. We confirmed by western blot analysis that LV-sh-RGS2 efficiently silenced endogenous mouse RGS2 in primary mouse neurons with no effect on endogenous LRRK2 levels (Supplementary Material, Fig. S7B). Primary cortical neurons were then infected with LV-sh-scramble and LV-sh-RGS2 at DIV3, followed by co-transfection at DIV5 with empty vector control or WT LRRK2 and GFP at a 10:1 molar ratio. WT LRRK2 rather than G2019S LRRK2 was used in this experiment in order to assess more subtle effects resulting from concomitant silencing of RGS2. Neurons were fixed at DIV20 and immunolabeled for GFP. Consistent with our previous observations (Figs 6B and C and 7A and B), overexpression of WT LRRK2 and silencing of RGS2 alone each led to significant decreases in axonal length relative to the control LV-sh-scramble group (∼12 and ∼16% respectively, Fig. 7C). However, when WT LRRK2 was expressed in the presence of reduced levels of RGS2, axonal length was reduced to an extent (∼44%) greater than would be expected from a simple additive effect of both insults (∼28%) (Fig. 7C). A synergistic effect between both factors was confirmed by two-way factorial ANOVA analysis (WT LRRK2 overexpression X RGS2 silencing, P < 0.001). These data therefore show that silencing of RGS2 has a significant synergistic effect on WT LRRK2-induced neurite shortening.
Next, to test whether the synergistic effect of RGS2 silencing on LRRK2-induced neurite shortening is dependent on LRRK2 kinase activity, we looked at the combined effects of RGS2 silencing with overexpression of a kinase-inactive K1906M LRRK2 variant, which is stable when overexpressed in primary neurons (Supplementary Material, Fig. S7C). Overexpression of K1906M LRRK2 by itself had no effect on axonal length relative to the control LV-sh-scramble group (Fig. 7D). Silencing of RGS2 alone reduced axonal length by ∼15% (Fig. 7D). No synergistic effects were produced by silencing RGS2 and overexpressing K1906M LRRK2 together (Fig. 7D). These findings therefore suggest that the synergistic effect of RGS2 silencing on LRRK2-induced neurite shortening is dependent on kinase activity of LRRK2.
Levels of RGS2 are reduced in mutant G2019S LRRK2 and sporadic PD brains
Since changes in expression levels of RGS2 (overexpression and silencing) modulate LRRK2-induced neurite shortening and neuronal toxicity, we investigated whether aberrant RGS2 expression could be found in the brains of LRRK2 patients. We observed that protein levels of RGS2 were significantly reduced in caudate striatal tissue of mutant G2019S LRRK2 PD patients compared with neurologic controls (Fig. 8A). We also observed reduced RGS2 expression levels in striatal tissue of sporadic PD patients compared with controls (Fig. 8B). Together, these data implicate RGS2 in the pathophysiology of human PD.
Figure 8.

Levels of RGS2 are reduced in mutant G2019S LRRK2 and sporadic PD brains. Immunoblots of endogenous RGS2 levels in caudate striatal lysates from human G2019S LRRK2 PD patients and neurologic controls (A) or human sporadic PD patients and neurologic controls (B). Normalization to actin levels reveals that RGS2 levels in PD patients are significantly reduced (*P < 0.05, **P < 0.01, Student's unpaired t-test) compared with controls. Bars represent mean ± SEM.
DISCUSSION
The regulatory mechanisms controlling the activity and toxicity of LRRK2 remain poorly understood. In this study, we implemented ‘top–down’ systems biology methods to human PD brain and blood transcriptomes to generate a PD LRRK2-centered gene regulatory network. This regulatory network provides a roadmap for understanding the signaling context of each LRRK2 interacting gene. This in silico approach has a high degree of face validity because it correctly highlights many of the genes/proteins previously shown to interact with LRRK2, including beta-tubulin (TUBB), FAS, proteins associated with MAPK signaling such as MKK7 (MAP2K7) or JIP1 (MAPK8IP1), and members of the Wnt signaling pathway. We also identify many novel regulators of LRRK2 function, positioned as ‘hubs’ in the network connected to large groups of secondary neighbors, including Actin (ACTA1) and the Wnt signaling members FZD1 and Tankyrase-1 (TNKS). Our network also includes members that are genetically linked to PD, including parkin, PINK1 and DJ-1. Further validation was evident through the knockdown studies in nematodes, which demonstrated functional effects on survival of dopaminergic neurons in C. elegans expressing LRRK2 for over 280 of the predicted network genes. These predictions included each of those mentioned above. By focusing on this sub-network of PD-linked genes, we identify the signaling gene RGS2, which encodes for a GAP protein, positioned as a regulatory ‘hub’ in the pathways linking LRRK2 with PINK1 and DJ-1 (Fig. 1B).
Our results allow an interesting comparison to a screen for LRRK2 interactors using a protein array performed by the Cookson group (55). Our work appears to identify many more kinases than the protein array work, while the protein array work identified more small GTPases (Rabs) (55). This might reflect difference in approaches. Transcriptome studies can identify interactions between proteins that bind only transiently, such as kinases, and interactions that are linked in similar pathways, but are physically indirect. Binding studies, though, will identify direct binding interactions that do not lead to changes in gene regulation. Combining the two approaches provides for a comprehensive overview of LRRK2 function.
RGS proteins are a family of proteins characterized by a GAP domain of ∼130 amino acids, termed RGS domain. They have been primarily described as regulators of GPCR-mediated signaling. GPCRs transduce extracellular signals into downstream intracellular effects via heterotrimeric G proteins, which shuttle between an active GTP-bound and inactive GDP-bound form. RGS proteins promote termination of the signal by accelerating the intrinsic GTPase activity of Gα subunits of G proteins, returning the G protein in its inactive GDP-bound form to the receptor (56). The specificity of the interaction of RGS2 with G proteins in cells is determined by selective recognition of the linked GPCR via its N-terminal domain (48). Here, we report the non-canonical interaction of RGS2 via its catalytic GAP domain with LRRK2 in vitro and in vivo. We identify RGS2 as a novel GAP for the GTPase activity of LRRK2 in vitro. RGS2 also inhibits the kinase activity of LRRK2. Analysis of the dose–response curves for RGS2 modulation of LRRK2 enzymatic activities shows that RGS2 achieves maximal inhibition of LRRK2 kinase activity (∼50% inhibition) at a dose of RGS2 that is at least one-tenth the dose required to maximally stimulate GTPase activity (i.e. RGS2:LRRK2 ratios of 1 versus >10, Fig. 5D versus Fig. 5A). These data suggest that RGS2 affects kinase activity of LRRK2 independently of its effects on GTPase activity. Our studies of LRRK2 autophosphorylation show results consistent with this hypothesis because co-expressing LRRK2 with RGS2 reduces phosphorylation of LRRK2 Serine 910. These findings are consistent with experiments showing that addition of guanine nucleotides directly to immunopurified LRRK2 has no effect on kinase activity (6,57). We further show in this study that the oligomerization of LRRK2 and stability of the kinase-active dimer is similar in the presence or absence of RGS2. We therefore propose a model in which binding of RGS2 to LRRK2 induces local conformational changes that directly inhibit kinase activity, independently of effects on GTP hydrolysis.
