

Abstract
Gain-of-function missense mutations in FGF receptor 2 (FGFR2) are responsible for a variety of craniosynostosis syndromes including Apert syndrome (AS), Pfeiffer syndrome (PS) and Crouzon syndrome (CS). Unlike the majority of FGFR2 mutations, S252W and P253R AS mutations and a D321A PS mutation retain ligand-dependency and are also associated with severe limb pathology. In addition, a recently identified ligand-dependent S252L/A315S double mutation in FGFR2 was shown to cause syndactyly in the absence of craniosynostosis. Here, we analyze the effect of the canonical AS mutations, the D321A PS mutation and the S252L/A315S double mutation on FGFR2 ligand binding affinity and specificity using surface plasmon resonance. Both AS mutations and the D321A PS mutation, but not the S252L/A315S double mutation, increase the binding affinity of FGFR2c to multiple FGFs expressed in the cranial suture. Additionally, all four pathogenic mutations also violate FGFR2c ligand binding specificity and enable this receptor to bind FGF10. Based on our data, we propose that an increase in mutant FGFR2c binding to multiple FGFs results in craniosynostosis, whereas binding of mutant FGFR2c to FGF10 results in severe limb pathology. Structural and biophysical analysis shows that AS mutations in FGFR2b also enhance and violate FGFR2b ligand binding affinity and specificity, respectively. We suggest that elevated AS mutant FGFR2b signaling may account for the dermatological manifestations of AS.
INTRODUCTION
Craniosynostosis, the premature fusion of one or more cranial sutures, occurs with a prevalence of 1 in 2500 births and is the hallmark of over 100 distinct syndromes, including Apert syndrome (AS) (MIM 101200), Pfeiffer syndrome (PS) (MIM 101600) and Crouzon syndrome (CS) (MIM 123500) (1–3). AS is also accompanied by severe syndactyly of the hands and feet, whereas traditionally, limb pathology in PS is moderate and not evident in CS. Other findings in AS that occur at high frequencies include dermatological abnormalities, central nervous system (CNS) malformations, dental anomalies and progressive synostosis of bones of the hands, feet and cervical spine (4–7).
AS, PS and CS result from activating missense mutations in fibroblast growth factor (FGF) receptor 2 (FGFR2) (8–12). The majority of these mutations result in constitutive (ligand-independent) receptor activation, although AS mutations activate receptor only in the presence of ligand (ligand-dependent). Almost all cases of AS result from one or the other of two mutations, S252W and P253R, which map to the invariant D2–D3 linker region (Fig. 1) and therefore can manifest in both mesenchymal ‘c’ and epithelial ‘b’ splice isoforms of FGFR2 (9). However, craniofacial pathology in AS is thought to be primarily mediated by mutant FGFR2c signaling since the majority of CS and PS mutations map specifically to the ‘c’ exon of FGFR2. Genotype–phenotype analyses of AS patients show that patients with the S252W mutation have a more severe craniofacial phenotype, while patients with the P253R mutation present with more severe syndactyly (13–15), although an earlier study did not observe these correlations (16). This has led to the suggestion that craniofacial and limb abnormalities in AS arise from distinct pathophysiological mechanisms (13). Interestingly, recent studies have also shown that although AS mutations are deleterious for human development, they paradoxically confer a selective advantage upon male germ cells (17,18).
Figure 1.
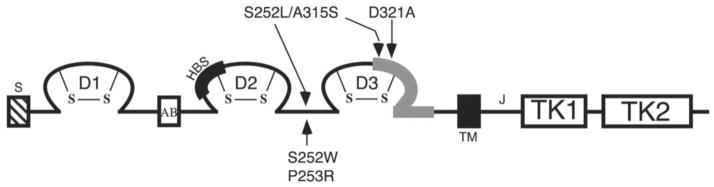
Mapping of pathogenic FGFR2 mutations. D1, D2, D3 represent immunoglobulin (Ig)-like domain 1, 2 and 3; S represents the signal peptide; AB represents the acid box; TM represents the transmembrane helix; TK1 and TK2 represent the split-kinase domain, which is interrupted by the kinase insert; J represents the juxtamembrane region. The heparin-binding site (HBS) in D2 is marked by a thickened black line. The alternatively spliced region (encoded by either exon ‘b’ or ‘c’ in a tissue specific manner) in D3 is represented by a thickened gray line. The location of AS S252W and P253R mutations, the D321A PS mutation and the S252L/A315S double mutation are indicated by arrows. The D321A PS mutation and the S252L/A315S double mutation manifest only in the ‘c’ isoform of FGFR2.
Anderson et al. (19) analyzed the effect of AS mutations on FGFR2c binding to FGF1, FGF2 and FGF4, using surface plasmon resonance (SPR), and found that both AS mutations enhance the binding affinity of FGFR2c for FGF2 only, with the S252W mutation causing a greater increase. Hence, it was proposed that craniofacial pathology is mediated by enhanced signaling of FGF2 through mutant FGFR2c and that the more severe craniofacial pathology in S252W AS patients reflects the greater enhancement in FGF2 binding. Analogously, it was postulated that the greater severity of syndactyly in P253R AS patients, relative to S252W AS patients, reflects the higher affinity of P253R FGFR2b for FGF7 or FGF10 (19).
Oldridge et al. (20) identified two AS and two PS patients harboring Alu-element insertions or nucleotide substitutions, which affected alternative splicing of FGFR2 and resulted in the ectopic expression of FGFR2b in the patients’ mesenchymal tissues. Notably, the severity of the limb phenotype correlated with the level of ectopic FGFR2b expression, as the AS patient had higher levels of ectopic FGFR2b expression compared to the levels of ectopic FGFR2b expression in PS patients. Hence, Oldridge et al. (20) proposed that autocrine activation of ectopic mesenchymal FGFR2b by mesenchymally expressed FGF7 or FGF10 leads to syndactyly. In contrast to the data of Anderson et al. (19), Yu et al. (21) did not observe increased binding of AS mutant FGFR2c to FGF2. Instead, they showed that AS mutations violate FGFR2 ligand binding specificity (21). Cell lines expressing S252W FGFR2c were stimulated by FGF7 and FGF10, ligands that have absolutely no activity on cell lines expressing wild-type FGFR2c. Conversely, cell lines expressing S252W FGFR2b were also shown to respond to FGF2, which normally has no activity on cell lines expressing wild type FGFR2b. Moreover, binding of FGF7 to both AS mutant FGFR2c was also demonstrated. Hence, it was suggested that AS results from autocrine signaling of mutant FGFR2c through FGF7 or FGF10 in the mesenchyme. The possibility of autocrine signaling of mutant FGFR2b by FGF2 or other epithelial ligands contributing to craniosynostosis and syndactyly was mentioned.
Crystallographic analysis of AS mutant FGFR2c in complex with FGF2 demonstrates that the S252W mutation results in additional contacts between the receptor and the N-terminal region of FGF2, whereas the P253R mutation results in additional interactions of the receptor with the β-trefoil core domain of FGF2 (22). Sequence alignment of the poorly conserved and flexible N-terminal region of FGFs suggested that the S252W mutation would enhance FGFR2c binding to a subset of FGFs. In contrast, the conserved nature of the gain-of-function interactions in the P253R FGFR2c-FGF2 crystal structure suggested that the P253R mutation will enhance FGFR2c binding to all FGFs. The distinct nature of the gain-of-function contacts mediated by the S252W and P253R mutations was proposed to reflect the phenotypic variability between the two subsets of AS patients.
Recently, a kindred remarkable for Apert-like syndactyly in the absence of craniosynostosis was found to segregate two mutations, S252L and A315S, in cis (23). These mutations respectively map to the D2–D3 linker region (Fig. 1) and the alternatively spliced βC′-βE loop (Fig. 1), a region that dictates the ligand binding specificity of FGFR2. Notably, the A315S substitution changes a FGFR2c specific residue to a FGFR2b specific residue, and led Oldridge et al. (20) to hypothesize that the double mutation results in syndactyly by enabling S252L/A315S FGFR2c to bind FGF7 or FGF10. Apert-like syndactyly has also been reported in a PS patient with a D321A mutation (24), which also maps to the alternatively spliced βC′-βE loop of FGFR2c (Fig. 1) and further implicates mutant FGFR2c signaling in the pathogenesis of craniosynostosis and syndactyly. Because S252L, A315S and D321A mutations map to key regions in the ligand binding site of FGFR2c (25) they are likely to be ligand-dependent mutations. Here, we examine the effect of the two canonical AS mutations, the D321A PS mutation and the S252L/A315S double mutation on FGFR2 ligand binding affinity and specificity in order to further understand how cranial and limb phenotypes dissociate in patients with different pathogenic FGFR2 mutations.
RESULTS
Effect of AS mutations on FGFR2c ligand binding affinity and specificity
Previous ligand binding analyses of wild-type FGFR2c are scattered throughout the literature and use a variety of techniques (19,26–34). To provide a coherent framework to characterize the effect of FGFR2c mutations, wild type FGFR2c was examined for binding to all FGFs, with the exception of FGF22, using SPR. The kinetic data are summarized in Table 1 and represent the most comprehensive ligand binding analysis of wild-type FGFR2c to date. Indeed, this is the first quantitative binding study of wild-type FGFR2c towards FGF8, FGF16, FGF17, FGF19 through FGF21 and FGF23.
Table 1.
