

Abstract
The glycomic profiling of purified glycoproteins and biological specimen is routinely achieved through different analytical methods, but mainly through MS and LC-MS. The enhanced ionization efficiency and improved tandem MS interpretation of permethylated glycans have prompted the popularity of this approach. This study focuses on comparing the glycomic profiling of permethylated N-glycans derived from model glycoproteins and human blood serum using MALDI-MS as well as RP-LC-MALDI-MS and RP-LC-ESI-MS. In the case of model glycoproteins, the glycomic profiles acquired using the three methods were very comparable. However, this was not completely true in the case of glycans derived from blood serum. RP-LC-ESI-MS analysis of reduced and permethylated N-glycans derived from 250 nl of blood serum allowed the confident detection of 73 glycans (the structures of which were confirmed by mass accuracy and tandem MS), while 53 and 43 structures were identified in the case of RP-LC-MALDI-MS and MALDI-MS analyses of the same sample, respectively. RP-LC-ESI-MS analysis facilitates automated and sensitive tandem MS acquisitions. The glycan structures that were detected only in the RP-LC-ESI-MS analysis were glycans existing at low abundances. This is suggesting the higher detection sensitivity of RP-LC-ESI-MS analysis, originating from both reduced competitive ionization and saturation of detectors, facilitated by the chromatographic separation. The latter also permitted the separation of several structural isomers; however, isomeric separations pertaining to linkages were not detected.
Keywords: Glycans, MALDI-MS, Permethylation, RP-LC-ESI-MS, RP-LC-MALDI-MS
1 Introduction
A common, intriguing, complex, and analytically challenging posttranslational modification of proteins is glycosylation which plays a major role in many biological functions, including protein folding, transport, and targeting [1]. Aberrant gly-cosylation has also been implicated in many human diseases, such as cancer, inflammation, and many immune diseases [2, 3]. Currently, correlation between aberrant glycosylation and disease development and progression is being investigated in search of reliable disease biomarkers that can be effectively employed for disease diagnosis and prognosis. As such, the characterization of protein glycosylation at high sensitivity in biological specimens remains an interesting field of research posing many significant analytical challenges.
Among the various analytical methods routinely employed for glycomics analysis, mass spectrometry (MS) is very effective and powerful, generating a plethora of reliable data. It also permits the effective characterization of glycans derived from purified glycoproteins or biological specimen. Although MS has recently been employed to elucidate glycan structures and their roles in disease progression [4–7], elucidation of glycan branching and linkages remains a challenge. Currently, the most reliable analytical tool for structural elucidation of glycans is tandem MS (MS2 and MSn), which provides structural information allowing unequivocal glycan structural assignment [8–10]. In many cases MS2 analysis is sufficient to deduce glycan structures, yet effective identification might not be attainable for many structures, since there are not enough fragments detected. This ambiguity can be somewhat resolved through MS3, or MSn analyses. However, MSn analysis is not possible for glycans existing at very low levels. Additionally, glycan concentration dynamic range within one sample can extend over several orders of magnitude, prompting saturation of detectors that potentially limit tandem MS use. For glycans existing at low concentrations, chromatographic or electrophoretic separations overcome such limitations and allow the acquisition of reliable MS2 and MSn spectra.
Combining liquid chromatography (LC) and MS to elucidate glycan structures offers a means by which glycans of complex samples are effectively identified and quantified. Chromatographic separation of native glycans have been achieved using different types of chromatography, including high-pH anion-exchange chromatography [11,12], HILIC using amid-based columns [13–19] and chips [20, 21], and more recently porous graphitized carbon columns [22–24] and chips [25]. However, the low ionization efficiencies of native glycan structures, especially sialylated glycans, remains a problem and makes it difficult to simultaneously detect both acidic and neutral glycans in positive-mode MS. The permethylation of N-glycans easily addresses this problem, permitting simultaneous detection of both types of glycans [26, 27].
Although permethylated glycans ionize and fragment better than native glycans [32, 33], permethylation of glycans has been considered by many as labor intensive and not highly reproducible when employing Ciucanu and Kerek original procedure [28]. However, these limitations were recently overcome by a modified permethylation procedure [29] and the introduction of solid-phase permethylation method [30, 31]. An attractive feature endured by glycans as a consequence of permethylation is the substantial increase in hydrophobicity. Delaney and Vouros were the first to demonstrate the ability to separate the hydrophobic permethylated glycans by reversed-phase chromatography [34]. They separated unlabeled permethylated maltooligomer ladder, a 2-aminobenzamide (2AB)-labeled maltooligomer ladder, a complex mixture of 2AB-labeled bi-, tri-, and tetra- antennary standards, and a mixture of recombinant glycoprotein carbohydrates from soluble CD4 with varying sialic acid content [34]. Six years later, Costello and coworkers reported the RP-LC-MS analysis of permethylated N-glycans derived from ribonuclease B (RNase B) and permethylated O-glycans derived from C. elegans [22]. Recently, permethylated N-glycans derived from human blood serum were separated using a reversed-phase microfluidic liquid chromatography, allowing the resolution of some closely related structures [35]; however, only a limited number of glycan structures (20 glycans) were reported in this study.
Recently, we have significantly enhanced the sensitive analysis of permethylated N-glycans through on-line purification prior to reversed-phase (RP)-LC-MS analysis employing conventional proteomic RP-LC-ESI-MS setting [36]. Here, we are qualitatively and quantitatively comparing and evaluating the glycomic profiles of permethylated N-glycans derived from both model glycoproteins (such as RNase B and porcine thyroglobulin) and pooled blood serum acquired using RP-LC-ESI-MS, RP-LC-MALDI-MS, and MALDI-MS.
