

Abstract
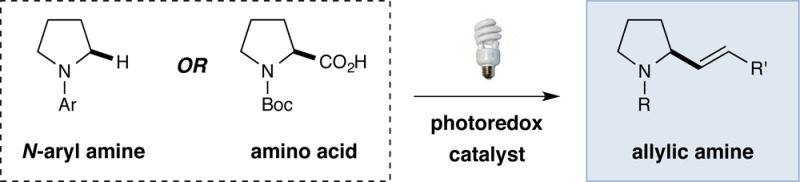
A new coupling protocol has been developed that allows the union of vinyl sulfones with photoredox-generated α-amino radicals to provide allylic amines of broad diversity. Direct C–H vinylations of N-aryl tertiary amines, as well as decarboxylative vinylations of N-Boc α-amino acids, proceed in high yield and with excellent olefin geometry control. The utility of this new allyl amine forming reaction has been demonstrated via the syntheses of several natural products and a number of established pharmacophores.
Visible light photoredox catalysis has recently emerged as a powerful activation strategy for the discovery and invention of new chemical transformations.1 The capacity of metal-to-ligand charge transfer (MLCT) catalysts to simultaneously act as strong reductants and oxidants under the influence of low-energy photon sources has enabled the design of many novel reaction mechanisms that can be chemoselectively triggered using visible light. One emerging application of photoredox catalysis is its use in the direct functionalization of unactivated sp3 C–H bonds,2 an area of research that has become a fundamental goal of modern organic chemistry.3 In this context, our laboratory has become interested in the activation of C–H bonds to generate α-amino radicals, which are versatile intermediates that can participate in radical–radical couplings4 to afford benzylic amines or be trapped with radical acceptors to forge α-N-alkylation adducts.5,6 Generation of α-amino radicals under photoredox conditions typically proceeds via single-electron oxidation of an amine and subsequent deprotonation of the resulting radical cation.4,5 Recently, our group has also reported an alternative strategy for α-amino radical formation via the decarboxylation of N-tert-butoxycarbonyl (N-Boc) α-amino acids,7,8 a CO2-extrusion mechanism that has implications for the use of biomass feedstocks in conjugate additions and organometallic couplings.
Allylic amines have long been attractive targets for new reaction development due to their importance as (i) functionalized building blocks9 and (ii) versatile intermediates in the production of medicinal agents.10 Indeed, over the last three decades, numerous methods have been developed for the preparation of allyl amines, typically proceeding through the addition of vinyl metal reagents to C=N bonds11 or the direct amination of allylic substrates.12 Moreover, deprotonation-based methods for the direct α-vinylation of nitrogen centers have been reported via the α-lithiation, transmetalation of tertiary carbamates, followed by metal-catalyzed cross-couplings with vinyl halides.3d,13
Taking inspiration from our previously developed photoredox arylation protocols,4,7 we sought to extend the utility of α-amino radicals to the direct synthesis of allylic amines from N-aryl amines or α-amino acids. Specifically, we hypothesized that allyl amines should be accessible via exposure of α-amino radicals to olefins that can generically participate in radical-based vinylation mechanisms (i.e., incorporate leaving groups that are susceptible to single-electron β-elimination pathways). As an overarching goal of this work we hoped to demonstrate that photoredox-generated α-amino radicals provide a complementary approach to allyl amine production in comparison to stoichiometric metal-based technologies. Herein, we describe the successful execution of these ideals and present the first direct C–H vinylation of N-aryl tertiary amines and the first decarboxylative olefination of N-Boc α-amino acids using vinyl sulfones in the presence of iridium-based MLCT catalysts.