Summary of wild-type and mutant FGFR2c bindin'g dataa
| FGF | WT | S252W | P253R | D321A | S252L/A315S | |
|---|---|---|---|---|---|---|
| FGF1 | ka (M −1 s−1)b | 3.76 × 105 | 1.24 × 106 | 7.21 × 105 | 1.30 × 105 | 3.77 × 105 |
| kd (s−1)b | 3.53 × 10−2 | 1.62 × 10−2 | 2.96 × 10−2 | 1.33 × 10−2 | 2.84 × 10−2 | |
| KD (M) c | 9.37 × 10−8 | 1.30 × 10−8 | 4.10 × 10−8 | 1.02 × 10−7 | 7.54 × 10−8 | |
| FGF2 | ka (M −1 s−1) | 6.76 × 105 | 2.73 × 105 | 7.19 × 105 | 2.61 × 104 | 5.81 × 105 |
| kd (s−1) | 7.02 × 10−3 | 6.05 × 10−4 | 3.20 × 10−3 | 1.68 × 10−2 | 1.95 × 10−2 | |
| KD (M) | 1.04 × 10−8 | 2.22 × 10−9 | 4.45 × 10−9 | 6.46 × 10−7 | 3.30 × 10−8 | |
| FGF3 | ka (M −1 s−1) | 1.11 × 104 | 5.07 × 104 | 2.98 × 104 | 1.17 × 104 | 1.9 × 104 |
| kd (s−1) | 1.33 × 10−2 | 7.37 × 10−3 | 8.71 × 10−3 | 6.08 × 10−3 | 7.14 × 10−3 | |
| KD (M) | 1.19 × 10−6 | 1.45 × 10−7 | 3.25 × 10−7 | 5.21 × 10−7 | 3.76 × 10−7 | |
| FGF4 | ka (M −1 s−1) | 1.14 × 106 | 9.38 × 105 | 1.32 × 106 | 1.05 × 106 | 1.21 × 105 |
| kd (s −1) | 3.02 × 10−2 | 5.12 × 10−3 | 7.33 × 10−3 | 3.03 × 10−2 | 2.62 × 10−2 | |
| KD (M) | 2.65 × 10−8 | 5.45 × 10−9 | 5.55 × 10−9 | 2.89 × 10−8 | 2.16 × 10−8 | |
| FGF5 | ka (M −1 s−1) | 1.10 × 104 | 1.68 × 105 | 9.22 × 104 | 2.36 × 104 | 1.86 × 105 |
| kd (s −1) | 5.76 × 10−3 | 1.39 × 10−2 | 2.18 × 10−2 | 4.67 × 10−3 | 4.64 × 10−2 | |
| KD (M) | 5.24 × 10−7 | 8.25 × 10−8 | 2.36 × 10−7 | 1.98 × 10−7 | 2.49 × 10−7 | |
| FGF6 | ka (M −1 s−1) | 1.05 × 106 | 3.81 × 105 | 1.14 × 106 | 9.03 × 105 | 8.95 × 105 |
| kd (s −1) | 3.84 × 10−2 | 2.34 × 10−3 | 1.39 × 10−2 | 5.10 × 10−2 | 7.23 × 10−2 | |
| KD (M) | 3.66 × 10−8 | 6.15 × 10−9 | 1.22 × 10−8 | 5.64 × 10−8 | 8.00 × 10−9 | |
| FGF8 | ka (M −1 s−1) | 7.17 × 104 | 8.79 × 104 | 3.26 × 105 | 3.80 × 104 | 3.72 × 104 |
| kd (s −1) | 5.93 × 10−3 | 4.61 × 10−3 | 2.58 × 10−3 | 6.40 × 10−3 | 7.05 × 10−3 | |
| KD (M) | 8.35 × 10−8 | 5.24 × 10−8 | 7.91 × 10−9 | 1.69 × 10−7 | 1.89 × 10−7 | |
| FGF9 | ka (M −1 s−1) | 8.00 × 104 | 6.04 × 105 | 5.78 × 105 | 9.31 × 104 | 1.02 × 105 |
| kd (s −1) | 9.94 × 10−2 | 4.85 × 10−2 | 5.90 × 10−2 | 1.16 × 10−1 | 8.71 × 10−2 | |
| KD (M) | 1.26 × 10−6 | 7.98 × 10−8 | 1.02 × 10−7 | 1.24 × 10−6 | 8.53 × 10−7 | |
| FGF10 | ka (M −1 s−1) | – | 3.97 × 104 | 5.86 × 104 | 2.14 × 104 | 1.99 × 104 |
| kd (s −1) | – | 5.71 × 10−2 | 5.51 × 10−2 | 6.05 × 10−2 | 5.85 × 10−2 | |
| KD (M) | NBd | 1.44 × 10−6 | 9.41 × 10−7 | 2.83 × 10−6 | 2.94 × 10−6 | |
| FGF16 | ka (M −1 s−1) | 7.98 × 103 | 2.43 × 105 | 2.67 × 105 | 2.13 × 104 | 8.37 × 103 |
| kd (s −1) | 1.54 × 10−2 | 1.30 × 10−2 | 1.48 × 10−2 | 1.32 × 10−2 | 1.24 × 10−2 | |
| KD (M) | 1.93 × 10−6 | 5.34 × 10−8 | 5.56 × 10−8 | 6.17 × 10−7 | 1.48 × 10−6 | |
| FGF17 | ka (M −1 s−1) | 7.72 × 104 | 1.09 × 105 | 7.34 × 105 | 3.33 × 104 | 4.39 × 104 |
| kd (s −1) | 4.90 × 10−2 | 2.57 × 10−2 | 1.00 × 10−1 | 5.19 × 10−3 | 2.52 × 10−2 | |
| KD (M) | 6.34 × 10−7 | 2.36 × 10−7 | 1.37 × 10−7 | 1.56 × 10−7 | 5.75 × 10−7 | |
| FGF18 | ka (M −1 s−1) | 1.17 × 105 | 1.22 × 105 | 3.44 × 105 | 2.32 × 105 | 2.14 × 104 |
| kd (s −1) | 5.81 × 10−2 | 2.14 × 10−2 | 4.95 × 10−2 | 4.93 × 10−2 | 1.64 × 10−2 | |
| KD (M) | 4.97 × 10−7 | 1.76 × 10−7 | 1.44 × 10−7 | 2.13 × 10−7 | 7.67 × 10−7 | |
| FGF19 | ka (M −1 s−1) | – | 5.32 × 104 | 9.78 × 104 | 3.61 × 104 | – |
| kd (s −1) | – | 5.49 × 10−3 | 5.23 × 10−2 | 9.22 × 10−3 | – | |
| KD (M) | NB | 1.03 × 10−7 | 5.35 × 10−7 | 2.55 × 10−7 | NB | |
| FGF20 | ka (M −1 s−1) | 9.09 × 103 | 4.76 × 104 | 7.77 × 104 | 1.35 × 104 | 1.06 × 104 |
| kd (s −1) | 1.25 × 10−2 | 7.62 × 10−3 | 1.47 × 10−2 | 4.54 × 10−3 | 1.05 × 10−2 | |
| KD (M) | 1.38 × 10−6 | 1.60 × 10−7 | 1.87 × 10−7 | 3.36 × 10−7 | 9.88 × 10−7 | |
| FGF23 | ka (M −1 s−1) | 1.30 × 104 | 4.98 × 104 | 5.23 × 104 | 2.74 × 104 | 7.46 × 103 |
| kd (s −1) | 4.39 × 10−3 | 3.97 × 10−3 | 5.32 × 10−3 | 3.11 × 10−3 | 4.26 × 10−3 | |
| KD (M) | 3.37 × 10−7 | 7.97 × 10−8 | 1.02 × 10−7 | 1.13 × 10−7 | 5.70 × 10−7 | |
Binding of wild-type and mutant FGFR2c constructs to FGF7 and FGF21 was not observed.
ka and kd were derived as described in Materials and Methods. χ2 was less than 10% of Rmax in all cases.
The apparent affinity, KD, is equal to kd/ka. Interactions where binding is enhanced (>1.5-fold relative to wild-type FGFR2c) are shown in bold, while interactions where binding is decreased (>1.5-fold relative to wild-type FGFR2c) are shown in italic.
NB, negligible binding.
Wild-type FGFR2c exhibits the highest affinity for FGF1, FGF2, FGF4, FGF6 and FGF8 (KD = 10–100 nM) (Table 1). Wild-type FGFR2c binds FGF5, FGF17, FGF18 and FGF23 with moderate affinity (KD = 300–700 nM) and binds FGF3, FGF9, FGF16 and FGF20 with weak affinity (KD = 1000–2000 nM) (Table 1). Injection of wild-type FGFR2c over sensor chips immobilized with FGF7, FGF10, FGF19 and FGF21 produces negligible RU responses (ΔRU < 13) over the background and is indicative of no binding (Table 1).
Both AS mutant FGFR2c display higher affinity, relative to wild-type FGFR2c, for nearly every FGF (Fig. 2A–H and Table 1). Interestingly, relative enhancements in affinity are similar within each FGF subfamily but differ between FGF subfamilies. Hence, it is likely that there are mechanistic similarities in the extent of gain-of-function contacts within each FGF subfamily, which vary between FGF subfamilies. The binding affinities of FGF9/16/20 subfamily members for S252W FGFR2c are most robustly enhanced (8.6–36-fold) (Fig. 2E). Increases in S252W FGFR2c affinity for other FGF subfamilies are also observed, including the FGF1/2 subfamily (4.7–7.2-fold) (Fig. 2A), the FGF3/5 subfamily (6.3–8.2-fold) (Fig. 2C), the FGF4/6 subfamily (4.9–6-fold) (Fig. 2B) and FGF23 (4.2-fold) (Fig. 2H). The enhancement in binding affinity of S252W FGFR2c for the FGF8/17/18 subfamily is less remarkable (1.6–2.8-fold) (Fig. 2F).