2 Materials and methods
2.1 Materials
Ammonia-borane complex, sodium hydroxide beads, DMSO, methyl iodide, 2,5-dihydroxybenzoic acid (DHB), TFA, MS-grade formic acid (FA), RNase B, porcine thyroglobulin (PTG), and pooled male human blood serum (HBS) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Empty microspin columns and active charcoal and C18 micro-spin columns were obtained from Harvard Apparatus (Holliston, MA, USA). Acetic acid and HPLC-grade solvents, including methanol and isopropanol, were procured from Fisher Scientific (Pittsburgh, PA, USA), while ACN was obtained from JT Baker (Phillipsburg, NJ, USA). HPLC grade water was acquired from Mallinckrodt Chemicals (Phillipsburg, NJ, USA). N-Glycosidase purified from Flavobacterium meningosepticum (PNGase F) was obtained from New England Biolabs (Ipswich, MA, USA).
2.2 Release of N-glycans from model glycoproteins
PNGase F was used to cleave asparagine-linked oligosaccharides (N-glycans) from RNase B and PTG as previously described [37]. Briefly, RNase B and PTG stock solutions were prepared at a concentration of 1 µg/µL. A 9-µL aliquot of the diluted G7 buffer solution (50mM sodium phosphate buffer, pH 7.5, diluted ten times in HPLC-grade water) was added into 1 µL of glycoproteins stock solution and mixed. Then, a 1.2-µ aliquot of PNGase F was added into the mixture prior to incubation at 37°C in a water bath for 18 h.
2.3 Release of N-glycans from pooled blood serum and purification
A 10-µL aliquot of blood serum was mixed with 90 µL G7 (10x diluted) solution. A 1.2 µL aliquot of PNGase F was then added to the sample mixture prior to overnight incubation at 37°C in a water bath. Next, charcoal macro spin-column was used to purify N-glycans released from the pooled blood serum sample. First, the active charcoal spin-column was washed with 400 µL of 100% ACN and followed with an additional wash with 85% ACN aqueous solution containing 0.1% TFA. A 5% ACN aqueous solution containing 0.1% TFA was then applied twice to condition the column prior to applying the sample. The released N-glycans were mixed with a 690-µL aliquot of 5% ACN aqueous solution containing 0.1% TFA and applied to the conditioned active charcoal spin column. Next, the spin column was washed five times with 400-µL aliquots of 5% ACN aqueous solution containing 0.1% TFA. Finally, N-glycans were eluted from the spin column using three 400-µL aliquots of 40% ACN aqueous solution containing 0.1% TFA. The collected eluents were then dried under vacuum.
2.4 Reduction of N-glycans
Ammonia-borane complex was used to reduce N-glycans to eliminate their anomeric forms as previously described [38]. Briefly, a 10-µL aliquot of freshly prepared ammonia-borane complex (10 µg/µL in water) was added to the N-glycans enzymatically released from RNase B and PTG. The same amount was also added to the purified N-glycans released from blood serum. Samples were then incubated at 65°C in a water bath for 1 h prior to the addition of 100-µL aliquot of 5% acetic acid aqueous solution. The samples were then dried under vacuum. Next, methanol was added to form the volatile methyl borate. This was repeated several times to ensure the removal of all borate salts. This was assessed by inspecting the bottom of the reaction vial for any white residue which is indicative of the presence of borate salts.
2.5 Permethylation of N-glycans
All N-glycans derived from RNase B, PTG, and HBS were permethylated as previously described using spin columns [30, 31]. Briefly, NaOH beads were packed into the empty micro spin columns to 3 cm prior to washing twice with 50 µL DMSO. Dried glycan samples were then resuspended in 7.5 µL DMSO and 1.2 µL water. A 20-µL aliquot of iodomethane (ICH3) was then added. The reaction mixtures were then applied to the NaOH micro spin columns and the permethylation reaction was allowed to proceed for 25 min. An additional 20-µL aliquot of ICH3 was added to each column and the permethylation was allowed to proceed for an additional 15 min. Next, the spin columns were subjected to centrifugation at a low speed (1600 rpm/min) for 30 sec prior to washing with a 50-µL aliquot of ACN. The collected eluents of each sample were then subjected to online purification as previously described [36] and briefly detailed.
2.6 Online solid-phase purification of N-glycans
The solutions collected at the end of permethylation consist of a mixture of DMSO, ACN, traces of water, excess iodomethane, reaction byproducts, and permethylated gly-cans. Evaporation of the reaction mixture under vacuum will result in the evaporation of all solvents, except DMSO (ca. 7.5 µL). In order to be able to effectively retain the permethylated glycan on the C18 trapping column of the LC-MS system, the total percentage of DMSO should not exceed 20%. Therefore, samples are resuspended in enough 2% aqueous ACN aqueous solution containing 0.1% FA solution to make 20% DMSO/ACN aqueous solution. Next, samples were loaded to Acclaims PepMap100 C18 nano-trap column (Dionex, Sunnyvale, CA, USA). The loaded sample was then washed using solvent A, consisting of 98% HPLC-grade water, 2% ACN, and 0.1% FA. This washing step was performed for 10 min at a flow rate of 3 µL/min. The equivalence of permethylated N-glycans derived from 0.1 µg of the model glycoproteins and 0.250 µL of human blood serum were loaded to the trap. For MALDI-MS analysis, the online purified permethylated N-glycans were eluted with 80% of solvent B at a flow rate of 3 (µL/min for 20 min.