We also identify RGS2 as a robust substrate for phosphorylation by LRRK2 in vitro. Phosphorylation of RGS2 has once previously been reported (58). Intriguingly, in that study, the authors demonstrate a phosphorylation-dependent inhibition of the GAP activity of RGS2 by Protein Kinase C, as part of the inositol lipid signaling pathway. Provided sufficient amounts of recombinant phosphorylated RGS2 can be obtained, future studies would aim to determine whether phosphorylation by LRRK2 also affects the GAP activity of RGS2, as part of a feedback mechanism. It will also be important to ascertain whether RGS2 represents an authentic physiological substrate of LRRK2 in vivo. Future mapping of phosphorylation sites and generation of phospho-mimic and phospho-deficient forms of RGS2 should clarify the role of LRRK2-mediated phosphorylation on RGS2 activities.
The discovery of RGS2 as modulator of LRRK2 activity may provide valuable clues about upstream signaling events potentially controlling LRRK2-dependent neuronal phenotypes in the brain. Indeed, very little is known about such events, especially in the striatum, which is particularly relevant to PD and where both LRRK2 and RGS2 are abundantly expressed (50,49). We observed significantly reduced protein levels of RGS2 in the striatum of both G2019S LRRK2 and sporadic PD patients. Previous experiments have shown that administration to rats of the 6-OHDA toxin, which causes a targeted loss of nigrostriatal dopaminergic projections, fails to induce changes in striatal RGS2 levels (59). Therefore, the reduction in striatal RGS2 levels in PD patients is unlikely to be due to the simple loss of dopaminergic projections that occurs in PD. Rather, striatal RGS2 expression has been shown to be dynamically responsive to neuronal activity with RGS2 mRNA being rapidly and selectively upregulated in the striatum in response to plasticity-inducing synaptic stimuli such as amphetamine (60) or haloperidol (61). This dynamic gene expression pattern is thought to be specifically mediated by dopamine D1 and D2 receptors (62). Changes in DA receptor responsiveness occur in PD as a consequence of striatal DA depletion and may explain the significantly reduced levels of RGS2 we measured in the striatum of PD patients. These findings therefore raise the intriguing possibility that LRRK2 activity may be regulated by dopamine-dependent mechanisms in the striatum, via its response to fluctuating cellular levels of RGS2. Interestingly, we show that knockdown of RGS2 leads to LRRK2-dependent neurite retraction in primary neurons. Reduced striatal levels of RGS2 may therefore lead to neurite shortening via LRRK2 and potentially contribute to exacerbate neurodegeneration in G2019S LRRK2 and sporadic PD.
Our study also identifies RGS2 as a potential therapeutic target for treatment of PD caused by mutations in LRRK2. Indeed, we show that overexpression of RGS2 protects against neuronal toxicity induced by mutant G2019S LRRK2 in primary neurons. As described below, we suggest that this protection is mediated by RGS2 inhibition of LRRK2 kinase activity, which would counteract the hyper-kinetic effects of the G2019S mutation (17,19). Because of the biological and pharmaceutical importance of GPCR signaling pathways (almost a third of drugs on the market target GPCR receptors), significant advances have already been made towards the development of allosteric activators of RGS protein action (64). If specifically tailored to target RGS2, our findings suggest that such compounds may be neuroprotective in PD.
In addition to showing that RGS2 overexpression protects against LRRK2-induced neurite shortening, we also find that silencing of RGS2 induces neurite shortening in a LRRK2-dependent manner and has a significant synergistic effect on LRRK2-induced neurite shortening. Taken together, these findings suggest that RGS2 and LRRK2 act in the same pathway to regulate neurite length in primary neurons. Furthermore, if RGS2 were acting downstream of LRRK2 in this pathway, then the effect of RGS2 silencing on neurite length would be independent of the presence of LRRK2. Rather, our results are consistent with RGS2 acting upstream to modulate LRRK2's function in the control of neurite length. We next sought to establish whether RGS2 required LRRK2 kinase activity to modulate LRRK2-induced neurite shortening. We show that RGS2 silencing has a significant synergistic effect on WT LRRK2-induced neurite shortening. In contrast, loss of RGS2 expression fails to produce a synergistic shortening of neurites in combination with the kinase-inactive K1906M LRRK2 mutant. Since recombinant RGS2 inhibits LRRK2 kinase activity in in vitro enzyme activity assays, by combining our biochemical and functional data, we propose a model in which RGS2 regulates LRRK2 function in the control of neurite length by modulating LRRK2 kinase activity (Fig. 9). This model is supported by our finding that neurite shortening induced by overexpression of WT LRRK2 is rescued by genetic inhibition (K1906M mutation) of LRRK2 kinase activity (WT LRRK2 versus K1906M LRRK2, Fig. 7C versus Fig. 7D). Previous observations have also shown that neurite shortening and toxicity induced by the hyper-kinetic G2019S mutant are dependent on kinase activity (17,19,53). We propose that overexpression of RGS2 protects against neurite shortening and toxicity induced by G2019S LRRK2 by inhibiting kinase activity, thereby counteracting the hyper-kinetic effects of the G2019S mutant (Fig. 9). From the analysis of our LRRK2 GTPase and kinase activity dose–response curves described above, we further showed that RGS2 regulates kinase activity independently of GTPase activity. Our model therefore suggests that the GTPase domain has no functional role in mediating the effects of RGS2 on the phenotypic output of LRRK2. This model is strengthened by the finding that RGS2 does not influence in any meaningful way the steady-state levels of GTP-bound LRRK2 in vivo (Fig. 5B). GTP hydrolysis activity of LRRK2 has indeed been shown to be very low compared with that of small GTPases (e.g. ras is ∼10-fold more active than LRRK2 under identical assay conditions) (14,16).
Figure 9.
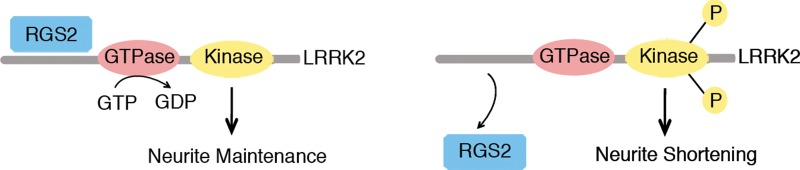
Model of the regulation by RGS2 of LRRK2 function in the control of neurite length. Binding of RGS2 to LRRK2 (i.e. RGS2 overexpression in our experimental set-up) inhibits LRRK2 kinase activity, protecting against neurite shortening induced by LRRK2 overexpression and resulting in maintenance of neurite length. In contrast, loss of RGS2 binding (i.e. RGS2 silencing) enhances kinase activity, inducing neurite retraction. For clarity purposes, LRRK2 kinase activity is illustrated here as autophosporylation.
In summary, we use a reverse-engineered gene regulatory network to identify the signaling gene RGS2, encoding for a GAP protein, as a ‘hub’ linking LRRK2 with other PD-associated genes. Our data show that RGS2 modulates both the GTPase and kinase activities of LRRK2. We identify a novel cellular function for RGS2 in the regulation of neuronal process length via modulation of LRRK2 activity. This non-canonical role of RGS2 is distinct from its function in regulating G-protein mediated signaling. We demonstrate a protective effect of RGS2 against toxicity induced by the prevalent PD-associated G2019S mutation in LRRK2. Our data support a model in which RGS2 regulates LRRK2 function and neuronal toxicity through its effects on kinase activity and independently of GTPase activity. This work strongly supports the hypothesis that the kinase activity is the primary enzymatic output of LRRK2 and reveals a novel mode of action for RGS proteins independently of their established GAP domain function. It also identifies RGS2 as a potential target for interfering with neurodegeneration due to LRRK2 mutations in PD patients.
