

Abstract
The immune response protects against Pneumocystis infection, but is also a key component of PcP-related immunopathogenesis. Signaling through MyD88 is critical for activation of immune pathways downstream of TLRs and IL-1 receptor. To determine whether MyD88 regulates normal host defense against Pneumocystis, non-immunosuppressed wild-type (WT) and MyD88 deficient mice were infected. MyD88−/− mice had higher early Pneumocystis burdens than WT mice, but mounted an effective adaptive immune response and cleared Pneumocystis similar to WT. However, MyD88−/− mice displayed a more intense and prolonged pulmonary immune response than WT mice. To determine the role of MyD88 in the development of PcP-related immunopathogenesis, WT and MyD88−/− mice were rendered susceptible to PcP by depletion of CD4+ T cells. At 4 weeks post-infection, CD4-depleted WT and MyD88−/− mice harbored similar organism burdens, but MyD88−/− mice were protected from the PcP-related respiratory impairment observed in WT mice. Improved pulmonary physiology in MyD88−/− mice correlated with lower lung CCL2 levels, and reduced cell recruitment. However, by 5 weeks post-infection the overall health of MyD88−/− mice began to deteriorate rapidly relative to WT, with accelerated weight loss, impaired lung function, and exacerbated alveolar inflammation. This physiological decline of MyD88−/− mice was associated with increased TNF-α and IFN-γ in the lung, and by the inability to control Pneumocystis burden. Thus, MyD88 is not required for resistance to Pneumocystis infection, but limits the adaptive immune response in immunocompetent mice. In the setting of active PcP, MyD88 signaling contributes to both immunopathogenesis and control of fungal burden.
Introduction
Pneumocystis is respiratory fungal pathogen which causes pneumonia (PcP) in immunocompromised individuals. PcP-related morbidity and mortality continues to be a major health concern for HIV patients, as well as for non-HIV patients who are undergoing immunosuppression as a consequence of chemotherapy or organ transplant (1, 2). New immunosuppressive therapies, such as anti-TNF-α therapy for Crohn’s disease and rheumatoid arthritis, are increasing the pool of “at risk” patients (3). In addition, Pneumocystis frequently colonizes COPD patients, which appears to exacerbate disease severity (4). Therefore, a better understanding of the mechanisms of PcP-related immunopathogenesis is key to improving upon current treatments.
Clinical observations and animal studies have indicated that lung injury during PcP is caused primarily by the host’s immune-mediated inflammatory response, and is not absolutely related to Pneumocystis burden (5–8). For example, in the CD4+ T cell-depleted model of PcP physiological deterioration is associated with an increase in lung chemokine and cytokine levels, and the recruitment of large numbers of CD8+ T cells and neutrophils to the lung. Interestingly, when CD4+ and CD8+ T cells are depleted simultaneously, there are fewer signs of inflammation, less cell recruitment, and improved lung function, suggesting that CD8+ T cells are responsible for lung injury and respiratory impairment in this model of PcP (9).
Recent studies have focused on characterizing the mechanisms involved in generating pathogenic immune and inflammatory responses that damage the lung and other tissues. The Toll-Like Receptor (TLR) system is one of the most important host defense machineries involved in recognition of invading pathogens. Upon recognition pathogens, TLRs activate downstream kinases and transcription factors that induce the expression of genes involved in innate and adaptive immune responses. All TLRs, with the exception of TLR3, signal through the adaptor molecule myeloid differentiation factor 88 (MyD88). MyD88 is also critical for signaling through cytokine receptors that belong to the IL-1 receptor (IL-1R) family (10). A protective role for MyD88 in the control fungal infections such as Candida albicans, Aspergillus fumigatus, Cryptococcus neoformans and Paracoccidioides brasiliensis has been reported (11–14). Moreover, our laboratory and others have demonstrated that MyD88-dependent signaling is required for optimal alveolar epithelial cell (AEC) and alveolar macrophage (AM) cytokine responses to Pneumocystis or Pneumocystis cell wall components (15, 16). TLRs, including TLR2 and TLR4, have also been linked to Pneumocystis-stimulated AM cytokine responses (17, 18) but neither TLR2 nor TLR4 are required for AEC chemokine responses to Pneumocystis. Rather, the IL-1 receptor is the upstream molecule required for the MyD88-dependent AEC response (16).
Although in vitro studies suggest that TLR-, IL-1R-, and MyD88-dependent responses are involved in the AEC and AM responses to Pneumocystis, the in vivo role of MyD88-dependent signaling events during active Pneumocystis infection remain undefined. In the current study, we utilized WT and MyD88 deficient mice to assess the role of MyD88 in host defense against Pneumocystis infection, and/or the immunopathogenesis of PcP.
Materials and Methods
Mice
CB.17 severe combined immunodeficient (SCID) and C57BL/6 wild type (WT) mice were bred at the University of Rochester. C57BL/6 MyD88−/− mice were generously provided by Dr. S. Akira (Osaka University, Japan) (19, 20). All animal protocols were approved by the University Committee for Animal Research at the University of Rochester Medical Center.
Isolation and enumeration of mouse Pneumocystis
Pneumocystis was isolated from the lungs of heavily infected SCID mice and enumerated by Gomori’s methenamine silver (GMS) staining as previously described (21). Each Pneumocystis preparation was tested to insure there was no bacterial contamination.
Mouse models of Pneumocystis infection
For the immunocompetent mouse model, 6 to 8 week old mice were anesthetized and 1e6 freshly isolated Pneumocystis (based on cyst count) were inoculated directly into the lungs via the trachea. For the immunosuppressed mouse model of PcP, CD4+ T cells were depleted by intraperitoneal injections of 250μg per mouse of monoclonal antibody specific for mouse CD4 (clone GK1.5, ATCC TIB207). Injections were given twice per week, starting one week prior to infection and continuing throughout the experiment. Depleted mice were inoculated with 5e5 freshly isolated Pneumocystis (based on cyst count).
Physiologic assessment of pulmonary function in live, ventilated mice
Dynamic lung compliance and lung resistance were measured in live ventilated mice using a whole body plethysmograph (BUXCO Electronics Inc., Wilmington, NC) connected to a Harvard rodent ventilator (Harvard Apparatus, Southnatic, MA) as previously described (22). Dynamic lung compliance was normalized to the peak body weight of each animal. Respiratory rates were measured using whole body unrestrained chambers (BUXCO Electronics Inc). Data was collected and analyzed using the Biosystems XA software package (BUCXO Electronics Inc.).
Lung tissue preparation and Bronchoalveolar lavage (23)
At indicated time points, mice were anesthesized, exanguinated and perfused through the heart. Right lung lobes were lavaged with three, one-ml aliquots of 1X Hank’s balanced salt solution (HBSS) (GIBCO®, Invitrogen), and then snap frozen in liquid nitrogen and stored at −80°C. The BAL fluid was centrifuged for 10 minutes at 150 × g at 4°C, and supernatants were frozen at −80°C. Recovered BAL cells were enumerated in the presence of trypan blue using a hemacytometer (Hausser Scientific) and centrifuged onto glass slides for differential staining (Diff-Quick). Differential counts were performed by microscopic examination of the slides. Multi-parameter flow cytometric analyses were also performed in BAL samples. Anti-CD4-Fluorescein (clone RM4–4) and anti-CD8a-Peridinin Chlorophyll-α Protein (clone 53–6.7), were purchased from BD Biosciences (San Diego, CA). The anti-CD4 clone RM4–4 was used to confirm CD4+ cell depletion in vivo in spleen samples because it is not blocked by the CD4-depleting antibody (clone GK1.5). Cells were analyzed on a FACSCalibur (BD Biosciences, San Jose, CA)
Quantification of Pneumocystis burden
Real-time quantitative PCR method was utilized to quantify lung Pneumocystis burden in lung homogenate preparations. Right lung lobes were homogenized, subjected to three freeze/thaw cycles, boiled for 20 min, and then centrifuged at max speeding a μfuge. The lung homogenate supernatants were utilized for quantification of Pneumocystis burden. Quantitative PCR using TaqMan primer/fluorogenic probe chemistry was performed with a primer/probe set specific for the single copy mouse Pneumocystis kexin gene as previously described (22). The Applied Biosystems Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA) was used to quantify Pneumocystis kexin gene copies.
Measurement of P. carinii-specific IgG and IgM
Pneumocystis-specific immunoglobulin G (24) and immunoglobulin M (IgM) in mouse serum was determined by enzyme-linked immunosorbent assay as described previously (25, 26). Flat-bottom microtitration plates (Flow Laboratories, McLean, Va.) were coated with mouse Pneumocystis soluble total protein from the lungs of infected SCID mice (10 mg of protein per ml), or protein from uninfected mouse lung. After blocking using 5% nonfat dry milk in phosphate-buffered saline, test sera were diluted 1:25 in phosphate-buffered saline–0.05% Tween 20. After washing the plates, goat anti-mouse IgG+IgM (H+L) alkaline phosphatase (Jackson ImmunoResearch Laboratories, Inc.) diluted 1:5,000 was used as secondary antibody. The plates were developed with p-nitrophenylphosphate as a substrate, and optical density (20) was measured at 405 nm. The background OD from uninfected lung protein coated plates was subtracted from Pneumocystis-infected mice lung protein coated plates.