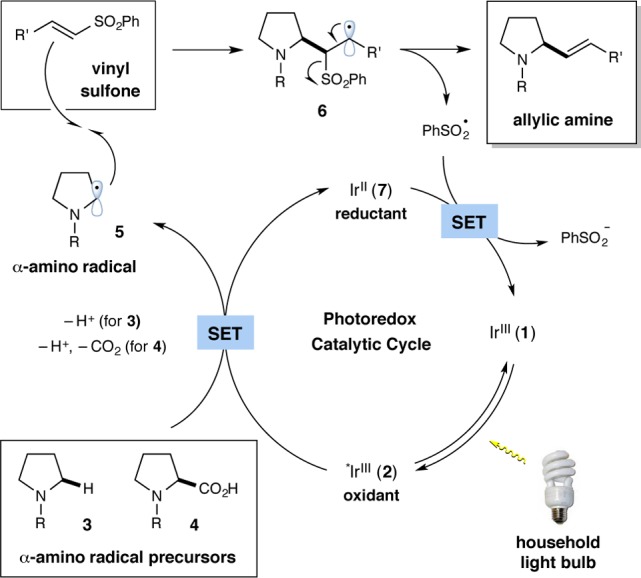
Critical to our proposal of generating allylic amines from α-amino radicals was the identification of an appropriate alkene SOMO-phile, an olefinic coupling partner that would be susceptible to radical addition yet could further participate in single-electron β-elimination to deliver the required vinylation adduct. In this context, Nozaki et al., among others, have ingeniously demonstrated that unsaturated sulfones can generically react with alkyl radicals to produce a wide variety of olefin containing products.14,15 For our purposes, we presumed that vinyl sulfones should be useful substrates for the proposed photoredox vinylation given that (i) they are electron-deficient olefins which should readily couple with nucleophilic α-amino radicals and (ii) the radical C–C bond forming step should be rapidly followed by β-sulfone elimination to produce a sulfinyl radical, a species that should rapidly undergo single-electron reduction via the reduced state of the photocatalyst.
The specific mechanistic details of our proposed photoredox vinylation reaction are outlined in Scheme 1. It has been established that irradiation of Ir[dF(CF3)ppy]2(dtbbpy)PF6 [dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-trifluoromethylpyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine] (1, depicted as IrIII in Scheme 1) with visible light will generate the excited-state IrIII species 2, which is a strong oxidant (E1/2 [*IrIII/II] = +1.21 V vs SCE in MeCN).17 This photoexcited complex should readily undergo single-electron transfer (SET) with a tertiary amine 3 (e.g., N-phenylpyrrolidine (R = Ph), E1/2red = +0.70 V vs SCE)18 to form a radical cation that upon deprotonation will deliver the α-amino radical 5 (for R = Ph). Similarly, the excited-state IrIII species 2 could also undergo SET with the carboxylate formed by deprotonation of α-amino acid 4 (e.g., Boc-Pro-OCs, E1/2red = +0.95 V vs SCE)7a to generate a carboxyl radical, which upon loss of CO2 would deliver α-amino radical 5 (for R = Boc). Reaction of 5 with a vinyl sulfone would then generate the β-sulfonyl radical 6, which after elimination of a sulfinyl radical would provide the allylic amine product. Completion of the photocatalytic cycle would then be accomplished via reduction of the sulfinyl radical (For PhSO2•/PhSO2Na, E1/2red = +0.50 V vs SCE)19 with IrII7 (E1/2 [IrIII/II] = −1.37 V vs SCE)17 to give a sulfinate anion while reconstituting the photocatalyst 1.
Scheme 1. Proposed Mechanism for the Vinylation Reaction.
With this mechanistic hypothesis in mind, we set out to explore the feasibility of our proposed vinylation reaction (Table 1). Our initial investigations began by exposure of N-phenylpyrrolidine (8) to (E)-(2-(phenylsulfonyl)vinyl)benzene (9), in the presence of Ir(ppy)3, NaOAc, and a 26 W household fluorescent light bulb. To our delight, the desired allylic amine product 10 was observed under these new photocatalytic conditions, albeit in modest yield and stereoselectivity (entry 1, 28%, 21:79 E:Z). The efficiency of this vinylation protocol was improved by changing to less polar solvents, with toluene providing higher yield and selectivity compared to N,N-dimethylacetamide (DMA) (entry 2, 57% yield, 91:9 E:Z). Moreover, evaluation of a number of different photocatalysts revealed that less reducing photocatalysts, such as Ir(ppy)2(dtbbpy)PF6 (11) and Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1), provided enhanced E-selectivities (entries 3 and 4, ≥95:5 E:Z).20,21 The choice of base was also found to have a dramatic effect on the yield while maintaining the high E-selectivity, with CsOAc proving to be optimal (entry 5, 81% yield). Finally, the use of 1,2-dichloroethane (DCE) as the reaction medium was found to provide slightly higher yields than toluene, delivering allyl amine 10 in 91% yield and with a 98:2 E:Z ratio (entry 7). The required participation of both the photocatalyst and light were confirmed via control experiments (entries 8 and 9).