MATERIALS AND METHODS
Systems biology analysis
PD patient microarray data were collected from the NCBI GEO databases (http://www.ncbi.nlm.nih.gov/geo/) entries GSE8397 and GSE6613. These data sets contain microarray expression data of 47 PD and control cases post mortem brain SNpc and frontal cortex tissue samples from GSE8397 (29) and 72 PD and control cases whole blood samples from GSE6613 (30), respectively. All the raw .CEL files were downloaded and gene expression levels were normalized with the robust multichip average (RMA) algorithm. Gene set enrichment analysis was performed to identify gene sets associated with PD (http://www.broadinstitute.org/gsea/index.jsp). The 2776 genes in these gene sets, which included LRRK2, were then used as input for reverse-engineering the PD gene regulatory network using the CLR algorithm, and the Mode of Action by Network Identification (MNI) algorithm (28,65). The CLR algorithm estimates a likelihood of the mutual information (MI) score for a particular pair of genes, i and j, by comparing the MI value for that pair of genes to a background distribution of MI values (the null model). The background distribution is constructed from two sets of MI values: (MIi), the set of all the mutual information values for gene i (in row or column i), and (MIj), the set of all the mutual information values for gene j (in row or column j) (28). Because biological regulatory networks are sparse, most MI scores in each row of the mutual matrix represent random background MI (e.g. due to indirect network relationships). CLR approximates this background MI as a joint normal distribution with MIi and MIj as independent variables, which provides a reasonable approximation to the empirical distribution of mutual information. Thus, the final form of the CLR likelihood estimate becomes , where Zi and Zj are the Z-scores of MIij from the marginal distributions, and f(Zi, Zj) is the joint likelihood measure. The likelihood of mutual information for a particular pair of genes is considered significant when f(Zi, Zj) ≥ 2.0. The CLR algorithm was implemented in Matlab code, and the software package can be downloaded at http://www.bu.edu/abl/data_and_software.html.
Expression plasmids, proteins and antibodies
Mammalian expression plasmids containing FLAG-tagged full-length human LRRK2 (WT, R1441C, Y1699C, G2019S and D1994N) and FLAG-tagged human LRRK2 deletion mutants were kindly provided by Dr Christopher Ross (Johns Hopkins University, Baltimore, MD, USA) (19). N-terminal 2X-Myc-tagged full-length human LRRK2 plasmids (WT, R1441C, G2019S and K1906M) were kindly provided by Dr Mark Cookson (National Institutes of Health, Bethesda, MD, USA) (17). Full-length N-terminal 3X-HA-tagged human RGS2 was purchased from Missouri S&T cDNA Resource Center (plasmid #RGS020TN00); C-terminal HA-tagged truncated human RGS2 lacking the first 78 amino acids (ΔN-RGS2) was kindly provided by Dr John Hepler (Emory University, Atlanta, GA, USA) (48) and the N-terminal HA-tagged full-length mouse RGS2 was kindly provided by Dr Luc de Vries (Pierre Fabre Research Institute, Castres, France) (64). Empty vector control pcDNA 3.1+ plasmid was obtained from Invitrogen, and pEGFP-N2 from Clontech (Mountain View, CA, USA). shRNA sequences in lentiviral plasmid pLKO.1 targeting mouse RGS2 (sh-RGS2 #1, TRCN0000034444; sh-RGS2 #2, TRCN0000034445; sh-RGS2 #3, TRCN0000034446; sh-RGS2 #4, TRCN0000054758; sh-RGS2 #5, TRCN0000054759) were obtained from Thermo Fisher Scientific (Open Biosystems, Huntsville, AL, USA). shRNA targeting mouse LRRK2 (sh-LRRK2, TRC0000022656) was obtained from Sigma-Aldrich (St Louis, MO, USA). This shRNA was previously validated for silencing of endogenous mouse LRRK2 in reference (54). A non-silencing scramble control shRNA sequence in lentiviral plasmid pLKO.1 was purchased from Addgene (plasmid #1864) (66). The GST-tagged full-length human RGS2 protein was obtained from Novus Biologicals (Littleton, CO, USA). LRRKtide peptide (RLGRDKYKTLRQIRQ) was obtained from Invitrogen. The following antibodies were employed: mouse monoclonal anti-FLAG (M2), anti-FLAG-(M2)-peroxidase (Sigma-Aldrich, Buchs, Switzerland); mouse monoclonal anti-c-myc (clone 9E10) (Roche Applied Science, Bradford, CO, USA); mouse monoclonal anti-HA (HA.11) (Covance, Princeton, NJ, USA); rabbit monoclonal anti-HA (#3724) (Cell Signaling Technology, Danvers, MA, USA); goat anti-actin (MAB1501) (Millipore, Billerica, MA, USA); rabbit polyclonal anti-RGS2 (sc-9103) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); rabbit polyclonal anti-RGS2 (GWB 68126D) (GenWay Biotech, San Diego, CA, USA); rabbit polyclonal anti-GFP (A11122) (Invitrogen); rabbit polyclonal anti-LRRK2 [MJFF2 (c41-2)] (ab133474) (Abcam, Cambridge, MA, USA); mouse monoclonal anti-pSer910 LRRK2 (UDD1, Epitomics, Burlingame, CA, USA), mouse monoclonal anti-pSer935 LRRK2 (UDD2, Epitomics, Burlingame, CA, USA), peroxidase-coupled anti-mouse and anti-rabbit IgG secondary antibodies, donkey anti-mouse and anti-rabbit IgG coupled to AlexaFluor-488, and -594, and donkey anti-mouse IgG coupled to DyLight-405 were all obtained from Jackson Immuno Research (West Grove, PA, USA).
Cell culture and transient transfection
HEK293FT cells were maintained at 37°C in a 5% CO2 atmosphere in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA, USA) and 1× penicillin/streptomycin. For transient transfection, cells were transfected with plasmid DNAs using FuGENE HD reagent (Roche Applied Science) or Lipofectemine (Invitrogen) according to the manufacturer's instructions. Cells were routinely harvested at 48–72 h post-transfection for western blot analysis and biochemical assays.
Lentivirus production
LV were produced in HEK-293T cells using a third-generation packaging system by calcium phosphate transfection with the following plasmids: pCMV-D8.92 (13 μg), pRSVRev (3.75 μg), pMD2.G (3 μg) and pLKO.1 vector containing shRNA sequence (13 μg) (67). After 72 h, the medium was collected and centrifuged in a SW32Ti ultracentrifuge rotor at 19 000 rpm for 90 min at 4°C. The pellet was resuspended in 1.5 ml of buffer containing 1× phosphate-buffered saline (PBS) pH 7.4 and 0.5% BSA for a 100× concentrated virus stock. Viral titer was determined using the HIV-1 p24 antigen ELISA kit (Zeptometrix Corp., Buffalo, USA). A p24 of 8 ng/ml media was used for infecting primary cortical neurons at a density of 133 000 cells in 1.5 ml media (per well of a 12-well dish).
