

Abstract

N-Enoxyphthalimides undergo a Rh(III)-catalyzed C–H activation initiated cyclopropanation of electron deficient alkenes. The reaction is proposed to proceed via a directed activation of the olefinic C–H bond followed by two migratory insertions, first across the electron-deficient alkene and then by cyclization back onto the enol moiety. A newly designed isopropylcyclopentadienyl ligand drastically improves yield and diastereoselectivity.
Rh(III) mediated C–H activation has proven to be a powerful approach for the synthesis of nitrogen containing heterocycles.1 Building on a foundation of reactivity first described by Miura and Satoh2 and Fagnou,3 we4 and others5 reported the coupling of benzamides with alkynes to form isoquinolones via Rh(III) C–H activation.6,7 Seminal work by Fagnou8 and Glorius9 demonstrated that an internal oxidant in the form of an N-pivaloxy substituent leads to efficient Rh(III)-catalyzed C–H activation giving dihydroisoquinolinones from benzamides and alkenes (eq 1, Scheme 1). A number of other internal oxidants10 have been advanced including oximes,11 hydrazines,12 and phenoxyamides (eq 2).13 With prominent exceptions (such as eqs 3 and 4), most of these examples functionalize arene C–H bonds. Reasoning that the discovery of novel activation reactions must entail developing new directing groups, we sought to develop a protocol to activate vinyl C–H bonds and considered enoxyamides as potential precursors. The directing group would bear the ubiquitous amide carbonyl moiety required for activation but, similar to phenoxyamide directing groups, would result in release of the amide in the N–O cleavage/Rh reoxidation step. Herein, we report the development of the first Rh(III)-catalyzed cyclopropanation via C–H activation (eq 5). Critical to the success of this reaction was the design of a monosubstituted isopropylcyclopentadienyl ligand.
Scheme 1. Reaction Discovery.

We selected phenyl-N-enoxyphthalimide 1a as a model substrate, readily accessible using Anderson’s approach (eq 6).14 With a suitable Rh(III)-catalyst, activation of the vinyl C–H bond could generate seven-membered rhodacycle I, allowing for further functionalization with a suitable electrophile. To our surprise, when alkene 2 was used as the coupling partner in initial screening, intermediate I acted as a one-carbon component in a [2 + 1] annulation giving the disubstituted trans-cyclopropane 3. Owing to their prevalence in biologically active natural and synthetic compounds, cyclopropane units are fundamental structures in organic chemistry.15 As a result, the development of new strategies for their synthesis is of foremost interest, and stimulated our efforts to improve and generalize this transformation.
Our discovery and initial development of this reaction is summarized in Table 1. The substrate 1a and ethyl acrylate 2a were submitted to various Rh(III) catalysis conditions. The initial discovery utilized a mixture of 5 mol % of [Cp*RhCl2]2 and 2 equiv of CsOAc in trifluoroethanol (TFE) at room temperature to provide trans-1,2-disubstituted cyclopropane 3aa in 63% isolated yield and 2:1 dr (entry 1). Attempts to optimize the reaction conditions using different bases, solvents, and Cp*Rh(III) complexes had only a marginal effect on the diastereoselectivity (data not shown). On the basis of previous reports by our group and others, we speculated that steric and electronic tuning of the cyclopentadienyl ligand could potentially increase selectivity and reactivity. Disappointingly, the known sterically hindered di-tert-butylcyclopentadienyl (Cpt)16,6d and electron deficient tetramethyl(trifluoromethyl)-(CpCF3)11e,11f or ethoxycarbonyl-(CpE)17 cyclopentadienylrhodium(III) complexes provide no significant improvement in the level of diastereocontrol (entries 2, 5, and 6). We then focused on the design of new cyclopentadienyl ligands. Using isopropyl-tetramethyl-(Cp1) and tetramethyl-(Cp2) cyclopentadienylrhodium(III) complexes failed to improve the overall efficiency of the reaction (entries 3 and 4). To our delight, we found the isopropylcyclopentadienyl ligand (CpiPr) affords a high degree of selectivity and reactivity, furnishing the desired trans-disubstituted cyclopropane 3aa in 79% yield and 12:1 dr (entry 7).
