

Abstract
High-throughput generation of bispecific molecules promises to expedite the discovery of new molecular therapeutics and guide engineering of novel multifunctional constructs. However, high synthesis complexity and cost have hampered the discovery of bispecific molecules in drug development and biomedical research. Herein we describe a simple solid-phase bioconjugation procedure for preparation of Protein A(G,L)-PEG-Streptavidin heterobifunctional adaptors (with 1:1:1 stoichiometry), which enable self-assembly of unmodified antibodies and biotinylated molecules into bispecific targeting ligands in a versatile mix-and-use manner. Utility of such adaptors is demonstrated by assembly of anti-CD3 and anti-Her2 antibodies into bispecific CD3xHer2 targeting ligands, which efficiently drive T-cell-mediated lysis of Her2-positive cancer cells. In comparison to bioconjugation in solution, the solid-phase procedure described here offers precise stoichiometry control, ease of purification, and high yield of functional conjugates. Simplicity and versatility should prove this methodology instrumental for preparation of bispecific ligands, as well as for high-throughput screening of bispecific combinations, before proceeding to synthesis of lead candidates via recombinant engineering or chemical cross-linking.
Introduction
Methods for engineering of composite multifunctional molecules with preserved biological activity of individual functional parts are highly sought after for preparation of bispecific antibodies,1−3 antibody–drug conjugates,4,5 and antibody-imaging agent probes.6,7 Toward this end, many strategies have been exploited, such as Dock-and-Lock,8,9 chemical cross-linking,10,11 peptide nucleic acid conjugation via unnatural amino acids,12 hybrid–hybridoma,13 assembly via short synthetic peptides,14 and genetic engineering.15,16 However, current methods suffer from high complexity and cost of engineering of individual constructs, hampering high-throughput production of bispecific molecules. Here we describe a versatile solid-phase bioconjugation platform that enables straightforward synthesis of a variety of homo- and heterobifunctional molecules. Further, we have employed this platform for assembly of universal heterobifunctional adaptors consisting of two strong binary affinity systems—Protein A(G,L)/Antibody and biotin/streptavidin—which facilitate simple preparation of antibody–antibody, antibody–drug, and antibody–reporter pairs via self-assembly in a mix-and-use manner (Figure 1a). Use of such universal molecular adaptors should prove instrumental in a high-throughput screening of bispecific constructs prior to costly and laborious synthesis of lead candidates via recombinant engineering or chemical cross-linking.
Figure 1.

Preparation of universal molecular adaptors for bispecific ligand self-assembly. (a) Schematic of a universal heterobifunctional adaptor molecule consisting of Streptavidin and Protein A(G,L) connected via PEG linker. Use of two versatile binary affinity systems—SA/biotin and PrA(G,L)/Antibody—enables assembly of a wide variety of ligands in a simple mix-and-use manner. Streptavidin and Protein A Z-domain structures adopted from NCBI MMDB. (b) Workflow of solid-phase bioconjugation of heterobifunctional PrA(G,L)-PEG-SA adaptors. Monomeric avidin column (with free amines blocked) is loaded with biotin-PEG-NH2. Following avidin–biotin binding, PrA(G,L) activated by EDC/NHS is added and allowed to conjugate to primary amine groups on PEG. Physical separation of conjugation sites (NH2 groups) ensures conjugation of PrA(G,L) to only one PEG molecule, yielding 1:1 PrA(G,L):PEG–biotin conjugates. Monofunctional PrA(G,L)-PEG-biotin is eluted with 2 mM d-biotin, further immobilized onto IgG agarose column and incubated with excess SA. Similarly, physical separation of biotin moieties on a solid surface ensures SA binding to only one biotin, forming column-bound heterobifunctional PrA(G,L)-PEG-SA adaptors. Finally, elution with 0.1 M pH 2.4 Glycine releases fully functional product from the column.