Co-IP assay and western blot analysis
For co- IP assays, HEK-293FT cells were transiently transfected with each combination of plasmids in 10 cm dishes. After 48 h, confluent cells were harvested in 1 ml of IP buffer [1× PBS pH 7.4, 1% Triton X-100, 1× phosphatase inhibitor cocktail 1 and 2 (Sigma-Aldrich), 1× Complete Mini protease inhibitor cocktail (Roche Applied Sciences)]. Cell lysates were rotated at 4°C for 1 h and soluble fractions were obtained by centrifugation at 17 500g for 15 min at 4°C.
For in vitro pull-down assays with anti-FLAG, soluble fractions were combined with 50 μl protein G-Dynabeads (Invitrogen) pre-incubated with mouse anti-FLAG (5 μg; Sigma-Aldrich) antibody followed by overnight incubation at 4°C. Dynabead complexes were sequentially washed once with IP buffer supplemented with 500 mm NaCl, twice with IP buffer and three times with PBS. Immunoprecipitates were eluted by heating at 70°C for 10 min in 2× Laemmli sample buffer (Bio-Rad AG, Reinach, Switzerland) with 5% 2-mercaptoethanol. IPs and inputs (1% total lysate) were resolved by SDS–PAGE, transferred to Protran nitrocellulose (0.2 μm; Perkin Elmer, Schwerzenbach, Switzerland), and subjected to western blot analysis with appropriate primary and secondary antibodies. Proteins were visualized by enhanced chemiluminescence (ECL; GE Healthcare, Glattbrugg, Switzerland) on a FujiFilm LAS-4000 Luminescent Image Analysis system.
For in vitro pull-down assays with anti-HA, pre-cleared soluble fractions were incubated overnight at 4°C with monoclonal mouse anti-HA (HA.11; Covance) followed by incubation for 2 h at 4°C with 50 μl rec-Protein G-Sepharose 4B beads (Invitrogen) previously blocked with 1% BSA. Sepharose bead complexes were sequentially washed three times with IP buffer. Immunoprecipitates were eluted by heating at 70°C for 10 min in 1× NuPAGE LDS Sample Buffer and 1× NuPAGE Sample Reducing Agent (Invitrogen). IPs and inputs were resolved by SDS–PAGE, transferred to PVDF membranes (Bio-Rad, Hercules, CA, USA), and subjected to western blot analysis with appropriate primary and secondary antibodies. Proteins were visualized by enhanced chemiluminescence (ECL; GE Healthcare, Pittsburgh, PA, USA) on a Bio-Rad ChemiDoc™ XRS+ Luminescent Image Analysis system (Hercules, CA, USA).
For in vivo co-IP, protein extracts were prepared from the cerebral cortex of adult wild-type mice by homogenization in TNE buffer [10 mm Tris–HCl pH 7.4, 150 mm NaCl, 5 mm EDTA, 0.5% NP-40, 1× phosphatase inhibitor cocktail 1 and 2 (Sigma-Aldrich), 1× Complete Mini protease inhibitor cocktail (Roche Applied Sciences)]. Protein concentration was determined by BCA assay (Pierce Biotechnology, Rockford, IL, USA). Brain extracts were incubated overnight at 4°C with polyclonal rabbit anti-LRRK2 (MJFF-2/c41-2; Abcam) or normal rabbit IgG (sc-2027; Santa Cruz), followed by incubation for 2 h at 4°C with 50 μl TrueBlot anti-Rabbit Ig IP Beads [Rabbit TrueBlot® Set (with IP beads)-88-1688-31, Rockland, Gilbertsville, PA, USA). Bead complexes were sequentially washed twice with TNE buffer and twice with TBS buffer (10 mm Tris–HCl pH 7.4, 150 mm NaCl). Immunoprecipitates were eluted by heating at 70°C for 10 min in 1× NuPAGE LDS Sample Buffer and 1× NuPAGE Sample Reducing Agent (Invitrogen). IPs and inputs were resolved by SDS–PAGE, transferred to PVDF membranes (Bio-Rad, Hercules, CA, USA), and subjected to western blot analysis as above except that anti-rabbit IgG HRP TrueBlot (Rockland) was used as a secondary antibody for detection of RGS2. TrueBlot preferentially detects the non-reduced form of rabbit IgG over the reduced, SDS-denatured form of IgG, enabling detection of immunoblotted RGS2 target protein band (∼25 kDa), without hindrance by interfering immunoprecipitating immunoglobulin light chains.
Native PAGE/SDS–PAGE analysis
HEK293T cells were transiently transfected with each combination of plasmids in six-well plates. After 48 h, confluent cells were gently washed once with ice-cold PBS, then harvested using a cell scraper in native lysis buffer [PBS pH 7.4, 1× protease and phosphatase inhibitors (Roche Applied Science)], lysed by four cycles of freezing and thawing, and centrifuged at 17 500 g for 10 min. Protein concentration of cleared lysates was determined by the BCA assay (Pierce, Thermo Scientific). Equal amounts of proteins for each sample were loaded on a NativePAGE Novex 3–12% Bis–Tris gel (Invitrogen) or combined with 1× NuPAGE LDS sample buffer and 1× NuPAGE Sample Reducing Agent (Invitrogen) before loading on a NuPAGE Novex SDS–PAGE Bis–Tris gel (Invitrogen). Overnight transfer to PVDF membranes at 4°C was carried out, followed by western blot analysis with anti-myc and anti-HA antibodies and visualization using enhanced chemoluminescence.
Postmortem human tissues samples
Postmortem human caudate tissue samples were provided by the UCL brain bank (London, UK). The average ages (years) and postmortem intervals (h) ± STDEV for each set of samples were: control PD: 82.7 ± 13.3, 31.2 ± 9.2, n = 6; PD: 77.8 ± 4.0, 29.2 ± 14.0, n = 6; control G2019S: 86.7 ± 9.3, 33.4 ± 10.1, n = 3; G2019S PD: 81.7 ± 2.1, 30.5 ± 14.8, n = 3. The samples were homogenized as 20% (w/v) homogenates in low salt 20 mm Tris pH 7.4 buffer, containing 0.5% Triton X-100, 1 mm EDTA, 1 mm trichostatin, 5 mm nicotinamide, 1× phosphatase inhibitor cocktail 1 and 2 (Sigma-Aldrich) and 1X Complete Mini protease inhibitor cocktail (Roche Applied Sciences). Total homogenates were subsequently cleared by centrifugation at 10 000 g for 10 min at 4°C.
GTP binding assay
HEK293T cells in 10 cm plates transiently expressing myc-tagged LRRK2 variants in the presence or absence of HA-RGS2 were lysed in 1 ml of lysis buffer G [1× PBS pH 7.4, 1% Triton X-100, 1× phosphatase inhibitor cocktail 1 and 2 (Sigma-Aldrich), 1× Complete Mini protease inhibitor cocktail (Roche Applied Sciences)], rotated for 1 h at 4°C and clarified by centrifugation at 17 500g for 10 min at 4°C. Soluble proteins were incubated with 50 μl γ-aminohexyl-GTP-sepharose bead suspension (Jena Bioscience, Jena, Germany) by rotating for 2 h at 4°C. Beads were washed three times with buffer G and once with PBS alone. For GTP competition assays, incubation was allowed to proceed for 60 min at 4°C, GTP was added to a final concentration of 2 mm and incubation was continued for a further 60 min at 4°C followed by washing. GTP-bound proteins were eluted in 1× NuPAGE LDS sample buffer and 1× NuPAGE Sample Reducing Agent (Invitrogen) by heating at 70°C for 10 min. GTP-bound proteins or input lysates (1% total lysate) were resolved by SDS–PAGE and subjected to western blotting with anti-myc and anti-HA antibodies.