Table 1. Cyclopentadienyl Ligands Screena.

| entry | Cpx | dr (trans:cis)b | yield 3aa (%)c |
|---|---|---|---|
| 1 | Cp* | 2:1 | 63d |
| 2 | CpCF3 | 3:1 | 56 |
| 3 | Cp1 | 1:1 | 27 |
| 4 | Cp2 | 2:1 | 40 |
| 5 | Cpt | 1:1 | 22e |
| 6 | CpE | 1:1 | 40 |
| 7 | CpiPr | 12:1 | 79d |
Conditions: 1a (1.0 equiv), 2a (1.2 equiv), CsOAc (2.0 equiv), [Rh] (5 mol %) in TFE (0.2 M), at 21 °C for 16 h.
Determined by analysis of the crude 1H NMR.
Determined by 1H NMR.
Isolated yield.
Low conversion.
With optimized reaction conditions in hand, the scope of the cyclopropanation reaction was investigated. Structural variations on the aryl unit of the N-enoxyphthalimide were examined (Table 2). Substituents at the para or meta positions, regardless of their electronic nature (methyl, tert-butyl, fluoro, and methoxy) exerted no substantial effect on the outcome of the reaction, providing the corresponding trans-cyclopropanes (3ba–da and 3fa–ga) in high yields and diastereoselectivities. The ortho-fluoro substrate was compatible with the reaction conditions and afforded 3ha in 41% yield and 11.4:1 dr. Because of their low solubility in the reaction medium, substrates 1e, 1i, and 1j required longer reaction times. In contrast with 1j, substrates 1e and 1i were not fully converted even after 48 h, giving the corresponding products 3ea and 3ia in 41 and 32% yields, respectively.
Table 2. N-Enoxyphthalimide Scope.

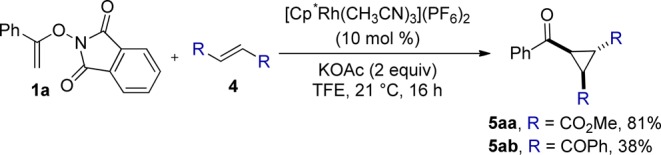
The reactivity of a range of alkenes was examined next (Table 3). The reaction proceeds smoothly with acrylates 2b–e and acrylamides 2j–k, all forming the expected trans-disubstituted cyclopropanes 3ab–ae, 3aj, and 3ak in good to excellent yields (64–89%) and high diastereoselectivities. Enones 2f–i are competent coupling partners in the reaction providing the desired trans-cyclopropanes 3af–ai as the major isomers in good yields, although with slightly decreased dr. Acrylonitrile 2l gives the desired cyclopropane 3al in 54% yield and 10.4:1 dr. We were pleased to find (E)-1,2-disubstituted alkenes 4 were tolerated under the reaction conditions when using [Cp*Rh(CH3CN)3](PF6)2 as the precatalyst.18 Methyl fumarate 4a and (E)-dibenzoylbutene 4b provide the corresponding 1,2,3-trisubstituted cyclopropanes 5aa and 5ab as a single diastereoisomer in 81 and 38% yields respectively (Scheme 2).
Table 3. Alkene Scope.

Scheme 2. Reaction with (E)-1,2-Disubstituted Alkenes.
After exploring the scope of the reaction, we then sought to study the mechanism of this transformation (Scheme 3). Conducting the reaction in TFE-d1 without 2a led to no deuterium incorporation on the alkene, suggesting the C–H functionalization is irreversible (eq 7, Scheme 3). In another experiment, treatment of 1a and 2a in TFE-d1 gives the cyclopropane 3aa′ with no deuteration observed on the phenyl ring, suggesting the C–H functionalization event is chemoselective for the double bond at the expense of the neighboring phenyl ring (eq 8). Since N-enoxyphthalimides (1) are only partially soluble in the reaction mixture, any attempts to probe the mechanism of the C–H functionalization step were rendered inconclusive. However, when experiments were run without base or with triethylamine in place of cesium acetate, cyclopropane 3aa is not formed (eq 9), suggesting a concerted-metalation deprotonation (CMD)19 mechanism is operative.