Table 1. Initial Studies Towards the C–H Vinylation Reaction.

| entry | photocatalyst | base | solvent | yielda | E:Z |
|---|---|---|---|---|---|
| 1 | Ir(ppy)3 | NaOAc | DMA | 28% | 21:79 |
| 2 | Ir(ppy)3 | NaOAc | toluene | 57% | 91:9 |
| 3 | 11 | NaOAc | toluene | 33% | >98:2 |
| 4 | 1 | NaOAc | toluene | 40% | 95:5 |
| 5 | 1 | CsOAc | toluene | 81% | 96:4 |
| 6b | 1 | CsOAc | toluene | 87% | 97:3 |
| 7b | 1 | CsOAc | DCE | 91% | 98:2 |
| 8b | none | CsOAc | DCE | <5% | – |
| 9b,c | 1 | CsOAc | DCE | 0% | – |
Determined by 1H NMR analysis using an internal standard.
Performed with 3.0 equiv base and at 0.1 M in solvent.
Performed in the absence of light.
Having identified optimal reaction conditions, we next examined the scope of the C–H vinylation reaction with respect to the amine substrate. As shown in Table 2, this protocol is successful with a range of tertiary N-aryl amines (products 10 and 12–24, 64–98% yield, 94:6 to >98:2 E:Z). A variety of cyclic amines are well tolerated, with five-, six-, and seven-membered rings giving their respective products in excellent yields (products 10, 12, and 13, 84–98% yield, ≥97:3 E:Z). This vinylation protocol is relatively unaffected by the nature of the N-aryl group, with substrates possessing electron donating or withdrawing functionalities demonstrating good efficiency (products 14–18). Heteroatom-containing cyclic amines were also found to be suitable (products 19 and 20) along with acyclic substrates (products 21 and 22). Vinylation of a number of pharmacologically important fused nitrogen heterocycles such as N-benzyl-protected indolines and tetrahydroquinolines was demonstrated to be possible, with excellent regioselectivity for the α-methylene ring position over the benzylic α-amino site (products 23 and 24).