Figure 2.

Surface plasmon resonance analysis of wild-type and mutant FGFR2c-FGF interactions. Sensorgrams of representative analyte injections of wild-type, S252W, P253R, D321A and S252L/A315S FGFR2c binding to (A) FGF2 (at 12.5 nM), (B) FGF4 (at 6.25 nM), (C) FGF5 (at 200 nM), (D) FGF10 (at 400 nM), (E) FGF16 (at 200 nM), (F) FGF18 (at 400 nM), (G) FGF19 (at 400 nM), and (H) FGF23 (at 400 nM). Analyte injections are colored as follows: wild-type FGFR2c in black, S252W FGFR2c in blue, P253R FGFR2c in green, D321A FGFR2c in yellow and S252L/A315S in red. The biosensor chip response is indicated on the y-axis (ΔRU) as a function of time (x-axis) at 25°C. Kinetic data are summarized in Table 1.
Importantly, injection of S252W FGFR2c on to FGF10 sensor chips results in a significant increase in RU, compared to wild-type FGFR2c, as shown in Figure 2D. Kinetic analysis of the S252W FGFR2c-FGF10 interaction yields a KD value of 1440 nM. Hence, the S252W mutation violates FGFR2c ligand binding specificity and agrees with the findings of Yu et al. (21). The S252W mutation has an even more profound effect on FGFR2c binding to FGF19, and enables high affinity binding (KD = 103 nM) (Fig. 2G). In contrast to the findings of Yu et al. (21), we were unable to detect any significant effect of the S252W mutation on the FGFR2c-FGF7 interaction. Binding of FGF21 to S252W FGFR2c is not observed.
The binding affinities of P253R FGFR2c for FGF9/16/20 subfamily members (7.4–34-fold) (Fig. 2E and Table 1) were most robustly enhanced, followed by FGF8/17/18 subfamily members (3.5–10.6-fold) (Fig. 2F and Table 1). For the remaining ligands, the P253R mutation enhances ligand binding affinity to a lesser degree than the S252W mutation does (Table 1). Binding of P253R FGFR2c was increased for the FGF1/2 subfamily (2.3-fold) (Fig. 2A), the FGF3/5 subfamily (2.2–3.7-fold) (Fig. 2C), the FGF4/6 subfamily (3–4.8-fold) (Fig. 2B) and FGF23 (3.3-fold) (Fig. 2H) to similar extents. Importantly, like the S252W mutation, the P253R mutation also violates FGFR2c ligand binding specificity and enables FGFR2c to bind FGF10 (Fig. 2D) and FGF19 (Fig. 2G) with KD values of 941 nM and 535 nM, respectively. Again, binding of P253R FGFR2c to FGF7 and FGF21 is not observed. The relative enhancement in AS mutant FGFR2c binding affinity to FGF2 is consistent with the data of Anderson et al. (19). However, unlike Anderson et al. (19), we also measure enhanced binding of AS mutant FGFR2c to nearly every FGF.
The exceptional increase in S252W FGFR2c and P253R FGFR2c binding affinity for the FGF9/16/20 subfamily is reminiscent of increases in FGF9 binding to P252R FGFR1c and P250R FGFR3c, which result in type I PS and Muenke syndrome (MIM 602849), respectively (35). We have previously shown that the FGF9/16/20 subfamily, unlike other FGFs, is subject to autoinhibitory regulation via homodimerization (36). The dimer interface involves the N-terminus and β8–β9 turn region, and precludes receptor binding. Because these regions are also involved in additional ligand–receptor contacts with AS mutations, it is possible that AS mutations shift the balance between FGF9/16/20 homodimerization and FGFR2c-FGF9/16/20 binding to favor the latter event.
Effect of D321A and S252L/A315S mutations on FGFR2c ligand binding affinity and specificity
Next, we examined the effect of the D321A mutation and the S252L/A315S double mutation, which manifest only in the FGFR2c splice isoform, on FGFR2c ligand binding affinity and specificity. Importantly, the S252L/A315S double mutation causes syndactyly in the absence of craniosynostosis and therefore provides an attractive opportunity to understand the mechanisms by which craniofacial and limb pathology arise in patients with pathogenic FGFR2 mutations. The binding of D321A FGFR2c to all FGFs, with the exception of FGF22, was examined. Unlike the AS mutations, which ubiquitously increase FGFR2c binding to nearly all FGFs, the D321A mutation increases the binding affinity of FGFR2c to a subset of FGFs including FGF3 (2.3-fold), FGF5 (2.7-fold) (Fig. 2C), FGF16 through FGF18 (3.1-, 4-and 2.3-fold, respectively) (Fig. 2E and F), FGF20 (4.1-fold), and FGF23 (3-fold) (Fig. 2H and Table 1). Importantly, like the AS mutations, the D321A mutation also enables FGFR2c to bind FGF10 and FGF19 (Fig. 2D and G) with KD values of 2830 and 255 nM, respectively. The D321A mutation had no major impact on the binding affinities of FGFR2c for FGF1, FGF4 (Fig. 2B) and FGF9 (Table 1). Notably, an exceptional loss in FGF2 binding (60-fold) (Fig. 2A) and mild decreases in FGF6 (1.5-fold) and FGF8 (2-fold) binding are observed for D321A FGFR2c (Table 1). Interestingly, the effect of the D321A mutation varies even for members within the same FGF subfamily.
The S252L/A315S double mutation also has a variable effect on FGFR2c binding to FGFs (Fig. 2A–H and Table 1). Increased binding of S252L/A315S FGFR2c to only FGF3 (3.2-fold), FGF5 (2.1-fold) (Fig. 2C), and FGF6 (4.6-fold) is observed (Table 1). Similar to the AS mutations and the D321A PS mutation, the S252L/A315S double mutation also violates FGFR2c ligand binding specificity by enabling FGFR2c to bind to FGF10 (Fig. 2D) with a KD value of 2940 nM. However, unlike these mutations, the S252L/A315S double mutation did not enable FGFR2c to bind FGF19 (Fig. 2G and Table 1). The S252L/A315S double mutation does not have a major impact on the binding of FGF1, FGF4 (Fig. 2B), FGF9, FGF16 (Fig. 2E), FGF17 and FGF20 to FGFR2c (Table 1). Notably, diminished binding of S252L/A315S FGFR2c to FGF2 (3.2-fold) (Fig. 2A), FGF8 (2.3-fold), FGF18 (1.5-fold) (Fig. 2F) and FGF23 (1.7-fold) (Fig. 2H) is observed (Table 1). As with the AS mutations, neither the D321A mutation nor the S252L/A315S double mutation permits FGFR2c to bind FGF7 and FGF21.
Effect of AS mutations on FGFR2b ligand binding affinity and specificity
Previous ligand binding analyses of wild-type FGFR2b have been reported and employed a variety of techniques (32,37–43). To provide a coherent framework to characterize the effect of AS mutations on FGFR2b, wild-type FGFR2b was examined for binding to FGF1 through FGF10. This set of ligands contains multiple specific and non-specific ligands for FGFR2b and therefore is sufficient to assess the effect of AS mutations on both FGFR2b ligand binding affinity and specificity. The data are summarized in Table 2 and represent the first binding analysis of AS mutant FGFR2b.
Table 2.
Summary of wild-type and mutant FGFR2b binding data
| FGF | WT | S252W | P253R | |
|---|---|---|---|---|
| FGF1 | ka (M −1 s−1)a | 3.31 × 105 | 1.53 × 105 | 3.16 × 105 |
| kd (s −1)a | 5.34 × 10−2 | 6.67 × 10−3 | 3.03 × 10−2 | |
| KD (M) b | 1.61 × 10−7 | 4.37 × 10−8 | 9.59 × 10−8 | |
| FGF2 | ka (M −1 s−1) | – | 1.97 × 105 | 8.20 × 105 |
| kd (s −1) | – | 8.56 × 10−2 | 3.44 × 10−1 | |
| KD (M) | NBc | 4.34 × 10−7 | 4.19 × 10−7 | |
| FGF3 | ka (M −1 s−1) | 1.32 × 105 | 8.32 × 104 | 4.14 × 104 |
| kd (s −1) | 4.77 × 10−2 | 1.52 × 10−2 | 1.26 × 10−2 | |
| KD (M) | 3.62 × 10−7 | 1.83 × 10−7 | 3.04 × 10−7 | |
| FGF4 | ka (M −1 s−1) | 4.79 × 104 | 1.05 × 105 | 1.18 × 105 |
| kd (s −1) | 2.56 × 10−2 | 2.65 × 10−2 | 3.38 × 10−2 | |
| KD (M) | 5.34 × 10−7 | 2.52 × 10−7 | 2.86 × 10−7 | |
| FGF5 | ka (M −1 s−1) | – | – | – |
| kd (s −1) | – | – | – | |
| KD (M) | NB | NB | NB | |
| FGF6 | ka (M −1 s−1) | 1.96 × 105 | 3.58 × 105 | 1.57 × 105 |
| kd (s −1) | 1.30 × 10−1 | 5.30 × 10−2 | 3.44 × 10−2 | |
| KD (M) | 6.64 × 10−7 | 1.48 × 10−7 | 2.19 × 10−7 | |
| FGF7 | ka (M −1 s−1) | – | – | – |
| kd (s −1) | – | – | – | |
| KD (M) | 1.04 × 10−5 | 1.19 × 10−6 | 2.13 × 10−6 | |
| FGF8 | ka (M −1 s−1) | – | 4.30 × 103 | 4.52 × 103 |
| kd (s −1) | – | 5.12 × 10−3 | 7.44 × 10−3 | |
| KD (M) | NB | 1.28 × 10−6 | 1.65 × 10−6 | |
| FGF9 | ka (M −1 s−1) | – | 6.00 × 104 | 5.93 × 104 |
| kd (s −1) | – | 4.57 × 10−2 | 4.73 × 10−2 | |
| KD (M) | NB | 7.62 × 10−7 | 7.98 × 10−7 | |
| FGF10 | ka (M −1 s−1) | 1.27 × 105 | 2.88 × 104 | 5.63 × 104 |
| kd (s −1) | 7.88 × 10−2 | 8.42 × 10−3 | 2.24 × 10−2 | |
| KD (M) | 6.22 × 10−7 | 2.92 × 10−7 | 3.97 × 10−7 | |
ka and kd were derived as described in Materials and Methods. χ2 was less than 10% of Rmax in all cases.
