

Abstract
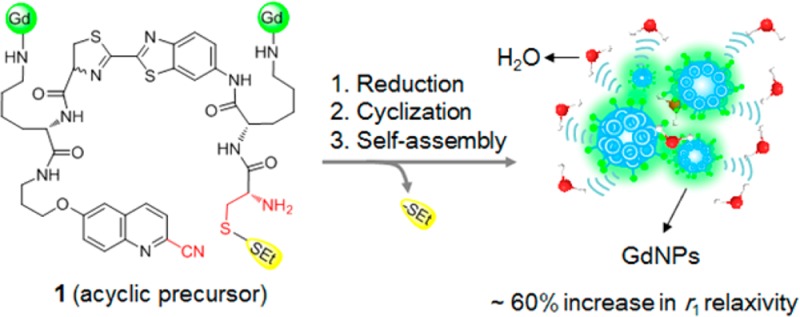
Controlled self-assembly of small molecule gadolinium (Gd) complexes into nanoparticles (GdNPs) is emerging as an effective approach to design activatable magnetic resonance imaging (MRI) probes and amplify the r1 relaxivity. Herein, we employ a reduction-controlled macrocyclization reaction and self-assembly to develop a redox activated Gd-based MRI probe for sensing a reducing environment. Upon disulfide reduction at physiological conditions, an acyclic contrast agent 1 containing dual Gd-chelates undergoes intramolecular macrocyclization to form rigid and hydrophobic macrocycles, which subsequently self-assemble into GdNPs, resulting in a ∼60% increase in r1 relaxivity at 0.5 T. Probe 1 has high r1 relaxivity (up to 34.2 mM–1 s–1 per molecule at 0.5 T) upon activation, and also shows a high sensitivity and specificity for MR detection of thiol-containing biomolecules.
Introduction
Over the past few decades, MRI has become one of the most powerful diagnostic tools available in both clinical and research settings. It can provide noninvasive, multidimensional visualization of the entire body with high spatial resolution.1 However, compared to other molecular imaging modalities, MRI has relatively poor sensitivity, and therefore contrast agents (CAs) are often used to enhance imaging contrast between pathological and normal tissues. Currently, more than 40% of clinical MRI protocols include the use of CAs.2
Gd-based CAs are able to shorten the longitudinal relaxation time (T1) of water protons to produce “positive” (bright) contrast and are most commonly used in MRI diagnosis. However, current clinically used CAs possess relatively low contrast efficacy (relaxivity, r1), and large amounts of CAs have to be administered to achieve sufficient contrast, which is often a safety concern. In addition, they are generally nonspecific and not capable of sensing biochemical activity.3 As a result, the design of molecular probes with higher relaxivity is crucial for MR molecular imaging. “Smart” or “activatable” MR probes are particularly attractive because they modulate their MR properties (relaxivity) leading to signal amplification upon molecule target interaction.3−5 These smart probes can greatly improve MR detection of specific molecular events arising from either receptor binding (i.e., RIME, receptor-induced magnetization enhancement)6−8 or molecular activation (i.e., pH, light, metal ions, and enzymes).9−14 A number of environmentally responsive MRI probes15−21 capable of specifically reporting on their local physiological or biochemical conditions (e.g., pH, redox, ions) have been actively explored, providing wide applications in diagnosis.
Thiol redox chemistry is central to the control of cell fate and is associated with various abnormal biochemical processes.17 Alternations in the levels of thiol-containing biomolecules (biothiols), including glutathione (GSH), cysteine (Cys), and homocysteine (Hcy), are directly linked with many diseases (e.g., cancer, AIDS, liver damage, psoriasis, and Alzheimer’s disease).22 Therefore, various methods for monitoring of their levels have been developed, including thiol-sensitive organic optical probes,23−25 inorganic quantum dots,26 and luminescent semiconductors.27 Recently, a few thiol-responsive Gd-based MRI probes have also been reported.28−34 These probes typically have a small value of r1 relaxivity (2.77 mM–1 s–1)31 or show a decrease in r1 relaxivity after thiol-activation that leads to a darker contrast.28,29,31 Here we report MR probes that give an increasing, high value (>10 mM–1 s–1) of r1 relaxivity upon biothiol activation, resulting in a brighter contrast.
