

Abstract
Light sheet microscopy is the ideal imaging technique to study zebrafish embryonic development. Due to minimal photo-toxicity and bleaching, it is particularly suited for long-term time-lapse imaging over many hours up to several days. However, an appropriate sample mounting strategy is needed that offers both confinement and normal development of the sample. Multilayer mounting, a new embedding technique using low-concentration agarose in optically clear tubes, now overcomes this limitation and unleashes the full potential of light sheet microscopy for real-time developmental biology.
Keywords: Developmental Biology, Issue 84, zebrafish, Danio rerio, light sheet microscopy, Selective Plane Illumination Microscopy, sample mounting, time lapse microscopy, long-term imaging
Introduction
To understand the complex events and interactions during embryogenesis, research in developmental biology is more and more moving from microscopy of single cells and organs to in vivo imaging of entire organisms. Traditional microscopy techniques typically fail to offer the spatial and temporal resolution to follow rapid events over a long period of time and often cause bleaching or phototoxic effects in the sample1. We and others found light sheet microscopy (Figure 1) to be the ideal technique for in vivo imaging of zebrafish2-4. The illumination of the sample with a thin sheet of laser light, orthogonal to the detection axis, offers fast optical sectioning with high spatial resolution as well as minimized photo-toxicity5. At the same time, due to this unique optical arrangement, traditional sample mounting techniques using Petri dishes or culture chambers are not suitable. In a typical light sheet microscope three translational motors and a rotational motor hold the sample, such that it can be precisely positioned, turned and viewed from any direction. In the past specimens for light sheet microscopy have often been embedded in 1-2% agarose inside a glass capillary6,7. In this case the solidified agarose cylinder with the embedded specimen is extruded out of the glass capillary into the sample chamber filled with water-like medium for imaging. Agarose has a refractive index close to water and is therefore perfectly suited for in vivo light sheet microscopy that facilitates water-corrected illumination- and detection optics. The sample is confined in the rigid agarose and can be imaged well for a short period of time, but morphogenetic changes and growth of the embryo are strongly impaired when imaging for several hours7,8. Although solutions for other applications have been developed9, the noninvasive long-term time-lapse zebrafish imaging potential of light sheet microscopy cannot be exploited using the conventional 1.5% agarose embedding.
We therefore developed a new mounting strategy for in vivo light sheet microscopy of zebrafish embryos10. The sample is mounted in 0.1% agarose with 200 mg/L Tricaine inside a tube made of the optically clear polymer Fluorinated Ethylene Propylene (FEP). The embedded embryo develops normally due to low viscosity of the medium. At the same time, the FEP tube confines the medium and the sample for the duration of the experiment. Here we provide a detailed protocol of the new mounting technique and present data reflecting its advantages over the conventional protocol. We demonstrate that with our method zebrafish development can be continuously imaged with high spatial and temporal resolution over a period of more than two days.
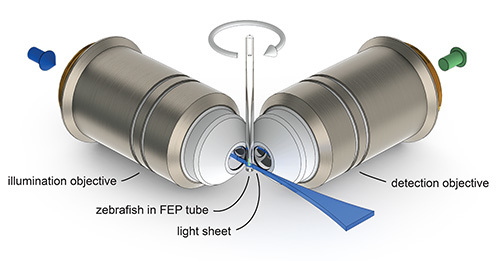 Figure 1. The principle of light sheet microscopy. A zebrafish mounted in a tube made of Fluorinated Ethylene Propylene (FEP) can be translated and rotated through the focal plane of the detection objective. A thin sheet of laser light (light sheet) is projected by the illumination objective into the focal plane of the detection objective and illuminates only a thin optical section of the specimen. The emitted fluorescence signal is collected by the detection objective and recorded on a camera chip.
Figure 1. The principle of light sheet microscopy. A zebrafish mounted in a tube made of Fluorinated Ethylene Propylene (FEP) can be translated and rotated through the focal plane of the detection objective. A thin sheet of laser light (light sheet) is projected by the illumination objective into the focal plane of the detection objective and illuminates only a thin optical section of the specimen. The emitted fluorescence signal is collected by the detection objective and recorded on a camera chip.
Protocol
1. Material Preparation
- Preparation of FEP tubes
- Clean the FEP tube (Figure 2A) by flushing liquids (in the steps noted below) through the tube and using a syringe attached with a blunt end cannula (Figure 2B). Note 1: Use gloves throughout the procedure. Note 2: We recommend cutting the tube in pieces of ca. 1 m in length and cleaning those in parallel.