The apparent affinity, KD, is equal to kd/ka. Interactions where binding is enhanced (>1.5-fold relative to wild-type FGFR2b) are shown in bold.
NB, negligible binding.
Wild-type FGFR2b exhibits the highest affinity for FGF1 (KD = 161 nM), followed in decreasing order by FGF3 (KD = 363 nM), FGF4 (KD = 534 nM), FGF10 (KD = 622 nM), FGF6 (KD = 664 nM) and FGF7 (KD = 10.4 μM) (Table 2). The sensorgrams for wild-type FGFR2b interactions with FGF2, FGF5, FGF8 and FGF9 show negligible RU responses (ΔRU < 13) over the background and are indicative of no binding (Table 2).
As in the case of FGFR2c, SPR analysis demonstrates that both AS mutations increase the binding affinity of FGFR2b for FGFs (Fig. 3C–F and Table 2), and the effect is consistently greater for the S252W mutation. Large increases in the binding affinities of S252W FGFR2b for FGF1 (3.7-fold) (Fig. 3D), FGF6 (4.5-fold) and FGF7 (8.7-fold) are observed. Increases in binding affinity of S252W FGFR2b for FGF3 (1.9-fold), FGF4 (2.1-fold) and FGF10 (2.1-fold) (Fig. 3C) are modest. In the case of P253R FGFR2b, a large increase in binding affinity is only observed for FGF7 (4.9-fold) and increases in binding affinity for FGF1 (1.7-fold) (Fig. 3D), FGF3 (1.19-fold), FGF4 (1.9-fold), FGF6 (3-fold) and FGF10 (1.6-fold) (Fig. 3C) are more modest. Consistent with the data of Yu et al. (21), the S252W mutation also violates FGFR2b ligand binding specificity by enabling binding to FGF2 (Fig. 3E and Table 2) and FGF9 (Table 2). Binding of S252W FGFR2b to FGF8 (Fig. 3F and Table 2) was also enabled and the P253R mutation also violates FGFR2b ligand binding specificity and enables binding to FGF2, FGF8 and FGF9 (Fig. 3E and F and Table 2). Hence, the SPR data show that AS mutations enhance and violate FGFR2b ligand binding affinity and specificity, respectively.
Figure 3.

Structural and biophysical analysis of wild-type and AS mutant FGFR2b-FGF interactions. (A) Gain-of-function contact in the S252W FGFR2b-FGF10 complex. D2 and D3 of FGFR2b are shown in green and cyan, respectively. The alternatively spliced region of D3 is colored purple. The D2–D3 linker is colored gray. FGF10 is shown in orange. (Right) View of whole structure in the exact orientation as the detailed view is shown, with the region of interest boxed. (B) Gain-of-function hydrogen bonds in the P253R FGFR2b-FGF1 complex. Coloring is as in (A). Dotted lines represent hydrogen bonds and the hydrogen-bonding distances are indicated. (Right) View of whole structure in the exact orientation as the detailed view is shown, with the region of interest boxed. (C, D, E and F) Sensorgrams of representative analyte injections of wild-type and AS mutant FGFR2b binding to (C) FGF10 (at 100 nM), (D) FGF1 (at 400 nM), (E) FGF2 (at 400 nM), and (F) FGF8 (at 800 nM). Analyte injections are colored as follows: wild-type FGFR2b in black, S252W FGFR2b in blue, and P253R FGFR2b in green. The biosensor chip response is indicated on the y-axis (ΔRU) as a function of time (x-axis) at 25°C. Kinetic data are summarized in Table 2.
Crystallographic analysis of AS mutant FGFR2b
To ascertain if AS mutations enhance FGFR2b binding to FGFs through additional receptor-ligand contacts, we crystallized S252W FGFR2b-FGF10 and P253R FGFR2b-FGF1 complexes. Data collection and refinement statistics are given in Table 3. As we previously reported for AS mutant FGFR2c-FGF2 structures (22), these mutations do not affect the D2–D3 linker region conformation and the overall structures of the S252W FGFR2b-FGF10 and P253R FGFR2b-FGF1 complexes are identical to the overall structures of respective wild-type FGFR2b-FGF complexes. Both S252W FGFR2b and P253R FGFR2b make additional contacts with FGF ligand. These gain-of-function interactions, albeit less extensive, are reminiscent of the additional receptor–ligand interactions observed in AS mutant FGFR2c-FGF2 and PS mutant FGFR1c-FGF2 crystal structures (22,35).
Table 3.
Summary of crystallographic analysis
| Data collection statistics | S252W FGFR2b-FGF10 | P253R FGFR2b-FGF1 |
|---|---|---|
| Resolution, Å | 30.0–2.8 | 30.0–2.1 |
| Reflections (total/unique) | 157104/16101 | 330055/27015 |
| Completeness, % | 99.5 (100)b | 99.5 (98.6)b |
| Rsym a | 5.3 (41.6)b | 5.4 (10.3)b |
| Signal (〈I/σI〉) | 35.0 | 40.5 |
| Refinement statisticsc | ||
| Resolution, Å | 25.0–2.8 | 25.0–2.1 |
| Reflections | 15301 | 26221 |
| Rcryst/Rfree d | 25.8/29.2 | 25.2/28.2 |
| Root-mean-square deviations | ||
| Bonds, Å | 0.008 | 0.008 |
| Angles, ° | 1.4 | 1.5 |
| B factorse | 1.33 | 0.2 |
Rsym = 100 X ΣhklΣI|II(hkl) − 〈I(hkl)〉| ΣhklΣIII(hkl).
Value in parentheses is for the highest resolution shell: S252W FGFR2b-FGF10 (2.9–2.8 Å), P252R FGFR2b-FGF1 (2.18–2.1 Å).
Atomic model: S252W FGFR2b-FGF10: 2678 protein atoms, one PEG-400 molecule and 2 sulfate ions; P253R FGFR2b-FGF1: 2683 protein atoms and 2 sulfate ions.
Rcryst/Rfree = 100 X hkl Fo(hkl)| |Fc(hkl)/hkl |Fo(hkl)|, where Fo (>0) and Fc are the observed and calculated structure factors, respectively. 10% of the reflections were used for calculation of Rfree.
For bonded protein atoms.
In the S252W FGFR2b-FGF10 structure, Trp252 of FGFR2b interacts hydrophobically with Leu73 in the αN helix of FGF10 (Fig. 3A). Unlike the S252W FGFR2c-FGF2 structure (22), where the gain-of-function interactions result in the additional ordering of the N-terminus of FGF2 (at the expense of entropy loss), the αN helix already interacts with the receptor in the wild type FGFR2b-FGF10 structure (44). The closest approach between Leu73 of FGF10 and Trp252 of FGFR2b is 4.22 Å and may explain why, despite the lack of entropic cost, the increase in affinity of S252W FGFR2b for FGF10 is less than expected. As in the S252W FGFR2c-FGF2 structure, Trp252 of FGFR2b also inserts into a shallow hydrophobic pocket on the surface of D3 between Ile257 and Tyr281 and probably contributes to the general enhancement in binding affinity.
In the P253R FGFR2b-FGF1 structure, the guanidinium group of Arg253 in FGFR2b makes one hydrogen bond with the backbone carbonyl oxygen of Glu90 in the β8-β9 turn of FGF1 (Fig. 3B) and accounts for the 1.7-fold increase in binding affinity. The number of additional hydrogen bond correlates with the relative increase in binding affinity as the P253R FGFR2c-FGF2 structure (22) shows three additional hydrogen bonds and a 2.3-fold increase in binding affinity. Taken together, the structural data demonstrate that the AS mutations introduce gain-of-function contacts for FGFR2b as well. However, these interactions are less extensive than in the case of FGFR2c and result in smaller enhancements in affinity.