We have previously described an activatable Gd-based CA containing a disulfide that undergoes reduction-triggered polymerization and self-assembly to generate enhanced r1 relaxivity after activation, by taking advantage of the increased molecular tumbling time (τR) arising from in situ self-assembly of GdNPs.35 The mechanism of polymerization is based on the reduction-triggered disulfide cleavage to yield 1,2-aminothiol, which further condenses with 2-cyanobenzothiazole (CBT) in another monomer to produce oligomers.36 A potential limitation of this system is the competition from the reaction between CBT in the monomer and the endogenous free cysteine―that stops polymerization and self-assembly. On the other hand, the r1 relaxivity of the probe after reduction activation is just ∼8.3 mM–1 s–1 (Table 1), which is smaller than that of other reported GdNPs-based CAs.37−39 The lower r1 value is presumably due to the flexibility of the linear oligomer of the scaffold after polymerization. In order to effectively out-compete free cysteine, we have optimized the self-assembly strategy by switching the concentration-dependent intermolecular polymerization to concentration-independent intramolecular macrocyclization chemistry, showing high efficacy to self-assemble fluorescence40,41 and PET probes42 into nanoparticles for imaging of enzyme activity in biological systems. This optimized self-assembly system can also be applied to develop the first example of caspase-3/7 activatable Gd-based MRI probe by introducing a Gd-DOTA amide chelate.43 The r1 relaxivity following activation is ∼15.6 mM–1 s–1 per molecule at 1.5 T, which is around 2-fold higher than that of polymerization-based probe 1a (∼8.3 mM–1 s–1). This suggests that, with the replacement of the flexible linear oligomer with a macrocyclic scaffold after activation, the CAs with more hydrophobic and rigid macrocyclic scaffold can further enhance the r1 relaxivity following activation according to the Slomon-Bloembergen-Morgan (SBM) theory.44 To further investigate the capability of the macrocyclization chemistry mediated self-assembly approach for activatable MRI probes with improved MR properties, we develop new redox activated CAs containing two Gd-DOTA amide chelates and a biothiol sensitive disulfide bond. The newly designed CAs upon reduction activation have high r1 relaxivities per molecule (up to ∼34.2 mM–1 s–1 at 0.5 T) and are specific to biothiols and capable of detecting GSH concentration with high sensitivity.
Table 1. Relaxivities of Gd-Based MR Probes before and after Activation with Reduced Agent (TCEP) at Different Magnetic Field Strengths.
| effective r1 relaxivitya (mM–1 s–1) |
||||||
|---|---|---|---|---|---|---|
| 0.5 T |
1.5 T |
3 T |
||||
| probe | per Gd | per molecule | per Gd | per molecule | per Gd | per molecule |
| Dotarem | 4.6 ± 0.3 | 4.6 ± 0.3 | 4.2 ± 0.2 | 4.2 ± 0.2 | 3.9 ± 0.1 | 3.9 ± 0.1 |
| 1ab,c | 3.9 | 3.9 | 3.8 | 3.8 | ||
| 1-Ic | 9.8 ± 1.0 | 19.6 ± 2.0 | 9.7 ± 0.3 | 19.4 ± 0.6 | 8.4 ± 0.2 | 16.8 ± 0.4 |
| 1-IIc | 9.0 ± 1.2 | 18.0 ± 2.4 | 8.7 ± 0.2 | 17.4 ± 0.4 | 7.5 ± 0.4 | 15.0 ± 0.8 |
| 1-Oc | 12.5 ± 1.1 | 25.0 ± 2.2 | 11.4 ± 0.3 | 22.8 ± 0.6 | 8.8 ± 0.1 | 17.6 ± 0.2 |
| 1a (+)b·d | 8.3 | 8.3 | 5.4 | 5.4 | ||
| 1-I (+)d | 14.9 ± 1.1 | 29.8 ± 2.2 | 13.9 ± 0.6 | 27.8 ± 1.2 | 9.5 ± 0.3 | 19.0 ± 0.6 |
| 1-II (+)d | 14.7 ± 0.9 | 29.4 ± 1.8 | 13.4 ± 0.4 | 26.8 ± 0.8 | 9.2 ± 0.1 | 18.4 ± 0.2 |
| 1-O (+)d | 17.1 ± 1.3 | 34.2 ± 2.6 | 15.9 ± 0.2 | 31.8 ± 0.4 | 10.5 ± 0.2 | 21.0 ± 0.4 |
The relaxation time T1 was measured at 37 °C in PBS buffer (10×) using the standard inversion recovery spin–echo sequence on 0.5, 1.5, and 3 T MR scanner. These concentration-dependent T1 values were plotted versus Gd3+ concentration, and the rising curve was fitted by linear regression to calculate molar relaxivity r1. The data are shown as mean ± SD.
Data from ref (35).
Before activation.
After activation.
Results and Discussion
Design of Redox Activated Gd-Based CAs
The conceptual design in Figure 1 was demonstrated with a biocompatiable intramolecular macrocyclization system derived from reaction between 2-cyano-6-hydroxyquinoline (CHQ) and free cysteine.40 Macrocycles are an important class of molecules which exhibit profound chemical and biological properties derived from the preorganized ring structures.45 In comparison to their acyclic analogues, macrocycles inherently possess a lower number of rotatable bonds, and are more conformationally restricted. Topologically, macrocycles also have larger surface areas.46,47 All these characteristics make macrocycles attractive scaffolds for drug and material design. For example, well-defined macrocycles can assemble to robust organic nanotubes48 or supramolecular barrels driven by noncovalent interactions.49 We envisioned that if two Gd-chelates were simultaneously incorporated into the acyclic precursor (1), the increasing number of Gd-chelates can result in a higher value of MR relaxivity upon formation of Gd-based macrocycle (3). Furthermore, the self-assembled GdNPs from these macrocycles will further increase τR to amplify the r1 relaxivity compared to that of the acyclic precursor (small molecule) with an identical Gd concentration.50,51
Figure 1.