Table 2. Direct C–H Vinylation: Amine Scopea.

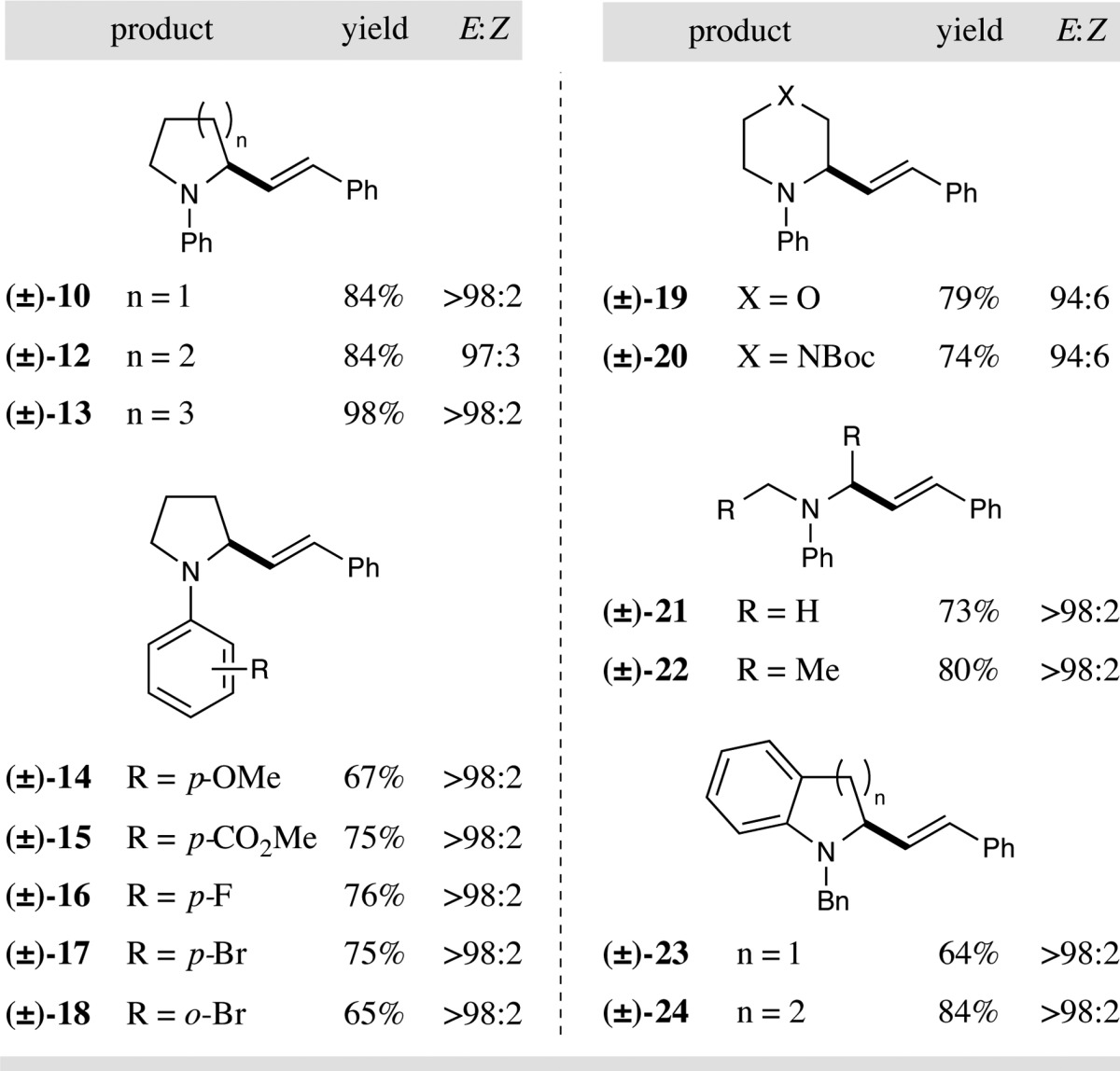
All reactions performed with 0.50 mmol vinyl sulfone, 2.5 equiv amine, 3.0 equiv CsOAc, and 1.0 mol % photocatalyst 1 in DCE (5.0 mL). Yields are of isolated products. E:Z ratio determined by 1H NMR analysis.
We next sought to establish the scope of the vinyl sulfone in this direct C–H olefination reaction. As shown in Table 3, β-sulfonyl styrenes were found to be excellent substrates regardless of the electronic nature of the aryl substituent (products 25–31, 74–85% yield, 92:8 to >98:2 E:Z). Moreover, vinyl sulfones that incorporate heteroaromatic substituents were found to be viable, including electron-deficient pyridines and electron-rich indoles (products 32–34, 68–83% yield, 93:7 to >98:2 E:Z). The reaction is not limited to styrenyl reagents, as exemplified by the use of a conjugated dienyl sulfone to generate an α-amino diene product (35, 60% yield). Trisubstituted alkene adducts could also be delivered via the use of β,β-disubstituted vinyl sulfones that incorporate alkyl–aryl and aryl–aryl groups (products 36 and 37, 84% yield). Perhaps most notable, unsaturated sulfones bearing a β-heteroatom were also successful in this protocol, allowing direct access to enamine functionality (product 38, 79% yield).
Table 3. Direct C–H Vinylation: Vinyl Sulfone Scopea.