Results and Discussion
In general terms, the solid-phase bioconjugation platform described here involves monofunctionalization of molecule A with a surface-bound cross-linker, release of an activated molecule A from the surface, and binding to a molecule B on another solid support at 1:1 molar ratio. Typically, due to availability of multiple potential conjugation sites on a single biomolecule, conventional liquid-phase bioconjugation procedures inevitably yield heterogeneous products with poorly controlled stoichiometry and require time-consuming laborious purification. In contrast, restricting chemical cross-linking to sparsely dispersed active sites on a solid support ensures monovalent conjugation, while aiding in quick and efficient purification.17,18 To demonstrate this concept, we assembled heterobifunctional Protein A(G,L)-PEG-Streptavidin (PrA(G,L)-PEG-SA, 1:1:1 molar ratio) adaptors using two commercially available solid supports, monomeric avidin resin and human IgG agarose, and employing 10 kDa PEG as a flexible spacer between PrA(G,L) and SA to prevent potential steric hindrance and loss of functionality. Monomeric avidin resin presents an optimal support for reversible immobilization of biotinylated molecules due to its requirement for mild elution conditions and compatibility with multiple regenerations (over 10 times). However, it is important to block exposed primary amine groups, should amine-based cross-linking chemistry be used. In this regard, we modified the resin with sulfo-NHS acetate to irreversibly protect all exposed primary amines that might interfere with further conjugation steps. Notably, protecting amine groups did not affect biotin–avidin binding, which may be attributed to a unique conformation of biotin binding site composed of a conserved Trp120, a hydrophobic pocket, and eight hydrogen bonds,19 while lacking exposed amines. Human IgG agarose, in turn, presents a suitable support for immobilization of all IgG-binding adaptor proteins (such as PrA, PrG, PrL used here), featuring efficient elution at low-pH conditions, which are sufficient for IgG/PrA(G,L) dissociation, but not for breaking a stronger SA–biotin bond or for irreversible protein denaturation.
The workflow for solid-phase adaptor bioconjugation includes five key steps (Figure 1b): (1) Monomeric avidin resin (with primary amine groups protected) is loaded with biotin-PEG-NH2. Then, EDC/NHS-activated PrA(G,L) is added onto the column and reacted with exposed primary amine groups on biotin-PEG, forming monovalent PrA(G,L)-PEG-biotin conjugates. (2) PrA(G,L)-PEG-biotin conjugates are eluted from the column using d-biotin and (3) immobilized onto human IgG column via noncovalent PrA(G,L) binding to IgG. (4) SA is then loaded onto the column and allowed to bind to exposed biotins on immobilized PrA(G,L)-PEG-biotin conjugates. (5) After washing, heterobifunctional PrA(G,L)-PEG-SA conjugates with precisely defined stoichiometry of 1:1:1 are eluted with 0.1 M Glycine (pH 2.4) buffer.
PEGylation of PrA on avidin resin produced monofunctional PrA-PEG-biotin conjugates at high purity of over 96%, whereas bioconjugation in solution yielded a mixture of PrA with 1, 2, 3, and 4+ PEG molecules, containing only 20–30% of mono-PEG conjugates (Figure 2a). Low-density distribution of biotin-PEG-NH2 on the column surface ensured that at most 1 PEG could be conjugated to an activated PrA molecule, whereas efficient column-based purification aided in quick removal of unconjugated PrA. Similarly, solid-phase PEGylation of PrG and PrL yielded monofunctional products at 94% and 97% purity, respectively, whereas liquid-phase reaction yielded 60% and 25% purity, respectively (Supporting Information Figure S1a,b). Taken together, these results support versatility and efficiency of monomeric avidin-based solid-phase bioconjugation procedure for monofunctionalization of various proteins with PEG-biotin.
Figure 2.

Characterization of PrA-PEG-SA adaptor assembly on a solid support and in solution. (a) Conjugation of PrA to biotin-PEG-NH2 was performed on a solid support (monomeric avidin resin) and in solution. SDS-PAGE characterization of products reveals high purity of mono-PEG-PrA conjugates produced via solid-phase bioconjugation, in contrast to a heterogeneous mixture of PrA with 1, 2, 3, 4+ PEG molecules produced by bioconjugation in solution. Molecular weight reference is shown in kDa. (b) Conjugation of PrA-PEG-biotin to SA on a solid support (IgG agarose) and in solution. Characterization with hybrid gel demonstrates 1:1:1 PrA-PEG-SA stoichiometry achieved with solid-phase procedure, whereas mixed SA conjugates with 0, 1, 2, 3+ PrA-PEG-biotin are produced via assembly in solution.