In the case of LC-MS analyses, the permethylated gly-cans purified by the C18 trapping columns were then eluted to a nano reverse phase Acclaim® PepMap capillary column (150 mm × 75 µm id) packed with 100 ×Å C18 bounded phase (Dionex). The separation was attained using a two-pump system with pump A delivering solvent A consisting of 2% ACN aqueous solution containing 0.1% FA, while pump B delivering solvent B consisting of ACN and 0.1% FA. The flow rate was set to 350 nL/min and the gradient separation conditions consisted of increasing solvent B percentage of the mobile phase from 38 to 45% over 32 min. Eluent from the capillary column was sprayed directly into LTQ Orbitrap Velos Hybrid FT Mass Spectrometer (Thermo Scientific, San Jose, CA, USA) for the LC-ESI-MS/MS analysis, or collected at each minute and spotted on the MALDI plate in the case of LC-MALDI MS analysis.
2.7 ESI-MS acquisitions
The LTQ Orbitrap Velos. Hybrid FT Mass Spectrometer was operated in an automated data-dependent acquisition mode. The scan mode switched between MS full (m/z from 500–2000) and CID MS/MS scans. The latter was performed on the eight most abundant ions with the following parameters, 0.250 Q-value, 20 ms activation time, and 35% normalized collision energy.
2.8 MALDI-MS acquisition
MALDI-TOF MS analyses were performed using 4800 plus MALDI TOF/TOF (Applied Biosystems, Foster, CA, USA). The instrument was operated in positive-ion reflector mode with 2000 laser shots employing a mass range of 1500–5000 m/z. The LC collected samples were directly spotted on the MALDI plate in the case of LC-MALDI-MS analysis, or collected in a single vial over 30 min before drying and re-suspending in 1.0 µL of an 80%/20%/0.1% water/methanol solution. A 0.5-µL aliquot of each of the resuspended samples was then spotted on the MALDI plate. To each spot, a 0.5-µL aliquot of the matrix solution consisting of 10 mg/mL 2,5-DHB prepared in a 50% methanol aqueous solution containing 1 mM sodium acetate, was added. The inclusion of the sodium acetate was necessary to ensure that all permethylated glycans form exclusively sodium adducts.
3 Results and discussion
Recently, we have demonstrated the ability to significantly enhance the sensitive analysis of permethylated N-glycans through online purification [36]. We have also demonstrated the use of conventional proteomic setting without any changes to acquire glycomic profile [36]. However, we only described LC-MS analyses. Here, we are qualitatively and quantitatively comparing RP-LC-ESI-MS, RP-LC-MALDI-MS, and MALDI-MS glycomic profiles of permethylated N-glycans derived from both model glycoproteins (such as RNase B and PTG) and pooled human blood serum.
The glycomic profiles of permethylated N-glycans derived from RNase B (model glycoprotein) is depicted in Fig. 1. The base peak intensity (BPI) chromatogram and 2D overlaid BPI plot of the permethylated N-glycans derived from RNase B and analyzed by LC-ESI-MS are shown in Fig. 1A, while the extracted ion chromatograms (EICs) of the same permethylated glycan generated by LC-MALDI-MS is depicted in Fig. 1B. RP-LC-MALDI-MS traces here and elsewhere were constructed using Microsoft excel for smooth line fitting of MALDI-MS ion counts data. Both traces exhibit the endogenous distributions of the different glycans commonly observed on RNase B. Some of the traces shown in Fig. 1B do not appear to resemble their counterpart in Fig. 1A, especially in the case of Man7. However, we believe that these differences could be partially attributed to the manual deposition that was employed as well as the “sweet spot” limitation of MALDI-MS analysis.
Figure 1.

(A) RP-LC-ESI-MS base peak intensity (BPI) chro-matogram and 2D overlaid BPI plot of reduced and permethy-lated N-glycans derived from 100 ng of RNase B. The insets are the mass spectra of each peak. (B) Extracted ion chromatograms (EICs) of the same sample analyzed by RP-LC-MALDI-MS. These traces were generated using Microsoft excel and smooth fitting of ion count data. Symbols: blue square, N-acetylglucosamine; green circle, mannose; red triangle, fucose; yellow circle, galac-tose, magenta diamond, N-acetylneuraminic acid.
Multiple ions are formed in the ESI source for each permethylated glycan structures as shown in the insets of Fig. 1A, including doubly protonated ([M+2H]2+), singly pro-tonated and ammoniated ([M+H+NH4]2+), and doubly am-moniated ([M+2NH4]2+). The ammoniated adducts are believed to originate from the reduction step employed to eliminate the anomers. Excessive washing of the loaded samples did not eliminate the formation of these adducts. Also, excessive treatment with methanol and evaporation did not seem to eliminate or reduce the abundance of these adducts. Interestingly, these adducts are more abundant in the case of high mannose and hybrid type of glycans and less abundant in the case of complex type. The formation of these adducts is currently under investigation. The formation of these adducts does not appear to reduce the sensitive analysis of permethylated glycans. The chromatograms shown in Fig. 1 represent permethylated N-glycans derived from 100-ng of RNase B. The five high mannose glycans of RNase B were also detected (>3 S/N) for permethylated glycans derived from a 10-ng aliquot of RNase using LC-ESI-MS or LC-MALDI-MS (data not shown). However, this amount was not sufficient to detect all five high mannose glycans of RNase B by MALDI-MS. Accordingly, LC analysis is offering higher sensitivity irrespective of the MS used. This is expected, since the byproducts and impurities associated with a sample are eluted at different retention times than the analytes. These interfering species are all present in the sample in the case of MALDI-MS analysis and adversely influencing the ionization process.