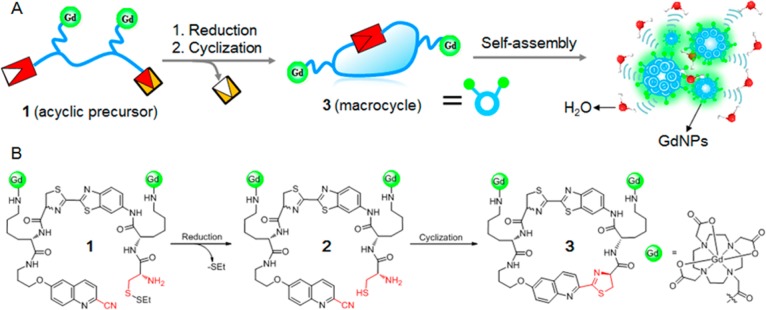
General design of redox activated MRI probe. (A) Illustration of reduction-controlled macrocyclization and self-assembly for redox activated MRI probe. An acyclic precursor 1 containing two Gd-chelates is reduced to form reactive intermediate 2, which subsequently undergoes intramolecular cyclization to generate a more rigid and hydrophobic macrocyclic product 3. The following self-assembly of 3 into GdNPs generates increased r1 relaxivities and brighter MRI contrast. (B) The chemical structure of probe 1 and the proposed chemical conversions following disulfide reduction.
To prove this concept, we designed probe 1 containing a disulfide that could be activated by reduction (Figure 1). The application of thiol/disulfide redox couple is well explored for biothiol sensing.23−34 The established chemical reaction is highly biocompatible, ensuring sufficient reaction to form more rigid and hydrophobic macrocycles and effectively self-assemble into GdNPs. The introduction of two Gd-DOTA amide chelates to the same molecule should increase the accumulation of Gd3+ ions in the GdNPs to further enhance r1 relaxivity of the activation product.52−54
Synthesis and Characterization of Gd-based CAs
The general synthesis of probe 1 is shown in Scheme 1, which afforded two isomers (1-I and 1-II) with a ratio of 1:2 on HPLC. Both isomers showed the same UV/vis absorption and fluorescence spectra (Supporting Information Figure S2 and Table S1), and HRMS characterization gave the same molecular weight (Supporting Information Figure S1). 2-D NMR spectroscopy revealed an identical NMR pattern of the precursors (11-I and 11-II) of both isomers. Interestingly, both isomers can undergo spontaneous structural isomerization to each other. When diastereomerically pure 1-I and 1-II were incubated in PBS buffer (pH 7.4) at r.t., they could gradually isomerize and the ratio of 1-I and 1-II in both solutions stabilized at around 2 after 60 h based on the HPLC analysis (Figure 2A and B). We estimated the isomerization rate constants of 1-I and 1-II at the first 25 h of incubation (Figure 2C and D). The pseudo-first-order rate constants are 0.0133 h–1 for 1-I and 0.0296 h–1 for 1-II. These results demonstrate that isomer 1-I is thermodynamically more stable than 1-II in PBS buffer, and the kinetics of transforming 1-I to 1-II is about 2-fold slower than that of transforming 1-II to 1-I. Furthermore, the spontaneous isomerization of 1-I and 1-II in solution is pH-dependent (see Supporting Information Figure S3). A much slower isomerization process was observed in aqueous solution with a lower pH value (<5.5). Since the chiral center at the C-4 position of thiazoline (4-(S) or 4-(R)) is reported to easily racemize/epimerize via a proposed keto–enol tautomerism process under basic condition (Supporting Information Figure S4),55 the same mechanism is likely responsible for the epimerization of 1-I and 1-II. Indeed, the enolization-based racemization of l-luciferin has been proposed for the biosynthesis of firefly d-luciferin.56 Based on the structure of precursor 11-II, which contains a 4-(S) chiral center at the thiazoline linker, we therefore assigned isomer 1-II the same 4-(S) chirality as that of 11-II, and 1-I the 4-(R) configuration (Scheme 1). To further verify this assumption, we used MnO2 to oxidize thiazoline to eliminate the chiral center at C-4 position (Scheme 1). As expected, both isomers 1-I and 1-II were converted to the same oxidation compound 1-O that contains a thiazole linker, with an identical peak on HPLC analysis. 1-O was stable in PBS buffer without any isomerization.