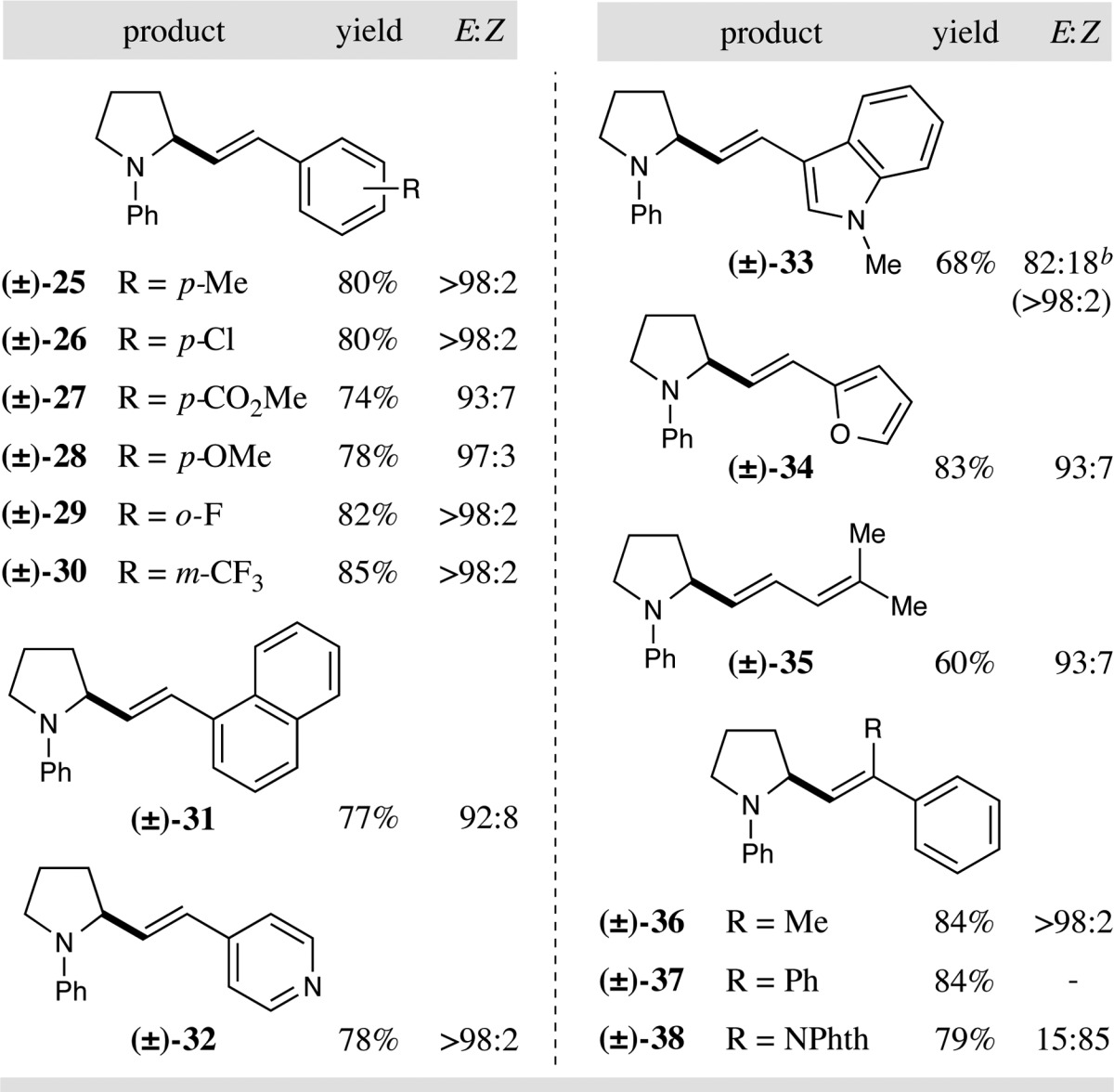
All reactions performed using the conditions described in Table 2. Yields are of isolated products. E:Z ratio determined by 1H NMR analysis.
Isomerization from 82:18 E:Z (crude ratio) to >98:2 E:Z occurred during the mildly acidic purification conditions. NPhth = phthalimidoyl.