The relative abundances of RNase B glycans acquired using the three methods employed in this study, as well as other analytical techniques such as NMR [39], LC-MS of gly-cans labeled with negatively charged reagent and separated on a reversed-phase column with ion pairing reagent [40], and capillary gel electrophoresis of APTS labeled glycans [41], are summarized in Table 1. Although there are differences between the results, such differences might be attributed to the source of RNase B samples. It is not expected that the different studies will have exactly the same numbers. Moreover, the differences shown in Table 1 might be partially contributed to the measurement uncertainties. Nevertheless, the data listed in Table 1 appears to be analytically comparable. The repro-ducibility of the LC-ESI-MS and MALDI-MS data appears to be substantially higher than that of LC-MALDI-MS. Moreover, the MALDI-MS and LC-ESI-MS data appears to be in a better agreement with the literature data. We believe that this discrepancy might be partially attributed to the manual spotting employed here or to “sweet spot” limitation of MALDI-MS. Although the glycomic profiles of RNase B N-glycans appear to be quantitatively comparable, LC-MS analyses are offering higher sensitivity for the profiling of N-glycans derived from a glycoprotein. This is expected since LC analysis reduced competitive ionization and saturation of detectors.
Table 1.
Relative abundances of the N-glycan structures commonly associated with RNase B acquired using different analytical techniques and methods (n = 3 for all methods with expressed uncertainties)
| Glycans | Relative abundance |
||||||
|---|---|---|---|---|---|---|---|
| NMRa) | CGEb) | LC-MSc) | MALDI-MS | MALDI-MS permethylated |
LC-ESI-MS MS permethylated |
LC-MALDI permethylated |
|
| Man 5 | 57 | 51.5 | 52 ± 2 | 51 ± 1 | 59.0 ± 0.6 | 53.5 ± 0.4 | 50 ± 6 |
| Man 6 | 31 | 30.3 | 32 ± 2 | 27.4 ± 0.7 | 29.0 ± 0.7 | 32.5 ± 0.6 | 30 ± 5 |
| Man 7 | 4 | 4 | 6.3 ± 0.6 | 7 ± 1 | 4.0 ± 0.2 | 8.0 ± 0.2 | 11 ± 4 |
| Man 8 | 7 | 8.5 | 7.2 ± 0.8 | 10.9 ± 0.6 | 7.0 ± 0.2 | 5.2 ± 0.3 | 8 ± 4 |
| Man 9 | 1 | 3.7 | 1.6 ± 0.1 | 3.5 ± 0.6 | 1.00 ± 0.05 | 0.82 ± 0.06 | 2 ± 1 |
Porcine thyroglobulin is a glycoprotein that has N-glycans representing all types, including high mannose complex and hybrid [42–46]. The LC-ESI-MS and LC-MALDI-MS glycomic profiles of PTG are depicted in Fig. 2. The extracted ion chro-matograms of the abundant glycans associated with PTG as well as the 2D overlaid BPI plot of LC-ESI-MS glycomic profiling are shown in Fig. 2A. This analysis permitted the detection of 11 glycan structures associated with PTG. The relative distribution of the different glycans observed in the LC-ESI-MS analysis was comparable to what is reported in the literature [42–46]. The LC-MALDI-MS profile of the same sample is shown in Fig. 2B. Less glycan structures were observed in the LC-MALDI profile. In the case of this glycoprotein which has a more heterogeneous glycan profile than that of the RNase B, LC-ESI-MS appears to permit better detection of permethylated glycans. The chromatographic peaks of the sialylated glycan depicted in Fig. 3 are broad with shouldering. We believe that these features are common for all sialylated glycans which might beattributed tothe presenceofmultiple isomers (different linkages and location on the antenna) that are not completely resolved under the chromatographic conditions employed here.
Figure 2.

(A) RP-LC-ESI-MS BPI chromatogram, EICs and 2D overlaid BPI plot of reduced and permethylated N-glycans derived from 100 ng of porcine thyroglubin (PTG). The BPI chro-matograms (B) RP-LC-MALDI-MS EICs of the same sample analyzed by. These traces were generated using Microsoft excel and smooth fitting of ion count data. Symbols: as in Fig. 1.
Figure 3.

(A) RP-LC-ESI-MS 2D overlaid BPI plot of reduced and permethylated N-glycans derived from 250 nl of human blood serum. (B) RP-LC-MALDI-MS EICs of the same sample analyzed by. (C) MALDI mass spectra and RP-LC-MALDI-MS EICs of N-glycans derived from blood serum. These traces were generated using Microsoft excel and smooth fitting of ion count data. The inset depicting the mass spectra associated with the RP-LC analysis. Symbols: as in Fig. 1.
The relative abundances of the glycan structures commonly associated with PTG and determined by different analytical techniques are listed in Table 2. Again as expected, better detection is observed in the case of the LC-MALDI-MS relative to those of MALDI-MS which is attributed to the reduced competitive ionization and saturation of detectors. Due to the high molecular weight of PTG (>650 kDa), the amount of N-glycans analyzed was substantially lower than that of RNase B. The same amount of glycoprotein (100-ng aliquots) was analyzed. Accordingly, a 100-ng aliquot is needed to allow quantitative profiling of PTG N-glycans. Although there is no absolute agreement between the different studies listed in Table 2, the overall results appear to be comparable with LC-ESI-MS permitting the detection of more structures than any of the other methods. All of the structures listed in Table 2 were detected only through RP-LC-ESI-MS.
Table 2.