GTP hydrolysis assay
The GTPase assay was conducted in buffer containing 20 mm Tris (pH7.4), 50 mm NaCl, 10 mm MgCl2, 1 mm DTT, 0.5 mg/ml BSA, 200 μm GTP and 3 μCi [α–33P]-GTP. Recombinant full-length human RGS2 (Abcam) was pre-incubated at various concentrations with 32 nm of immunopurified full-length WT LRRK2 (18) to give ratios of [RGS2]/[LRRK2]: 10, 5, 2.5, 1.3 and 0. The reactions were conducted in triplicate, initiated by the addition of LRRK2 and incubated at room temperature for 20 min. The reaction was stopped by the addition of 20 mm EDTA, and the product [α–33P]-GDP was separated from [α–33P]-GTP by PEI-cellulose thin layer chromatography (Sigma-Aldrich), developed by 0.5 m KH2PO4 (pH 3.4) developing buffer and analyzed by scintillation counter. Background reaction was conducted in the absence of LRRK2.
Kinase activity assay
The kinase assay for LRRK2-catalyzed LRRKtide (RLGRDKYKTLRQJRQ) phosphorylation was conducted in buffer containing 20 mm HEPES (pH 7.4), 50 mm NaCl, 10 mm MgCl2, 1 mm DTT, 0.5 mg/ml BSA, 1 mm beta-Gly-PO4, 100 μm LRRKtide, 100 μm ATP and 1 μCi [γ–33P]-ATP. Recombinant full-length human RGS2 (Abcam) was pre-incubated at various concentrations with 100 nm of immunopurified full-length WT LRRK2 (18) to give ratios of [RGS2]/[LRRK2]: 5, 2.5, 1.3, 0.6 and 0. The reactions were conducted in duplicate, initiated by the addition of LRRK2 and incubated at room temperature for 45 min. The reaction was stopped by the addition of 20 mm EDTA, and the mixtures were transferred to a multiscreen PH filtration plate (Millipore) and washed six times with 75 mm H3PO4. The plate was dried, filters were removed and the samples were counted with a scintillation counter. Background reaction was conducted in the absence of LRRK2.
RGS2 phosphorylation assay
HEK-293T cells were plated at high density in 10 cm dish plates and transfected with (10 µg) FLAG-tagged LRRK2 DNA (WT, G2019S and D1994A) and a mock control (pcDNA3.1). Forty-eight hours post-transfection, cells were harvested and lysed in 1 ml IP buffer [Tris–HCl 20 mm pH 7.5, NaCl 150 mm, EDTA 1 mm pH 8, Tween-20 1%, phosphatase inhibitors 2 and 3 (Sigma-Aldrich) and 1× Complete protease inhibitor cocktail (Roche Applied Sciences)] and centrifuged at 17 500g for 15 min. Cleared lysates were incubated over-night with 60 μl of M2-FLAG resin and complexes were washed five times with Tris–HCl 20 mm pH 7.5, Tween-20 0.5% and NaCl from 500 to 150 mm. This procedure was repeated twice. Finally, M2-FLAG complexes were re-suspended in 1× kinase buffer (25 mm Tris–HCl pH 7.5, 5 mm β-glycerophosphate, 2 mm dithiothreitol, 0.1 mm Na3VO4, 10 mm MgCl2). LRRK2 proteins were eluted with 3xFLAG peptide as per manufacturer's instructions (Cat.N.F4799) from M2-FLAG resin. Recombinant commercially purified full-length GST-tagged human RGS2 (Novus Biologicals) was used in this experiment and for the kinase reactions 1 μg of purified RGS2 protein was combined together with equal amount of eluted LRRK2. In vitro kinase reactions were then performed in kinase buffer (5 mm EGTA and 20 mm β-glycerol phosphate in PBS) in the presence of [33P]-γ-ATP (2 µCi/reaction; Perkin Elmer, MA, USA) and 5 µm cold ATP (Sigma-Aldrich) at 30°C for 1 h in a final volume of 25 µl. The assays were terminated using 4× Laemmli buffer and by boiling at 70°C for 10 min. Complexes were resolved on 4–16% SDS–PAGE pre-cast gels (Invitrogen) and transferred to PVDF membranes. Incorporated radioactivity was detected by autoradiography and the same membranes were probed with anti-GST and anti-FLAG antibodies for protein loading control.
Immunocytochemistry and confocal microscopy
For co-localization of overexpressed myc-LRRK2 and HA-RGS2, HEK293T cells or primary cortical neurons transiently expressing myc-LRRK2 and HA-RGS2 were fixed in 4% paraformaldehyde (PFA) for 10 min at room temperature and immunolabeled with mouse anti-myc (Roche) and rabbit anti-HA (Cell Signaling) antibodies, followed by donkey anti-mouse AlexaFluor-594 and donkey anti-rabbit AlexaFluor-488 secondary antibodies (Jackson Immuno Research). Confocal microscopy was performed on an upright Carl Zeiss LSM 510 META confocal microscope (Carl Zeiss Microscopy) using 63× high numerical-aperture oil immersion objective lenses. Image size was set to 1024 × 1024 pixels. Between 12–15 and 18–20 optical slices for primary neurons and HEK293T cells respectively were collected through the z plane with a 0.5 μm step between each slice. All images were processed using ImageJ software. Images shown are taken from a single z-plane (XY slice) with additional orthogonal views of XZ and YZ slices along the indicated lines of the z-stack obtained using the Stacks → Orthogonal Views option on ImageJ.
For co-immunolabeling of triple-transfected neurons, fixed GFP-positive transfected neurons were immunolabeled with mouse anti-myc and rabbit anti-HA, followed by anti-rabbit IgG-AlexaFluor-488 and anti-mouse IgG-DyLight-405 antibodies. Images were acquired using an upright Carl Zeiss LSM 510 META confocal microscope.
Primary neuronal cultures
Whole brains were dissected from CD-1 P0 mice and the cerebral cortices were stereoscopically isolated and dissociated in media containing papain (20 U/ml; Sigma). The cells were grown in 12-well plates on glass cover slips pre-coated with mouse laminin (33 μg/ml; Invitrogen) and poly-d-lysine (20 ng/ml; BD Biosciences, San Jose, CA, USA) in media consisting of Neurobasal (Invitrogen), B27 supplement (2% w/v), l-glutamine (500 mm) and penicillin/streptomycin (100 U/ml). At DIV3, cortical cultures were treated with cytosine β-d-arabinofuranoside (AraC, 2 μm) to inhibit glial cell division. No change of medium was made during the course of the experiment.
Neurite length assay
For co-expression experiments, primary cortical cultures at DIV5 were co-transfected with pcDNA3.1(Control) or myc-LRRK2, pcDNA3.1 or HA-RGS2 and GFP plasmids at a 10:10:1 molar ratio (2 μg total DNA per well) using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's recommendations. At DIV20, cultures were fixed with 4% PFA and processed for immunocytochemistry with rabbit anti-GFP and mouse anti-MAP2 antibodies, followed by anti-rabbit IgG-AlexaFluor-488 and anti-mouse IgG-AlexaFluor-594 antibodies.