Histological examination of lung sections
Left lung lobes were inflated with 15 cm gravity flow-pressure of 10% formalin (Sigma, St. Louis, MO). The lungs were fixed in situ for 10 minutes, removed from the mouse and placed in fixative solution for 16 h at 4°C. Lung tissue was embedded in paraffin and 4μM sections were cut. Lung sections were stained with hematoxylin and eosin to visualize tissue.
Cytokine and chemokine enzyme linked immunosorbent assay (ELISA)
BAL fluid was collected and centrifuged at 12,000 × g for 10 min to remove cell debris. BAL fluids were stored at −80°C. Chemokine levels of CCL2 and CXCL2 and cytokine levels of TNF-α, IL-5, IL-17, IFN-γ, IL-1α and IL-1β were determined by commercially available ELISA kits (R&D Systems), and utilized according to manufacturer’s instructions.
Generation of chimeras by bone marrow transplantation (BMT)
Bone marrow transplant (BMT) chimeras were generated as previously described (27). Briefly, (donor → recipient) WT→WT, KO→ WT, WT→KO and KO→KO (WT=C57BL/6; KO= C57BL/6 MyD88−/−) BMT chimeras were prepared by radioablation by total body irradiation of female recipients (6 Gy × 2 doses), followed by reconstitution with male donor bone marrow (Shepherd irradiator, 6000 Ci 137Cs source). To extract bone marrow from donors of the respective mouse strains, femurs and tibias were flushed into HBSS with 1% FCS. Cells were dispersed through a 21-gauge needle, and pooled by strain. Erythrocytes were removed by hypotonic lysis. The cells were counted, resuspended, and delivered to recipient mice by tail vein injection of 1 ×107 cells per mouse in a volume of 100μL. After BMT, animals were allowed to reconstitute for 8–10 weeks under microisolator conditions, supplied with high efficiency particulate filtered air, sterilized food, acid water, and bedding. Fluorescent in situ hybridization (FISH) to detect the Y chromosome, was used on BAL cells from some mice to confirm the male origin of cells transplanted to female recipients, as previously described (28).
Statistical Analysis
Data are presented as mean ± 1 standard error measurement (SE). In some experiments differences between strains at different time points were analyzed using two-way ANOVA and Bonferroni’s Multiple Comparison Test as a post test. Differences between infected animals and control uninfected were analyzed using one-way analysis of variance (ANOVA) using Tukey’s Multiple Comparison Test as a post test. Differences were considered significant at P <0.05. All data were analyzed using GraphPad Prism (version 5.00) software (GraphPad Software, San Diego, CA).
Results
MyD88 deficient mice maintain effective host defense against Pneumocystis infection
The MyD88 adaptor molecule functions in the generation and maintenance of innate and adaptive immune responses. However, the specific contribution of MyD88 to host defense against respiratory infection with Pneumocystis is unknown. To address this question, non-immunosuppressed C57BL/6 WT and MyD88 deficient mice were infected with freshly isolated Pneumocystis organisms, and organism burden over time was determined. As expected, immunocompetent WT mice harbored Pneumocystis organisms in the lung for approximately 3 weeks before an adaptive immune response cleared the infection (Fig. 1A, B). However, MyD88 deficient mice had significantly higher Pneumocystis burden than WT mice on day 14 post-infection (Fig. 1A). At later time points there was no difference between WT and MyD88 deficient mice with respect to lung burden, and the MyD88 deficient mice were competent to mount an effective immune response, which included the production of anti-Pneumocystis antibody (Fig. 1B). These results demonstrated that MyD88 plays a role in the control of Pneumocystis growth early after infection, but that MyD88 is not essential for effective adaptive immunity, the production of anti-Pneumocystis antibody, or the clearance of Pneumocystis from the lung.
Figure 1. MyD88 deficient mice maintain effective host defense against Pneumocystis infection.
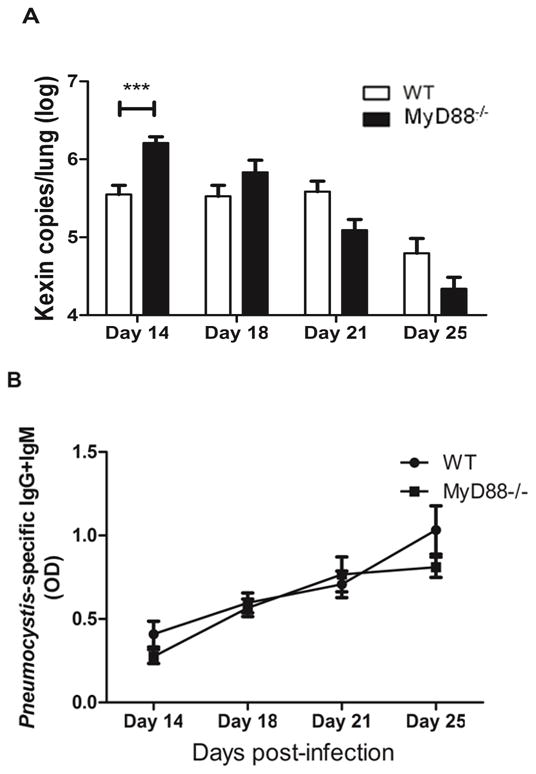
Pneumocystis burden was determined by quantitative real-time PCR for Pc kexin gene copies in WT and MyD88−/− mice on days 14, 18, 21 and 25 post-Pneumocystis infection (A). Pneumocystis specific IgG and IgM in the serum of mice were measured by ELISA using soluble Pneumocystis antigens from infected mouse lung homogenates as target (B). Values are mean ± 1 standard error measurement (n=4 to 8/timepoint/group). ***, P<0.001 in WT compared to MyD88−/−. Mean represents combined data from two independent experiments.
In addition to monitoring fungal growth and clearance, pulmonary function was also measured to determine whether loss of MyD88 affected PcP-related lung disease independent of changes in Pneumocystis burden. As expected, immunocompetent WT mice showed little evidence of respiratory impairment during Pneumocystis growth and immune-mediated clearance. Furthermore, lung compliance and lung resistance measurements were very similar in WT and MyD88 deficient mice at all time points examined, and were not statistically different from uninfected control mice (Fig. 2A and B). In addition, neither group lost weight during Pneumocystis infection, which is another indicator of PcP-related respiratory disease (data not shown). These findings confirm that MyD88 is not required for effective host defense against Pneumocystis, and that loss of MyD88 does not render mice more susceptible to lung injury associated with clearance of Pneumocystis by the normal immune system.
Figure 2. MyD88 deficiency does not alter lung function during Pneumocystis infection in the immunocompetent host.
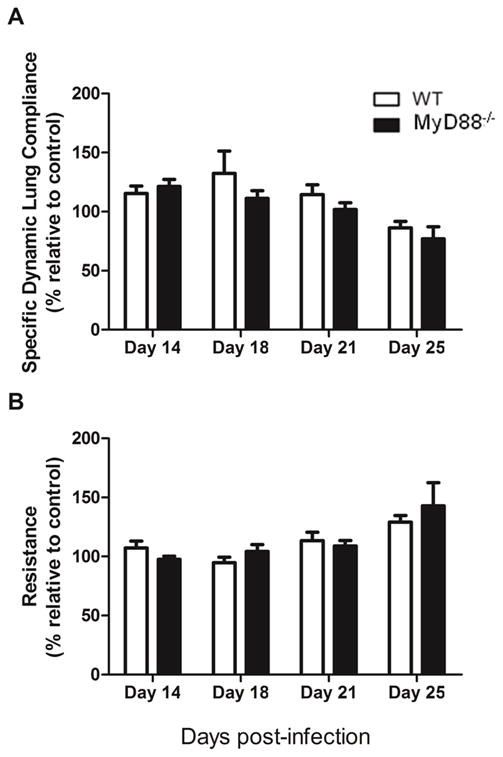
Dynamic lung compliance and lung resistance were measured in C57BL/6 (WT) and MyD88−/− mice on days 14, 18, 21 and 25 after Pneumocystis infection. Uninfected mice were used as a control. Values are mean ± 1 standard error measurement (n=4 to 8/timepoint/group) of combined data from two independent experiments (P>0.05).
MyD88-dependent signals regulate the normal immune response to Pneumocystis
Although there were not obvious respiratory consequences of Pneumocystis infection in MyD88 deficient mice, it remained possible that the nature or magnitude of the immune response to Pneumocystis was altered by the absence of MyD88. Total cells recovered in the BAL fluid of WT and MyD88 deficient mice were determined on days 14, 18, 21 and 25 post-infection. Pneumocystis-infected WT mice had a significantly greater number of total BAL cells than uninfected mice at day 14 post-infection, and this number decreased over time coincident with Pneumocystis clearance (Fig. 3A). Interestingly, Pneumocystis-infected MyD88 mice showed a similar increase in the number of total BAL cells following infection. However, in contrast to WT mice, the number of BAL cells recovered from MyD88 mice did not diminish over the course of the study (Fig. 3A). Pneumocystis-infected MyD88 mice had more lymphocytes, macrophages, eosinophils and PMNs than Pneumocystis-infected WT mice at all time points. These differences reached statistical significance on day 21 post-infection for all cell types, and additionally on day 25 for PMNs (Fig. 3B–E).