To attach the second functional component, SA, to PrA(G,L)-PEG-biotin conjugates in a 1:1 stoichiometry-controlled manner, PrA(G,L)-PEG-biotin was first reversibly immobilized onto a human IgG agarose column via noncovalent PrA(G,L)/IgG binding, and SA was then added and allowed to bind to surface-immobilized biotins. Similarly to mono-PEGylation, restricting SA-biotin binding to distant sites on agarose bead surface prevents cross-linking of multiple PrA(G,L)-PEG-biotin molecules by a single SA (which has 4 biotin-binding sites and can bind up to 4 biotinylated molecules in solution), while use of SA excess ensures complete functionalization of all immobilized PrA(G,L)-PEG-biotin conjugates. Elution with 0.1 M pH 2.4 Glycine buffer achieved efficient release of fully functional PrA(G,L)-PEG-SA adaptors into solution without disrupting the SA-biotin bond. The large product size and potential PEG–SDS interaction20 interfered with characterization by conventional SDS-PAGE, which failed to distinguish SA containing multiple copies of PrA(G,L)-PEG-biotin (data not shown). To overcome this issue, we employed a hybrid gel (0.5% agarose + 2.5% polyacrylamide), which was previously used for separation of large nanoparticle–protein conjugates.21,22 As shown in Figure 2b, hybrid gel successfully fractionated the conjugates produced by simple mixing of SA with PrA-PEG-biotin in solution, yielding distinct bands corresponding to unreacted SA, as well as SA with 1, 2, and 3+ copies of PrA-PEG-biotin. In contrast, solid-phase assembly on IgG agarose column produced a single pure product—1PrA-1PEG-1SA (Figure 2b, lane 2). Similar 1:1:1 stoichiometry was achieved with solid-phase assembly of adaptors based on PrG (Supporting Information Figure S1c) and PrL (Supporting Information Figure S1d). Therefore, the solid-phase platform presented here has proven instrumental for preparation of composite heterobifunctional molecules with defined 1:1 stoichiometry.
Preserved functionality of adaptor components (PrA(G,L) and SA) was confirmed by performing enzyme-linked immunosorbent assay (ELISA, Figure 3a). First, IgG molecules were immobilized on a 96-well plate and incubated with PrA(G,L)-PEG-SA adaptors, resulting in PrA(G,L)-PEG-SA binding to the surface via preserved PrA(G,L)/IgG binding. Then, a solution of biotin-HRP (biotinylated horseradish peroxidase) was added, depositing HRP to the surface via preserved SA/biotin binding. As expected, fully functional PrA(G,L)-PEG-SA adaptors successfully linked surface-bound IgG and biotin-HRP, yielding IgG concentration-dependent positive signal, whereas only the background signal was detected in a control experiment with a simple mixture of PrA(G,L), biotin-PEG, and SA (Figure 3b, Supporting Information Figure S2). The results further confirmed proper PrA(G,L)-PEG-SA adaptor structure and preserved dual functionality.
Figure 3.
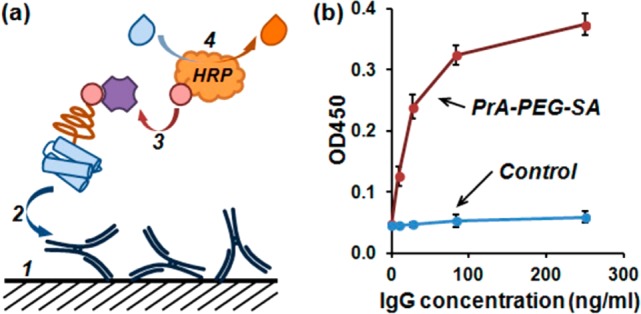
Characterization of adaptor binding functionality with ELISA. (a) Test schematic. IgG-coated surface was incubated with PrA(G,L)-PEG-SA, then labeled with biotinylated HRP via binding to available SA sites on IgG-bound PrA(G,L)-PEG-SA adaptors. (b) IgG concentration-dependent signal detected with PrA-PEG-SA adaptors confirms preserved functionality of both PrA and SA blocks, whereas only background signal detected in control (simple mixture of adaptor components) shows lack of nonspecific binding.