For assays using shRNA plasmids, primary cortical cultures at DIV5 were co-transfected with sh-scramble (pLKO.1) or sh-RGS2 #3, sh-scramble or sh-RGS2 #3 or sh-LRRK2, and GFP plasmids at a 10:10:1 molar ratio (1.6 μg total DNA per well) using Lipofectamine 2000 reagent (Invitrogen). At DIV20, cultures were fixed and immunolabeled as above.
For lentivirus silencing experiments, lentiviruses based on pLKO.1 (LV-sh-scramble) and sh-RGS2 #3 (LV-sh-RGS2) were used. Primary cortical cultures at DIV3 were infected with LV-sh-scramble or LV-sh-RGS2 at a final dose of 8 ng of p24 antigen per ml of media, followed at DIV5 by co-transfection of pcDNA3.1 or myc-LRRK2 and GFP plasmids at a 10:1 ratio using Lipofectamine 2000 (Invitrogen). At DIV20, cultures were fixed and immunolabeled as above. For each experiment, n = 30–45 individual GFP-positive neurons were randomly selected. Fluorescent images were acquired using an upright Olympus BX-60 equipped with epifluorescence optics and a digital camera (AxioCam MRm; Carl Zeiss Microscopy) with a 10× objective. The length of the longest MAP2-negative neurite, corresponding to the axon, of each selected neuron was measured using the line tool function of the NIH ImageJ software by an investigator blinded to each condition. Only neurons with processes at least twice the length of the cell body diameter were selected. Camera lucida drawings were traced using the NeuroJ plug-in of ImageJ (NIH). In cases where the length of the longest neurite exceeded the dimensions of a single 10× photograph, figures were reconstructed using Adobe Photoshop software. Three independent experiments were carried out.
Neuronal toxicity assay
Fluorescent images of randomly selected fields of view were acquired using an upright Olympus BX-60 equipped with epifluorescence optics and a digital camera (AxioCam MRm; Carl Zeiss Microscopy) with a 10× objective. For each experiment, n = 150–200 GFP-positive transfected neurons were counted by an investigator blinded to each condition. Neurons with no obvious processes or processes shorter than twice the size of the cell body were counted as non-viable. The number of viable transfected neurons was expressed as a percentage of total transfected neurons counted in each experiment/culture. Three independent experiments were carried out.
Statistical analysis
Analyses included one-way ANOVA with Newman–Keuls post hoc testing for comparison of multiple data groups, and two-tailed unpaired Student's t-test for pair-wise comparisons, using the GraphPad Prism program. Two-way factorial ANOVA with the Newman–Keuls post hoc analysis was performed to demonstrate the synergistic effect of RGS2 silencing on WT LRRK2 overexpression, using the Statistica software (Statsoft). P < 0.05 was considered significant. Error bars reflect SEM values.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Alzheimer Association (to B.W.); the NIH (ES15567 and NS060872 to B.W.) the Howard Hughes Medical Institute (to J.J.C.); the Swiss National Science Foundation (Fellowship for Advanced Researchers to J.D., 31003A_144063 to D.J.M.); the EPFL (to D.J.M.); the Mayo Clinic (to H.L.); the Michael J Fox Foundation (to R.B. and A.M.); the Harvard NeuroDiscovery Center (to M.L. and M.A.G.); and the National Institutes of Health (R21 NS 072519 to M.L. and M.A.G.).
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Fabienne Pidoux for production of lentiviruses (Brain Mind Institute, EPFL) and Dr John Hepler (Emory University) and Dr Luc de Vries (Pierre Fabre Research Institute) for providing truncated RGS2-HA and mouse HA-RGS2 plasmids, respectively. The rrf1 line was provided by the CGC (U. Minn.).
Conflict of Interest statement. None declared.
REFERENCES
- 1.Lesage S., Durr A., Tazir M., Lohmann E., Leutenegger A.L., Janin S., Pollak P., Brice A. LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. N. Engl. J. Med. 2006;354:422–423. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- 2.Healy D.G., Falchi M., O'Sullivan S.S., Bonifati V., Durr A., Bressman S., Brice A., Aasly J., Zabetian C.P., Goldwurm S., et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol. 2008;7:583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gasser T. Mendelian forms of Parkinson's disease. Biochim. Biophys. Acta. 2009;1792:587–596. doi: 10.1016/j.bbadis.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Dusonchet J., Kochubey O., Stafa K., Young S.M., Jr, Zufferey R., Moore D.J., Schneider B.L., Aebischer P. A rat model of progressive nigral neurodegeneration induced by the Parkinson's disease-associated G2019S mutation in LRRK2. J. Neurosci. 2011;31:907–912. doi: 10.1523/JNEUROSCI.5092-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee B.D., Shin J.H., VanKampen J., Petrucelli L., West A.B., Ko H.S., Lee Y.I., Maguire-Zeiss K.A., Bowers W.J., Federoff H.J., et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat. Med. 2010;16:998–1000. doi: 10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.West A.B., Moore D.J., Choi C., Andrabi S.A., Li X., Dikeman D., Biskup S., Zhang Z., Lim K.L., Dawson V.L., et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 7.Imai Y., Gehrke S., Wang H.-Q., Takahashi R., Hasegawa K., Ooata E., Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillardon F. Leucine-rich repeat kinase 2 phosphorylates brain tubulinbeta isoforms and modulates microtubule stability—a point of convergence in parkinsonian neurodegeneration? J. Neurochem. 2009;110:1514–1522. doi: 10.1111/j.1471-4159.2009.06235.x. [DOI] [PubMed] [Google Scholar]
- 9.Parisiadou L., Xie C., Cho H.J., Lin X., Gu X.L., Long C.X., Lobbestael E., Baekelandt V., Taymans J.M., Sun L., et al. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J. Neurosci. 2009;29:13971–13980. doi: 10.1523/JNEUROSCI.3799-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stafa K., Trancikova A., Webber P.J., Glauser L., West A.B., Moore D.J. GTPase activity and neuronal toxicity of Parkinson's disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet. 2012;8:e1002526. doi: 10.1371/journal.pgen.1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong Y., Yuan C., Chen R., Dawson T.M., Dawson V.L. ArfGAP1 is a GTPase activating protein for LRRK2: reciprocal regulation of ArfGAP1 by LRRK2. J. Neurosci. 2012;32:3877–3886. doi: 10.1523/JNEUROSCI.4566-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trancikova A., Mamais A., Webber P.J., Stafa K., Tsika E., Glauser L., West A.B., Bandopadhyay R., Moore D.J. Phosphorylation of 4E-BP1 in the mammalian brain is not altered by LRRK2 expression or pathogenic mutations. PLoS ONE. 2012;7:e47784. doi: 10.1371/journal.pone.0047784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar A., Greggio E., Beilina A., Kaganovich A., Chan D., Taymans J.M., Wolozin B., Cookson M.R. The Parkinson's disease associated LRRK2 exhibits weaker in vitro phosphorylation of 4E-BP compared to autophosphorylation. PLoS ONE. 2010;5:e8730. doi: 10.1371/journal.pone.0008730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis P.A., Greggio E., Beilina A., Jain S., Baker A., Cookson M.R. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem. Biophys. Res. Commun. 2007;357:668–671. doi: 10.1016/j.bbrc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daniels V., Vancraenenbroeck R., Law B.M., Greggio E., Lobbestael E., Gao F., De Maeyer M., Cookson M.R., Harvey K., Baekelandt V., et al. Insight into the mode of action of the LRRK2 Y1699C pathogenic mutant. J. Neurochem. 2011;116:304–315. doi: 10.1111/j.1471-4159.2010.07105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito G., Okai T., Fujino G., Takeda K., Ichijo H., Katada T., Iwatsubo T. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. 2007;6:1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- 17.Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M.P., Beilina A., Blackinton J., Thomas K.J., et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Li X., Tan Y.C., Poulose S., Olanow C.W., Huang X.Y., Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J. Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith W.W., Pei Z., Jiang H., Dawson V.L., Dawson T.M., Ross C.A. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- 20.Gehrke S., Imai Y., Sokol N., Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466:637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan D., Citro A., Cordy J.M., Shen G.C., Wolozin B. Rac1 protein rescues neurite retraction caused by G2019S leucine-rich repeat kinase 2 (LRRK2) J. Biol. Chem. 2011;286:16140–16149. doi: 10.1074/jbc.M111.234005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piccoli G., Condliffe S.B., Bauer M., Giesert F., Boldt K., De Astis S., Meixner A., Sarioglu H., Vogt-Weisenhorn D.M., Wurst W., et al. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J. Neurosci. 2011;31:2225–2237. doi: 10.1523/JNEUROSCI.3730-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saha S., Guillily M.D., Ferree A., Lanceta J., Chan D., Ghosh J., Hsu C.H., Segal L., Raghavan K., Matsumoto K., et al. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J. Neurosci. 2009;29:9210–9218. doi: 10.1523/JNEUROSCI.2281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plowey E.D., Cherra S.J., Liu Y.-J., Chu C.T. Role of autophagy in G2019S- LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 2008;105:1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gómez-Suaga P., Luzón-Toro B., Churamani D., Zhang L., Bloor-Young D., Patel S., Woodman P.G., Churchill G.C., Hilfiker S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2011;21:511–525. doi: 10.1093/hmg/ddr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsika E., Moore D.J. Contribution of GTPase activity to LRRK2-associated Parkinson disease. Small GTPases. 2013;4:164–170. doi: 10.4161/sgtp.25130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudenko I.N., Chia R., Cookson M.R. Is inhibition of kinase activity the only therapeutic strategy for LRRK2-associated Parkinson's disease? BMC Med. 2012;10:20. doi: 10.1186/1741-7015-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faith J.J., Hayete B., Thaden J.T., Mogno I., Wierzbowski J., Cottarel G., Kasif S., Collins J.J., Gardner T.S. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moran L.B., Duke D.C., Deprez M., Dexter D.T., Pearce R.K., Graeber M.B. Whole genome expression profiling of the medial and lateral substantia nigra in Parkinson's disease. Neurogenetics. 2006;7:1–11. doi: 10.1007/s10048-005-0020-2. [DOI] [PubMed] [Google Scholar]
- 30.Scherzer C.R., Eklund A.C., Morse L.J., Liao Z., Locascio J.J., Fefer D., Schwarzschild M.A., Schlossmacher M.G., Hauser M.A., Vance J.M., et al. Molecular markers of early Parkinson's disease based on gene expression in blood. Proc. Natl Acad. Sci. USA. 2007;104:955–960. doi: 10.1073/pnas.0610204104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mutez E., Larvor L., Lepretre F., Mouroux V., Hamalek D., Kerckaert J.P., Perez-Tur J., Waucquier N., Vanbesien-Mailliot C., Duflot A., et al. Transcriptional profile of Parkinson blood mononuclear cells with LRRK2 mutation. Neurobiol. Aging. 2011;32:1839–1848. doi: 10.1016/j.neurobiolaging.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 32.Carballo-Carbajal I., Weber-Endress S., Rovelli G., Chan D., Wolozin B., Klein C.L., Patenge N., Gasser T., Kahle P.J. Leucine-rich repeat kinase 2 induces alpha-synuclein expression via the extracellular signal-regulated kinase pathway. Cell Signal. 2010;22:821–827. doi: 10.1016/j.cellsig.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ho C.C., Rideout H.J., Ribe E., Troy C.M., Dauer W.T. The Parkinson disease protein leucine-rich repeat kinase 2 transduces death signals via Fas-associated protein with death domain and caspase-8 in a cellular model of neurodegeneration. J. Neurosci. 2009;29:1011–1016. doi: 10.1523/JNEUROSCI.5175-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu C.H., Chan D., Greggio E., Saha S., Guillily M.D., Ferree A., Raghavan K., Shen G.C., Segal L., Ryu H., et al. MKK6 binds and regulates expression of Parkinson's disease-related protein LRRK2. J. Neurochem. 2010;12:1593–1604. doi: 10.1111/j.1471-4159.2010.06568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsu C.H., Chan D., Wolozin B. LRRK2 and the stress response: interaction with MKKs and JNK-interacting proteins. Neurodegener. Dis. 2010;7:68–75. doi: 10.1159/000285509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sancho R.M., Law B.M., Harvey K. Mutations in the LRRK2 Roc-COR tandem domain link Parkinson's disease to Wnt signalling pathways. Hum. Mol. Genet. 2009;18:3955–3968. doi: 10.1093/hmg/ddp337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venderova K., Kabbach G., Abdel-Messih E., Zhang Y., Parks R.J., Imai Y., Gehrke S., Ngsee J., Lavoie M.J., Slack R.S., et al. Leucine-rich repeat kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson's disease. Hum. Mol. Genet. 2009;18:4390–4404. doi: 10.1093/hmg/ddp394. [DOI] [PubMed] [Google Scholar]
- 38.Harrington A.J., Knight A.L., Caldwell G.A., Caldwell K.A. Caenorhabditis elegans as a model system for identifying effectors of alpha-synuclein misfolding and dopaminergic cell death associated with Parkinson's disease. Methods. 2011;53:220–225. doi: 10.1016/j.ymeth.2010.12.036. [DOI] [PubMed] [Google Scholar]
- 39.Wolozin B., Gabel C., Ferree A., Guillily M., Ebata A. Watching Worms Whither Modeling Neurodegeneration in C. elegans. Prog. Mol. Biol. Transl. Sci. 2011;100:499–514. doi: 10.1016/B978-0-12-384878-9.00015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gloeckner C.J., Schumacher A., Boldt K., Ueffing M. The Parkinson disease-associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. J Neurochem. 2009;109:959–968. doi: 10.1111/j.1471-4159.2009.06024.x. [DOI] [PubMed] [Google Scholar]