Scheme 3. Mechanistic Experiments.

We believed the insertion of the alkene occurs with complete retention of stereochemistry since only one diastereomer is formed when (E)-1,2-disubstituted alkenes (4) are used (Scheme 2). In agreement with our hypothesis, (E)-d1-ethyl acrylate (E)-2a-d1 gives cyclopropane 3aa″ as the sole product, preserving the anti relationship of the deuterium atom and ester (eq 10). Of mechanistic relevance, the reaction of 1a-d7 under standard reaction conditions gives the trans-cycloadduct with an important loss of deuterium on the cyclopropane moiety (eq 11). Control experiments performed demonstrate epimerization does not take place after the formation of the cycloadduct 3aa in the reaction medium (see Supporting Information). Thus, we reasoned the deuterium extrusion must occur before the formation of the final product. Interestingly, the same reaction with [Cp*RhCl2]2 exhibits a significant loss of deuterium for both trans and cis-cyclopropanes, suggesting both diastereomers are formed via similar mechanisms.
On the basis of these observations, we propose the following mechanism (Scheme 4). After formation of the active Rh(III) catalyst I, irreversible C–H activation at the β-position of the double bond and ligand exchange leads to rhodacycle II. Subsequent migratory insertion of the alkene 2a generates σ-alkylrhodium(III) complex III. The latter can then undergo an intramolecular carborhodation through a 3-exo-trig cyclization mode to form the intermediate IV. According to the deuterium labeling experiments (eq 11, Scheme 3), after C–C bond rotation, β-hydride elimination could give the Rh–hydride complex V. Then, two distinct pathways can be envisioned. First, the Rh(III)–hydride V could collapse to form a Rh(I) complex VI. Oxidative addition of the N–O bond into Rh(I) following by protonation/tautomerization could liberate the cycloadduct 3aa and regenerate the active catalyst. Alternatively, the Rh(III)–hydride V could reversibly reinsert into the double bond and undergo a β-elimination with N–O bond cleavage to close the catalytic cycle.20
Scheme 4. Proposed Mechanism.

To summarize, we have discovered a Rh(III)-catalyzed cyclopropanation reaction using N-enoxyphthalimides and alkenes. This reaction is a rare example of Rh(III)-catalyzed carbocycle synthesis. Through ligand development, we found a new monosubstituted isopropylcyclopentadienyl ligand enables a high degree of diastereocontrol in the reaction. The process allows the synthesis of a wide range of trans 1,2-disubstituted cyclopropanes. Mechanistic studies revealed an unconventional reactivity for Rh(III)-catalysis and allows for insight into the diastereodetermining step of the reaction.
Acknowledgments
We thank NIGMS (GM80442) for generous support and Johnson Matthey for a generous loan of Rh salts.