Figure 3. MyD88-dependent signals regulate cell recruitment during normal immune response to Pneumocystis.
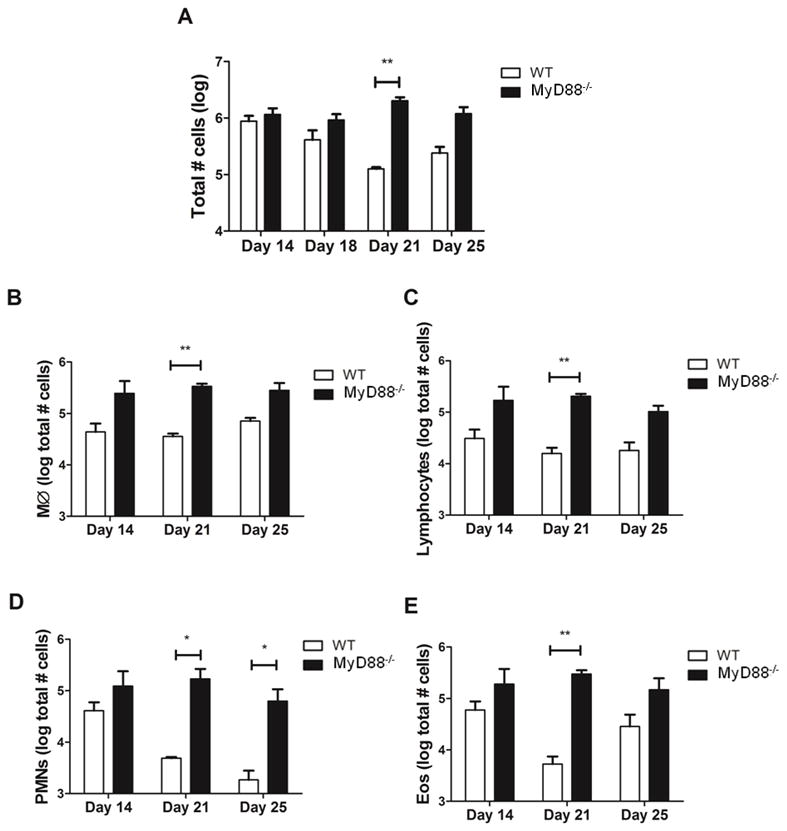
C57BL/6 (WT) and MyD88−/− mice were infected with Pneumocystis. On days 14, 18, 21 and 25 post-infection total cells, macrophages (MØ), lymphocytes, neutrophils (PMNs) and eosinophils (Eos) were enumerated from BAL. Values are mean ± 1 standard error measurement (n=4 to 8/timepoint/group) of combined data from two independent experiments (*, P<0.05; **, P<0.01).
To determine whether the altered immune response in Pneumocystis-infected MyD88 deficient mice was associated with an altered pattern of cytokine and chemokine production, protein levels were measured in BAL fluid of experimental mice. MyD88 deficient mice had statistically higher lung levels of the chemokine CCL2 than WT mice on days 14, 18 and 21 post-infection, although on day 25 both strains have similar baseline levels of the chemokine (Fig. 4A). These results correlated with increased numbers of lymphocytes and macrophages in lungs of Pneumocystis-infected MyD88 deficient mice (Fig. 3B, C). IL-4 levels in the BAL of MyD88 deficient mice were also significantly elevated as compared to WT mice on days 14, 18 and 21 post-infection, but decreased to basal levels on day 25 (Fig. 4B), suggesting a Th2-associated immune response. IL-5 levels were also elevated relative to WT mice in the lungs of MyD88 deficient mice and peaked at day 21 (Fig. 4C), coincident with the peak number of eosinophils in the lungs (Fig. 3E). Furthermore, IL-17 levels were also elevated in the lungs of Pneumocystis-infected MyD88 deficient mice compared to WT (Fig. 4D), and was associated with increased numbers of neutrophils (Fig. 3D). There was no significant difference in TNF-α levels, although there was a trend towards higher TNF-α levels in MyD88 deficient mice on days 14, 18 and 21 post-infection (Fig. 4E). IFN-γ levels in the BAL fluid were very low and nearly at the limit of detection in both strains, and there was no difference at any time point (Fig. 4F). These results further confirm that loss of MyD88 signaling alters the normal immune response to Pneumocystis, including more CCL2, more Th2-associated IL-4 and IL-5, and Th17-associated IL-17. These results suggest that MyD88 helps to regulate the degree of inflammation during a normal immune response to Pneumocystis infection, and that MyD88 deficiency results in exacerbated Th2 and Th17 responses following Pneumocystis infection.
Figure 4. MyD88-deficiency alters chemokine and cytokine responses to Pneumocystis.

C57BL/6 (WT) and MyD88−/− mice were infected with 5 × 105 freshly isolated Pneumocystis cysts. On days 14, 18, 21 and 25 post-infection CCL2 (A), IL-4 (B), IL-5 (C),IL-17 (D), TNF-α (E) and IFNγ (F) were measured by ELISA from BAL. Values are mean ± 1 standard error measurement (n=4 to 8/timepoint/group) of combined data from two independent experiments (*, P<0.05; **, P<0.01).
MyD88 signaling contributes to PcP-related immunopathogenesis during early stages of infection, but is protective during later stages of disease
MyD88 plays a role in the early inflammatory response to Pneumocystis (16). To investigate the role of MyD88 in the immunopathogenesis of PcP, a model of AIDS-related immunosuppression was utilized to induce susceptibility to PcP. For this, WT and MyD88 deficient mice were depleted of CD4+ T cells and infected with freshly isolated Pneumocystis organisms. During the progression of the infection, body weights and respiratory rates were measured non-invasively to assess overall health and disease progression. WT and MyD88 deficient mice exhibited no weight loss over the first 4 weeks of infection, but both began to show weight loss after 4 weeks of infection. However, MyD88 deficient mice displayed a much more rapid weight loss than WT mice at this time (Fig. 5A). There were also differences in respiratory rates. WT mice displayed increased respiratory rates compared to MyD88 deficient mice during the first 3 weeks of infection (Fig. 5B). However, between 3 and 5 weeks post-infection, the MyD88 deficient mice displayed a much more dramatic elevation in respiratory rate than WT mice. The respiratory rates of MyD88 deficient mice rose approximately 100%, compared to a 12% increase observed in WT mice over this period. By day 31 post-infection MyD88 deficient mice had higher respiratory rates than WT mice, with 514 ± 31 and 465 ± 46 breaths per minute, respectively.
Figure 5. MyD88 signaling contributes to severity of PcP and PcP-associated respiratory impairment during early stage of infection, but is protective during later stage of disease.
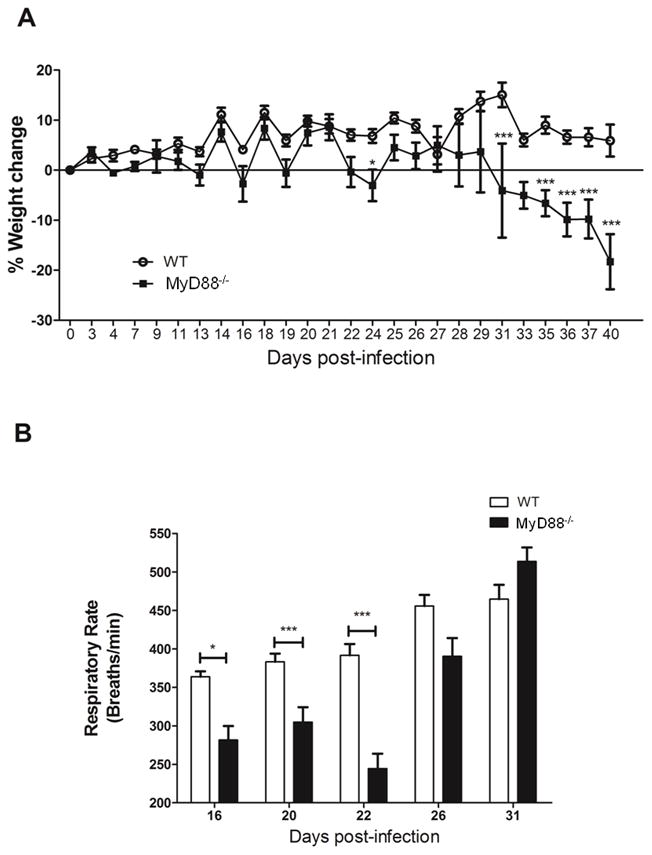
CD4+ T cell depleted C57BL/6 (WT) and MyD88−/− mice were infected with 5 × 105 freshly isolated Pneumocystis cysts. Mice were weighed from two to three times a week, and changes in body weight (% weight change) were calculated. Respiratory rates were measured non-invasively on days 16, 20, 22, 26 and 31 post-infection. Values are mean ± 1 standard error measurement (n=6 to 11/timepoint/group) of combined data from two independent experiments (*, P<0.05; ***, P<0.001).