DISCUSSION
The importance of FGF signaling in human skeletal development is highlighted by the numerous mutations in FGFR1-3 that result in craniosynostosis (1–3) and chondrodysplasia syndromes (45). Elucidation of the molecular basis for AS and other ligand-dependent craniosynostosis syndromes is an important avenue to assess the physiological and pathological roles of various FGFs in craniofacial and limb development. In this study, we examined the effect of AS mutations, the D321A PS mutation and the S252L/A315S double mutation on FGFR2 ligand binding affinity and specificity using SPR. These data show that each of the pathogenic FGFR2 mutations elicit distinct changes in FGFR2 ligand binding affinity and specificity, which correlate to the craniofacial and limb phenotypes observed in patients harboring these mutations.
Widespread enhancement of FGF binding correlates with craniosynostosis
It is presently unclear which FGF ligands mediate cranial suture closure. RT–PCR analysis suggests a complex picture in the calvaria as all FGFs, except for FGF3, FGF4, FGF5, FGF6 and FGF8, are expressed in mouse cranial sutures (46). Our data show that both AS mutations increase FGFR2c binding affinity to all FGFs, with the exception of FGF7 and FGF21. The S252W mutation, which is associated with more severe craniofacial pathology, results in greater enhancement in FGFR2c binding to most FGF ligands, relative to the P253R mutation. The PS D321A mutation also enhances FGFR2c binding affinity to FGF3, FGF5, FGF10, FGF16, FGF17, FGF18, FGF19, FGF20 and FGF23. Based on these data, we suggest that craniosynostosis in AS and D321A PS patients arises from the ability of these mutations to enhance FGFR2c signaling by several FGFs expressed in the cranial suture. A similar model in which elevated mutant FGFR2c signaling by FGF2 results in craniosynostosis was previously proposed (19,23). However, the ability of the D321A mutation to result in craniosynostosis without enhancing FGFR2c-FGF2 binding (Table 1), suggests that other FGFs, besides FGF2, contribute to craniosynostosis. The ability of the AS mutations and the D321A PS mutation to cause widespread enhancement of FGF binding will lead to a global elevation of mutant FGFR2c signaling that is parallel to the ligand-independent increase in FGFR signaling causing CS and nearly all cases of PS.
Based on this model, the lack of craniosynostosis in S252L/ A315S patients is due to the inability of the S252L/A315S double mutation to result in widespread enhancement of FGF binding to FGFR2c. Of the FGFs expressed in the cranial suture, only notable enhancement of S252L/A315S FGFR2c binding to FGF10 is observed. The involvement of several FGFs in cranial suture fusion also accounts for why gain-of-function mutations in FGF ligands have not been detected in craniosynostosis patients, as well as why most FGF knockout mice do not display delayed suture closure. FGF18 null mice embryos do have wider cranial sutures, relative to wild-type mice embryos, at 16.5 and 18.5 days postcoitum (47,48). However, this phenotype is suggested to be secondary to a generalized decrease in calvarial bone size, rather than a disruption in cranial suture maturation (47).
Structural basis for the generalized increase in FGF binding affinity by AS mutations
Notably, the general increase in P253R FGFR2c ligand binding affinity is entirely consistent with the structural mode of gain-of-function observed in the P253R FGFR2c-FGF2 structure (22). We have previously shown that the P253R mutation results in three additional hydrogen bonds between receptor and the β8–β9 turn in the core homology region of FGF2 (22). Two of these hydrogen bonds involve backbone carbonyl atoms of FGF2, and therefore should occur in every P253R FGFR2c-FGF interaction. The third hydrogen bond is mediated by the side chain of Asn111 in FGF2. Interestingly, this Asn is conserved in the FGF8/17/ 18 subfamily and may account for the larger increase in affinity of the mutant receptor for this subfamily.
The ability of the S252W mutation to ubiquitously increase FGFR2c ligand binding affinity is not completely predicted by the S252W FGFR2c-FGF2 structure (22). The S252W mutation was shown to result in an additional hydrogen bond and hydrophobic contact between FGFR2c and Phe21 in the N-terminus of FGF2. These interactions cause the ordering of additional residues in the otherwise flexible N-terminus of FGF2. Crystal structures and solution structures of other FGF ligands also demonstrate that the N-termini of FGFs are generally disordered (49–51), but become ordered in a receptor specific manner upon FGFR binding (25,44,52–54). Thus, the adaptable nature and poor sequence homology of FGF N-termini makes structure-based predictions on how the S252W mutation affects the interaction of receptor with the N-termini of other FGFs unreliable. Despite this caveat, we predicted that only a subset of FGFs, which possess a hydrophobic residue at a location corresponding to Phe21 of FGF2, would exhibit higher affinity for S252W FGFR2c (22). The enhancement of S252W FGFR2c binding to nearly every FGF highlights that the flexibility of FGF N-termini can allow nearby hydrophobic residues to substitute for Phe21 in FGF2 and interact with S252W FGFR2c. Furthermore, in the S252W FGFR2c-FGF2 structure, Trp252 inserts into a hydrophobic pocket on D3 between Ile257 and Tyr281. This interaction likely keeps the receptor in a conformation that is more favorable for ligand binding and may also contribute to the generalized increase in FGF binding for S252W FGFR2c.
Binding of FGFR2c to FGF10 correlates with syndactyly
All four pathogenic mutations in this study violate FGFR2c ligand binding specificity by enabling FGFR2c to bind FGF10, a ligand that normally does not bind to wild-type FGFR2c. The KD values for mutant FGFR2c-FGF10 interactions are still 1.5–4.9-fold weaker than the KD value of the wild-type FGFR2b-FGF10 interaction (Tables 1 and 2). Nevertheless, these gain of interactions are probably pathological because FGF10 and FGFR2c are both mesenchymally expressed, and the high local concentrations of FGF10 would allow pathological autocrine signaling to take place. Therefore, we suggest that the illegitimate binding and signaling of FGFR2c by FGF10 is responsible for syndactyly. Indeed, FGF10 has been shown to stimulate BaF3 cells expressing S252W FGFR2c, whereas it has no activity on BaF3 cells expressing wild-type FGFR2c (21). The greater binding of FGF10 to P253R FGFR2c, relative to S252W FGFR2c, explains the more severe syndactyly in P253R AS patients. The inability of analogous Pro → Arg mutations in FGFR1c and FGFR3c to confer FGF10 binding is also harmonious with the lack of syndactyly in type I PS and Muenke syndrome patients (35). Additionally, both S252L and A315S mutations have been singly detected in normal individuals (55,56), and suggests that the S252L/A315S double mutation results in syndactyly by enhancing FGFR2c binding to FGF10 in a synergistic fashion.
These findings bear similarity to the findings of Yu et al. (21), who were the first to show that AS mutations violate FGFR2 specificity. However, we did not observe binding of FGF7 to either AS mutant FGFR2c, although this is not surprising given the fact that FGF7 binds to FGFR2b nearly 20 times weaker than FGF10 (Table 2). Based on our SPR data, we believe that the majority of limb pathology is due to autocrine signaling of mutant FGFR2c through FGF10. This is consistent with genetic studies showing that FGF10 is critical for limb development and outgrowth (57,58), whereas FGF7 is dispensible for proper limb development (59,60).
Molecular mechanisms by which pathogenic FGFR2c mutations affect ligand binding specificity
The variable effect of the D321A mutation and the S252L/ A315S double mutation on ligand binding is consistent with the crystal structures of highly specific FGFR2c-FGF2 and FGFR2b-FGF10 complexes, which reveal that interactions between FGF ligand and the alternatively spliced βC′-βE loop are major determinants of binding specificity (25,44). These structures also provide convincing explanations by which pathogenic mutations in the βC′-βE loop reduce FGFR2c binding to FGF2 and concomitantly enhance FGFR2c binding to FGF10 (Fig. 2A and D). The dramatic loss of FGF2 binding to D321A FGFR2c can readily be explained by the crystal structure of the wild-type FGFR2c-FGF2 complex. In this structure, Asp321 makes three hydrogen bonds with FGF2 (25) and the huge loss in FGF2 binding by D321A FGFR2c reflects the elimination of these interactions. The loss of FGF2 binding to S252L/A315S FGFR2c can also be accounted for by the crystal structure of the wild-type FGFR2c-FGF2 complex. In this structure, Ala315 makes intramolecular interactions that facilitate a βC′-βE loop conformation optimal for FGF2 binding, thus indirectly contributing to ligand binding affinity. It is noteworthy that the A315S mutation causes a greater loss in FGF2 binding, compared to the S252L/A315S double mutation, and suggests that the S252L mutation partially rescues the loss of FGF2 binding (data not shown).
The available structural data also provide molecular explanations for the ability of the D321A mutation and the S252L/ A315S double mutation to confer FGF10 binding. In a hypothetical FGFR2c-FGF10 model, the side chain of Asp321 is in major steric clash as well as in charge repulsion with Asp78 in the N-terminus of FGF10, and explains why wild-type FGFR2c rejects FGF10. Removal of both the steric and electrostatic conflicts by the D321A mutation likely accounts for the large gain in FGF10 binding by D321A FGFR2c. The molecular basis for the ability of the S252L/A315S double mutation to confer FGF10 binding upon FGFR2c can be inferred from available structural data. In the wild-type FGFR2b-FGF10 structure (44), Ser315, a residue that is highly conserved in FGFR2b, makes two hydrogen bonds with Asp78, a residue unique to the FGF7/10/22 subfamily. Hence, the A315S substitution should enable FGFR2c to make hydrogen bonds with Asp78 of FGF10 and contribute to a gain in FGF10 binding. The S252L mutation likely enhances the interaction of FGFR2c with FGF10 by introducing a similar, but weaker, hydrophobic contact as described for the S252W mutation (see below). It is likely that these two mutations synergize to enable FGFR2c to bind FGF10.