Scheme 1. Synthesis of Probes 1 and 1-O.

(a) (I) N-Boc-Nε-Fmoc-l-Lys-OH, i-BuO-COCl, NMM, THF, 0 °C to r.t., 72%; (II) 20% TFA/DCM, r.t., 2 h, 98%; (III) S-Trt-N-Boc-d-Cys-OH, HBTU, DIPEA, THF, 3 h, 82%; (b) 5% Piperidine/DMF, r.t., 10 min, 63%; (c) Tri-(t-BuO)-DOTA-NHS, DMF, DIPEA, 2 h; 75%; (d) (I) Py·HCl, 200 °C, 2h, 35%; (II) K2CO3, KI, DMF, BocNH(CH2)3Cl, 80 °C, overnight, 90%; (III) 20% TFA/DCM, 99%; (e) (I) S-Trt-N-Boc-l-Cys-Lys-(Nε-Fmoc)-OH, HBTU, DIPEA, DMF, 3 h, 91%; (II) 5% Piperidine/DMF, r.t., 10 min, 65%; (f) (I) Tri-(t-BuO)-DOTA-COOH, HBTU, DIPEA, DMF, 3 h, 90%; (II) TFA/TIPSH/DCM (95/2.5/2.5), r.t., 3 h, 85%; (g) TCEP, DIPEA, MeOH, r.t. 1 h, 60%; (h) (I) TFA/TIPSH/DCM (95/2.5/2.5); (II) (EtS)S-Py, MeOH, r.t. 1 h, 79% for two steps; (i) PBS buffer (pH 7.4), Ar, r.t., 24 h, 43% yield with 45% recovery of 11-II; (j) GdCl3, NaHCO3, r.t. overnight, 93%; (k) MnO2, DIPEA, DCM/MeOH, 67%.
Figure 2.

Spontaneous isomerization of 1-I and 1-II in PBS solution. (A) Isomer 1-I (100 μM) is slowly converted to 1-II, and gives rise to a final ratio of ∼2:1 (1-I: 1-II) after 60 h incubation at r.t. (B) Isomer 1-II (100 μM) is slowly converted to 1-I, and gives rise to a final ratio of ∼2:1 (1-I: 1-II) after 72 h incubation at r.t. (C, D) Plots of the HPLC data estimate the pseudo-first-order rate constant for isomerization of 1-I (C) and 1-II (D) at the first 25 h incubation time (1-I: k1 = 0.0133 h–1; 1-II: k1 = 0.0296 h–1).
To evaluate whether disulfide reduction of probe 1 could trigger intramolecular cyclization, isomer 1-I was treated with reducing agent TCEP at pH 3 to afford reduced intermediate 2-I (Figure 3A). Subsequent adjustment of pH value to 7.4 resulted in intramolecular cyclization to give two macrocyclic isomers 3-I and 3-II as observed before40 (Supporting Information Figure S5). The ratio of 3-I to 3-II is about 7:1 based on the HPLC analysis. Similarly, treatment of 1-II with TCEP followed by adjustment of pH value to 7.4 provided the same macrocyclic products 3-I and 3-II but at a ratio of 1:4 (Figure 3B). This difference may arise from different chirality at the C-4 position of thiazoline linker in 1-I and 1-II and their different epimerization rates. Macrocyclization of 1-O provided just one product 3-O based on HPLC analysis (Figure 3C). Furthermore, incubation of 1-I, 1-II, and 1-O in lysates of human breast cancer cell line MDA-MB-468 gave the same macrocyclic products as those in the buffer (Figure 3D). Similar results were obtained when incubating 1-I and 1-II in HeLa cell lysates (Supporting Information Figure S5). These results suggest that the reduction-triggered macrocyclization of our designed probes can take place efficiently in a complex cellular environment.
Figure 3.

Characterization of disulfide reduction-triggered macrocyclization in vitro. (A, B) HPLC traces of the incubation of 1-I and 1-II (20 μM) with reducing agent TCEP (200 μM) in aqueous solution. Bottom: 1-I (A) or 1-II (B) alone; middle: reduced intermediate 2-I (A) or 2-II (B) formed following 20 min TCEP reduction at pH 3; top: two cyclized diastereoisomeric products 3-I and 3-II formed following incubation of 1-I (A) or 1-II (B) with TCEP at pH 7.4 for 8 h. (C) HPLC traces of the incubation of 1-O (20 μM) with TCEP (200 μM) in aqueous solution. Bottom: 1-O alone; middle: reduced intermediate 2-O formed following 20 min TCEP reduction at pH 3; top: only one cyclized product 3-O formed following incubation of 1-O with TCEP at pH 7.4 overnight. (D) HPLC traces of the incubation solutions of 1-I, 1-II, and 1-O in cell lysates. Bottom to top: blank MDA-MB-468 cell lysates; 1-I, 1-II, or 1-O (100 μM) incubated in the MDA-MB-468 cell lysates overnight, respectively.