- First flush the tube with 1 M NaOH repeatedly. Then transfer the tube to a 50 ml centrifuge tube filled with 0.5 M NaOH and ultrasonicate it for 10 min.
- Transfer the polymer tube to a small basin with ddH2O. Flush the tube first with ddH2O and then with 70% EtOH.
- Subsequently transfer the FEP tube to a fresh centrifuge tube with 70% EtOH. Again ultrasonicate this centrifuge tube for 10 min.
- Cut the cleaned FEP tube into pieces of about 3 cm (a typical length for use in zebrafish mounting and imaging) and straighten them if necessary. Store the cleaned FEP tubes in a fresh 50 ml centrifuge tube filled with ddH2O (Figure 2C).
- Preparation of Tricaine stock solution
- Prepare 0.4% Tricaine stock solution by dissolving 400 mg Tricaine (MS-222) in 97.9 ml ddH2O. After the powder has completely dissolved, adjust the pH to 7.0 using 2.1 ml 1M Tris (pH 9.0). Protect the solution from light. Store aliquots at -20 °C.
- Preparation of 3% methylcellulose
- Prepare 3% methylcellulose solution for the inner coating of the FEP tubes as described below. Heat up E311 in a glass bottle to approximately 60-70 °C and add the appropriate amount of methylcellulose powder. Add a stirring bar and stir at 4 °C. As soon as the solution is cooled down to below 30 °C, add Tricaine solution to a final concentration of 100 mg/L. Keep stirring at 4 °C overnight.
- Preparation of 1.5% and 0.1% agarose in E3
- Dissolve the appropriate amount of low melting point agarose powder in a defined volume of E3 in a glass flask. Heat up the mixture in a microwave and shake it once in a while, until the agarose solution appears homogeneous, without any remaining crystals.
- Prepare 1 ml aliquots of 0.1% agarose solution in 1.5 ml reaction tubes. The aliquots can be stored at 4 °C.
- Use the 1.5% solution to coat a plastic Petri dish. The thickness of the agarose layer should be ca. 2 mm. After the agarose is solidified, add E3 medium on top of the layer to prevent it from drying. The coated dish can be stored at 4 °C.
- Preparation of zebrafish
- Keep Zebrafish (Danio rerio) adults and embryos at 28.5 °C and handle them according to the established protocols11,12.
- Set up male and female fish in the afternoon and separate them using a divider. Remove the divider the following morning to time the mating of the fish.
- Collect the embryos in a dish filled with E3 and examine them using a stereomicroscope.
- At 24 hpf (or the developmental stage of your interest), select the zebrafish embryo for fluorescence expression and transfer the positive ones to a new dish with fresh E3.
- Carefully dechorionate the embryos using two sharp forceps (Figures 2D and 2E). Use the first forceps to grab and hold the chorion and the second forceps to tear it open and pull it off. Note 1: Perform this step with the help of a stereomicroscope.
2. Multilayer Mounting
- Coating the FEP tube
- Attach a blunt end cannula to a syringe. Insert the cannula carefully for about 3 mm into one end of a cleaned and cut piece of FEP tube (Figures 2F and 2G). Note 1: Use gloves and avoid any bending or squeezing of the tube.
- Dip the free end of the FEP tube into 3% methylcellulose and fill the tube with the solution.
- Slowly release the methylcellulose by pushing on the syringe. Discard the solution.
- Repeat steps 2.1.2 and 2.1.3 using E3. Note 1: The coated tubes need to be used for mounting immediately.
- Transferring the zebrafish to the agarose
- Heat up one aliquot of 0.1% agarose to 70°C in a heat block. Vortex the reaction tube briefly and transfer it to a heat block set to 38 °C (Figure 2H).
- Take the reaction tube out from the heat block and let it cool down briefly. Add Tricaine to a final concentration of 133-200 mg/L and vortex again (Figure 2I).
- Select one of the dechorionated zebrafish embryos and transfer it to the reaction tube with the preheated 0.1% agarose using a glass Pasteur pipette (Figures 2J and 2K). Note 1: Try to transfer as little additional E3 as possible.
- Mounting the zebrafish
- Take up the embryo with the syringe-attached FEP tube (Figure 2L). The tube should be filled completely with 0.1% agarose , with the embryo positioned close to either end of the tube and with the head pointing towards the opening of the tube. Note 1: Make sure to take up some agarose before taking up the fish. Note 2: Pull very gently on the syringe to avoid bubbles. Note 3: Keep the tube in a horizontal orientation to avoid leakage of its content (Figures 2M and 2N).