Relative abundances of the N-glycan structures derived from porcine thyroglobulin acquired using different analytical techniques
According to the above results, LC-ESI-MS analyses offer higher sensitivity as the heterogeneity of the glycomic profile increases. This increase in sensitivity is seen through an increase in the number of glycan structures that are detected with S/N ratios better than 3. Additionally, LC of permethylated glycan enhanced the MALDI analysis by reducing competitive ionization and minimizing saturation of detectors. This was further supported by the glycomic profile of human blood serum sample.
Alley et al. have recently reported the separation of reduced and permethylated N-glycans derived from blood serum on a reversed-phase chip interfaced to an ion trap mass spectrometer [35]. Only 20 N-glycan structures were confidently detected. This limited number of glycan structures might be due to the limited loading capacity of the chip as well as to the mass spectrometer employed. The 2D overlaid base peak intensity plot of LC-ESI-MS analysis of 35 reduced and permethylated N-glycans derived from the equivalence of 250 nl of blood serum is depicted in Fig. 3A, while the EIC of ten of these structures observed in the LC-MALDI-MS analysis is shown in Fig. 3B. In total, 73 glycan structures were confidently detected in the LC-ESI-MS analysis of250 nl ofblood serum, while53structures were detected in the LC-MALDI-MS analysis of the same amount of blood serum (Table 3). All of the detected glycan structures exhibited an S/N ratio better than 3. On the other hand, only 42 glycans structures were reported in the case of the MALDI-MS analysis of the same amount of sample with S/N ratio of 4.7 or better (Table 3). The chromatographic separation substantially enhanced the S/N ratios observed in MALDI-MS analyses as shown in Fig. 3C for three representative glycan structures. The S/N ratio increased between 13- and 16-folds as a result of the chromatographic separation. This substantial increase is expected, and is mainly originating from the reduction in both competitive ionization and saturation of detectors.
Table 3.
Signal-to-noise ratios and relative intensities of human blood serum permethylated N-glycans analyzed by RP-LC-ESI-MS, RP-LC-MALDI-MS, and MALDI-MS
| Structuresa) | S/N |
Intensity |
|||
|---|---|---|---|---|---|
| LC-ESI | LC-MALDI | MALDI | LC-ESI | LC-MALDI | |
| 871.9 | 2020.8 | 24.0 | 0.8 | 5.1 | |
| 7.7 | 57.4 | ND | 0.04 | 0.1 | |
| 3.4 | 33.9 | ND | 0.04 | 0.2 | |
| 42.8 | 62.2 | ND | 0.4 | 0.3 | |
| 58.0 | 708.2 | 17.2 | 0.3 | 1.7 | |
| 29.9 | 119.1 | 9.7 | 0.2 | 0.4 | |
| 1510.0 | 7554.6 | 172.8 | 14.1 | 18.2 | |
| 140.8 | 198.0 | 12.8 | 0.9 | 2.6 | |
| 15.4 | 49.8 | ND | 0.1 | 0.2 | |
| 9.1 | 84.0 | 6.7 | 0.1 | 0.2 | |
| 12.1 | 346.7 | 11.0 | 0.08 | 0.9 | |
| 30.0 | 53.3 | ND | 0.2 | 0.1 | |
| 1482.6 | 9109.7 | 478.5 | 16.4 | 21.9 | |
| 92.7 | 129.5 | 30.4 | 0.9 | 1.0 | |
| 394.6 | 963.3 | 56.6 | 2.2 | 2.6 | |
| 51.0 | 220.6 | 8.7 | 0.4 | 0.4 | |
| 32.3 | 70.4 | 8.0 | 0.2 | 0.2 | |
| 27.7 | 228.7 | ND | 0.1 | 0.5 | |
| 32.2 | 162.9 | 10.5 | 0.2 | 0.5 | |
| 9.72 | ND | ND | 0.08 | ND | |
| 1012.1 | 3863.0 | 199.5 | 8.5 | 11.4 | |
| 391.8 | 643.1 | 97.4 | 2.9 | 1.7 | |
| 28.5 | 34.3 | 11.9 | 0.2 | 0.07 | |
| 114.2 | 276.8 | 27.0 | 0.7 | 0.8 | |
| 297.8 | 1904.9 | 194.2 | 2.5 | 4.1 | |
| 120.3 | ND | ND | 0.2 | ND | |
| (Hex)5(HexNAc)+(Man)3(GlcNAc)2 | 181.5 | ND | 14.3 | 1.5 | ND |
| 126.0 | 187.3 | ND | 1.0 | 0.5 | |
| ND | 28.0 | ND | ND | 0.07 | |
| 126.8 | 321.2 | 35.3 | 1.0 | 0.7 | |
| 217.3 | 1006.4 | 165.2 | 1.8 | 2.7 | |
| 21.5 | ND | ND | 0.1 | ND | |
| 10.9 | 70.7 | 15.4 | 0.1 | 0.2 | |
| 3.7 | 15.2 | ND | 0.04 | 0.05 | |
| ND | ND | 31.3 | ND | ND | |
| 1292.5 | 5243.7 | 1493.3 | 14.2 | 12.2 | |
| 555.0 | 365.0 | ND | 6.1 | 1.2 | |
| (Hex)4(HexNAc)2 (Deoxyhexose)2 + (Man)3(GlcNAc)2 | 224.5 | ND | ND | 2.5 | ND |
| 355.3 | 703.9 | 163.6 | 1.5 | 1.2 | |
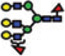 |
79.7 | ND | ND | 0.3 | ND |
| 12.7 | 99.2 | 29.5 | 0.2 | 0.3 | |
| 832.8 | 1141.1 | 212.8 | 3.9 | 2.0 | |
| (Hex)3(HexNAc)2 (Deoxyhexose)2 + (Man)3(GlcNAc)2 | 26.8 | 38.5 | 13.2 | 0.09 | 0.08 |
| 39.1 | 37.6 | 15.5 | 0.2 | 0.07 | |
| 34.8 | ND | ND | 0.2 | ND | |
| 639.1 | 356.6 | 100 | 2.00 | 0.6 | |
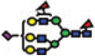 |
273.2 | 41.5 | 24.5 | 1.5 | 0.1 |