To further extend the utility of this photoredox-mediated vinylation reaction, we next turned our attention to the decarboxylative olefination of N-carbamoyl α-amino acids. Importantly, the use of readily cleavable carbamate protecting groups provides a valuable synthetic handle for further elaboration. To our delight, we found that Ir(ppy)2(dtbbpy)PF6 (11), in the presence of 26 W light and CsHCO3, rapidly promoted the decarboxylative coupling of Boc-Pro-OH with sulfone 9 to generate the desired allylic amine 39 in 76% yield and with excellent olefin geometry control (Table 4, 94:6 E:Z).22 Intriguingly, we discovered that while this CO2-extrusion, vinylation mechanism is efficiently catalyzed by photocatalyst 1, the use of photocatalyst 11 was necessary to achieve high levels of E-olefin selectivity. As demonstrated in Table 4, these optimized conditions could be applied to a wide range of α-amino acids, to generate allylic amines in high yield and olefin geometry control (products 39–45, 68–77% yield, 92:8 to >98:2 E:Z). Modification of the α-amino acid protecting group is well tolerated, allowing incorporation of different carbamate systems (products 39 and 40, 76 and 75% yield, respectively). Unnatural cyclic amino acids are readily employed to create the corresponding α-amino olefins (product 42, 74% yield, >98:2 E:Z). Importantly, this decarboxylative vinylation may also be applied to a range of acyclic α-amino acids that incorporate a variety of functional groups including aliphatic, aromatic, sulfide, and indolic functionalities (products 41 and 43–45, 68–77% yield, >98:2 E:Z).
Table 4. Decarboxylative Vinylation: Amino Acid Scopea.
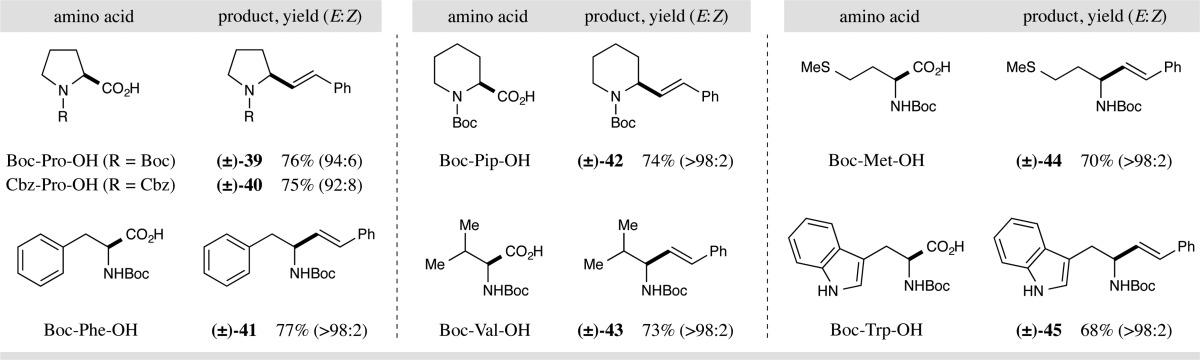
All reactions performed with 0.50 mmol vinyl sulfone, 1.2 equiv amino acid, 2.0 equiv CsHCO3, and 0.5 mol % photocatalyst 11 at 50 °C in 1,4-dioxane (30 mL). Yields are of isolated products. E:Z ratio determined by 1H NMR analysis.
We next examined the scope of the vinyl sulfone coupling partner in this decarboxylative olefination (Table 5). It is clear that a range of vinyl sulfones can be successfully employed in this CO2-extrusion mechanism without loss in yield or olefin geometry control (entries 1–4, 69–84% yield, 94:6–97:3 E:Z). Again, unsaturated sulfones bearing electron-withdrawing functional groups react efficiently (entries 1 and 2, 79 and 84% yield, 94:6 E:Z), as do those possessing electron-donating groups (entry 3, 69% yield, 97:3 E:Z). Finally, heteroaryl rings are also readily tolerated in this coupling protocol, as demonstrated by the synthesis of drug-like pyridinyl-pyrrolidine adduct 49 (entry 4, 69% yield, 95:5 E:Z).
Table 5. Decarboxylative Vinylation: Vinyl Sulfone Scopea.
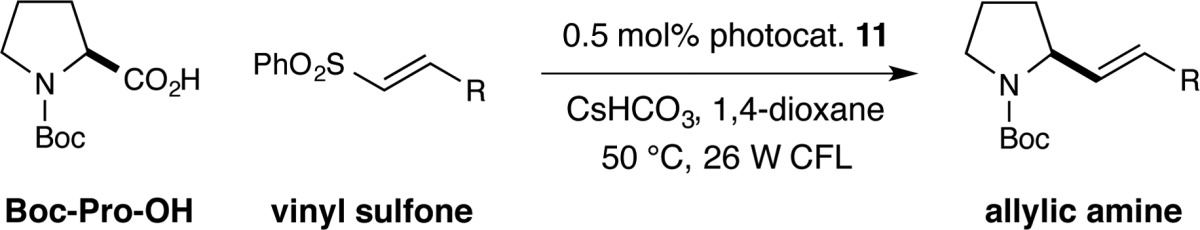
All reactions performed using the conditions described in Table 4. Yields are of isolated products. E:Z ratio determined by 1H NMR analysis.