To directly assess the contribution of MyD88 to PcP-related respiratory impairment, pulmonary function was measured on experimental mice at 4 and 5 weeks post-infection. We chose two time points that reflected the distinct stages of disease in the MyD88 mice; prior to weight loss and respiratory rate increase (Week 4), and when MyD88 deficient mice displayed signs of exacerbated disease compared to WT (Week 5). At weeks 4 and 5 post-infection WT mice showed significantly lower dynamic lung compliance and higher lung resistance relative to uninfected mice, which is characteristic of PcP (Fig. 6A and B). In contrast, at 4 weeks post-infection MyD88 deficient mice were protected from the PcP-related lung function deficits observed in WT mice. The Pneumocystis-infected MyD88 deficient mice had significantly higher dynamic lung compliance than WT infected mice, which was similar to lung compliance measurements in uninfected control mice (Fig. 6A). Similarly, MyD88 deficient mice also showed comparable lung resistance measurements to control uninfected mice at week 4 post-infection (Fig. 6B). However, between 4 and 5 weeks post-infection, MyD88 deficient mice displayed a 50% drop in dynamic lung compliance and a 25% increase in lung resistance, signifying a dramatic acceleration in the severity of PcP in these mice over this period. These values were significantly different from uninfected mice, and worse than infected WT mice (Fig. 6A and B). Together these data demonstrate that MyD88 signaling contributes to PcP-associated respiratory impairment early during infection, but protects at later stages of disease. Thus, MyD88 signaling likely plays distinct roles during different stages of PcP.
Figure 6. MyD88 signaling contributes to PcP-related lung function deficits during the early stage of infection, but is protective during the later stage of disease.
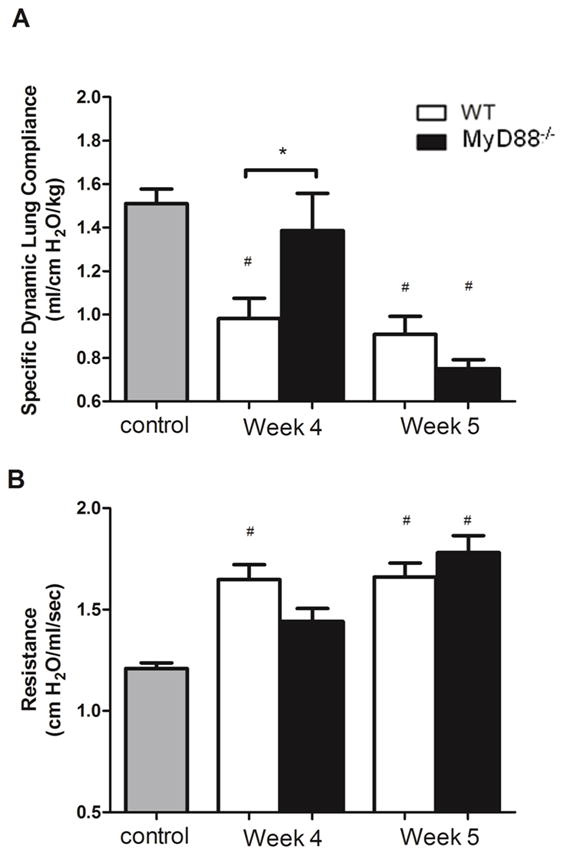
CD4+ T cell depleted C57BL/6 (WT) and MyD88−/− mice were infected with 5 × 105 freshly isolated Pneumocystis cysts. Dynamic lung compliance and lung resistance were measured on week 4 and week 5 post-infection. Uninfected mice were used as a control. Values are mean ± 1 standard error measurement (n=6 to 11/timepoint/group) of combined data from two independent experiments (*, P<0.05; #, P<0.05 compared to control).
MyD88-dependent mechanisms limit fungal burden during active PcP
To determine whether MyD88 plays a role in controlling Pneumocystis growth in a CD4+ T cell-depleted model of PcP, lungs of WT and MyD88 deficient mice were tested for Pneumocystis burden using quantitative real-time PCR as we have described (22). There was no difference in Pneumocystis burden between WT and MyD88 deficient mice at 4 weeks post-infection (Fig. 7). However, by 5 weeks post-infection the lungs of MyD88 deficient mice contained nearly 2-fold more Pneumocystis organisms than WT mice (Fig. 7). These data show that MyD88-dependent mechanisms help to control fungal burden in CD4+ T cell-depleted mice.
Figure 7. MyD88-dependent mechanisms limit fungal burden during active PcP.
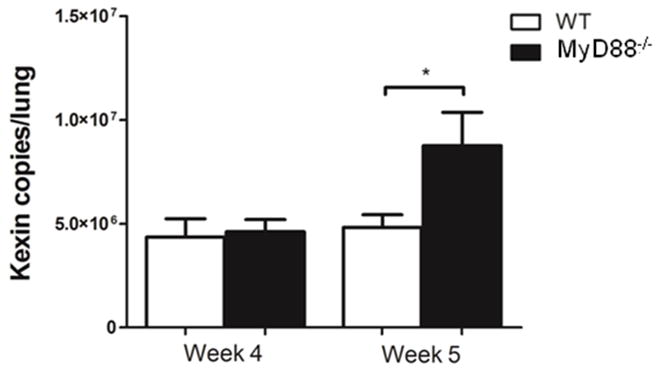
Pneumocystis burden was determined by quantitative real-time PCR of kex1 gene copies in CD4-depleted WT and MyD88−/− mice on week 4 and week 5 post-Pneumocystis infection. Values are mean ± 1 standard error measurement (n=6 to 11/timepoint/group) *, P<0.05.Mean represents combined data from two independent experiments.
MyD88 contributes to lung inflammation early during Pneumocystis infection
To determine whether the differences observed in overall health and lung function between Pneumocystis-infected MyD88 deficient and WT mice correlated with the degree of pulmonary inflammation, total cell counts in BAL fluid were performed. Pneumocystis-infected MyD88 deficient mice had significantly fewer BAL cells than infected WT mice at week 4 (Table I). Significantly fewer numbers of macrophages and CD8+ T cells were present in the lung of MyD88 deficient mice at this time. Both of these cell types are effectors of PcP-related immunopathogenesis, and reduced numbers in the lungs of MyD88 deficient mice is likely a factor in the preserved lung function observed in these mice. At week 5 post-infection, there was not a significant difference in total cells or any specific cell type recruited to the lungs of Pneumocystis-infected WT or MyD88 deficient mice, suggesting that mice can compensate for the loss of MyD88 at least with respect to cell recruitment to the lung.
Table I.
Cellular composition of BAL fluid from Pneumocystis-infected, CD4-depleted WT and MyD88−/− mice
| Week 4 | Week 5 | |||
|---|---|---|---|---|
| Cell Type (105) | WT | MyD88−/− | WT | MyD88−/− |
| Total cells | 4.87 ± 0.51 | 2.85 ± 0.59* | 3.34 ± 0.37 | 4.08 ± 0.54 |
| Macrophages | 1.46 ± 0.12 | 0.97 ± 0.19* | 1.01 ± 0.25 | 0.92 ± 0.28 |
| PMNs | 1.78 ± 0.39 | 0.84 ± 0.29 | 1.27 ± 0.22 | 1.49 ± 0.28 |
| Lymphocytes | 1.59 ± 0.20 | 0.96 ± 0.24 | 1.05 ± 0.16 | 1.63 ± 0.27 |
| CD8+ T cells | 1.30 ± 0.33 | 0.23 ± 0.14* | 0.81 ± 0.20 | 0.49 ± 0.17 |
BAL fluid was isolated from one lung and cell populations were distinguished by differential counts or flow cytometry.
, p<0.05 as compared with Pneumocystis-infected WT mice at the same time point. Values are mean ± 1 standard error measurement (n=6 to 11/time point/group). Data is pooled from two independent experiments.
Histological examination of lung sections from experimental mice demonstrated increased cellular infiltration and inflammation in the alveolar region of Pneumocystis-infected WT mice at 4 weeks post-infection, as compared with uninfected lungs (Fig. 8A and B). In contrast, the lungs of Pneumocystis-infected MyD88 deficient mice displayed fewer alveolar infiltrates and less inflammation than WT mice at this time. Furthermore, while the cell infiltrates appeared dispersed throughout the alveoli in WT mice, they were localized mainly to peribronchial regions of the MyD88 deficient lungs, with less involvement of the alveolar regions. At week 5 post-infection, the lungs of Pneumocystis-infected MyD88 deficient mice showed more signs of inflammation and cell recruitment throughout the lung, which was similar to that observed in WT mice (Fig. 8C). Interestingly, the lungs of MyD88 deficient mice displayed more foamy exudates filling the alveolar spaces than were evident in WT mice (Fig. 8C, D). These results demonstrate that MyD88 regulates cell recruitment and inflammation in the lung during PcP.
Figure 8. MyD88 contributes to lung inflammation early during Pneumocystis infection.