Supporting Information Available
Experimental procedures, compound characterization, and additional experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For reviews, see:; a Colby D. A.; Bergman R. G.; Ellman J. A. Chem. Rev. 2010, 110, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Satoh T.; Miura M. Chem.—Eur. J. 2010, 16, 11212. [DOI] [PubMed] [Google Scholar]; c Patureau F. W.; Wencel-Delord J.; Glorius F. Aldrichim. Acta 2012, 45, 31. [Google Scholar]; d Song G.; Wang F.; Li X. Chem. Soc. Rev. 2012, 41, 3651. [DOI] [PubMed] [Google Scholar]
- a Ueura K.; Satoh T.; Miura M. Org. Lett. 2007, 9, 1407. [DOI] [PubMed] [Google Scholar]; b Umeda N.; Tsurugo H.; Satoh T.; Miura M. Angew. Chem., Int. Ed. 2008, 47, 4019. [DOI] [PubMed] [Google Scholar]; c Fukutani T.; Umeda N.; Hirano K.; Satoh T.; Miura M. Chem. Commun. 2009, 5141. [DOI] [PubMed] [Google Scholar]; d Morimoto K.; Hirano K.; Satoh T.; Miura M. Org. Lett. 2010, 12, 2068. [DOI] [PubMed] [Google Scholar]
- Stuart D. R.; Bertrand-Laperle M.; Burgess K. M. N.; Fagnou K. J. Am. Chem. Soc. 2008, 130, 16474. [DOI] [PubMed] [Google Scholar]
- Hyster T. K.; Rovis T. J. Am. Chem. Soc. 2010, 132, 10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Guimond N.; Gouliaras C.; Fagnou K. J. Am. Chem. Soc. 2010, 132, 6908. [DOI] [PubMed] [Google Scholar]; b Mochida S.; Umeda N.; Hirano K.; Satoh T.; Miura M. Chem. Lett. 2010, 39, 744. [Google Scholar]; c Song G.; Chen D.; Pan C.-L.; Crabtree R. H.; Li X. J. Org. Chem. 2010, 75, 7487. [DOI] [PubMed] [Google Scholar]; d Rakshit S.; Patureau F. W.; Glorius F. J. Am. Chem. Soc. 2010, 132, 9585. [DOI] [PubMed] [Google Scholar]
- This approach has been expanded by many workers to form other heterocycles; for selected recent examples, see:; a Stuart D. R.; Alsabeh P.; Kuhn M.; Fagnou K. J. Am. Chem. Soc. 2010, 132, 18326. [DOI] [PubMed] [Google Scholar]; b Huestis M. P.; Chan L.; Stuart D. R.; Fagnou K. Angew. Chem., Int. Ed. 2011, 50, 1338. [DOI] [PubMed] [Google Scholar]; c Davis T. A.; Hyster T. K.; Rovis T. Angew. Chem., Int. Ed. 2013, 52, 14181. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hyster T. K.; Rovis T. Synlett 2013, 1842. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Neely J. M.; Rovis T. J. Am. Chem. Soc. 2014, 136, 2735. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Boultadakis-Arapinis M.; Hopkinson M. N.; Glorius F. Org. Lett. 2014, 16, 1630. [DOI] [PubMed] [Google Scholar]
- Recently, the concept of asymmetric Rh(III)-catalyzed reactions was validated using metallo-enzymatic conditions or chiral cyclopentadienyl ligands; see:; a Hyster T. K.; Knorr L.; Ward T. R.; Rovis T. Science 2012, 338, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ye B.; Cramer N. Science 2012, 338, 504. [DOI] [PubMed] [Google Scholar]; c Ye B.; Cramer N. J. Am. Chem. Soc. 2013, 135, 636. [DOI] [PubMed] [Google Scholar]; d Ye B.; Donets P. A.; Cramer N. Angew. Chem., Int. Ed. 2014, 53, 507. [DOI] [PubMed] [Google Scholar]; e Ye B.; Cramer N. Angew. Chem., Int. Ed. 2014, 53, 7896. [DOI] [PubMed] [Google Scholar]
- Guimond N.; Gorelsky S. I.; Fagnou K. J. Am. Chem. Soc. 2011, 133, 6449. [DOI] [PubMed] [Google Scholar]