Incorporation of two versatile adaptor components, PrA(G,L) and SA, within a single heterobifunctional structure should enable high-throughput assembly of a variety of bispecific ligands and bifunctional probes in a simple mix-and-use procedure. For example, we have employed PrA-PEG-SA adaptors for preparation of reporter probes for one-step immunoassays. By eliminating intermediate steps, direct target labeling with unique reporters is advantageous for multiplexed detection and accurate quantitative analysis of the target expression. However, modification of primary antibodies with bulky reporters (such as nanoparticles and enzymes) is often complex and cost-prohibitive. Self-assembly via heterobifunctional adaptors presents an attractive alternative for small-scale on-demand preparation of targeting probes, featuring all the benefits of one-step immunoassays, while offering simplicity and flexibility of the mix-and-use methodology. As shown in Figure 4, self-assembled probes composed of biotinylated fluorescent nanoparticles (quantum dots, QDs) and unmodified primary antibodies targeting androgen receptor (AR) produced a characteristic nuclear staining in AR-positive formalin-fixed permeabilized LNCap cells via a one-step immunofluorescence procedure (Figure 4a,b), while anti-Her2 antibody-HRP assembly specifically detected abundance of Her2 protein in a lysate of Her2-positive SKBR3 cells via a one-step ELISA (Figure 4c,d).
Figure 4.
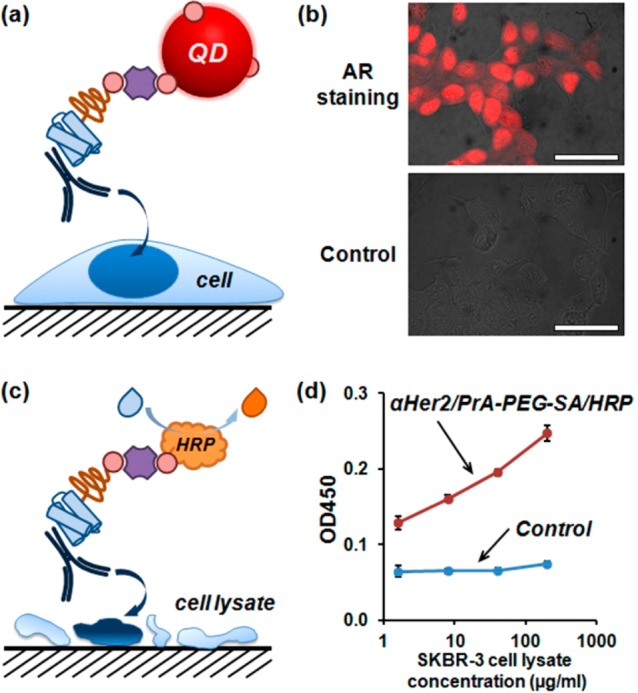
Self-assembly of detection probes via PrA-PEG-SA adaptors for direct target labeling in bioassays. (a) Schematic of a one-step immunofluorescence staining with self-assembled antibody-QD probes. (b) Androgen receptor (AR) in formalin-fixed permeabilized LNCap prostate cancer cells was labeled with red-emitting QDs preassembled with anti-AR primary antibodies via a one-pot 2 h incubation. Clear characteristic nuclear AR staining is observed with αAR/PrA-PEG-SA/QD605 probes, whereas only minimal background fluorescence is detected in a control lacking anti-AR antibody (i.e., PrA-PEG-SA/QD605), confirming specificity and sensitivity of target labeling with self-assembled probes. Scale bar, 50 μm. (c) Schematic of a one-step ELISA with self-assembled antibody-HRP probes. (d) Abundance of Her2 antigen in a lysate of Her2-positive SKBR3 breast cancer cells was measured using biotinylated HRP preassembled with anti-Her2 primary antibodies via a one-pot 2 h incubation. The expected dose-dependent response was achieved with fully functional αHer2/PrA-PEG-SA/HRP probes, while a control composed of a simple mixture of anti-Her2 antibodies and biotinylated HRP failed to produce signal above background, supporting the utility of self-assembled probes for ELISA applications.