- Plugging the tube
 Figure 2. Multilayer mounting for zebrafish embryos.
A) Fluorinated Ethylene Propylene (FEP) tubes on a cable drum before preparation. B) A syringe with a blunt end cannula attached. C) A cleaned and cut FEP tube. D, E) Dechorionation of 24 hpf zebrafish embryos. F) Attaching the tube to the blunt end cannula. G) The syringe with cannula and tube attached. H) An aliquot of 0.1% agarose inside a reaction tube while transferring it to a heat block. I) Vortexing of the melted agarose. J) Uptake of a dechorionated embryo using a glass pipette. K) Transfer of the embryo into the melted agarose. L) Intake of the embryo with surrounding agarose into the FEP tube. M, N) The zebrafish embryo inside the FEP tube. O) Cutting the tube between specimen and cannula. P) Plugging the tube by the use of an agarose-coated dish. Q) The embryo inside the plugged tube. R) The final sample preparation in a metal holder. Click here to view larger image.
Figure 2. Multilayer mounting for zebrafish embryos.
A) Fluorinated Ethylene Propylene (FEP) tubes on a cable drum before preparation. B) A syringe with a blunt end cannula attached. C) A cleaned and cut FEP tube. D, E) Dechorionation of 24 hpf zebrafish embryos. F) Attaching the tube to the blunt end cannula. G) The syringe with cannula and tube attached. H) An aliquot of 0.1% agarose inside a reaction tube while transferring it to a heat block. I) Vortexing of the melted agarose. J) Uptake of a dechorionated embryo using a glass pipette. K) Transfer of the embryo into the melted agarose. L) Intake of the embryo with surrounding agarose into the FEP tube. M, N) The zebrafish embryo inside the FEP tube. O) Cutting the tube between specimen and cannula. P) Plugging the tube by the use of an agarose-coated dish. Q) The embryo inside the plugged tube. R) The final sample preparation in a metal holder. Click here to view larger image.
- Use a razor blade to cut the tube off the cannula (Figure 2O).
- Slowly stick the end of the tube containing the fish through the entire layer of 1.5% agarose in the plastic dish to plug the tube (Figure 2P). Note 1: Avoid the formation of bubbles and try to obtain a straight plug surface by holding the tube exactly perpendicular to the agarose surface. Note 2: Rotate the tube slightly to obtain a nice plug.
- Keep the mounted specimen in a 1.5 ml reaction tube with E3 to avoid drying. The sample is now ready to be imaged. Before imaging, check whether the fish is sitting right on top of the plug (Figures 2Q and 2R). Make sure to also add Tricaine to the medium inside the imaging chamber at the same concentration as in the mounting medium.
Representative Results
Multiday Imaging of Zebrafish Vasculature Development
We use 1 dpf Tg(kdrl:GFP)13 zebrafish to demonstrate the advantages of the multilayer mounting strategy for long-term light sheet microscopy. The aim is to image the development of the vasculature in the entire zebrafish over a course of 2 days. The zebrafish embryo was mounted in 0.1% agarose inside a methylcellulose coated polymer tube. Here, the zebrafish is not restricted by the mounting medium and can develop normally. Its head raises, the tail extends and vascular sprouting appears unhindered (Figure 3).
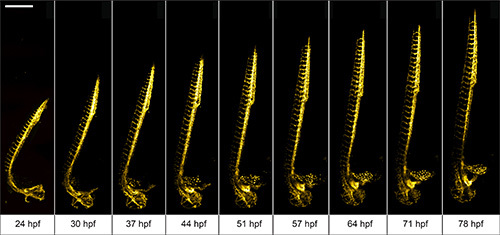 Figure 3. Multiday imaging of zebrafish vasculature development. The vasculature of a multilayer mounted 1 dpf Tg(kdrl:GFP)
13 zebrafish develops unhindered in a light sheet microscope over the course of two days. A 488 nm laser light sheet excited the fluorescence and the signal was detected using 10X/0.3 W objective and an EMCCD camera. Z-stacks were acquired on four adjacent positions every 10 min and subsequently stitched with Fiji 15. Maximum intensity projections of a subset of time-points are shown. Scale bar: 500 μm. Click here to view larger image.