- Rakshit S.; Grohmann C.; Besset T.; Glorius F. J. Am. Chem. Soc. 2011, 133, 2350. [DOI] [PubMed] [Google Scholar]
- For a review, see:Patureau F. W.; Glorius F. Angew. Chem., Int. Ed. 2011, 50, 1977. [DOI] [PubMed] [Google Scholar]
- a Too P. C.; Wang Y.-F.; Chiba S. Org. Lett. 2010, 12, 5688. [DOI] [PubMed] [Google Scholar]; b Too P. C.; Chua S. H.; Wong S. H.; Chiba S. J. Org. Chem. 2011, 76, 6159. [DOI] [PubMed] [Google Scholar]; c Too P. C.; Noji T.; Lim Y. J.; Li X.; Chiba S. Synlett 2011, 2789. [Google Scholar]; d Zhang X.; Chen D.; Zhao M.; Zhao J.; Jia A.; Li X. Adv. Synth. Catal. 2011, 353, 719. [Google Scholar]; e Hyster T. K.; Rovis T. Chem. Commun. 2011, 47, 11846. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Neely J. M.; Rovis T. J. Am. Chem. Soc. 2013, 135, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhao D.; Shi Z.; Glorius F. Angew. Chem., Int. Ed. 2013, 52, 12426. [DOI] [PubMed] [Google Scholar]; b Liu B.; Song C.; Sun C.; Zhou S.; Zhu J. J. Am. Chem. Soc. 2013, 135, 16625. [DOI] [PubMed] [Google Scholar]; c Wang C.; Huang Y. Org. Lett. 2013, 15, 5294. [DOI] [PubMed] [Google Scholar]; d Zheng L.; Hua R. Chem.—Eur. J. 2014, 20, 2352. [DOI] [PubMed] [Google Scholar]; e Chuang S.-C.; Gandeepan P.; Cheng C.-H. Org. Lett. 2013, 15, 5750. [DOI] [PubMed] [Google Scholar]
- a Liu G.; Shen Y.; Zhou Z.; Lu X. Angew. Chem., Int. Ed. 2013, 52, 6033. [DOI] [PubMed] [Google Scholar]; b Shen Y.; Liu G.; Zhou Z.; Lu X. Org. Lett. 2013, 15, 3366. [DOI] [PubMed] [Google Scholar]; c Hu F.; Xia Y.; Ye F.; Liu Z.; Ma C.; Zhang Y.; Wang J. Angew. Chem., Int. Ed. 2014, 53, 1364. [DOI] [PubMed] [Google Scholar]; d Li X. G.; Liu K.; Zou G.; Liu P. N. Adv. Synth. Catal. 2014, 356, 1496. [Google Scholar]
- N-Enoxyphthalimide 1a was synthesized in one step from commercially available N-hydroxyphthalimide and 1-phenylvinylboronic acid; see:Patil A. S.; Mo D.-L.; Wang H.-Y.; Mueller D. S.; Anderson L. L. Angew. Chem., Int. Ed. 2012, 51, 7799. [DOI] [PubMed] [Google Scholar]
- For reviews, see:; a Salaun J. Chem. Rev. 1989, 89, 1247. [Google Scholar]; b Li A.-H.; Dai L.-X. Chem. Rev. 1997, 97, 2341. [DOI] [PubMed] [Google Scholar]; c Pfalz A. In Comprehensive, Asymmetric Catalysis II; Jacobsen E. N., Pfaltz A., Yamamoto H., Eds.; Springer: Berlin, 1999, 513. [Google Scholar]; d Davies H. M. L.; Antoulinakis E. G. Organic Reactions 2004, 57, 1. [Google Scholar]
- Hyster T. K.; Rovis T. Chem. Sci. 2011, 2, 1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shibata Y.; Tanaka K. Angew. Chem., Int. Ed. 2011, 50, 11109. [DOI] [PubMed] [Google Scholar]; b Hoshino Y.; Shibata Y.; Tanaka K. Adv. Synth. Catal. 2014, 356, 1577. [Google Scholar]; c Fukui M.; Hoshino Y.; Satoh T.; Miura M.; Tanaka K. Adv. Synth. Catal. 2014, 356, 1638. [Google Scholar]
- When [CpiPrRhCl2]2 was used only a trace of compound 5aa was observed.
- Lapointe D.; Fagnou K. Chem. Lett. 2010, 39, 1118. [Google Scholar]
- Alkyl enoxyphthalimides as substrates provide an inseparable mixture of products under these conditions that do not show the presence of a cyclopropane. Efforts at illuminating this process are ongoing.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.