With the scope of the C–H and decarboxylative vinylation reactions shown to be broad across a range of substrates, we next turned our attention to synthetic applications of this methodology. As shown in Figure 1, a variety of pharmacophores were readily accessible via an intramolecular Heck reaction (50), an acid-promoted cyclization (51), and an alkene reduction (52). Furthermore, the decarboxylative and C–H vinylation reactions were applied to enable the concise total syntheses of the natural products (±)-norruspoline (53)24 and (±)-galipinine (54),25 respectively (see Supporting Information for details).
Figure 1.
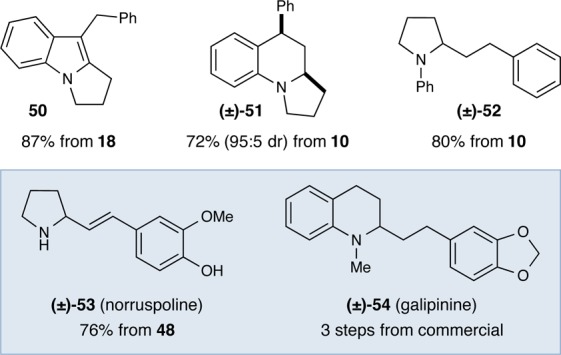
Derivatization of Allylic Amine Products.
Acknowledgments
Financial support was provided by NIHGMS (R01 GM103558-03) and kind gifts from Merck and Amgen.
Supporting Information Available
Experimental procedures and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For recent reviews, see:; a Tucker J. W.; Stephenson C. R. J. J. Org. Chem. 2012, 77, 1617. [DOI] [PubMed] [Google Scholar]; b Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Xie J.; Jin H.; Xu P.; Zhu C. Tetrahedron Lett. 2014, 55, 36. [Google Scholar]; d Schultz D. M.; Yoon T. P. Science 2014, 343, 1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pirnot M. T.; Rankic D. A.; Martin D. B. C.; MacMillan D. W. C. Science 2013, 339, 1593. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Petronijević F. R.; Nappi M.; MacMillan D. W. C. J. Am. Chem. Soc. 2013, 135, 18323. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Qvortrup K.; Rankic D. A.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 626. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Terrett J. A.; Clift M. D.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bergman R. G. Nature 2007, 446, 391. [DOI] [PubMed] [Google Scholar]; b Godula K.; Sames D. Science 2006, 312, 67. [DOI] [PubMed] [Google Scholar]; c Labinger J. A.; Bercaw J. E. Nature 2002, 417, 507. [DOI] [PubMed] [Google Scholar]; d Campos K. R. Chem. Soc. Rev. 2007, 36, 1069. [DOI] [PubMed] [Google Scholar]
- McNally A.; Prier C. K.; MacMillan D. W. C. Science 2011, 334, 1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kohls P.; Jadhav D.; Pandey G.; Reiser O. Org. Lett. 2012, 14, 672. [DOI] [PubMed] [Google Scholar]; b Miyake Y.; Nakajima K.; Nishibayashi Y. J. Am. Chem. Soc. 2012, 134, 3338. [DOI] [PubMed] [Google Scholar]; c Zhou H.; Lu P.; Gu X.; Li P. Org. Lett. 2013, 15, 5646. [DOI] [PubMed] [Google Scholar]; d Miyake Y.; Nakajima K.; Nishibayashi Y. Chem.—Eur. J. 2012, 18, 16473. [DOI] [PubMed] [Google Scholar]
- For a recent review, see:; Hu J.; Wang J.; Nguyen T. H.; Zheng N. Beilstein J. Org. Chem. 2013, 9, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zuo Z.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 5257. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zuo Z.; Ahneman D.; Chu L.; Terrett J.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a photoredox-mediated generation of α-amino radicals from N-aryl α-amino acids, see:Chen L.; Chao C. S.; Pan Y.; Dong S.; Teo Y. C.; Wang J.; Tan C.-H. Org. Biomol. Chem. 2013, 11, 5922. [DOI] [PubMed] [Google Scholar]