- 41.Liou A.K., Leak R.K., Li L., Zigmond M.J. Wild-type LRRK2 but not its mutant attenuates stress-induced cell death via ERK pathway. Neurobiol. Dis. 2008;32:116–124. doi: 10.1016/j.nbd.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosen E.Y., Wexler E.M., Versano R., Coppola G., Gao F., Winden K.D., Oldham M.C., Martens L.H., Zhou P., Farese R.V., Jr., et al. Functional genomic analyses identify pathways dysregulated by progranulin deficiency, implicating Wnt signaling. Neuron. 2011;71:1030–1042. doi: 10.1016/j.neuron.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aladzsity I., Toth M.L., Sigmond T., Szabo E., Bicsak B., Barna J., Regos A., Orosz L., Kovacs A.L., Vellai T. Autophagy genes unc-51 and bec-1 are required for normal cell size in Caenorhabditis elegans. Genetics. 2007;177:655–660. doi: 10.1534/genetics.107.075762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Furuya N., Yu J., Byfield M., Pattingre S., Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1:46–52. doi: 10.4161/auto.1.1.1542. [DOI] [PubMed] [Google Scholar]
- 45.Larminie C., Murdock P., Walhin J.P., Duckworth M., Blumer K.J., Scheideler M.A., Garnier M. Selective expression of regulators of G-protein signaling (RGS) in the human central nervous system. Brain Res. Mol. Brain Res. 2004;122:24–34. doi: 10.1016/j.molbrainres.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 46.Stanwood G.D., Parlaman J.P., Levitt P. Genetic or pharmacological inactivation of the dopamine D1 receptor differentially alters the expression of regulator of G-protein signalling (Rgs) transcripts. Eur. J. Neurosci. 2006;24:806–818. doi: 10.1111/j.1460-9568.2006.04970.x. [DOI] [PubMed] [Google Scholar]
- 47.Dachsel J.C., Behrouz B., Yue M., Beevers J.E., Melrose H.L., Farrer M.J. A comparative study of Lrrk2 function in primary neuronal cultures. Parkinsonism Relat. Disord., 2010;16:650–655. doi: 10.1016/j.parkreldis.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernstein L.S., Ramineni S., Hague C., Cladman W., Chidiac P., Levey A.I., Hepler J.R. RGS2 binds directly and selectively to the M1 muscarinic acetylcholine receptor third intracellular loop to modulate Gq/11alpha signaling. J. Biol. Chem. 2004;279:21248–21256. doi: 10.1074/jbc.M312407200. [DOI] [PubMed] [Google Scholar]
- 49.Bodenstein J., Sunahara R.K., Neubig R.R. N-terminal residues control proteasomal degradation of RGS2, RGS4, and RGS5 in human embryonic kidney 293 cells. Mol. Pharmacol. 2007;71:1040–1050. doi: 10.1124/mol.106.029397. [DOI] [PubMed] [Google Scholar]
- 50.Higashi S., Biskup S., West A.B., Trinkaus D., Dawson V.L., Faull R.L., Waldvogel H.J., Arai H., Dawson T.M., Moore D.J., et al. Localization of Parkinson's disease-associated LRRK2 in normal and pathological human brain. Brain Res. 2007;1155:208–219. doi: 10.1016/j.brainres.2007.04.034. [DOI] [PubMed] [Google Scholar]
- 51.Biosa A., Trancikova A., Civiero L., Glauser L., Bubacco L., Greggio E., Moore D.J. GTPase activity regulates kinase activity and cellular phenotypes of Parkinson's disease-associated LRRK2. Hum. Mol. Genet. 2013;22:1140–1156. doi: 10.1093/hmg/dds522. [DOI] [PubMed] [Google Scholar]
- 52.Sen S., Webber P.J., West A.B. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J. Biol. Chem. 2009;284:36346–36356. doi: 10.1074/jbc.M109.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.MacLeod D., Dowman J., Hammond R., Leete T., Inoue K., Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 54.Nichols R.J., Dzamko N., Hutti J.E., Cantley L.C., Deak M., Moran J., Bamborough P., Reith A.D., Alessi D.R. Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson's disease. Biochem. J. 2009;424:47–60. doi: 10.1042/BJ20091035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beilina A., Rudenko I.N., Kaganovich A., Civiero L., Chau H., Kalia S.K., Kalia L.V., Lobbestael E., Chia R., Ndukwe K., et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl Acad. Sci., USA. 2014;111:2626–2631. doi: 10.1073/pnas.1318306111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hollinger S., Hepler J.R. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol. Rev. 2002;54:527–559. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- 57.Taymans J.M., Vancraenenbroek R., Ollikainen P., Beilina A., Lobbestael E., De Maeyer M., Baekelandt V., Cookson M.R. LRRK2 kinase activity is dependent on LRRK2 GTP binding capacity but independent of LRRK2 GTP binding. PLoS ONE. 2011;6:e2320. doi: 10.1371/journal.pone.0023207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cunningham M.L., Waldo G.L., Hollinger S., Hepler J.R., Harden T.K. Protein kinase C phosphorylates RGS2 and modulates its capacity for negative regulation of Galpha 11 signaling. J. Biol. Chem. 2001;23:5438–5444. doi: 10.1074/jbc.M007699200. [DOI] [PubMed] [Google Scholar]
- 59.Taymans J.M., Wintmolders C., Te Riele P., Jurzak M., Groenewegen H.J., Leysen J.E., Langlois X. Detailed localization of regulator of G protein signaling 2 messenger ribonucleic acid and protein in the rat brain. Neuroscience. 2002;114:39–53. doi: 10.1016/s0306-4522(02)00260-9. [DOI] [PubMed] [Google Scholar]
- 60.Burchett S.A., Bannon M.J., Granneman J.G. RGS mRNA expression in rat striatum: modulation by dopamine receptors and effects of repeated amphetamine administration. J. Neurochem. 1999;72:1529–1533. doi: 10.1046/j.1471-4159.1999.721529.x. [DOI] [PubMed] [Google Scholar]
- 61.Robinet E.A., Wurch T., Pauweis P.J. Different regulation of RGS2 mRNA by haloperidol and clozapine. Neuroreport. 2001;12:1731–1735. doi: 10.1097/00001756-200106130-00043. [DOI] [PubMed] [Google Scholar]
- 62.Taymans J.M., Leysen J.E., Langlois X. Striatal gene expression of RGS2 and RGS4 is specifically mediated by dopamine D1 and D2 receptors: clues for RGS2 and RGS4 functions. J. Neurochem. 2003;84:1118–1127. doi: 10.1046/j.1471-4159.2003.01610.x. [DOI] [PubMed] [Google Scholar]
- 63.Kimple A.J., Bosch D.E., Giguiere P.M., Siderovski D.P. Regulators of G-protein signaling and their Ga substrates: promises and challenges in their use as drug discovery targets. Pharmacol. Rev. 2011;63:728–749. doi: 10.1124/pr.110.003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langer I., Tikhonova I.G., Boulegue C., Esteve J.P., Vatinel S., Ferrand A., Moroder L., Robberecht P., Fourmy D. Evidence for a direct and functional interaction between the regulators of G protein signaling-2 and phosphorylated C-terminus of cholecystokinin-2 receptor. Mol. Pharm. 2009;75:502–513. doi: 10.1124/mol.108.051607. [DOI] [PubMed] [Google Scholar]
- 65.di Bernardo D., Thompson M.J., Gardner T.S., Chobot S.E., Eastwood E.L., Wojtovich A.P., Elliott S.J., Schaus S.E., Collins J.J. Chemogenomic profiling on a genome-wide scale using reverse-engineered gene networks. Nat. Biotechnol. 2005;23:377–383. doi: 10.1038/nbt1075. [DOI] [PubMed] [Google Scholar]
- 66.Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 67.Deglon N., Tseng J.L., Bensadoun J.C., Zurn A.D., Arsenijevic Y., Pereira de Almeida L., Zufferey R., Trono D., Aebischer P. Self-inactivating lentiviral vectors with enhanced transgene expression as potential gene transfer system in Parkinson's disease. Hum. Gene Ther. 2000;11:179–190. doi: 10.1089/10430340050016256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.