One area of active research that might substantially benefit from such a methodology is cancer immunotherapy with bispecific antibodies capable of simultaneously binding two different molecular targets to direct effector cells (e.g., NK cell, T cells) against pathogenic target cells.15,23,24 Since T cells cannot be directly recruited by monoclonal antibodies due to the lack of Fcγ receptors, bispecific antibodies, such as CD3xCD1925 and CD3xEpCAM,26 have been employed to direct T cells against cancer cells, showing potential to eradicate tumors.27 To overcome many limitations of chemical cross-linking methodologies for preparation of bispecific antibodies, we have employed heterobifunctional PrA-PEG-SA adaptors for straightforward assembly of unmodified anti-Her2 and biotinylated anti-CD3 antibodies into bispecific CD3xHer2 targeting ligands (Figure 5a). First, PrA-PEG-SA was incubated with unmodified anti-Her2 antibody at 1:1 molar ratio and followed by blocking the excess binding sites with human IgG, resulting in assembly of Her2-targeting IgG/PrA-PEG-SA ligands. Then, biotinylated anti-CD3 antibody was added at 1:1 antibody-to-adaptor molar ratio, producing CD3xHer2 bispecific ligands featuring an anti-Her2 antibody bound to the PrA end and anti-CD3 antibody bound to the SA end of a heterobifunctional PrA-PEG-SA adaptor. In order to control the orientation of a biotinylated antibody upon binding to SA and prevent cross-linking of multiple adaptors, we performed site-specific biotinylation to an IgG hinge region, which introduced a single accessible SA binding site on an antibody, while preserving the accessibility and functionality of antigen-binding sites.21 Targeting activity and specificity of resulting bispecific ligands was tested on Her2-positive human breast cancer SKBR3 cells and CD3-positive human Peripheral Blood Mononuclear Cells (PBMCs) with flow cytometry. Cells were incubated with CD3xHer2 targeting ligands and then labeled with FITC-conjugated anti-PrA antibodies for fluorescence detection. Consistent with predicted targeting profile, bispecific ligands efficiently labeled both SKBR3 cells and PBMCs, whereas ligands missing a single targeting antibody failed to label respective cell populations, confirming lack of nonspecific binding (Figure 5b).
Figure 5.
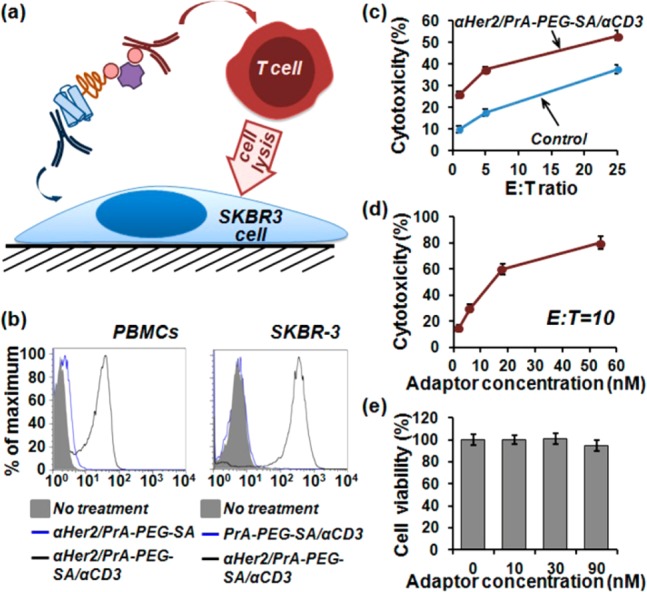
Assembly of bispecific CD3xHer2 antibodies via PrA-PEG-SA adaptors for enhanced T cell-mediated lysis of cancer cells. (a) Schematic of T cell-mediated lysis of cancer cells directed via cross-linking with self-assembled bispecific antibodies. (b) Characterization of target-binding functionality of CD3xHer2 bispecific antibodies with flow cytometry. Both CD3-positive PBMCs and Her2-positive SKBR3 cells were specifically labeled with bispecific antibodies, whereas ligands missing a single targeting antibody failed to bind to respective cells. (c) PBMCs (effector, E) were co-cultured with SKBR3 cells (target, T) at different E:T ratios in the presence of 10 nM CD3xHer2 ligands or equal amount of control (mixed anti-CD3 and anti-Her2 antibodies), exhibiting enhanced cell lysis mediated by CD3xHer2 bispecific ligands. (d) PBMCs were co-cultured with SKBR3 at a fixed E:T = 10 in the presence of different concentrations of CD3xHer2, showing further enhancement of cell lysis with increasing ligand concentration. (e) PrA-PEG-SA molecular adaptor alone showed a lack of cytotoxicity in a concentration range up to 90 nM.