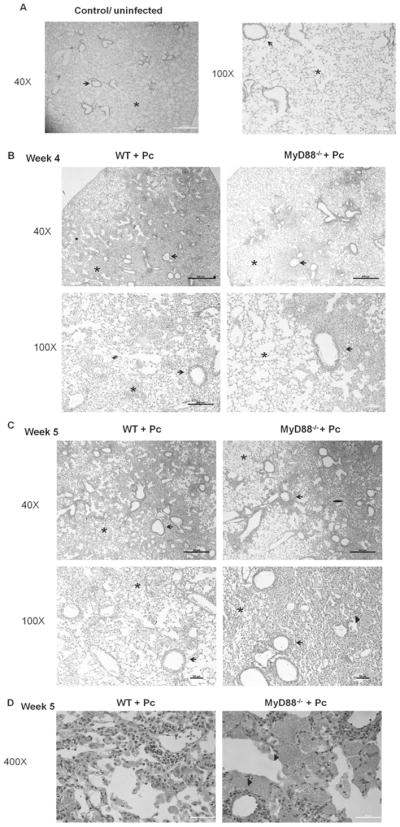
CD4+ T cell depleted C57BL/6 (WT) and MyD88−/− mice were infected with 5e5 freshly isolated Pneumocystis cysts. Lung sections from uninfected control mice (A), or Pneumocystis-infected mice at week 4 (B), or week 5 post-infection (C, D) were stained with Hematoxylin and Eosin. Representative pictures were taken by microscopy under 40×, 100× (A–C) or 400× (D) magnification as indicated. Arrows denote peribronchiolar regions in each section, while asterisks denote alveolar regions. In C and D arrow heads indicate foamy exudates in alveolar spaces.
MyD88 regulates chemokine and cytokine production during PcP
PcP-related immunopathogenesis is characterized by an increase in cytokine and chemokine production in the lungs. To determine whether the reduced cell-mediated pulmonary inflammation and preserved pulmonary physiology in the Pneumocystis-infected MyD88 deficient mice early during infection is associated with reduced chemokine and/or cytokine production in the lung, ELISA was used to evaluate BAL fluid from experimental mice. As expected, CCL2 levels were increased in the lungs of Pneumocystis-infected WT mice compared to uninfected controls. Also consistent with our earlier work (16), CCL2 levels were significantly reduced in the lungs of MyD88 deficient mice compared to WT mice at 4 weeks post-infection (Fig. 8A), which likely explains our finding of fewer macrophages and CD8+ T cells in the lungs of MyD88 deficient mice at this time (Table I). However, by 5 weeks post-infection CCL2 levels were similar in the lungs of MyD88 deficient and WT mice (Fig. 9A), indicating that CCL2 production can proceed via a MyD88-independent mechanism at later time points. Surprisingly, CXCL2, TNF-α, and IFN-γ levels were higher in the lungs of Pneumocystis-infected MyD88 deficient mice compared to WT lungs (Fig. 9B, C, and D), and elevated TNF-α and IFN-γ in the lungs at 5 weeks post-infection likely contributes to the MyD88-independent inflammation observed at this time. IL-1α and IL-1β levels were also higher in infected MyD88 deficient mice compared to WT mice (Fig. 9E and F), which is likely related to lack of negative feedback regulation due to impaired IL-1 receptor signaling. These data demonstrate that deletion of MyD88 causes dysregulated cytokine and chemokine responses in the lung during PcP.
Figure 9. MyD88 regulates chemokine and cytokine production during PcP.
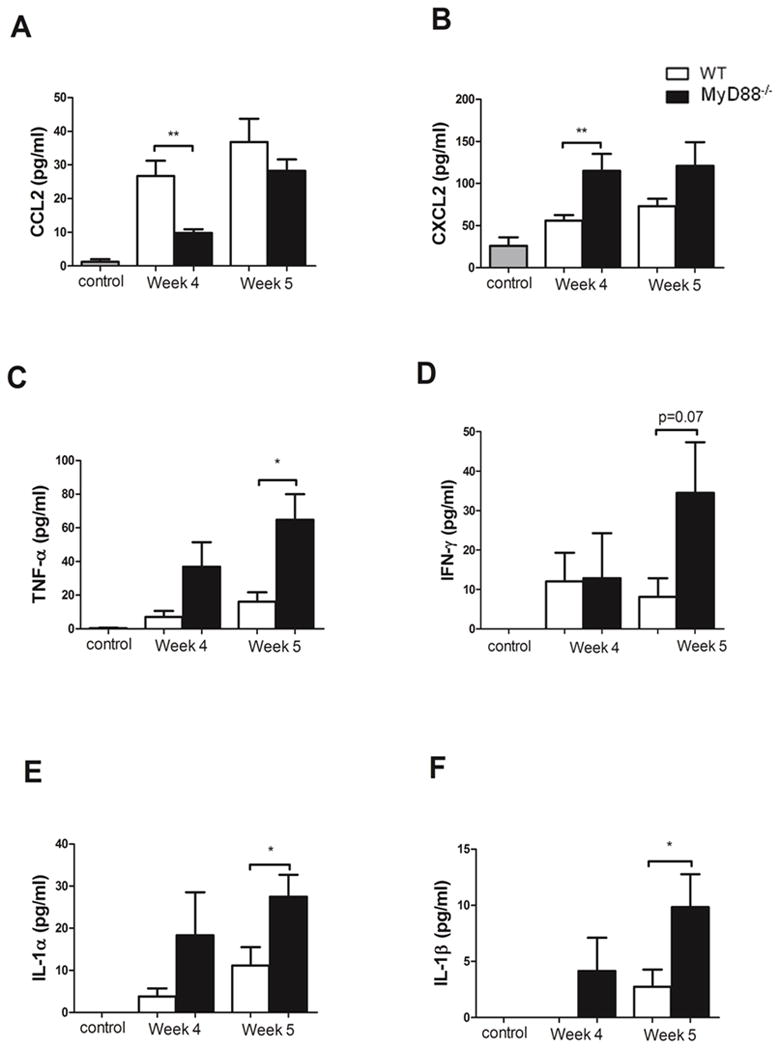
CD4+ T cell depleted C57BL/6 (WT) and MyD88−/− mice were infected with 5 × 105 freshly isolated Pneumocystis cysts. CCL2 (A), CXCL2 (B), TNF-α (C), IFN-γ (D), IL-1α (E) and IL-1β (E) levels were measured in the BAL fluid from Pneumocystis-infected mice and control uninfected mice. Values are mean ± 1 standard error measurement (n=6 to 11/timepoint/group) of combined data from two independent experiments (*, P<0.05; **, P<0.01).
MyD88-dependent hematopoietic cell responses control fungal burden during active PcP
To determine whether the MyD88-dependent mechanisms required for the control of Pneumocystis burden rely on lung resident cells or hematopoietic cells, we generated bone-marrow chimera mice. WT and MyD88−/− mice were lethally irradiated and reconstituted with WT or MyD88−/− bone marrow cells. After 8 to 10 weeks post-reconstitution, mice were CD4-depleted and infected with Pneumocystis. At week 4 post-infection, there were not statistically significant differences in Pneumocystis lung burden between any of the groups, although WT mice reconstituted with MyD88−/− cells (KO→WT) showed higher organism burden (Fig. 10). On week 5 post-infection, irradiated WT mice that were reconstituted with WT bone-marrow derived cells (WT→WT) presented with similar Pneumocystis counts as MyD88−/− mice reconstituted with WT cells (WT→KO). However, WT mice that received MyD88−/− bone marrow-derived cells (KO→WT) were unable to control Pneumocystis growth, as demonstrated by significant increase in organism burden in the lungs of these mice (Fig. 10). MyD88−/− mice reconstituted with MyD88−/− (KO→KO) bone marrow cells showed slightly higher Pneumocystis burden than WT→WT mice, but significantly lower burden than KO→WT (Fig. 10), suggesting that MyD88 signaling in hematopoietic and parenchymal cells may have opposing effects on control of Pneumocystis infection. These data suggest that the MyD88 signaling pathway in hematopoietic derived cells is important for control of organism burden during Pneumocystis infection in immunocompromised host.
Figure 10. MyD88-dependent signals in hematopoietic cells control fungal burden during active PcP.
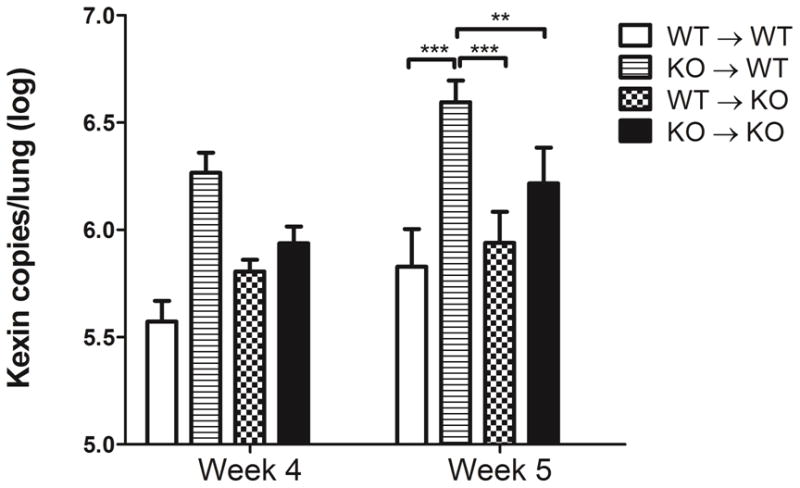
Pneumocystis burden was determined by quantitative real-time PCR for kex1 gene copies in CD4-depleted WT and MyD88−/−(KO) bone marrow chimeras (BMC) at weeks 4 and week 5 post-Pneumocystis infection. BMC mice were prepared by lethal irradiation of either WT or MyD88−/− mice (KO) as described in Materials and Methods. Irradiated WT mice reconstituted with WT bone marrow cells (WT→WT), irradiated WT mice reconstituted with MyD88−/− bone marrow cells (KO→WT), irradiated MyD88−/− mice reconstituted with WT bone marrow cells (WT→KO), irradiated MyD88−/− mice reconstituted with MyD88−/− bone marrow cells (KO→KO). Values are mean ± 1 standard error measurement (n=6 to 11/timepoint/group) **, P<0.01; and ***, P<0.001.Mean represents combined data from two independent experiments.