The mechanism by which the S252W mutation enables FGFR2c to bind FGF10 can be gleaned from the crystal structure of the S252W FGFR2b-FGF10 complex reported in this article. Based on this structure, we predict that Leu73 in αN of FGF10 will engage in analogous hydrophobic contact with S252W FGFR2c. Finally, the gain in binding of P253R FGFR2c to FGF10 is fully expected based on the highly conserved nature of the gain-of-function hydrogen bonds mediated by the P253R mutation.
A potential role for FGFR2b in AS
The SPR data show that AS mutations enhance and violate FGFR2b ligand binding affinity and specificity, respectively, although the relative increases in FGF binding are generally less remarkable than for FGFR2c. Anderson et al. (19) suggest that syndactyly in AS is mediated by elevated FGFR2b signaling by FGF7 or FGF10. However, the greater enhancement of S252W FGFR2b binding to FGF7 and FGF10, relative to P253R FGFR2b, is not consistent with the relative severity of syndactyly between the two AS mutations (13–15). Additionally, missense mutations that selectively manifest in FGFR2c can cause syndactyly, and therefore, it is unclear if elevated FGFR2b signaling plays a major role in mediating syndactyly. FGFR2b is expressed on epithelial cells of ectodermal and endodermal origin and is critical for skin development (61–63). It is possible that enhanced signaling by AS mutant FGFR2b accounts for unique dermatological phenotypes in AS, such as hyperhidrosis, hypopigmentation and severe widespread acne (5). Indeed, an otherwise normal patient who presented for severe localized acne was found to have the AS Ser252Trp mutation in FGFR2b from lesional epidermal cells, whereas unaffected areas were found to contain wild-type FGFR2b (64).
CONCLUSIONS
In summary, analyses of pathogenic ligand-dependent FGFR2 mutations by several laboratories have established that these mutations violate the rules governing FGFR ligand binding affinity and specificity. Based on these data, we suggest that craniosynostosis is mediated by the enhancement of FGFR2c signaling by a number of FGFs, whereas syndactyly is caused by illegitimate signaling of mutant FGFR2c through FGF10. Genetic studies are needed to substantiate these binding data and will likely require the development of more advanced genetic tools. Additional biochemical and crystallographic characterization of these mutant receptors should provide further insight into the mechanism by which ligand binding specificity is bypassed and will facilitate the design of antagonists capable of alleviating the deleterious effect of these mutations.
MATERIALS AND METHODS
Protein expression and purification
Recombinant full length human FGFs, FGF1 through FGF10 and FGF16 through FGF23, with the exception of FGF22, were expressed in Escherichia coli. FGF homologous factor 1b (FHF1b) (formally known as FGF12b) was also produced in E. coli. Soluble FGFs (FGF1, FGF2, and FGF10) and FHF1b were purified by heparin affinity, ion exchange and size exclusion chromatography as previously described (35). FGF7 was generously provided by Amgen (Amgen Inc.). Insoluble FGFs (FGF3, FGF5, FGF6, FGF8, FGF16 through FGF21) were refolded in vitro and subsequently purified in a similar manner to soluble FGFs. FGF4 and FGF9 were purified through salt extraction and ammonium sulfate precipitation, respectively, and then purified as soluble FGFs. The expression and purification of FGF23 is described elsewhere. The ‘b’ isoforms of FGF8 and FGF17 were used in this study.
S252W, P253R, D321A and S252L/A315S point mutations were introduced into the FGF-binding portion of human FGFR2c (residues 147–366) and S252W and P253R point mutations were introduced into the FGF-binding portion of human FGFR2b (residues 140–369) by using the Quik Change site-directed mutagenesis kit (Stratagene, La Jolla, CA). Wild-type and mutant FGFRs were expressed in E. coli and refolded in vitro using a previously described protocol (25). Briefly, cells were induced with IPTG for 5 h, centrifuged, and the bacterial pellet was lysed in 25 mM K-Na phosphate buffer (pH 7.5) containing 150 mM NaCl, 2 mM EDTA and 10% glycerol using a French press. Following centrifugation, the pellets were dissolved in 6 M guanidinium hydrochloride and 10 mM DTT in 100 mM Tris-HCl buffer (pH 8.0). The solubilized wild-type and mutant FGFRs were refolded by dialysis against 25 mM HEPES buffer (pH 7.5) containing 150 mM NaCl, 10% glycerol and 1 mM L-cysteine. Wild-type and mutant FGFRs were subsequently purified using heparin affinity and size exclusion chromatography.
Surface plasmon resonance analysis of FGFR-FGF interactions
Kinetic data for wild-type and mutant FGFR2-FGF interactions were characterized using a BIAcore 3000 instrument (Biacore AB, Uppsala, Sweden) as previously described (35). Briefly, FGF ligands were immobilized on research grade CM5 chips and FHF1b, a protein that is structurally homologous to FGFs but incapable of binding FGFR, was immobilized on reference flow cells as a control. Ten different concentrations of analyte (wild-type or mutant FGFR2), prepared through serial dilutions ranging from 1.5625 nM to 800 nM, in HBS-EP buffer [0.01 M HEPES, 0.15 M NaCl, 3 mM EDTA, 0.005% polysorbate 20 (v/v), pH = 7.4] were injected over the FGF sensor chips at a flow rate of 50 μl/min. At the end of each sample injection (180 s), HBS-EP buffer was passed over the sensor surface to monitor the dissociation phase. Following 180 s of dissociation, the sensor surface was fully regenerated by injection of 50 μl of 2 M NaCl in 100 mM sodium acetate buffer (pH = 4.5).
SPR data analysis
Reference responses from the control flow cell, containing FHF1b, were subtracted from FGF flow cells for each analyte injection using BiaEvaluation software (Biacore AB). The resulting sensorgrams, with the exception of wild-type and mutant FGFR2b interactions with FGF7, were used for kinetic parameter determination by globally fitting the entire association and dissociation phases to a 1:1 Languimir binding model using BiaEvaluation software (Biacore AB). Dissociation constants (KD) for wild-type and mutant FGFR2b interactions with FGF7 were obtained by globally fitting the data to a steady state binding model, although association and dissociation constants (ka and kd, respectively) cannot be obtained using this binding model. A minimum of four different analyte concentrations was used in the fitting of each FGFR–FGF interaction. Following curve fitting, each sensorgram was manually examined for data quality and closeness of fit. Chi2 was less than 10% for each fit. The standard error (SE) values for the association and dissociation rates were 1–2 orders of magnitude smaller than the respective association and dissociation constant values.
Crystallization and data collection
The S252W FGFR2b-FGF10 complex was crystallized under similar conditions as described for the wild-type FGFR2b-FGF10 complex (44). S252W FGFR2b-FGF10 crystals are isomorphous to wild-type FGFR2b-FGF10 crystals and contain a single copy of the S252W FGFR2b-FGF10 complex in the asymmetric unit. These crystals belong to the hexagonal space group P6422 with unit cell dimension as follows: a = b = 114.35 Å and c = 163.43 Å. A 2.8 Å data set for the S252W FGFR2b-FGF10 complex was collected on a CCD detector at beamline X4A at the National Synchrotron Light Source, Broo-khaven National Laboratory using a flash frozen crystal in a dry nitrogen stream. Data were processed using DENZO and SCA-LEPACK (65).
Crystals of P253R FGFR2b in complex with FGF1 were grown by mixing 2 μl of protein solution [14 mg/ml in 25 mM HEPES-NaOH (pH 7.5) and 150 mM NaCl] mixed with 2 μl of the crystallization buffer consisting of 22% mPEG-5000, 0.2 M ammonium sulfate in 0.1 M HEPES-NaOH (pH 7.5) at 20°C. These crystals belong to the orthorombic space group P212121 with unit cell dimensions a = 66.673 Å, b = 72.403 Å and c = 91.144 Å, α = β = γ = 90°. P253R FGFR2b-FGF1 crystals were flash-frozen in dry nitrogen stream using mother liquor containing 25% glycerol as cryoprotectant. A 2.1 Å data set for the P253R FGFR2b-FGF1 complex was collected on a CCD detector at beamline X4A at the National Synchrotron Light Source, Brookhaven National Laboratory. Data were processed using DENZO and SCALEPACK (65).
Structure determination and refinement
Rigid body refinement was used to place one copy of the wild-type FGFR2b-FGF10 complex (PDB ID: 1NUN) (44) into the unit cell of S252W FGFR2b-FGF10 crystal. The crystal structure of the P253R FGFR2b-FGF1 complex was solved using molecular replacement (66) with the wild-type FGFR2c-FGF1 complex as the search model (PDB ID: 1DJS) (53). Rigid body, positional and B factor refinements and simulated annealing were preformed with CNS (67). Model building into 2Fo−Fc and Fo−Fc electron density maps was performed with program O (68). The refined S252W FGFR2b-FGF10 model is composed of FGF10 residues 69–207, FGFR2b residues 151–359, one PEG-400 molecule and two sulfate ions. The refined P253R FGFR2b-FGF1 model is composed of FGF1 residues 18–154, FGFR2b residues 153–359 and two sulfate ions.