Finally, we evaluated the capability of CD3xHer2 bispecific ligands to drive T cell-mediated lysis of Her2-positive breast cancer cells. Unstimulated CD3-positive PBMCs were used as effector (E) cells, and SKBR3 breast cancer cells served as Her2-positive targets (T). Binding of CD3xHer2 bispecific ligand to both CD3 on PBMCs and Her2 on SKBR3 cells should bridge effector and target cells, stimulating cell lysis. PBMCs and SKBR3 cells were co-cultured for 20 h at 37 °C at different E:T ratios in 96-well plates in the presence of either 10 nM CD3xHer2 bispecific ligands or equal concentration of anti-CD3 and anti-Her2 antibodies without PrA-PEG-SA adaptors as a control. The efficacy of cell lysis was evaluated by lactate dehydrogenase (LDH) release assay. In comparison to the control, CD3xHer2 bispecific ligands increased T cell-mediated cytotoxicity by 15–20% throughout the range of E:T ratios from 1:1 to 25:1 (Figure 5c). To test the effect of CD3xHer2 ligand concentration on cell lysis activity, we fixed E:T ratio at 10 and incubated cells with a range of CD3xHer2 concentrations. As shown in Figure 5d, lysis of SKBR3 breast cancer cells in the presence of PBMCs happened in a CD3xHer2 ligand dose-dependent manner. Importantly, heterobifunctional PrA(G,L)-PEG-SA adaptors alone did not exhibit any cytotoxicity over a concentration range up to 90 nM (Figure 5e and Supporting Information Figure S3), further supporting specificity of CD3xHer2 ligand-driven cancer cell lysis by effector T cells. In an analogous manner, a number of bispecific antibodies can be easily assembled for a variety of immunotherapy studies, highlighting the versatility of PrA(G,L)-PEG-SA adaptors described here. It should be noted, however, that multivalency of adaptor components (i.e., availability of up to 5 IgG-binding sites on PrA(G,L) and up to 3 biotin-binding sites on SA) allows for formation of highly multivalent bispecific assemblies, featuring more than 1 antibody on each adaptor terminal. A higher antibody load typically results in increased binding avidity and prolonged circulation time in vivo, while it might also lead to steric hindrance to target binding and reduced tissue penetration.28 Therefore, the antibody-to-adaptor stoichiometry should be characterized and taken into account when assessing therapeutic efficacy of self-assembled bispecific ligands.
In summary, we have developed a versatile solid-phase bioconjugation platform that can be readily adapted for preparation of a variety of hetero- and homobifunctional molecules. We have further employed this platform for synthesis of PrA(G,L)-PEG-SA adaptors with precisely controlled stoichiometry of 1:1:1 and demonstrated the utility of adaptors for assembly of detection probes for cell staining and ELISA, as well as bispecific targeting ligands for ligand-driven cancer cell lysis by effector T cells. In general, incorporation of two binary affinity systems, Protein A(G, L)/Antibody and biotin/streptavidin, within a single adaptor construct offers a straightforward route for assembly of target-specific antibodies with other functional biotinylated ligands, such as antibodies, reporter molecules, and therapeutic moieties, without requiring extensive customization. This makes PrA(G,L)-PEG-SA an instrumental preclinical tool for high-throughput screening of bifunctional combinations before proceeding to synthesis of lead candidates via recombinant engineering or chemical cross-linking.
Acknowledgments
This work was supported in part by NIH (R01CA140295). H.Y.L thanks the NIH for an F32 fellowship (CA150301) and Georgia Regents University for the new faculty start-up funding. P.Z. thanks the National Cancer Institute for a T32 fellowship (T32CA138312).