Discussion
The MyD88 signaling pathway plays an important role in the generation of effective innate and adaptive immune responses to a variety of pathogens. In vitro studies have determined that MyD88 is required for alveolar epithelial cell and alveolar macrophage chemokine and cytokine responses to Pneumocystis (15), (16). However, the role of MyD88-dependent signaling during Pneumocystis infection and immunity in vivo has not been determined. In this study we expanded upon our previous work by; 1) identifying a regulatory role for MyD88 during the normal adaptive immune response against Pneumocystis; and 2) demonstrating that MyD88 signaling participates in PcP-related immunopathogenesis. While MyD88 signaling is not required for resistance to Pneumocystis infection in immunocompetent mice, it does help control fungal burden early during infection and limits the subsequent protective immune response. In the CD4-depleted AIDS-related model of active PcP, MyD88 contributes to inflammation, cell recruitment, and impaired pulmonary function during the early stages of PcP. However, at later stages of disease, MyD88 signaling limits inflammation and lung injury, and helps control Pneumocystis burden. These results suggest that MyD88 regulates the intensity of the immune response to Pneumocystis in immunocompetent hosts. However, MyD88 plays a dual role during active PcP by contributing to immunopathogenesis at early stages, but protecting against lung injury and uncontrolled growth of the organism at later stages.
Our results suggest that MyD88-dependent mechanisms control Pneumocystis burden early during the CD4+ T cell-mediated immune response. Even though there were slightly higher numbers of macrophages in the lungs of MyD88−/− mice early at day 14 post-infection, it seems that the MyD88 deficient macrophages were less efficient in controlling the fungal burden. MyD88 deficiency may prevent effective killing of Pneumocystis by AMs, either by defects in phagocytosis, or through impaired production of antimicrobial agents such as reactive oxygen or nitrogen species. Other studies have shown similar increased susceptibility of Pneumocystis infection in dectin-1 or TLR2 deficient mice, and have suggested a role for reactive oxygen and nitrogen species in clearance of Pneumocystis (29, 30). Interestingly, a similar pattern of increased fungal burden early during infection followed by effective resolution has been observed in MyD88−/− mice infected with Aspergillus (12). Importantly, MyD88−/− mice were able to clear Pneumocystis infection similar to WT mice, suggesting that MyD88 is not required for host defense against this infection. The ability of MyD88−/− mice to clear Pneumocystis is probably due to an effective adaptive response and production of Pneumocystis-specific antibody that was comparable to WT mice. Production of Pneumocystis specific IgG and IgM antibody has been shown to correlate with clearance of Pneumocystis organisms (25, 31, 32).
In addition, we found that MyD88 regulates the intensity and resolution of the Pneumocystis-specific immune response in non-immunosuppressed mice. Although MyD88−/− mice were able to clear the infection, the mice displayed an exacerbated immune response characterized by elevated CCL2 which correlated with higher numbers of lymphocytes and macrophages recruited to the lung, and an increase in the Th2-related cytokines IL-4 and IL-5 which may explain increased lung eosinophilia. Interestingly, MyD88−/− mice that were infected with the pathogens Legionella pneumophila, Salmonella tyhimurium, Chlamidya muridarum or Leishmania major all presented polarized Th2 responses that were associated with enhanced disease pathology (33–36). In addition, levels of the Th17 cytokine, IL-17, were also elevated in MyD88−/− mice, which correlated with higher numbers of neutrophils recruited to the lungs. Increased inflammation and lung pathology in Pneumocystis infected nude IFNγ−/− mice correlated with higher levels of IL-17 in the lung (37). It should be noted that despite displaying an exacerbated immune response against Pneumocystis, non-immunosuppressed MyD88−/− mice did not exhibit reduced pulmonary function relative to WT mice. We speculate that the recruited MyD88 deficient inflammatory cells are less activated than WT cells, and thus do not have profound affects on lung function.
Recent studies have suggested a role for TLR2 and TLR4 in the in vivo responses to Pneumocystis in a CD4-depleted, AIDS-related model of PcP (18, 29). However, both studies suggest that either TLR2 or TLR4 are not the sole pathways responsible for the immunopathogenesis during Pneumocystis infection. Our results using infected MyD88−/− mice show an increase in lung TNF-α and CXCL2 compared to WT mice, which is consistent with results reported using a model of pulmonary Aspergillus infection (12). Conversely, our results show a decrease in CCL2 in the BAL of CD4-depleted MyD88−/− mice with PcP. CCL2 (or MCP-1), is a member of the CC chemokine family which recruits macrophages and T cells to sites of infection (23, 38–40). Macrophages and CD8+ T cells contribute to PcP-related immunopathogenesis, and reduced CCL2 in MyD88−/− mice correlated with fewer macrophages and CD8+ T cells recruited to the lungs and preserved lung function. (9, 41, 42). These results suggest that MyD88 mediates the inflammatory response and lung injury early during infection through CCL2-dependent recruitment of CD8+ T cells and macrophages. MyD88 also regulates other chemokines and cytokines such as CXCL2, TNF-α, IFN-γ, IL-1α and IL-1β (Fig. 8). Our data also suggests that additional MyD88-dependent receptors other than TLR2 or TLR4 may be involved in the response to Pneumocystis. For example, IL-1R was found to mediate the chemokine response to Pneumocystis in primary AECs, and may play a role in the immunopathogenesis of PcP (16). Interestingly, IL-1β is required for effective resolution of important and perhaps somewhat redundant roles in the immunopathogenesis of PcP. For this reason, our study attempted to determine the role during PcP of the adaptor molecule MyD88, which participates in the TLR/IL-1 pathways.
MyD88 is required for optimal alveolar macrophage TNF-α responses to Pneumocystis β-glucan (15), and the MyD88-dependent pattern recognition receptors TLR2 and TLR4 also contribute to AM chemokine and cytokine responses to Pneumocystis (18, 29). In vivo studies found that CD4-depleted TLR2−/− mice harbored higher Pneumocystis burdens than WT mice, similar to our findings in MyD88−/− mice (29). AECs also respond to Pneumocystis, and we recently reported that the alveolar epithelial cell chemokine response is dependent on MyD88 and IL-1 receptor, but independent of TLR2 or TLR4 (16). It is interesting that our chimeric studies determined that loss of AEC MyD88 signaling did not affect fungal burden in CD4-depleted mice, whereas loss of hematopoietic cell MyD88 caused increased burden. These findings indicate that AEC responses may not directly contribute to control of Pneumocystis burden during active PcP in immunosuppressed mice. However, it remains possible that AEC responses contribute to inflammatory cell recruitment and PcP-related immunopathogenesis through MyD88-dependent mechanisms. Further studies are required to explore this possibility.
MyD88-independent pathways may also limit the inflammatory response during active PcP in CD4-depleted mice, especially at later stages of disease. At 5 weeks post-infection, when MyD88−/− mice developed severe inflammation and exhibited signs of impaired lung function, BAL TNF-α levels were significantly higher than in WT mice. Excessive TNF-α is associated with inflammatory cell recruitment, uncontrolled activation of pro-inflammatory pathways, and endothelial leakiness. A previous study from our group showed that TNF receptor signaling drives pulmonary inflammation and respiratory impairment in Pneumocystis-infected CD4-depleted mice (22). While this study found that the presence of CD8+ T cells is required for maximal TNF production during PcP in CD4-depleted mice (22), CD8+ T cell-independent mechanisms of TNF production may also contribute to elevated lung levels in MyD88−/− mice. For example, excessive lung injury in MyD88−/− mice due to higher organism burdens at 5 weeks post-infection could cause elevated TNF-α release. A similar outcome was observed in a study in which lymphocyte deficient SCID mice were infected with Pneumocystis. These mice produced very little lung TNF-α until the very late stages of disease when lungs were injured and the Pneumocystis burden was very high (43). It is possible that various redundant pathways work in cooperation to detect and respond to Pneumocystis. For example, dectin-1 has been found to cooperate with TLR2 to stimulate TNF-α production in macrophages exposed to mycobacteria infection (44). In addition, macrophage mannose receptor (MR) deficient mice infected with Pneumocystis showed increased total protein, albumin and inflammatory cells in lungs compared to WT mice (45). Recently, a role for intracellular osteopontin in the control of Pneumocystis burden in immunocompromised mice was reported. The authors suggested that osteopontin clusters TLR2, dectin-1 and MR in macrophages exposed to Pneumocystis (46). It is possible that MyD88-independent pathways cooperate with the MyD88 pathway during the host response to Pneumocystis, although more studies are required to confirm these interactions.