Acknowledgments
We thank X. Yang and R. Abramowitz for synchrotron beam-line assistance and A. Gurney for FGF19 cDNA. The authors acknowledge A. Joyner, C. Loomis, A. Mansukhani, D. Moscatelli and S. Orlow for comments and helpful discussions. Beamline X4A at the National Synchrotron Light Source, a Department of Energy facility, is supported by the New York Structural Biology Center. This work is supported by National Institute of Health grants DE13686 (M.M.) and HL52622 (R.J.L.).
References
- 1.Cohen MM., Jr . Craniosynostosis, Diagnosis, Evaluation, and Management. Oxford Univeristy Press; New York: 2000. [Google Scholar]
- 2.Muenke M, Wilkie AO. Craniosynostosis syndromes. In: Scriver CR, Beaudet AL, Valle D, Sly WS, Childs Kinzler K, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. IV. McGraw-Hill; New York, NY: 2001. pp. 6117–6146. [Google Scholar]
- 3.Jabs EW. Genetic etiologies of craniosynostosis. In: Mooney MK, Siegel MI, editors. Understanding Craniofacial Anomalies: The Etiopathogenesis of Craniosynostoses and Facial Clefting. Wiley-Liss; New York: 2002. pp. 125–146. [Google Scholar]
- 4.Cohen MM, Jr, Kreiborg S. The central nervous system in the Apert syndrome. Am J Med Genet. 1990;35:36–45. doi: 10.1002/ajmg.1320350108. [DOI] [PubMed] [Google Scholar]
- 5.Cohen MM, Jr, Kreiborg S. Cutaneous manifestations of Apert syndrome. Am J Med Genet. 1995;58:94–96. doi: 10.1002/ajmg.1320580119. [DOI] [PubMed] [Google Scholar]
- 6.Kaloust S, Ishii K, Vargervik K. Dental development in Apert syndrome. Cleft Palate Craniofac J. 1997;34:117–121. doi: 10.1597/1545-1569_1997_034_0117_ddias_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 7.Thompson DN, Slaney SF, Hall CM, Shaw D, Jones BM, Hayward RD. Congenital cervical spinal fusion: a study in Apert syndrome. Pediatr Neurosurg. 1996;25:20–27. doi: 10.1159/000121091. [DOI] [PubMed] [Google Scholar]
- 8.Jabs EW, Li X, Scott AF, Meyers G, Chen W, Eccles M, Mao JI, Charnas LR, Jackson CE, Jaye M. Jackson-Weiss and Crouzon syndromes are allelic with mutations in fibroblast growth factor receptor 2. Nat Genet. 1994;8:275–279. doi: 10.1038/ng1194-275. [DOI] [PubMed] [Google Scholar]
- 9.Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- 10.Schell U, Hehr A, Feldman GJ, Robin NH, Zackai EH, de Die-Smulders C, Viskochil DH, Stewart JM, Wolff G, Ohashi H, et al. Mutations in FGFR1 and FGFR2 cause familial and sporadic Pfeiffer syndrome. Hum Mol Genet. 1995;4:323–328. doi: 10.1093/hmg/4.3.323. [DOI] [PubMed] [Google Scholar]
- 11.Passos-Bueno MR, Wilcox WR, Jabs EW, Sertie AL, Alonso LG, Kitoh H. Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat. 1999;14:115–125. doi: 10.1002/(SICI)1098-1004(1999)14:2<115::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 12.Kan SH, Elanko N, Johnson D, Cornejo-Roldan L, Cook J, Reich EW, Tomkins S, Verloes A, Twigg SR, Rannan-Eliya S, et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet. 2002;70:472–486. doi: 10.1086/338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slaney SF, Oldridge M, Hurst JA, Moriss-Kay GM, Hall CM, Poole MD, Wilkie AO. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet. 1996;58:923–932. [PMC free article] [PubMed] [Google Scholar]
- 14.Lajeunie E, Cameron R, El Ghouzzi V, de Parseval N, Journeau P, Gonzales M, Delezoide AL, Bonaventure J, Le Merrer M, Renier D. Clinical variability in patients with Apert’s syndrome. J Neurosurg. 1999;90:443–447. doi: 10.3171/jns.1999.90.3.0443. [DOI] [PubMed] [Google Scholar]
- 15.von Gernet S, Golla A, Ehrenfels Y, Schuffenhauer S, Fairley JD. Genotype-phenotype analysis in Apert syndrome suggests opposite effects of the two recurrent mutations on syndactyly and outcome of craniofacial surgery. Clin Genet. 2000;57:137–139. doi: 10.1034/j.1399-0004.2000.570208.x. [DOI] [PubMed] [Google Scholar]
- 16.Park WJ, Theda C, Maestri NE, Meyers GA, Fryburg JS, Dufresne C, Cohen MM, Jr, Jabs EW. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet. 1995;57:321–328. [PMC free article] [PubMed] [Google Scholar]
- 17.Goriely A, McVean GAT, Rojmyr M, Ingemarsson B, Wilkie AOM. Evidence for Selective Advantage of Pathogenic FGFR2 Mutations in the Male Germ Line. Science. 2003;301:643–646. doi: 10.1126/science.1085710. [DOI] [PubMed] [Google Scholar]
- 18.Glaser RL, Broman KW, Schulman RL, Eskenazi B, Wyrobek AJ, Jabs EW. The paternal-age effect in Apert syndrome is due, in part, to the increased frequency of mutations in sperm. Am J Hum Genet. 2003;73:939–947. doi: 10.1086/378419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson J, Burns HD, Enriquez-Harris P, Wilkie AO, Heath JK. Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. Hum Mol Genet. 1998;7:1475–1483. doi: 10.1093/hmg/7.9.1475. [DOI] [PubMed] [Google Scholar]
- 20.Oldridge M, Zackai EH, McDonald-McGinn DM, Iseki S, Morriss-Kay GM, Twigg SR, Johnson D, Wall SA, Jiang W, Theda C, et al. De novo alu-element insertions in FGFR2 identify a distinct pathological basis for Apert syndrome. Am J Hum Genet. 1999;64:446–461. doi: 10.1086/302245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu K, Herr AB, Waksman G, Ornitz DM. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc Natl Acad Sci USA. 2000;97:14536–14541. doi: 10.1073/pnas.97.26.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibrahimi OA, Eliseenkova AV, Plotnikov AN, Yu K, Ornitz DM, Mohammadi M. Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc Natl Acad Sci USA. 2001;98:7182–7187. doi: 10.1073/pnas.121183798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilkie AO, Patey SJ, Kan SH, van den Ouweland AM, Hamel BC. FGFs, their receptors, and human limb malformations: clinical and molecular correlations. Am J Med Genet. 2002;112:266–278. doi: 10.1002/ajmg.10775. [DOI] [PubMed] [Google Scholar]
- 24.Nagase T, Nagase M, Hirose S, Ohmori K. Mutations in fibroblast growth factor receptor 2 gene and craniosynostotic syndromes in Japanese children. J Craniofac Surg. 1998;9:162–170. doi: 10.1097/00001665-199803000-00015. [DOI] [PubMed] [Google Scholar]
- 25.Plotnikov AN, Hubbard SR, Schlessinger J, Mohammadi M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell. 2000;101:413–424. doi: 10.1016/s0092-8674(00)80851-x. [DOI] [PubMed] [Google Scholar]
- 26.Johnson DE, Lee PL, Lu J, Williams LT. Diverse forms of a receptor for acidic and basic fibroblast growth factors. Mol Cell Biol. 1990;10:4728–4736. doi: 10.1128/mcb.10.9.4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mansukhani A, Dell’Era P, Moscatelli D, Kornbluth S, Hanafusa H, Basilico C. Characterization of the murine BEK fibroblast growth factor (FGF) receptor: activation by three members of the FGF family and requirement for heparin. Proc Natl Acad Sci USA. 1992;89:3305–3309. doi: 10.1073/pnas.89.8.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clements DA, Wang JK, Dionne CA, Goldfarb M. Activation of fibroblast growth factor (FGF) receptors by recombinant human FGF-5. Oncogene. 1993;8:1311–1316. [PubMed] [Google Scholar]
- 29.Hecht D, Zimmerman N, Bedford M, Avivi A, Yayon A. Identification of fibroblast growth factor 9 (FGF9) as a high affinity, heparin dependent ligand for FGF receptors 3 and 2 but not for FGF receptors 1 and 4. Growth Factors. 1995;12:223–233. doi: 10.3109/08977199509036882. [DOI] [PubMed] [Google Scholar]
- 30.Mathieu M, Chatelain E, Ornitz D, Bresnick J, Mason I, Kiefer P, Dickson C. Receptor binding and mitogenic properties of mouse fibroblast growth factor 3. Modulation of response by heparin. J Biol Chem. 1995;270:24197–24203. doi: 10.1074/jbc.270.41.24197. [DOI] [PubMed] [Google Scholar]
- 31.LaRochelle WJ, Dirsch OR, Finch PW, Cheon HG, May M, Marchese C, Pierce JH, Aaronson SA. Specific receptor detection by a functional keratinocyte growth factor-immunoglobulin chimera. J Cell Biol. 1995;129:357–366. doi: 10.1083/jcb.129.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 33.Igarashi M, Finch PW, Aaronson SA. Characterization of recombinant human fibroblast growth factor (FGF)-10 reveals functional similarities with keratinocyte growth factor (FGF-7) J Biol Chem. 1998;273:13230–13235. doi: 10.1074/jbc.273.21.13230. [DOI] [PubMed] [Google Scholar]