Figure 3. Multiday imaging of zebrafish vasculature development. The vasculature of a multilayer mounted 1 dpf Tg(kdrl:GFP)
13 zebrafish develops unhindered in a light sheet microscope over the course of two days. A 488 nm laser light sheet excited the fluorescence and the signal was detected using 10X/0.3 W objective and an EMCCD camera. Z-stacks were acquired on four adjacent positions every 10 min and subsequently stitched with Fiji 15. Maximum intensity projections of a subset of time-points are shown. Scale bar: 500 μm. Click here to view larger image.
Imaging of Early Zebrafish Embryogenesis
Here, we demonstrate the capability of FEP tubes as a mounting support for light sheet microscopy of early zebrafish embryogenesis during the first day after fertilization. The 8 hpf Tg(H2A:GFP)14 zebrafish embryo was mounted with its intact chorion inside an E3-filled tube with an inner diameter of 1 mm. That results in a slight clamping of the chorion. All developmental processes during embryogenesis are unaffected by the mounting and can be optimally visualized using light sheet microscopy. In this example, we imaged the specimen over a course of 12 hr (Figure 4). Typical shape changes during early embryogenesis, such as the increasing elongation of the yolk and thickening of tissue along the midline, would be oppressed when using the traditional mounting without chorion in 1.5% agarose.
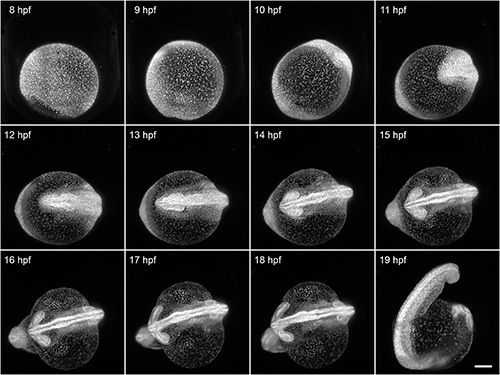 Figure 4. Imaging of early zebrafish embryogenesis. A Tg(H2A:GFP)14 zebrafish passes through early embryogenesis. A 488 nm laser light sheet excited GFP and the fluorescence signal was detected with a 10X/0.3 W objective and an EMCCD camera. Z-stacks from two angles were acquired every 3 min over a course of 12 hr. Normalized maximum intensity projections of a subset of the time-lapse are shown. Visible motion blur at 19 hpf results from the embryo twitching during acquisition, as no anesthetic were used. Scale bar: 150 μm. Click here to view larger image.
Figure 4. Imaging of early zebrafish embryogenesis. A Tg(H2A:GFP)14 zebrafish passes through early embryogenesis. A 488 nm laser light sheet excited GFP and the fluorescence signal was detected with a 10X/0.3 W objective and an EMCCD camera. Z-stacks from two angles were acquired every 3 min over a course of 12 hr. Normalized maximum intensity projections of a subset of the time-lapse are shown. Visible motion blur at 19 hpf results from the embryo twitching during acquisition, as no anesthetic were used. Scale bar: 150 μm. Click here to view larger image.
Discussion
Advanced microscopy techniques such as light sheet microscopy enable us to record image data with high temporal and spatial resolution over the course of several days, but also require sample mounting techniques to improve. We present a detailed protocol of multilayer mounting for light sheet microscopy of zebrafish development. The method is straightforward to implement and we use it in our lab on a daily basis.
The FEP tubes
Key components of our mounting strategy are tubes made of the polymer Fluorinated Ethylene Propylene (FEP). We decided to use FEP primarily because of its refractive index of 1.338, which is similar to water and therefore ideally suited for light sheet microscopy with water-dipping objectives and specimen requiring aqueous media. The tubes with 0.8 mm inner diameter and 0.4 mm wall thickness fit the developing zebrafish very well, can be used with typical sample holders suited for 1.6 mm capillaries and are our first choice for the presented mounting method. We tested them thoroughly in our home-built setups as well as in the commercial system "Lightsheet Z.1" by Carl Zeiss Microscopy and found that FEP tubes offer long-term stability, have only minimal impact on optical performance and are nonfluorescent10. They are, however, not suited for applications with samples that require a different refractive index, e.g. cleared specimen.
Tubes as well as foils made of FEP are available in a variety of diameters and thicknesses. In addition, FEP can be melted at 260 °C and thus offers virtually endless possibilities for custom-made sample mounting supports. The polymer tubes tend to be supplied nonsterile, which is why the cleaning protocol incorporates several steps to ensure best optical quality. The coating with methylcellulose ensures that no part of the growing zebrafish embryo sticks to the polymer.