Our data demonstrate that MyD88 signaling in hematopoietic cells is required to control Pneumocystis infection during active PcP in CD4-depleted mice. These results agree with recent findings showing that MyD88 signaling in the hematopoietic compartment was required for control of Legionella pneumophila (47). This appears to be dependent on the specific pathogen, since MyD88 expression in both resident and hematopoietic cells contribute to control of Klebsiella pneumoniae infection. In contrast, resident non-hematopoietic cells were important for control of Pseudomonas aeruginosa growth (24, 48). While we have clearly shown that MyD88 signaling modulates pulmonary immunity and contributes to PcP-related immunopathogenesis, many questions remain. For example, we have neither identified the exact cell types that utilize MyD88 signaling during Pneumocystis infection, nor defined the specific MyD88-dependent mechanisms that regulate host defense and immunopathogenesis. MyD88-dependent responses are known to regulate both innate and adaptive immunity, and many cell types, including epithelial cells, macrophages, neutrophils, lymphocytes, and DCs utilize the MyD88 signaling pathway during the course of an immune response. More in-depth follow-up studies are needed to define the cell type-specific role of MyD88 in the complicated network of immune interactions that occur in response to Pneumocystis in vivo.
The blockade of MyD88 signaling is being explored as a possible therapeutic strategy to attenuate inflammatory disease (49, 50). Our preliminary findings suggest that MyD88 may represent a potential target for immunotherapy in Pneumocystis infected patients. Since lack of MyD88 early during PcP limits inflammation and promotes better lung function, it is conceivable that blocking the MyD88 pathway in combination with effective antibiotic treatment may improve disease outcome. MyD88 represents a more specific pathway that might be blocked temporarily while antibiotic treatment to Pneumocystis is administered. This is in contrast to corticosteroids that sometimes cause undesired systemic side effects. However, we also found that loss of MyD88 signaling could have negative effects on the severity of PcP at later stages of disease by increasing fungal burden, which raises concerns regarding the therapeutic blockade of MyD88 during PcP. In addition, the MyD88 and TLR pathways have been found to protect against lung injury through recognition of extracellular matrix components such as hyaluronan. Hyaluronan degradation products present during lung injury promote lung repair and protect the lung epithelium through interactions with TLRs (51, 52). Thus, we speculate that the later deterioration in CD4-depleted MyD88 deficient mice with active PcP may be partly related to loss of MyD88-dependent protective mechanisms that function during lung injury. Further studies utilizing MyD88 blocking agents in vivo are required to thoroughly evaluate the potential of targeting MyD88 for anti-inflammatory therapy during PcP.
Acknowledgments
The authors would like to thank Dr. Samir Bhagwat, Dr. Nabilah Khan, and Jane Malone for expert technical assistance.
This work was supported by Public Health Service grants HL083761 and HL113495.
References
- 1.Wang EH, Partovi N, Levy RD, Shapiro RJ, Yoshida EM, Greanya ED. Pneumocystis pneumonia in solid organ transplant recipients: not yet an infection of the past. Transpl Infect Dis. 2012;14:519–525. doi: 10.1111/j.1399-3062.2012.00740.x. [DOI] [PubMed] [Google Scholar]
- 2.Morris A, Norris KA. Colonization by Pneumocystis jirovecii and its role in disease. Clin Microbiol Reviews. 2012;25:297–317. doi: 10.1128/CMR.00013-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaur N, Mahl TC. Pneumocystis jiroveci (carinii) pneumonia after infliximab therapy: a review of 84 cases. Dig Dis Sci. 2007;52:1481–1484. doi: 10.1007/s10620-006-9250-x. [DOI] [PubMed] [Google Scholar]
- 4.Morris A, Sciurba FC, Norris KA. Pneumocystis: a novel pathogen in chronic obstructive pulmonary disease? COPD. 2008;5:43–51. doi: 10.1080/1541255070181756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith RL, Mel-Sadr W, Lewis ML. Correlation of bronchoalveolar lavage cell populations with clinical severity of Pneumocystis carinii pneumonia. Chest. 1988;93:60–64. doi: 10.1378/chest.93.1.60. [DOI] [PubMed] [Google Scholar]
- 6.Mason GR, Hashimoto CH, Dickman PS, Foutty LF, Cobb CJ. Prognostic implications of bronchoalveolar lavage neutrophilia in patients with Pneumocystis carinii pneumonia and AIDS. Am Rev Respir Dis. 1989;139:1336–1342. doi: 10.1164/ajrccm/139.6.1336. [DOI] [PubMed] [Google Scholar]
- 7.Limper AH, Offord KP, Smith TF, Martin WJ., 2nd Pneumocystis carinii pneumonia. Differences in lung parasite number and inflammation in patients with and without AIDS. Am Rev Respir Dis. 1989;140:1204–1209. doi: 10.1164/ajrccm/140.5.1204. [DOI] [PubMed] [Google Scholar]
- 8.Norris KA, Morris A, Patil S, Fernandes E. Pneumocystis colonization, airway inflammation, and pulmonary function decline in acquired immunodeficiency syndrome. Immunol Res. 2006;36:175–187. doi: 10.1385/IR:36:1:175. [DOI] [PubMed] [Google Scholar]
- 9.Wright TW, Gigliotti F, Finkelstein JN, McBride JT, An CL, Harmsen AG. Immune-mediated inflammation directly impairs pulmonary function, contributing to the pathogenesis of Pneumocystis carinii pneumonia. J Clin Invest. 1999;104:1307–1317. doi: 10.1172/JCI6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi O, Akira S. MyD88 as a bottle neck in Toll/IL-1 signaling. Curr Top Microbiol Immunol. 2002;270:155–167. doi: 10.1007/978-3-642-59430-4_10. [DOI] [PubMed] [Google Scholar]
- 11.Villamon E, Gozalbo D, Roig P, Murciano C, O’Connor JE, Fradelizi D, Gil ML. Myeloid differentiation factor 88 (MyD88) is required for murine resistance to Candida albicans and is critically involved in Candida-induced production of cytokines. Eur Cytokine Netw. 2004;15:263–271. [PubMed] [Google Scholar]
- 12.Bretz C, Gersuk G, Knoblaugh S, Chaudhary N, Randolph-Habecker J, Hackman RC, Staab J, Marr KA. MyD88 signaling contributes to early pulmonary responses to Aspergillus fumigatus. Infect Immun. 2008;76:952–958. doi: 10.1128/IAI.00927-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biondo C, Midiri A, Messina L, Tomasello F, Garufi G, Catania MR, Bombaci M, Beninati C, Teti G, Mancuso G. MyD88 and TLR2, but not TLR4, are required for host defense against Cryptococcus neoformans. Eur J Immunol. 2005;35:870–878. doi: 10.1002/eji.200425799. [DOI] [PubMed] [Google Scholar]
- 14.Loures FV, Pina A, Felonato M, Feriotti C, de Araujo EF, Calich VL. MyD88 signaling is required for efficient innate and adaptive immune responses to Paracoccidioides brasiliensis infection. Infect Immun. 2011;79:2470–2480. doi: 10.1128/IAI.00375-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lebron F, Vassallo R, Puri V, Limper AH. Pneumocystis carinii cell wall beta-glucans initiate macrophage inflammatory responses through NF-kappaB activation. J Biol Chem. 2003;278:25001–25008. doi: 10.1074/jbc.M301426200. [DOI] [PubMed] [Google Scholar]
- 16.Bello-Irizarry SN, Wang J, Olsen K, Gigliotti F, Wright TW. The Alveolar Epithelial Cell Chemokine Response to Pneumocystis Requires Adaptor Molecule MyD88 and Interleukin-1 Receptor but Not Toll-Like Receptor 2 or 4. Infect Immun. 2012;80:3912–3920. doi: 10.1128/IAI.00708-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang C, Wang SH, Lasbury ME, Tschang D, Liao CP, Durant PJ, Lee CH. Toll-like receptor 2 mediates alveolar macrophage response to Pneumocystis murina. Infect Immun. 2006;74:1857–1864. doi: 10.1128/IAI.74.3.1857-1864.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding K, Shibui A, Wang Y, Takamoto M, Matsuguchi T, Sugane K. Impaired recognition by Toll-like receptor 4 is responsible for exacerbated murine Pneumocystis pneumonia. Microbes Infect. 2005;7:195–203. doi: 10.1016/j.micinf.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 19.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 20.Werts C, Tapping RI, Mathison JC, Chuang TH, Kravchenko V, Saint Girons I, Haake DA, Godowski PJ, Hayashi F, Ozinsky A, Underhill DM, Kirschning CJ, Wagner H, Aderem A, Tobias PS, Ulevitch RJ. Leptospiral lipopolysaccharide activates cells through a TLR2-dependent mechanism. Nat Immunol. 2001;2:346–352. doi: 10.1038/86354. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Gigliotti F, Maggirwar S, Johnston C, Finkelstein JN, Wright TW. Pneumocystis carinii activates the NF-kappaB signaling pathway in alveolar epithelial cells. Infect Immun. 2005;73:2766–2777. doi: 10.1128/IAI.73.5.2766-2777.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright TW, Pryhuber GS, Chess PR, Wang Z, Notter RH, Gigliotti F. TNF receptor signaling contributes to chemokine secretion, inflammation, and respiratory deficits during Pneumocystis pneumonia. J Immunol. 2004;172:2511–2521. doi: 10.4049/jimmunol.172.4.2511. [DOI] [PubMed] [Google Scholar]