- 34.Hoshikawa M, Yonamine A, Konishi M, Itoh N. FGF-18 is a neuron-derived glial cell growth factor expressed in the rat brain during early postnatal development. Brain Res Mol Brain Res. 2002;105:60–66. doi: 10.1016/s0169-328x(02)00393-5. [DOI] [PubMed] [Google Scholar]
- 35.Ibrahimi OA, Zhang F, Eliseenkova AV, Linhardt RJ, Mohammadi M. Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through enhancement of FGF binding affinity. Hum Mol Genet. 2004;13:69–78. doi: 10.1093/hmg/ddh011. [DOI] [PubMed] [Google Scholar]
- 36.Plotnikov AN, Eliseenkova AV, Ibrahimi OA, Shriver Z, Sasisekharan R, Lemmon MA, Mohammadi M. Crystal structure of fibroblast growth factor 9 reveals regions implicated in dimerization and autoinhibition. J Biol Chem. 2001;276:4322–4329. doi: 10.1074/jbc.M006502200. [DOI] [PubMed] [Google Scholar]
- 37.Miki T, Fleming TP, Bottaro DP, Rubin JS, Ron D, Aaronson SA. Expression cDNA cloning of the KGF receptor by creation of a transforming autocrine loop. Science. 1991;251:72–75. doi: 10.1126/science.1846048. [DOI] [PubMed] [Google Scholar]
- 38.Bottaro DP, Fortney E, Rubin JS, Aaronson SA. A keratinocyte growth factor receptor-derived peptide antagonist identifies part of the ligand binding site. J Biol Chem. 1993;268:9180–9183. [PubMed] [Google Scholar]
- 39.Orr-Urtreger A, Bedford MT, Burakova T, Arman E, Zimmer Y, Yayon A, Givol D, Lonai P. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2) Dev Biol. 1993;158:475–486. doi: 10.1006/dbio.1993.1205. [DOI] [PubMed] [Google Scholar]
- 40.Zimmer Y, Givol D, Yayon A. Multiple structural elements determine ligand binding of fibroblast growth factor receptors. Evidence that both Ig domain 2 and 3 define receptor specificity. J Biol Chem. 1993;268:7899–7903. [PubMed] [Google Scholar]
- 41.Igarashi M, Finch PW, Aaronson SA. Characterization of recombinant human fibroblast growth factor (FGF)-10 reveals functional similarities with keratinocyte growth factor (FGF-7) J Biol Chem. 1998;273:13230–13235. doi: 10.1074/jbc.273.21.13230. [DOI] [PubMed] [Google Scholar]
- 42.Hsu YR, Nybo R, Sullivan JK, Costigan V, Spahr CS, Wong C, Jones M, Pentzer AG, Crouse JA, Pacifici RE, et al. Heparin is essential for a single keratinocyte growth factor molecule to bind and form a complex with two molecules of the extracellular domain of its receptor. Biochemistry. 1999;38:2523–2534. doi: 10.1021/bi9821317. [DOI] [PubMed] [Google Scholar]
- 43.Lu W, Luo Y, Kan M, McKeehan WL. Fibroblast growth factor-10. A second candidate stromal to epithelial cell andromedin in prostate. J Biol Chem. 1999;274:12827–12834. doi: 10.1074/jbc.274.18.12827. [DOI] [PubMed] [Google Scholar]
- 44.Yeh BK, Igarashi M, Eliseenkova AV, Plotnikov AN, Sher I, Ron D, Aaronson SA, Mohammadi M. Structural basis by which alternative splicing confers specificity in fibroblast growth factor receptors. Proc Natl Acad Sci USA. 2003;100:2266–2271. doi: 10.1073/pnas.0436500100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilkin DJ, Hecht JT, Francomano CA. Achondroplasia and pseudoachondroplasia. In: Scriver CR, Beaudet AL, Valle D, Sly WS, Childs Kinzler K, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. IV. McGraw-Hill; New York, NY: 2001. pp. 5379–5395. [Google Scholar]
- 46.Hajihosseini MK, Heath JK. Expression patterns of fibroblast growth factors-18 and -20 in mouse embryos is suggestive of novel roles in calvarial and limb development. Mech Dev. 2002;113:79–83. doi: 10.1016/s0925-4773(01)00656-6. [DOI] [PubMed] [Google Scholar]
- 47.Liu Z, Xu J, Colvin JS, Ornitz DM. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 2002;16:859–869. doi: 10.1101/gad.965602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohbayashi N, Shibayama M, Kurotaki Y, Imanishi M, Fujimori T, Itoh N, Takada S. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 2002;16:870–879. doi: 10.1101/gad.965702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu X, Komiya H, Chirino A, Faham S, Fox GM, Arakawa T, Hsu BT, Rees DC. Three-dimensional structures of acidic and basic fibroblast growth factors. Science. 1991;251:90–93. doi: 10.1126/science.1702556. [DOI] [PubMed] [Google Scholar]
- 50.Osslund TD, Syed R, Singer E, Hsu EW, Nybo R, Chen BL, Harvey T, Arakawa T, Narhi LO, Chirino A, et al. Correlation between the 1.6 A crystal structure and mutational analysis of keratinocyte growth factor. Protein Sci. 1998;7:1681–1690. doi: 10.1002/pro.5560070803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bellosta P, Iwahori A, Plotnikov AN, Eliseenkova AV, Basilico C, Mohammadi M. Identification of receptor and heparin binding sites in fibroblast growth factor 4 by structure-based mutagenesis. Mol Cell Biol. 2001;21:5946–5957. doi: 10.1128/MCB.21.17.5946-5957.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Plotnikov AN, Schlessinger J, Hubbard SR, Mohammadi M. Structural basis for FGF receptor dimerization and activation. Cell. 1999;98:641–650. doi: 10.1016/s0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- 53.Stauber DJ, DiGabriele AD, Hendrickson WA. Structural interactions of fibroblast growth factor receptor with its ligands. Proc Natl Acad Sci USA. 2000;97:49–54. doi: 10.1073/pnas.97.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olsen SK, Ibrahimi OA, Raucci A, Zhang F, Eliseenkova AV, Yayon A, Basilico C, Linhardt RJ, Schlessinger J, Mohammadi M. Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc Natl Acad Sci USA. 2004;101:935–940. doi: 10.1073/pnas.0307287101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oldridge M, Lunt PW, Zackai EH, McDonald-McGinn DM, Muenke M, Moloney DM, Twigg SR, Heath JK, Howard TD, Hoganson G, et al. Genotype-phenotype correlation for nucleotide substitutions in the IgII-IgIII linker of FGFR2. Hum Mol Genet. 1997;6:137–143. doi: 10.1093/hmg/6.1.137. [DOI] [PubMed] [Google Scholar]
- 56.Johnson D, Wall SA, Mann S, Wilkie AO. A novel mutation, Ala315Ser, in FGFR2: a gene-environment interaction leading to craniosynostosis? Eur J Hum Genet. 2000;8:571–577. doi: 10.1038/sj.ejhg.5200499. [DOI] [PubMed] [Google Scholar]
- 57.Min H, Danilenko DM, Scully SA, Bolon B, Ring BD, Tarpley JE, DeRose M, Simonet WS. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev. 1998;12:3156–3161. doi: 10.1101/gad.12.20.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, et al. Fgf10 is essential for limb and lung formation. Nat Genet. 1999;21:138–141. doi: 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- 59.Guo L, Degenstein L, Fuchs E. Keratinocyte growth factor is required for hair development but not for wound healing. Genes Dev. 1996;10:165–175. doi: 10.1101/gad.10.2.165. [DOI] [PubMed] [Google Scholar]
- 60.Qiao J, Uzzo R, Obara-Ishihara T, Degenstein L, Fuchs E, Herzlinger D. FGF-7 modulates ureteric bud growth and nephron number in the developing kidney. Development. 1999;126:547–554. doi: 10.1242/dev.126.3.547. [DOI] [PubMed] [Google Scholar]
- 61.Celli G, LaRochelle WJ, Mackem S, Sharp R, Merlino G. Soluble dominant-negative receptor uncovers essential roles for fibroblast growth factors in multi-organ induction and patterning. EMBO J. 1998;17:1642–1655. doi: 10.1093/emboj/17.6.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li C, Guo H, Xu X, Weinberg W, Deng CX. Fibroblast growth factor receptor 2 (Fgfr2) plays an important role in eyelid and skin formation and patterning. Dev Dyn. 2001;222:471–483. doi: 10.1002/dvdy.1205. [DOI] [PubMed] [Google Scholar]
- 63.Petiot A, Conti FJ, Grose R, Revest JM, Hodivala-Dilke KM, Dickson C. A crucial role for Fgfr2-IIIb signalling in epidermal development and hair follicle patterning. Development. 2003;130:5493–5501. doi: 10.1242/dev.00788. [DOI] [PubMed] [Google Scholar]
- 64.Munro CS, Wilkie AO. Epidermal mosaicism producing localised acne: somatic mutation in FGFR2. Lancet. 1998;352:704–705. doi: 10.1016/S0140-6736(05)60820-3. [DOI] [PubMed] [Google Scholar]
- 65.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 66.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 67.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–1021. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 68.Jones TA, Zou JY, Cowan SW, Kjeldgaard G. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]