

Discovering useful new drugs to treat tuberculosis (TB) is not an exercise for the faint of heart: screening for growth inhibitors produces very few hits, and optimizing those hits into drug-like lead molecules is exceptionally difficult due to the impermeable waxy cell envelope the mycobacterium constructs. Even armed with a promising lead molecule, animal models and early stage clinical trials have little predictive ability, and safety hurdles for the currently long duration of therapy required to treat TB are therefore much higher than that for most bacterial infections. Repurposing an existing antibiotic class that is already known to be safe and effective in other infections but that has not demonstrated anti-tubercular activity is the attractive strategy at the heart of a recent report in Nature Medicine by Lee et al1.
These authors focus their efforts on spectinomycin, a natural product isolated from Streptomyces with a long history of safe use as second-line treatment for gonorrhea in patients allergic (or resistant) to penicillin. Despite being an agent that targets the highly conserved bacterial ribosome, spectinomycin itself shows little activity against Mycobacterium tuberculosis. Other translational inhibitors like streptomycin, kanamycin and amikacin are widely used in anti-TB chemotherapy but these have a binding site on the ribosome distinct from that occupied by spectinomycin. Armed with the crystal structure of spectinomycin bound to the ribosome of E. coli2 and a mutant strain of M. tuberculosis defective in an efflux pump that showed increased susceptibility to spectinomycin, these authors produced derivatives of spectinomycin, called spectinamides, that show potent anti-TB activity both in vitro and in mice.
Drug developers have long had a love-hate relationship with natural products. On the one hand the shear complexity and three dimensionality of natural products provides much more opportunity for highly specific tight binding to potential targets than do the small synthetic flat molecules typical of the large libraries amassed by most pharmaceutical companies. Natural products have evolved as the weapons of mass destruction deployed, often, but not always, by bacteria to kill other bacteria and taking advantage of that evolutionary war has obvious advantages. On the other hand such compounds rarely have ideal pharmacological properties to use directly as drugs and therefore require chemical transformation, a process called semi-synthesis, to make them suitable for use as medicines. Historically, such semi-syntheses can be extraordinarily complex and require an enormous investment in understanding the chemical properties of the parent molecule to allow a systematic exploration of where derivatives can be made. Current wisdom among scientists working in TB drug discovery lies heavily towards approaches that rely on screening and optimizing small synthetic lead molecules. One such molecule, named Bedaquiline, was recently the first drug in forty years to receive FDA approval for use in treating TB. Nonetheless over the past 40 years about three-quarters of all approved antibacterial drugs have been the results of se mi-synthetic efforts from natural product starting points3.
In fact, the biggest advance in TB chemotherapy achieved to date was unarguably the addition of rifampin in the 1970s to multidrug cocktails to treat the disease. Prior to the introduction of rifampin, the duration of therapy required to achieve a sterile cure of TB with two or three different agents in combination was 18-24 months. Following the landmark clinical trials of the British Medical Research Council in Africa, the standard therapy for TB infections became combinations of four drugs including rifampin for a mere six months4. The starting point for rifampin was a complex mixture of metabolites from Nocardia mediterranei isolated by the French pharmaceutical company Lepetit in 1957. It took 8 years of effort for this company (partnering with Ciba-Geigy in Switzerland) to understand the various rifampin related metabolites and their chemistry sufficiently to enable the discovery of an orally available analog that became the basis of these new “Short-Course Chemotherapy” regimens.
The molecule described in the paper by Lee et al1 is not a new rifampin but there are two exciting features in the paper that should encourage more effort in TB drug discovery by semi-synthetic modification of natural products. First, they took full advantage of recent advances in crystallography of macromolecular protein complexes, in this case the bacterial ribosome, to guide their chemical strategy for modification of the natural product. This allowed them to focus on a limited set of chemical modifications rather than the inefficient empirical approaches used in the past (several hundred rifampin analogs had to be explored before it even became clear which positions could be modified without losing biological activity completely). Second, they were able to utilize our expanding understanding of the role of drug efflux in TB6 to assess separately changes in on-target activity (inhibition of ribosomal protein synthesis) and whole cell activity against an efflux mutant of M. tuberculosis. The successful proof of concept achieved in this study will nudge the pendulum of interest back towards natural products as viable starting points for TB drug developers.