| 248.5 | 164.2 | 57.3 | 1.3 | 0.3 | |
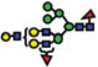 |
16.8 | ND | ND | 0.1 | ND |
| 9.1 | ND | ND | 0.1 | ND | |
| 64.8 | 31.5 | 21.0 | 0.3 | 0.07 | |
| 248.47 | ND | ND | 1.3 | ND | |
| 43.1 | ND | ND | 0.3 | ND | |
| 6.6 | ND | ND | 0.04 | ND | |
| 110.8 | 757.9 | 233.6 | 0.6 | 1.3 | |
| 66.4 | 46.4 | 18.7 | 0.4 | 0.08 | |
| 27.4 | ND | ND | 0.2 | ND | |
| 707.2 | 310.7 | 89.0 | 2.6 | 0.5 | |
| (Hex)5(HexNAc)3 (Deoxyhexose)2 (NeuAc) + (Man)3 (GlcNAc)2 | 51.8 | ND | ND | 0.2 | ND |
| 27.2 | 13.2 | 4.7 | 0.1 | 0.02 | |
 |
71.1 | 14.1 | 10.9 | 0.1 | 0.03 |
| 4.5 | ND | ND | 0.03 | ND | |
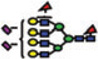 |
4.5 | ND | ND | 0.05 | ND |
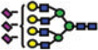 |
217.7 | 62.7 | 17.2 | 0.3 | 0.1 |
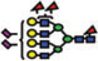 |
13.3 | ND | ND | 0.01 | ND |
| 5.4 | 22.8 | ND | 0.01 | 0.02 | |
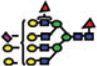 |
22.4 | ND | ND | 0.04 | ND |
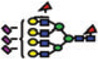 |
14.7 | 7.9 | 6.8 | 0.01 | 0.05 |
| 530.5 | 182.8 | 20.5 | 0.5 | 0.3 | |
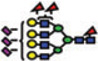 |
50.9 | 29.3 | ND | 0.05 | 0.02 |
| 204.7 | 29 | 9.4 | 0.2 | 0.07 | |
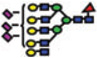 |
9.1 | ND | ND | 0.01 | ND |
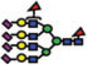 |
72.4 | ND | ND | 0.1 | ND |
 |
56.4 | ND | ND | 0.08 | ND |
ND, not detected.
Mass accuracy is better than 2 ppm.
Of the structures detected in the three analyses, 40 were common among all. Forty structures and 52 structures were common among the MALDI-MS and RP-LC-MALDI-MS analyses, and RP-LC-ESI-MS and RP-LC-MALDI-MS analyses, respectively. A single structure was unique for the MALDI-MS, while a different one was unique to the RP-LC-MALDI-MS analyses. Twenty N-glycan structures were only observed in the RP-LC-ESI-MS analysis, which is equal to the total number of structures reported by Alley et al. [35]. The confident detection of more structures in this study, relative to the previous study (ca. 4-fold), might be attributed to sample purification (liquid–liquid extraction versus online purification in this study), loading capacities of the traps, and the mass accuracy and resolution of mass spectrometers (ion trap versus Orbitrap this study). The N-glycan structures that were detected only as a result of LC-separation prior to MS were at low abundance (Table 3). The inability to see these structures in MALDI spectrum might be attributed to competitive ionization and in some cases might be a combination of both competitive ionization and saturation of detectors (structures with low m/z values).
The reverse-phase chromatographic separation of the permethylated N-glycans derived from model glycoproteins or blood serum did not only allow the detection of low abundant structures, but also resolved some structural isomers as shown in Fig. 4. The reversed-phase separation permitted the separation of permethylated glycans with the same molecular weight but different structures. The separation is based on the overall hydrophobicity of the permethylated structures; however, the branching of the glycan structures also dictates their overall hydrophobicity. The latter explains the higher retention of hybrid glycans on a reversed-phase media relative to that of complex-type glycans (Fig. 4A). Complex glycans are more compact with intramolecular interactions between the different methoxy groups on the different antenna. This intramolecular interaction reduces the overall hy-drophobicity of the molecule and subsequently reduces interactions with the chromatographic media. On the other hand, the hybrid structure shown in Fig. 4A appears to have less intramolecular interaction between the different methoxy groups. This limited intramolecular interaction is prompted by the bisecting GlcNAc. Accordingly, this hybrid glycan structure has more methoxy groups available for interaction with the chromatographicmedia andsubsequentlyisretained more on the reversed-phase column. The separation of the two structures shown in Fig. 4B could be also explained in the same way. Tandem MS data was essential to unequivocally assign the different structures (data not shown). Glycans that did not have tandem MS data are listed in Table 3 only in term of composition.
Figure 4.

RP-LC-ESI-MS EICs of some N-glycan isomers derived from human blood serum. Symbols: as in Fig. 1.