Glossary
Abbreviations
- PrA
protein A
- PrG
protein G
- PrL
protein L
- PEG
poly(ethylene glycol)
- SA
streptavidin
- IgG
immunoglobulin G
- ELISA
enzyme-linked immunosorbent assay
- HRP
horseradish peroxidase
- QD
quantum dot
- PBMCs
peripheral blood mononuclear cells
- FITC
fluorescein isothiocyanate
- LDH
lactate dehydrogenase
Supporting Information Available
Experimental details and supporting figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
Hong Yan Liu, Center for Biotechnology and Genomic Medicine, Medical College of Georgia, Georgia Regents University, Augusta, Georgia 30912, United States
Author Contributions
H.Y.L, P.Z., and X.G. contributed to the experiment design and data analysis. H.Y.L. performed the experiments. H.Y.L., P.Z., and X.G. wrote the paper.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Baeuerle P. A.; Reinhardt C. (2009) Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 69, 4941–4944. [DOI] [PubMed] [Google Scholar]
- Metz S.; Haas A. K.; Daub K.; Croasdale R.; Stracke J.; Lau W.; Georges G.; Josel H. P.; Dziadek S.; Hopfner K. P.; Lammens A.; Scheuer W.; Hoffmann E.; Mundigl O.; Brinkmann U. (2011) Bispecific digoxigenin-binding antibodies for targeted payload delivery. Proc. Natl. Acad. Sci. U. S. A. 108, 8194–8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes D. (2011) Buy buy bispecific antibodies. Nat. Rev. Drug Discovery 10, 798–800. [DOI] [PubMed] [Google Scholar]
- Chari R. V.; Miller M. L.; Widdison W. C. (2014) Antibody-drug conjugates: an emerging concept in cancer therapy. Angew. Chem., Int. Ed. Engl. 53, 3796–3827. [DOI] [PubMed] [Google Scholar]
- Hughes B. (2010) Antibody-drug conjugates for cancer: poised to deliver?. Nat. Rev. Drug Discovery 9, 665–667. [DOI] [PubMed] [Google Scholar]
- Ding H.; Carlton M. M.; Povoski S. P.; Milum K.; Kumar K.; Kothandaraman S.; Hinkle G. H.; Colcher D.; Brody R.; Davis P. D.; Pokora A.; Phelps M.; Martin E. W. Jr.; Tweedle M. F. (2013) Site specific discrete PEGylation of (124)I-labeled mCC49 Fab’ fragments improves tumor MicroPET/CT imaging in mice. Bioconjugate Chem. 24, 1945–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey R. M.; Cardillo T. M.; Rossi E. A.; Chang C. H.; Karacay H.; McBride W. J.; Hansen H. J.; Horak I. D.; Goldenberg D. M. (2005) Signal amplification in molecular imaging by pretargeting a multivalent, bispecific antibody. Nat. Med. 11, 1250–1255. [DOI] [PubMed] [Google Scholar]
- Backer M. V.; Patel V.; Jehning B. T.; Backer J. M. (2006) Self-assembled ″dock and lock″ system for linking payloads to targeting proteins. Bioconjugate Chem. 17, 912–919. [DOI] [PubMed] [Google Scholar]
- Rossi E. A.; Goldenberg D. M.; Chang C. H. (2012) The dock-and-lock method combines recombinant engineering with site-specific covalent conjugation to generate multifunctional structures. Bioconjugate Chem. 23, 309–323. [DOI] [PubMed] [Google Scholar]
- Brennan M.; Davison P. F.; Paulus H. (1985) Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin G1 fragments. Science 229, 81–83. [DOI] [PubMed] [Google Scholar]
- Glennie M. J.; McBride H. M.; Worth A. T.; Stevenson G. T. (1987) Preparation and performance of bispecific F(ab’ gamma)2 antibody containing thioether-linked Fab’ gamma fragments. J. Immunol. 139, 2367–2375. [PubMed] [Google Scholar]
- Kazane S. A.; Axup J. Y.; Kim C. H.; Ciobanu M.; Wold E. D.; Barluenga S.; Hutchins B. A.; Schultz P. G.; Winssinger N.; Smider V. V. (2013) Self-assembled antibody multimers through peptide nucleic acid conjugation. J. Am. Chem. Soc. 135, 340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindhofer H.; Mocikat R.; Steipe B.; Thierfelder S. (1995) Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J. Immunol. 155, 219–225. [PubMed] [Google Scholar]