- 23.Balamayooran G, Batra S, Balamayooran T, Cai S, Jeyaseelan S. Monocyte chemoattractant protein 1 regulates pulmonary host defense via neutrophil recruitment during Escherichia coli infection. Infect Immun. 2011;79:2567–2577. doi: 10.1128/IAI.00067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hajjar AM, Harowicz H, Liggitt HD, Fink PJ, Wilson CB, Skerrett SJ. An essential role for non-bone marrow-derived cells in control of Pseudomonas aeruginosa pneumonia. Am J Respir Cell Mol Biol. 2005;33:470–475. doi: 10.1165/rcmb.2005-0199OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harmsen AG, Chen W, Gigliotti F. Active immunity to Pneumocystis carinii reinfection in T-cell-depleted mice. Infect Immun. 1995;63:2391–2395. doi: 10.1128/iai.63.7.2391-2395.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garvy BA, Wiley JA, Gigliotti F, Harmsen AG. Protection against Pneumocystis carinii pneumonia by antibodies generated from either T helper 1 or T helper 2 responses. Infect Immun. 1997;65:5052–5056. doi: 10.1128/iai.65.12.5052-5056.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pryhuber GS, Huyck HL, Bhagwat S, O’Reilly MA, Finkelstein JN, Gigliotti F, Wright TW. Parenchymal cell TNF receptors contribute to inflammatory cell recruitment and respiratory failure in Pneumocystis carinii-induced pneumonia. J Immunol. 2008;181:1409–1419. doi: 10.4049/jimmunol.181.2.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theise ND, Badve S, Saxena R, Henegariu O, Sell S, Crawford JM, Krause DS. Derivation of hepatocytes from bone marrow cells in mice after radiation-induced myeloablation. Hepatology. 2000;31:235–240. doi: 10.1002/hep.510310135. [DOI] [PubMed] [Google Scholar]
- 29.Wang SH, Zhang C, Lasbury ME, Liao CP, Durant PJ, Tschang D, Lee CH. Decreased inflammatory response in Toll-like receptor 2 knockout mice is associated with exacerbated Pneumocystis pneumonia. Microbes Infect. 2008;10:334–341. doi: 10.1016/j.micinf.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saijo S, Fujikado N, Furuta T, Chung SH, Kotaki H, Seki K, Sudo K, Akira S, Adachi Y, Ohno N, Kinjo T, Nakamura K, Kawakami K, Iwakura Y. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Iimmunol. 2007;8:39–46. doi: 10.1038/ni1425. [DOI] [PubMed] [Google Scholar]
- 31.Gigliotti F, Haidaris CG, Wright TW, Harmsen AG. Passive intranasal monoclonal antibody prophylaxis against murine Pneumocystis carinii pneumonia. Infect Immun. 2002;70:1069–1074. doi: 10.1128/IAI.70.3.1069-1074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gigliotti F, Hughes WT. Passive immunoprophylaxis with specific monoclonal antibody confers partial protection against Pneumocystis carinii pneumonitis in animal models. J Clin Invest. 1988;81:1666–1668. doi: 10.1172/JCI113503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trunk G, Oxenius A. Innate instruction of CD4+ T cell immunity in respiratory bacterial infection. J Immunol. 2012;189:616–628. doi: 10.4049/jimmunol.1200924. [DOI] [PubMed] [Google Scholar]
- 34.Iweala OI, Smith DW, Matharu KS, Sada-Ovalle I, Nguyen DD, Dekruyff RH, Umetsu DT, Behar SM, Nagler CR. Vaccine-induced antibody isotypes are skewed by impaired CD4 T cell and invariant NKT cell effector responses in MyD88-deficient mice. J Immunol. 2009;183:2252–2260. doi: 10.4049/jimmunol.0804011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen L, Lei L, Chang X, Li Z, Lu C, Zhang X, Wu Y, Yeh IT, Zhong G. Mice deficient in MyD88 Develop a Th2-dominant response and severe pathology in the upper genital tract following Chlamydia muridarum infection. J Immunol. 2010;184:2602–2610. doi: 10.4049/jimmunol.0901593. [DOI] [PubMed] [Google Scholar]
- 36.Muraille E, De Trez C, Brait M, De Baetselier P, Leo O, Carlier Y. Genetically resistant mice lacking MyD88-adapter protein display a high susceptibility to Leishmania major infection associated with a polarized Th2 response. J Immunol. 2003;170:4237–4241. doi: 10.4049/jimmunol.170.8.4237. [DOI] [PubMed] [Google Scholar]
- 37.Hu T, Takamoto M, Hida S, Tagawa Y, Sugane K. IFN-gamma deficiency worsen Pneumocystis pneumonia with Th17 development in nude mice. Immunol Lett. 2009;127:55–59. doi: 10.1016/j.imlet.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Gigliotti F, Bhagwat SP, Maggirwar SB, Wright TW. Pneumocystis stimulates MCP-1 production by alveolar epithelial cells through a JNK-dependent mechanism. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1495–1505. doi: 10.1152/ajplung.00452.2006. [DOI] [PubMed] [Google Scholar]
- 39.Arias MA, Jaramillo G, Lopez YP, Mejia N, Mejia C, Pantoja AE, Shattock RJ, Garcia LF, Griffin GE. Mycobacterium tuberculosis antigens specifically modulate CCR2 and MCP-1/CCL2 on lymphoid cells from human pulmonary hilar lymph nodes. J Immunol. 2007;179:8381–8391. doi: 10.4049/jimmunol.179.12.8381. [DOI] [PubMed] [Google Scholar]
- 40.Morrison BE, Park SJ, Mooney JM, Mehrad B. Chemokine-mediated recruitment of NK cells is a critical host defense mechanism in invasive aspergillosis. J Clin Invest. 2003;112:1862–1870. doi: 10.1172/JCI18125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhagwat SP, Gigliotti F, Xu H, Wright TW. Contribution of T cell subsets to the pathophysiology of Pneumocystis-related immunorestitution disease. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1256–1266. doi: 10.1152/ajplung.00079.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gigliotti F, Crow EL, Bhagwat SP, Wright TW. Sensitized CD8+ T cells fail to control organism burden but accelerate the onset of lung injury during Pneumocystis carinii pneumonia. Infect Immun. 2006;74:6310–6316. doi: 10.1128/IAI.00668-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wright TW, Johnston CJ, Harmsen AG, Finkelstein JN. Analysis of cytokine mRNA profiles in the lungs of Pneumocystis carinii-infected mice. Am J Respir Cell Mol Biol. 1997;17:491–500. doi: 10.1165/ajrcmb.17.4.2851. [DOI] [PubMed] [Google Scholar]
- 44.Yadav M, Schorey JS. The beta-glucan receptor dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood. 2006;108:3168–3175. doi: 10.1182/blood-2006-05-024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swain SD, Lee SJ, Nussenzweig MC, Harmsen AG. Absence of the macrophage mannose receptor in mice does not increase susceptibility to Pneumocystis carinii infection in vivo. Infect Immun. 2003;71:6213–6221. doi: 10.1128/IAI.71.11.6213-6221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inoue M, Moriwaki Y, Arikawa T, Chen YH, Oh YJ, Oliver T, Shinohara ML. Cutting edge: critical role of intracellular osteopontin in antifungal innate immune responses. J Immunol. 2011;186:19–23. doi: 10.4049/jimmunol.1002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Archer KA, Ader F, Kobayashi KS, Flavell RA, Roy CR. Cooperation between multiple microbial pattern recognition systems is important for host protection against the intracellular pathogen Legionella pneumophila. Infect Immun. 2010;78:2477–2487. doi: 10.1128/IAI.00243-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Lieshout MH, Blok DC, Wieland CW, de Vos AF, van’t Veer C, van der Poll T. Differential roles of MyD88 and TRIF in hematopoietic and resident cells during murine gram-negative pneumonia. Journal of Infect Dis. 2012;206:1415–1423. doi: 10.1093/infdis/jis505. [DOI] [PubMed] [Google Scholar]
- 49.Loiarro M, Capolunghi F, Fanto N, Gallo G, Campo S, Arseni B, Carsetti R, Carminati P, De Santis R, Ruggiero V, Sette C. Pivotal Advance: Inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J Leuk Biol. 2007;82:801–810. doi: 10.1189/jlb.1206746. [DOI] [PubMed] [Google Scholar]
- 50.Bartfai T, Behrens MM, Gaidarova S, Pemberton J, Shivanyuk A, Rebek J., Jr A low molecular weight mimic of the Toll/IL-1 receptor/resistance domain inhibits IL-1 receptor-mediated responses. PNAS. 2003;100:7971–7976. doi: 10.1073/pnas.0932746100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 52.Noble PW, Jiang D. Matrix regulation of lung injury, inflammation, and repair: the role of innate immunity. Proc Am Thorac Soc. 2006;3:401–404. doi: 10.1513/pats.200604-097AW. [DOI] [PMC free article] [PubMed] [Google Scholar]