- Nag S.; Batra S. Tetrahedron 2011, 67, 8959. [Google Scholar]
- a Skoda E. M.; Davis G. C.; Wipf P. Org. Process Res. Dev. 2012, 16, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stütz A. Angew. Chem., Int. Ed. Engl. 1987, 26, 320. [Google Scholar]
- a Skucas E.; Ngai M.-Y.; Komanduri V.; Krische M. J. Acc. Chem. Res. 2007, 40, 1394. [DOI] [PubMed] [Google Scholar]; b Candeias N. R.; Montalbano F.; Cal P. M. S. D.; Gois P. M. P. Chem. Rev. 2010, 110, 6169. [DOI] [PubMed] [Google Scholar]
- a Cheikh R. B.; Chaabouni R.; Laurent A.; Mison P.; Nafti A. Synthesis 1983, 685. [Google Scholar]; b Johannsen M.; Jørgensen K. A. Chem. Rev. 1998, 98, 1689. [DOI] [PubMed] [Google Scholar]; c Ramirez T. A.; Zhao B.; Shi Y. Chem. Soc. Rev. 2012, 41, 931. [DOI] [PubMed] [Google Scholar]
- For selected examples, see:; a Dieter R. K.; Li S. J. Org. Chem. 1997, 62, 7726. [Google Scholar]; b Dieter R. K.; Oba G.; Chandupatla K. R.; Topping C. M.; Lu K.; Watson R. T. J. Org. Chem. 2004, 69, 3076. [DOI] [PubMed] [Google Scholar]; c Beng T. K.; Gawley R. E. Org. Lett. 2011, 13, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples, see:; a Miyamoto N.; Fukuoka D.; Utimoto K.; Nozaki H. Bull. Chem. Soc. Jpn. 1974, 47, 503. [Google Scholar]; b Russell G. A.; Ngoviwatchai P.; Tashtoush H.; Pla-Dalmau A.; Khanna R. K. J. Am. Chem. Soc. 1988, 110, 3530. [Google Scholar]; c Xiang J.; Jiang W.; Gong J.; Fuchs P. L. J. Am. Chem. Soc. 1997, 119, 4123. [Google Scholar]; d Schaffner A.-P.; Darmency V.; Renaud P. Angew. Chem., Int. Ed. 2006, 45, 5847. [DOI] [PubMed] [Google Scholar]
- After submission of this article, a related photochemical vinylation was reported:Amaoka Y.; Nagatomo M.; Watanabe M.; Tao K.; Kamijo S.; Inoue M. Chem. Sci. 2014, 10.1039/c4sc01631a. [DOI] [Google Scholar]
- Lowry M. S.; Goldsmith J. I.; Slinker J. D.; Rohl R.; Pascal R. A. Jr.; Malliaras G. G.; Bernhard S. Chem. Mater. 2005, 17, 5712. [Google Scholar]
- Liu W.; Ma Y.; Yin Y.; Zhao Y. Bull. Chem. Soc. Jpn. 2004, 79, 577. [Google Scholar]
- Persson B. Acta Chem. Scand. 1977, 31B, 88. [Google Scholar]
- The lower E-selectivities with Ir(ppy)3 were found to be a result of post-reaction isomerization. For a related isomerization process, see:Singh K.; Staig S. J.; Weaver J. D. J. Am. Chem. Soc. 2014, 136, 5275. [DOI] [PubMed] [Google Scholar]
- For discussions regarding the stereoselectivity of radical additions to vinyl sulfones see refs (14b) and (14c).
- See Supporting Information for optimization studies.
- Roessler F.; Ganzinger D.; Johne S.; Schöpp E.; Hesse M. Helv. Chim. Acta 1978, 61, 1200. [Google Scholar]
- Jacquemond-Collet I.; Hannedouche S.; Fabre N.; Fouraste I.; Moulis C. Phytochemistry 1999, 51, 1167. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.