The mounting medium
The decision for 0.1% agarose is based on extensive tests of growth rate and immobility in different concentrations of agarose and methylcellulose10. Agarose concentrations above 0.1% already showed a severe impact on the growth rate of 24 hpf zebrafish. Additionally we use low doses of the anesthetizing drug Tricaine. Tricaine blocks action potentials, thus inhibits the signal transmission between brain and muscles and suppresses muscle contractions16. A concentration of 133-200 mg/L ensures sufficient immobilization of zebrafish between 24-72 hpf. However, Tricaine and other anesthetizing drugs we tested show dose dependent side effects such as cardiac edema. Additionally the required dose of Tricaine varies with the developmental stage of the embryo. We therefore recommend reducing the Tricaine concentration to the amount needed for the developmental stages that are imaged. To ensure best performance and prevent the formation of toxic by-products, freshly prepared or defrosted Tricaine stock solution should be used, protected from light and not heated above 30 °C.
The embedding in FEP tubes also allows for easy modification of the mounting medium to account for specific needs. For time-lapse imaging of the heart, we recommend using 3% methylcellulose and 100 mg/L Tricaine instead of 0.1% agarose and 200 mg/L for mounting. Methylcellulose is more viscous than 0.1% agarose thus providing higher immobility on its own and less Tricaine needs to be used to ensure confinement of the embryo.
Tricaine is sensitive to temperatures above 30 °C and can also be diluted by the medium in the imaging chamber, leading to decreased performance and possible twitching of the embryo. Therefore, make sure to use Tricaine also in the imaging medium in which the mounted specimen is immersed. If your experiments require the use of higher temperatures, we recommend exchanging the medium in the imaging chamber either constantly by using a perfusion system or manually with a tube and a syringe between two time-points.
Critical steps
The critical steps during the process of sample mounting are the intake of the specimen into the tube and the insertion of the agarose plug. The intake defines the initial position of the embryo, which is cumbersome to change afterwards. Multiple specimens should be available to mount and the mounting quality should be monitored with a stereomicroscope. If the sample orientation is not satisfactory, altering the speed of drawing up the specimen and avoiding subsequent movements of the plunger may help as this often twists the embryo. The 1.5% agarose plug is necessary to avoid leakage of the 0.1% agarose. The surface of the plug inside the tube should be flat to not influence the sample orientation during development. To achieve a good surface, different thicknesses of the agarose layers need to be tested and the tube needs to be put into the agarose normal to the surface. Rotating the tube a little bit and pulling out the tube carefully releases the plug from the dish. The quality of the mounting should be checked with a stereomicroscope before imaging.
Multiview imaging
One major advantage of light sheet microscopy is multiview imaging, the ability to rotate the specimen, acquire z-stacks from multiple angles and subsequently register and fuse them. An established method to register the z-stacks in 3D space is bead-based registration using fluorescent beads surrounding the sample as fiduciary markers 17. This method however relies on a solid mounting medium such as 1.5% agarose and thus seems to be incompatible with the multilayer mounting presented here. But several alternative solutions work depending on the application. First, the fluorescent beads can be incorporated into the agarose plug, which has to be in the field of view during the acquisition. Second, the stage positions can be calibrated before the actual experiments using a 1.5% agarose column with fluorescent beads. Third, alternative markers such as fluorescent nuclei can be used to identify the position of the z-stacks relative to each other.
Evaporation
Typical light sheet microscopy setups use an open sample chamber, which inevitable leads to evaporation of the medium. Since the medium is essential for the survival of the specimen, to restrict evaporation of the mounting medium and for the correct refractive index between optics and sample, we recommend taking one or more of the following measures to prevent evaporation. First, the medium in the chamber can be constantly exchanged by a perfusion system. Second, the medium can be manually refilled. Third, the chamber can be covered with a lid or a flexible foil.
Oxygen supply
The polymer FEP was developed to withstand a large variety of chemicals and is therefore robust, chemically inert and nonpermeable to gas. In the case of multilayer mounting oxygen can only penetrate through the agarose plug and the agarose-air interface, but it remains to be shown if the oxygen supply in multilayer mounting is severely lower when compared to mounting in 1.5% agarose without supporting polymer tube. However, as mounting in 1.5% agarose can only be used for about two hours before the zebrafish morphology is affected multilayer mounting seems to be the overall superior solution for long-term imaging.