Figure.
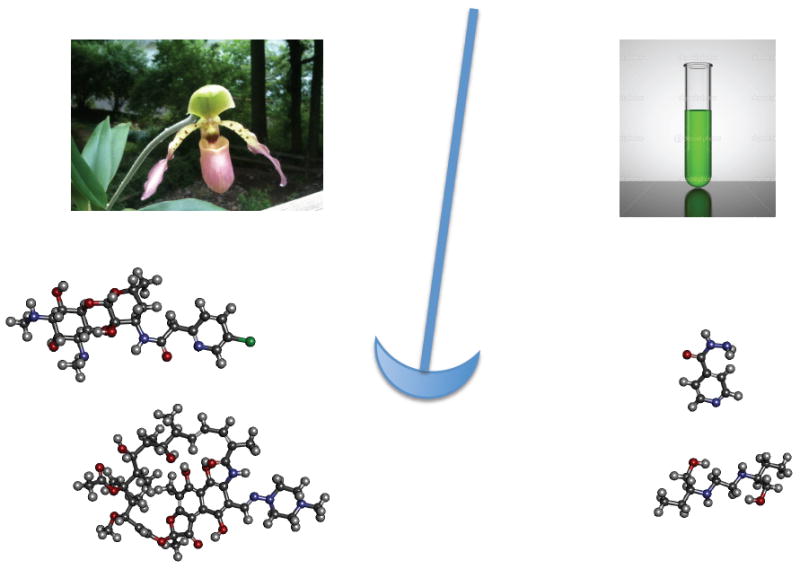
meant to represent a pendulum swinging between the beautifully complex natural products on the left and the simple synthetics. Over the past several decades interest in the pharmaceutical companies has tended more towards the synthetics because they are easier to make and cheap, 75% of approved antibacterials over the past 40 years have been natural products though and the N&V covers a semi-synthetic approach using some modern methodology.
References
- 1.Lee RE, Hurdle JG, Liu J, Bruhn DF, Matt T, Scherman MS, Vaddady PK, Zheng Z, Qi J, Akbergenov R, Das S, Madhura DB, Rathi C, Trivedi A, Villellas C, Lee RB, Rakesh, Waidyarachchi SL, Sun D, McNeil MR, Ainsa JA, Boshoff HI, Gonzalez-Juarrero M, Meibohm B, Böttger EC, Lenaerts AJ. Spectinamides: a new class of semisynthetic antituberculosis agents that overcome native drug efflux. Nat Med. 2014 Jan 26; doi: 10.1038/nm.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borovinskaya MA, Shoji S, Holton JM, Fredrick K, Cate JH. A steric block in translation caused by the antibiotic spectinomycin. ACS Chem Biol. 2007;2:545–552. doi: 10.1021/cb700100n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fox W, Ellard GA, Mitchison DA. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946-1986, with relevant subsequent publications. Int J Tuberc Lung Dis. 1999 Oct;3(10 Suppl 2):S231–79. [PubMed] [Google Scholar]
- 5.Sensi P. Rev Infect Dis. 1983 Jul-Aug;5(Suppl 3):S402–6. doi: 10.1093/clinids/5.supplement_3.s402. History of the development of rifampin. [DOI] [PubMed] [Google Scholar]
- 6.Balganesh M, et al. Efflux pumps of Mycobacterium tuberculosis play a significant role in antituberculosis activity of potential drug candidates. Antimicrob Agents Chemother. 2012;56:2643–2651. doi: 10.1128/AAC.06003-11. [DOI] [PMC free article] [PubMed] [Google Scholar]