Nanocharacterization of Self-Assembled GdNPs
We further confirmed the formation of GdNPs after macrocyclization of probe 1 upon reduction. DLS showed the particles with a mean diameter of 100–300 nm from both isomers 1-I and 1-II upon TCEP reduction in water at pH 7.4 (Supporting Information Figure S6). SEM (Figure 4A and B) and TEM (Figure 4C) images revealed the shapes of separated GdNPs with diameters ranging from 100 nm to a few hundred nanometers. The presence of Gd in the particles was confirmed by energy-dispersive X-ray (EDX) spectroscopy (Supporting Information Figure S6). The formation of nanoparticles from the macrocyclic products of 1-O was also observed based on the DLS analysis (Supporting Information Figure S6).
Figure 4.

Nanocharacterization of the reduction-triggered self-assembled GdNPs. (A, B) SEM image of self-assembled nanostructure of 1-I (500 μM, A) or 1-II (500 μM, B) following incubation with TCEP (2 mM) at pH 7.4 in water overnight. (C) TEM image of self-assembled nanostructure from the incubation solution of 1-I and 1-II (1:1, 500 μM) following TCEP (2 mM) reduction at pH 7.4 in water; 24 000× magnification.
Characterization of MR Properties
Water proton relaxivities for isomers 1-I and 1-II and the oxidized 1-O before and after disulfide reduction were measured on 0.5, 1.5, and 3 T clinical MRI scanners (Supporting Information Figure S7). The relaxivity values of all these probes were summarized in Table 1. Before disulfide reduction, all the three di-Gd-chelate based CAs exhibited higher relaxivity per molecule at all three magnetic field strengths compared to single-Gd chelate containing agents such as Dotarem or our previously reported probe 1a.35 The larger r1 relaxivities are in line with their larger molecular size and also more Gd-chelates in the probes.57 Upon disulfide reduction, they all produced enhanced relaxivities, which resulted in a 1.5-, 1.6-, and 1.4-fold increase in r1 for 1-I, 1-II, and 1-O at 0.5 T, respectively. These results indicate that, after macrocyclization, the relaxivities are further enhanced owing to the formation of GdNPs. When measured at a higher field strength (1.5 T), similar increases in r1 were also observed for 1-I (1.4-fold), 1-II (1.5-fold), and 1-O (1.4-fold), and even at 3T, more than 1.2-fold increase in r1 was observed for 1-II. Among all three probes, 1-O gave the highest r1 values per molecule upon activation at all three field strengths: 34.2 (0.5 T), 31.8 (1.5 T), and 21.0 mM–1 s–1 (3 T). The value of 1-O at 1.5 T is ∼8 times greater than that of Dotarem and ∼4 times greater than that of our previous probe 1a upon activation (Table 1), which demonstrates that the self-assembly of CAs with more Gd chelates can effectively increase the r1 relaxivity.
T1-weighted MR images of a series of solutions containing Dotarem, 1-I, 1-II, and 1-O in PBS buffer were obtained at 0.5 T (Figure 5) using a fast spin–echo pulse sequence with TE = 19 ms and TR = 117 ms. As expected, the solution of di-Gd probes 1-I, 1-II, and 1-O are clearly brighter than that of Dotarem at the same Gd concentration (800 μM). Moreover, upon disulfide reduction, the solutions containing di-Gd probes in PBS buffer exhibited much higher signal intensity than those before reduction. These results confirm that higher r1 associated with the self-assembled GdNPs can result in faster T1 relaxation, leading to a significant enhancement in MR contrast.
Figure 5.

T1-weighted MR images of Dotarem, 1-I, 1-II, and 1-O at the same Gd3+ concentration (800 μM) in PBS buffer (10×, pH 7.4) before and after incubation with TCEP (2 mM) at r.t. overnight. The images were acquired at a 0.5 T MR scanner (Signa HDx, GE Healthcare, Waukesha, Wisconsin) at 37 °C, using a fast spin–echo pulse sequence (TE/TR = 19/117 ms).
MR Detection of Reducing Environment
We first evaluated the capability for MR detection of GSH levels by measuring the T1 value of representative probe 1-II solution (200 μM) upon incubation with varying concentrations of GSH (0–10 mM) in PBS buffer at 0.5 T. As shown in Figure 6A, the T1 value of the solution reduced gradually as the GSH concentration was increased. At GSH concentration as low as 10 μM, a significant reduction in T1 value (ΔT1 = −17 ± 4 ms) was observed for 1-II, and at ≥1 mM GSH, 1-II was extensively activated in solution after 6 h, leading to the maximum reduction in T1 value (ΔT1 = −74 ± 5 ms). A plot of the change of relaxation rate (Δ(1/T1)) in solution shows a GSH level-dependent increase of relaxation rate for 1-II (Figure 6B). These results indicate that 1-II has a good sensitivity for MR detection of GSH levels. Since the GSH is present at millimolar levels in cells, the sensitivity of 1-II is sufficient for MR detection of cellular reducing environment.