Imaging earlier stages of development
Multilayer mounting has now become our standard embedding technique for time-lapse light sheet microscopy of zebrafish older than 24 hpf. Besides that, we also use the polymer tubes for imaging of earlier stage embryos. The embryos remain in their chorions and are mounted in a FEP tube with an inner diameter of 1 mm and filled with E3. The zebrafish can freely develop inside the chorion and can still be imaged well using light sheet microscopy.
Outlook
The presented multilayer sample mounting unleashes the full potential of light sheet microscopy for real-time developmental biology with zebrafish. It can easily be adopted for other microscopy techniques such as optical projection tomography (OPT) and other organisms. We believe that the standard-sized FEP tubes are just the first steps towards sample mounting techniques tailored for specific needs.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank the Max Planck Society, the Human Frontier Science Program (HFSP) and the Boehringer Ingelheim Fonds for funding and support.
References
- Huisken J, Stainier DYR. Selective plane illumination microscopy techniques in developmental biology. Development. 2009;136(12):1963–1975. doi: 10.1242/dev.022426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrens MB, Orger MB, Robson DN, Li JM, Keller PJ. Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat. Methods. 2013;10(5):413–420. doi: 10.1038/nmeth.2434. [DOI] [PubMed] [Google Scholar]
- Swoger J, Muzzopappa M, López-Schier H, Sharpe J. 4D retrospective lineage tracing using SPIM for zebrafish organogenesis studies. J. Biophoton. 2011;4(1-2):122–134. doi: 10.1002/jbio.201000087. [DOI] [PubMed] [Google Scholar]
- Jemielita M, Taormina MJ, DeLaurier A, Kimmel CB, Parthasarathy R. Comparing phototoxicity during the development of a zebrafish craniofacial bone using confocal and light sheet fluorescence microscopy techniques. J. Biophoton. 2012. pp. 1–11. [DOI] [PMC free article] [PubMed]
- Weber M, Huisken J. Light sheet microscopy for real-time developmental biology. Curr. Opin. Genet. Dev. 2011;21(5):566–572. doi: 10.1016/j.gde.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Huisken J, Swoger J, Del Bene F, Wittbrodt J, Stelzer EHK. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 2004;305(5686):1007–1009. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- Keller PJ, Stelzer EHK. Digital scanned laser light sheet fluorescence microscopy. Cold Spring Harb. Prot. 2010;2010(5) doi: 10.1101/pdb.top78. [DOI] [PubMed] [Google Scholar]
- Keller PJ, Stelzer EHK. Quantitative in vivo imaging of entire embryos with Digital Scanned Laser Light Sheet Fluorescence Microscopy. Curr. Opin. Neurobiol. 2008;18(6):624–632. doi: 10.1016/j.conb.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Desmaison A, Lorenzo C, Rouquette J, Ducommun B, Lobjois V. A versatile sample holder for single plane illumination microscopy. J. Microsc. 2013;10 doi: 10.1111/jmi.12051. [DOI] [PubMed] [Google Scholar]
- Kaufmann A, Mickoleit M, Weber M, Huisken J. Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Development. 2012;139(17):3242–3247. doi: 10.1242/dev.082586. [DOI] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Zebrafish Dahm R. Oxford University Press; 2002. [Google Scholar]
- Avdesh A, et al. Regular care and maintenance of a zebrafish (Danio rerio) laboratory: an introduction. J. Vis. Exp. 2012. [DOI] [PMC free article] [PubMed]
- Jin S-W, Beis D, Mitchell T, Chen J-N, Stainier DYR. Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development. 2005;132(23):5199–5209. doi: 10.1242/dev.02087. [DOI] [PubMed] [Google Scholar]
- Pauls S, Geldmacher-Voss B, Campos-Ortega JA. A zebrafish histone variant H2A.F/Z and a transgenic H2A.F/Z:GFP fusion protein for in vivo studies of embryonic development. Dev. Genes Evol. 2001;211(12):603–610. doi: 10.1007/s00427-001-0196-x. [DOI] [PubMed] [Google Scholar]
- Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier DT, Narahashi T. Tricaine (MS-222): effects on ionic conductances of squid axon membranes. Eur. J. Pharmacol. 1975;33(2):313–317. doi: 10.1016/0014-2999(75)90175-2. [DOI] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, Schindelin J. Software for bead-based registration of selective plane illumination microscopy data. Nat. Methods. 2010;7:418–419. doi: 10.1038/nmeth0610-418. [DOI] [PubMed] [Google Scholar]