- Ferrari E.; Soloviev M.; Niranjan D.; Arsenault J.; Gu C.; Vallis Y.; O’Brien J.; Davletov B. (2012) Assembly of protein building blocks using a short synthetic peptide. Bioconjugate Chem. 23, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Sereno A.; Aivazian D.; Langley E.; Miller B. R.; Snyder W. B.; Chan E.; Cantele M.; Morena R.; Joseph I. B.; Boccia A.; Virata C.; Gamez J.; Yco G.; Favis M.; Wu X.; Graff C. P.; Wang Q.; Rohde E.; Rennard R.; Berquist L.; Huang F.; Zhang Y.; Gao S. X.; Ho S. N.; Demarest S. J.; Reff M. E.; Hariharan K.; Glaser S. M. (2011) A stable IgG-like bispecific antibody targeting the epidermal growth factor receptor and the type I insulin-like growth factor receptor demonstrates superior anti-tumor activity. MAbs 3, 273–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack M.; Riethmuller G.; Kufer P. (1995) A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. U. S. A. 92, 7021–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. K.; Kwon J. S.; Kim H. J.; Yamamoto S.; Lee E. K. (2007) Solid-phase PEGylation of recombinant interferon alpha-2a for site-specific modification: process performance, characterization, and in vitro bioactivity. Bioconjugate Chem. 18, 1728–1734. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Ye C.; Liu Z.; Wang X.; Chen H.; Liu Y.; Tang L.; Zhao H.; Wang J.; Feng W.; Li X. (2012) Solid-phase N-terminus PEGylation of recombinant human fibroblast growth factor 2 on heparin-sepharose column. Bioconjugate Chem. 23, 740–750. [DOI] [PubMed] [Google Scholar]
- DeChancie J.; Houk K. N. (2007) The origins of femtomolar protein-ligand binding: hydrogen-bond cooperativity and desolvation energetics in the biotin-(strept)avidin binding site. J. Am. Chem. Soc. 129, 5419–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C.; Ma G.; Su Z. (2007) Native PAGE eliminates the problem of PEG-SDS interaction in SDS-PAGE and provides an alternative to HPLC in characterization of protein PEGylation. Electrophoresis 28, 2801–2807. [DOI] [PubMed] [Google Scholar]
- Liu H. Y.; Gao X. (2011) Engineering monovalent quantum dot-antibody bioconjugates with a hybrid gel system. Bioconjugate Chem. 22, 510–517. [DOI] [PubMed] [Google Scholar]
- Liu H. Y.; Vu T. Q. (2007) Identification of quantum dot bioconjugates and cellular protein co-localization by hybrid gel blotting. Nano Lett. 7, 1044–1049. [DOI] [PubMed] [Google Scholar]
- Scheffold C.; Kornacker M.; Scheffold Y. C.; Contag C. H.; Negrin R. S. (2002) Visualization of effective tumor targeting by CD8+ natural killer T cells redirected with bispecific antibody F(ab’)(2)HER2xCD3. Cancer Res. 62, 5785–5791. [PubMed] [Google Scholar]
- Xie Z.; Shi M.; Feng J.; Yu M.; Sun Y.; Shen B.; Guo N. (2003) A trivalent anti-erbB2/anti-CD16 bispecific antibody retargeting NK cells against human breast cancer cells. Biochem. Biophys. Res. Commun. 311, 307–312. [DOI] [PubMed] [Google Scholar]
- Bargou R.; Leo E.; Zugmaier G.; Klinger M.; Goebeler M.; Knop S.; Noppeney R.; Viardot A.; Hess G.; Schuler M.; Einsele H.; Brandl C.; Wolf A.; Kirchinger P.; Klappers P.; Schmidt M.; Riethmuller G.; Reinhardt C.; Baeuerle P. A.; Kufer P. (2008) Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 321, 974–977. [DOI] [PubMed] [Google Scholar]
- Brischwein K.; Schlereth B.; Guller B.; Steiger C.; Wolf A.; Lutterbuese R.; Offner S.; Locher M.; Urbig T.; Raum T.; Kleindienst P.; Wimberger P.; Kimmig R.; Fichtner I.; Kufer P.; Hofmeister R.; da Silva A. J.; Baeuerle P. A. (2006) MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol. Immunol. 43, 1129–1143. [DOI] [PubMed] [Google Scholar]
- Chames P.; Baty D. (2009) Bispecific antibodies for cancer therapy: the light at the end of the tunnel?. MAbs 1, 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta A. M.; Sainz-Pastor N.; Bonet J.; Oliva B.; Alvarez-Vallina L. (2010) Multivalent antibodies: when design surpasses evolution. Trends Biotechnol. 28, 355–362. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.