Figure 6.

MR detection of reducing environment with 1-II. (A) T1 values (0.5 T) of 1-II (200 μM) in PBS buffer (10×, pH 7.4) following incubation with varying concentrations of GSH at 37 °C for 6 h. (B) Plot of the change of relaxation rate (Δ(1/T1)) calculated from a) versus GSH concentrations between 0–10 mM. Δ(1/T1) = 1/T1 (with GSH) – 1/T1 (without GSH). The inset is a linear fitting line in the region of 10–100 μM GSH. (C) T1 values (0.5 T) of 1-II (200 μM) in PBS buffer (10×, pH 7.4) following incubation with different agents at 37 °C overnight. 1 mM of TCEP, various biothiols (GSH, Cys, and Hcy) and representative natural amino acids (Gly, Glu, Lys, His, Met, Ser, Phe) were used for the studies. HeLa cell lysates (10 million cells per mL PBS buffer) with or without 2 mM NEM pretreatment were studied. T1 value (0.5 T) for each solution was measured with Bruker Minispec (mq20 NMR analyzer) at 37 °C, using the standard inversion recovery program. Data represent mean values ± standard deviation; n = 4.
The selectivity of our CAs for biothiols against other representative natural amino acids was next examined according to T1 value measurement (Figure 6C). GSH, Cys, and Hcy, the three major biothiols, all caused remarkable reduction in the T1 value like TCEP. No apparent response to other amino acids including sulfur-containing methionine was observed in the test system. The response to Cys is of importance, suggesting that 1-II can efficiently outcompete free endogenous Cys in biological samples for macrocyclization. As shown in Figure 6C, the incubation of 1-II (200 μM) in healthy HeLa cell lysates resulted in a similar T1 value (T1 = 178 ± 4 ms) as that observed with GSH (1 mM) activation in PBS buffer, which was inhibited by a thiol scavenger, N-ethylmaleimide (NEM).58 Therefore, 1-II was specific to biothiols over other analytes existing in cells.
Since our probe will produce an enhanced MR contrast upon reductive activation, it would be more suitable for detection of pathology associated with higher cellular GSH levels as compared with normal tissues such as malignant and drug resistant tumors.59 In cells with oxidative stress that have lower GSH levels (e.g, Parkinson’s disease),60 our approach would result in a decrease in the MR contrast relative to healthy cells; in this case, other previously reported MRI probes that show a decrease in the r1 relaxivity after thiol-activation28,29,31 may be applied in order to generate a positively increased contrast. One potential limitation of our current MRI probe is its irreversibility after activation, and in the future a reversible MRI probe may be designed to image the dynamics of the cellular reducing environment if assembled nanoparticles can be disassembled and then reassembled.
Conclusions
In summary, we have described the development of novel redox activated Gd-based CAs using an optimized macrocyclization chemistry-mediated self-assembly approach. These CAs can be activated by disulfide reduction to form rigid and hydrophobic macrocycles, which can further assemble into GdNPs, resulting in enhanced relaxivity (∼60% amplified at 0.5 T). The developed CAs containing two Gd-chelates exhibit a high value of r1 (up to 34.2 mM–1 s–1 per molecule at 0.5 T) upon activation, and also show high specificity and sensitivity to biothiols with MR detection, indicating a feasible approach for sensing a reducing environment.
Experimental Procedures
Materials and Methods
All chemicals were purchased from commercial sources (such as Aldrich, Fluka, Anaspec, and Novabiochem). Analytical TLC was performed with 0.25 mm silica gel 60F plates with fluorescent indicator (254 nm). Plates were visualized by ultraviolet light. The 1H and 13C NMR spectra were taken on a Bruker 400 MHz magnetic resonance spectrometer. Data for 1H NMR spectra are reported as follows: chemical shifts are reported as δ in units of parts per million (ppm) relative to chloroform-d (δ 7.26, s); multiplicities are reported as follows: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), m (multiplet), or br (broadened); coupling constants are reported as a J value in Hertz (Hz); the number of protons (n) for a given resonance is indicated nH, and based on the spectral integration values. Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) and high-resolution mass spectrometry (HRMS) analyses were performed at the Mass Spectrometry Facility of Stanford University. Higher-performance liquid chromatography (HPLC) was performed on a Dionex HPLC System (Dionex Corporation) equipped with a GP50 gradient pump and an inline diode array UV–vis detector. A reversed-phase C18 (Phenomenax, 5 μm, 10 × 250 mm or Dionex, 5 μm, 4.6 × 250 mm) column was used with a MeCN (B)/H2O (A) gradient mobile phase containing 0.1% trifluoroacetic acid (TFA) at a flow of 1 or 3 mL/min for the analysis. Scan electron microscope (SEM) micrographs were obtained on a FEI Nova NanoSEM microscope. Transmission electron microscopy (TEM) micrographs were obtained on a JEM 1230 Electron Microscope. Dynamic light scattering (DLS) experiment was operated on a 90 plus particle size analyzer (Brookhaven Instruments Corporation). Inductively coupled plasma mass spectrometry (ICP-MS) measurement was conducted on Nu Plasma Attom equipment.