4 Concluding Remarks
To our knowledge, thisis thefirst study that highlights the advantages and disadvantages of LC-ESI-MS analysis of permethylated N-glycans. Reduced and permethylated N-glycans derived from both model glycoproteins and human blood serum were analyzed by different analytical techniques, including MALDI-MS and LC-MS with MALDI and ESI sources. Per-methylation of glycans made these structures hydrophobic enough to be retained and separated by reverse-phase nano-column. The chromatographic separation helped reduce the competitive ionization and saturation of detector in the case of MALDI-MS. The results of the analyses of N-glycans derived from model glycoproteins using three methods compared here were very comparable with a slight edge for LC-ESI-MS. However, the analysis of N-glycans derived from blood serum demonstrated the major advantage of LC-ESI-MS over the other methods. In conjunction with our recent online purification of permethylated glycans, LC-ESI-MS of permethylated N-glycans derived from blood serum allowed the confident detection of 73 glycan structures through tandem MS and the high mass accuracy offered by the mass spectrometer used. Although tandem MS can be acquired in the case of LC-MALDI-MS, it is more automated and requires a lot less sample in the case of an ion trap mass spectrometer. Although multiple ions and adducts are formed in LC-ESI-MS, this technique offers unmatched simplicity, convenience, high sensitivity, and better tandem MS data. Currently, we are routinely utilizing LC-ESI-MS for the analyses of permethylated glycans derived from biological specimens, including human blood serum, fluids, and tissues.
Acknowledgments
This work was supported by the office of the vice president for research at Texas Tech University and partially by an NIH grant (1R01 GM093322-01).
Abbreviations
- BPI
base peak intensity
- DHB
2,5-dihydroxybenzoic acid
- EIC
extracted ion chromatogram
- FA
formic acid
- PTG
porcine thyroglobin
- PNGase F
N-glycosidase from F. meningosepticum
- RNase B
ribonuclease B
Footnotes
Colour online: See the article online to view Figs. 1–7 and Tables 2 and 3 in colour.
The authors have declared no conflict of interest.
References
- 1.Helenius A, Aebi M. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 2.Dube DH, Bertozzi CR. Nat. Rev. Drug Discovery. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 3.Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Science. 2001;291:2370–2376. doi: 10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]
- 4.Goetz JA, Mechref Y, Kang P, Jeng M-H, Novotny MV. Glycoconj. J. 2009;26:117–131. doi: 10.1007/s10719-008-9170-4. [DOI] [PubMed] [Google Scholar]
- 5.Stanta JL, Sadova R, Struwe WB, Byrne JC, Leweke FM, Rothermund M, Rahmoune H, Levin Y, Guest PC, Bahn SMRP. J. Proteome Res. 2010;9:4476–4489. doi: 10.1021/pr1002356. [DOI] [PubMed] [Google Scholar]
- 6.Mechref Y, Hussein A, Bekesova S, Pungpapong V, Zhang M, Dobrolecki LE, Hickey RJ, Hammoud ZT, Novotny MV. J. Proteome Res. 2009;8:2656–2666. doi: 10.1021/pr8008385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen C, Schmilovitz-Weiss H, Liu X, Pappo O, Marisa Halpern O, Sulkes J, Braun M, Cohen M, Barak N, Tur-Kaspa R, Vanhooren V, Van Vlierberghe H, Libert C, Contreras R, Ben-Ari Z. J. Proteome Res. 2009;8:463–470. doi: 10.1021/pr800656e. [DOI] [PubMed] [Google Scholar]
- 8.Babu P, North SJ, Jang-Lee J, Chalabi S, Macker-ness K, Stowell SR, Cummings RD, Rankin S, Dell A, Haslam SM. Glycoconj. J. 2009;26:975–986. doi: 10.1007/s10719-008-9146-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paschinger K, Razzazi-Fazeli E, Furukawac K, Wilsona IBH. J. Mass Spectrom. 2011;46:561–567. doi: 10.1002/jms.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Emmett MR, Marshall AG. Anal. Chem. 2010;82:6542–6548. doi: 10.1021/ac1008833. [DOI] [PubMed] [Google Scholar]
- 11.Bruggink C, Wuhrer M, Koeleman CA, Barreto V, Liu Y, Pohl C, Ingendoh A, Hokke CH, Deelder AM. J. Chromatogr. B. 2005;829:136–143. doi: 10.1016/j.jchromb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Chataigne G, Couderc F, Poinsot V. J. Chromatogr. A. 2008;1185:241–250. doi: 10.1016/j.chroma.2008.01.065. [DOI] [PubMed] [Google Scholar]
- 13.Wuhrer M, Koeleman CA, Deelder AM, Hokke CH. Anal Chem. 2004;76:833–838. doi: 10.1021/ac034936c. [DOI] [PubMed] [Google Scholar]
- 14.Bereman MS, Williams TI, Muddiman DC. Anal. Chem. 2009;81:1130–1136. doi: 10.1021/ac802262w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mauko L, Nordborg A, Hutchinson JP, Lacher NA, Hilder EF, Haddad PR. Anal. Biochem. 2011;10:235–241. doi: 10.1016/j.ab.2010.09.035. [DOI] [PubMed] [Google Scholar]