Synthesis of Gd3+ Probes 1 and 1-O (Scheme 1)
Synthesis of Intermediate 5
Isobutyl chloroformate (i-BuO-COCl) (156 μL, 1.3 mmol) was added to a solution of Nα-Boc-Nε-Fmoc-l-Lys-OH (515 mg, 1.1 mmol) and N-methylmorpholine (NMM) (143 μL, 1.3 mmol) in dry tetrahydrofuran (THF) (5.0 mL) at 0 °C under Ar. After 10 min, the reaction was allowed to warm to room temperature (r.t.), and kept stirring at r.t. for 4–5 h. A solution of 2-cyano-6-amino benzothiazole (NH2-CBT) (175 mg, 1.0 mmol) in dry THF (2.0 mL) was then added dropwise to the reaction mixture at 0 °C. After further reaction at r.t. overnight, the solvent was removed under vacuum. The residue was dissolved in ethyl acetate (EtOAc) (50 mL) and washed with water (2 × 10 mL) and brine (10 mL). The organic layer was dried over anhydrous Na2SO4, and EtOAc was removed under vacuum. The residue was purified by silica gel chromatography (hexane:EtOAc = 1.5:1) to give a white solid in 72% yield, which was directly dissolved in dry dichloromethane (DCM) (8 mL). Trifluoroacetic acid (TFA) (2 mL) was then added to remove the Boc group. After removal of TFA under vacuum, the residue was dissolved in dry THF (15 mL), to which S-Trt-N-Boc-d-Cys-OH (1.05 equiv), O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU, 1.1 equiv), and N,N-diisopropylethylamine (DIPEA, 3 equiv) were added. After reaction at r.t. for 2 h, THF was removed, and compound 4 was obtained as a light yellow solid in 82% yield after purification by silica gel chromatography (Hexane:EtOAc = 1:1). The following deprotection of the Fmoc group in compound 4 with 5% piperidine in dimethylformamide (DMF) at r.t. for 10 min produced intermediate 5 in 63% yield after HPLC purification. 5: 1H NMR (400 MHz, CD3OD) δ 8.60 (s, 1H), 7.99 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 8.9 Hz, 1H), 7.34 (m, 6H), 7.26 (m, 6H), 7.20 (m, 3H), 4.55 (dd, J = 9.6, 4.3 Hz, 1H), 3.96 (t, J = 6.8 Hz, 1H), 2.88–2.76 (m, 2H), 2.54 (m, 2H), 2.05 (s, br, 1H), 1.83–1.24 (m, 14H); 13C NMR (101 MHz, DMSO-d6) δ 171.57, 171.18, 159.52 (TFA), 155.79, 148.43, 144.93, 139.83, 137.28, 135.77, 129.65, 128.71, 127.44, 125.40, 121.63 (TFA), 118.63, 115.70, 114.19, 112.21, 79.26, 66.61, 54.16, 54.02, 34.44, 31.74, 28.73, 27.29, 23.00. MS: calcd. for C41H45N6O4S2+ [(M+H)+]: 749.3; LC-MS found: m/z 749.7.
Synthesis of Intermediate 6
To a solution of 5 (0.2 mmol) in dry DMF (3 mL) was added tri-tert-butyl 2,2′,2″-(10-(2-((2,5-dioxopyrrolidin-1-yl)oxy)-2-oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate (Tri-(t-BuO)-DOTA-NHS) (0.21 mmol) and DIPEA (0.4 mmol). Two hours later, EtOAc (30 mL) was added, and the solution was washed with water (2 × 10 mL) and brine (10 mL). The organic layer was dried over anhydrous Na2SO4, and EtOAc was removed under vacuum. The residue was purified by HPLC to give 6 as white powder in 75% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 8.74 (s, 1H), 8.54 (s, 1H), 8.29 (m, 1H), 8.16 (d, J = 8.8 Hz, 1H), 7.79 (d, J = 8.9 Hz, 1H), 7.29–7.22 (m, 15H), 7.09 (m, 1H), 4.41 (s, br, 1H), 4.13 (s, br, 1H), 4.03 (m, 1H), 3.93 (s, br, 2H), 3.64–3.24 (m, 11H), 3.20–2.64 (m, 12H), 2.34 (s, br, 2H), 1.76 (s, 1H), 1.60 (m, 1H), 1.51–1.22 (m, 40H). 13C NMR (101 MHz, DMSO-d6) δ 171.60, 171.10, 170.22, 162.96, 159.68, 159.34 (TFA), 155.78, 148.40, 144.93, 139.94, 137.28, 135.68, 129.74, 128.67, 128.44, 128.15, 127.41, 125.41, 121.53 (TFA), 118.44, 115.52, 114.16, 112.59, 112.02, 83.97, 81.74, 79.24, 66.61, 55.25, 54.99, 54.17, 53.94, 51.50, 48.90, 48.49, 36.40, 34.41, 32.13, 31.38, 29.30, 28.70, 28.28, 23.40. MS: calcd. for C69H95N10O11S2+ [(M+H)+]: 1303.7; MALDI-MS found: m/z 1304.1.