- 16.Wohlgemuth J, Karas M, Jiang W, Hendriks R, An-drecht S. J. Sep. Sci. 2010;33:880–890. doi: 10.1002/jssc.200900771. [DOI] [PubMed] [Google Scholar]
- 17.Lam MP, Siu SO, Lau E, Mao X, Sun HZ, Chiu PC, Yeung WS, Cox DM, Chu IK. Anal. Bioanal. Chem. 2010;398:791–804. doi: 10.1007/s00216-010-3991-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bereman MS, Young DD, Deiters A, Muddiman DC. J. Proteome Res. 2009;8:3764–3770. doi: 10.1021/pr9002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo Q, Rejtar T, Wu SL, Karger BL. J. Chromatogr. A. 2009;1216:1223–1231. doi: 10.1016/j.chroma.2008.09.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Staples GO, Bowman MJ, Costello CE, Hitchcock AM, Lau JM, Leymarie N, Miller C, Naimy H, Shi X, Zaia J. Proteomics. 2009;9:686–695. doi: 10.1002/pmic.200701008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dreyfuss JM, Jacobs C, Gindin Y, Benson G, Staples GO, Zaia J. Anal. Bioanal. Chem. 2011;399:727–735. doi: 10.1007/s00216-010-4235-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costello CE, Contado-Miller JM, Cipollo JF. J. Am. Soc. Mass Spectrom. 2007;18:1799–1812. doi: 10.1016/j.jasms.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawasaki N, Itoh S, Ohta M, Hayakawa T. Anal. Biochem. J. 2003;316:15–22. doi: 10.1016/s0003-2697(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 24.Pabst M, Bondili JS, Stadlmann J, Mach L, Alt-mann F. Anal. Chem. 2007;79:5051–5057. doi: 10.1021/ac070363i. [DOI] [PubMed] [Google Scholar]
- 25.Ninonuevo M, An H, Yin H, Killeen K, Grimm R, Ward R, German B, Lebrilla C. Electrophoresis. 2005;26:3641–3649. doi: 10.1002/elps.200500246. [DOI] [PubMed] [Google Scholar]
- 26.Kang P, Mechref Y, Klouckova I, Novotny MV. Rapid Commun. Mass Spectrom. 2005;19:3421–3428. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mechref Y, Kang P, Novotny MV. Methods Mol. Biol. 2009;534:53–64. doi: 10.1007/978-1-59745-022-5_4. [DOI] [PubMed] [Google Scholar]
- 28.Ciucanu I, Kerek F. Carbohydrate Res. 1984;131:209–217. [Google Scholar]
- 29.Ciucanu I, Costello CE. J. Am. Chem. Soc. 2003;125:16213–16219. doi: 10.1021/ja035660t. [DOI] [PubMed] [Google Scholar]
- 30.Kang P, Mechref Y, Novotny MV. Rapid Commun. Mass Spectrom. 2008;22:721–734. doi: 10.1002/rcm.3395. [DOI] [PubMed] [Google Scholar]
- 31.Mechref Y, Kang P, Novotny MV. Methods Mol. Biol. 2009;534:53–64. doi: 10.1007/978-1-59745-022-5_4. [DOI] [PubMed] [Google Scholar]
- 32.Dell A, Morris HR. Carbohydr. Res. 1991;209:33–50. doi: 10.1016/0008-6215(91)80143-b. [DOI] [PubMed] [Google Scholar]
- 33.Dell A, Jang-Lee J, Pangs P-C, Parry S, Sutton-Smith M, Tissot B, Morris HR, Panico M, Haslam SM. In: Gly-coscience - Chemistry and Chemical Biology. Fraser-Reid BO, Tatsuta K, Thiem J, editors. Heidelberg: Springer; pp. 2191–2240. [Google Scholar]
- 34.Delaney J, Vouros P. Rapid Commun. Mass Spectrom. 2001;15:325–334. doi: 10.1002/rcm.230. [DOI] [PubMed] [Google Scholar]
- 35.Alley WR, Madera M, Mechref Y, Novotny MV. Anal. Chem. 2010;82:5095–5106. doi: 10.1021/ac100131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Desantos-Garcia JL, Khalil SI, Hussein A, Hu Y, Mechref Y. Electrophoresis. 2011;32:3516–3525. doi: 10.1002/elps.201100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mechref Y, Novotny MV. Anal. Chem. 1998;70:455–463. doi: 10.1021/ac970947s. [DOI] [PubMed] [Google Scholar]
- 38.Huang Y, Mechref Y, Novotny MV. Anal. Chem. 2001;73:6063–6069. doi: 10.1021/ac015534c. [DOI] [PubMed] [Google Scholar]
- 39.Fu D, Chen L, O′Neill RA. Carbohydr. Res. 1994;261:173–186. doi: 10.1016/0008-6215(94)84015-6. [DOI] [PubMed] [Google Scholar]
- 40.Gennaro LA, Harvey DJ, Vouros P. Rapid Commun. Mass Spectrom. 2003;17:1528–1534. doi: 10.1002/rcm.1079. [DOI] [PubMed] [Google Scholar]
- 41.Guttman A, Pritchett T. Electrophoresis. 1995;16:1906–1911. doi: 10.1002/elps.11501601314. [DOI] [PubMed] [Google Scholar]
- 42.Charlwood J, Birrell H, Organ A, Camilleri P. Rapid Commun. Mass Spectrom. 1999;13:716–723. doi: 10.1002/(sici)1097-0231(19990430)13:8<716::aid-rcm547>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 43.deWaard P, Koorevaar A, Kamerling JP, Vliegenthart JFG. J. Biol. Chem. 1991;266:4237–4243. [PubMed] [Google Scholar]
- 44.Tsuji T, Yamamoto K, Irimura T, Osawa T. Biochem. J. 1981;195:691–699. doi: 10.1042/bj1950691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamoto K, Tsuji T, Irimura T, Osawa T. Biochem. J. 1981;195:701–713. doi: 10.1042/bj1950701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wheeler SF, Domann P, Harvey DJ. Rapid Commun. Mass Spectrom. 2009;23:303–312. doi: 10.1002/rcm.3867. [DOI] [PubMed] [Google Scholar]