Synthesis of Intermediate 8
Starting from 2-cyano-6-methoxyquinoline (MeO-CHQ), intermediate 7 was obtained according to the method reported previously.40 To a solution of 7 in dry DMF (3 mL) was added a dipeptide of S-Trt-N-Boc-l-Cys-Lys (Nε-Fmoc)-OH (1.05 equiv), HBTU (1.1 equiv), and DIPEA (2.0 equiv). The reaction mixture was stirred at r.t. for 2 h. EtOAc (30 mL) was then added, and the mixture was washed with water (2 × 10 mL) and brine (10 mL). The organic layer was dried with Na2SO4. After removal of the solvent under vacuum, the residue was purified by silica gel chromatography (hexane:EtOAc = 1:2), which was followed by deprotection of the Fmoc group with 5% piperidine in DMF at r.t. for 10 min. Compound 8 was obtained in 65% yield after purification by HPLC. 1H NMR (300 MHz, DMSO-d6) δ 8.45 (d, J = 8.4 Hz, 1H), 8.08–7.89 (m, 3H), 7.80 (d, J = 7.5 Hz, 1H), 7.62 (s, br, 2H, NH2), 7.53 (d, J = 9.3 Hz, 1H), 7.44 (s, 1H), 7.37–7.16 (m, 15H), 7.07 (d, J = 7.5 Hz, 1H), 4.28–4.04 (m, 3H), 3.94 (d, J = 6.6 Hz, 1H), 3.26 (m, 2H), 2.70 (m, 2H), 2.38 (m, 2H), 1.92 (m, 2H), 1.65 (s, br, 1H), 1.56–1.13 (m, 16H). 13C NMR (75 MHz, DMSO-d6) δ 171.70, 170.56, 159.37, 155.70, 144.93, 144.35, 137.22, 131.44, 131.03, 130.62, 129.75, 128.72, 127.45, 125.37, 124.82, 118.65, 106.82, 79.85, 79.30, 66.59, 54.26, 52.94, 46.44, 39.41, 36.23, 34.27, 32.24, 29.23, 29.13, 28.78, 27.33, 22.70. MS: calcd. for C46H53N6O5S+ [(M+H)+]: 801.4; LC-MS found: m/z 801.1.
Synthesis of Intermediate 9
A solution of 8 (0.5 mmol), tri-tert-butyl 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetate (Tri-(t-BuO)-DOTA-COOH) (1.05 equiv), HBTU (1.1 equiv), and DIPEA (2.0 equiv) in dry DMF (5 mL) was stirred at r.t. for 3 h. After reaction, EtOAc (30 mL) was added. The solution was washed with water (2 × 15 mL), brine (10 mL), and dried with Na2SO4. After removal of the solvent under vacuum, the residue was purified by silica gel chromatography (DCM:MeOH = 1:20) to obtain coupling product in 90% yield, which was followed by treatment with a cocktail solution of TFA/DCM/triisopropylsilane (TIPSH) (95/2.5/2.5, 5 mL) to remove all of the protected groups after stirring at r.t. for 3 h. The solvent was then removed under vacuum, and the residue was precipitated out with diethyl ether (Et2O). Compound 9 was obtained in 85% yield after HPLC purification. 1H NMR (400 MHz, CD3OD) δ 8.35 (d, J = 8.5 Hz, 1H), 7.94 (d, J = 9.3 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.50 (dd, J = 9.3, 2.7 Hz, 1H), 7.31 (d, J = 2.7 Hz, 1H), 4.29 (dd, J = 8.3, 5.9 Hz, 1H), 4.18 (m, 2H), 4.13 (t, J = 5.4 Hz, 1H), 3.90–3.60 (m, 8H), 3.54–2.95 (m, 23H), 2.08 (m, 2H), 1.85–1.63 (m, 2H), 1.57–1.31 (m, 4H). 13C NMR (101 MHz, CD3OD) δ 172.97, 167.36, 161.46 (TFA), 159.56, 144.24, 136.72, 130.93, 130.50, 130.32, 125.01, 123.87, 118.19, 117.67, 115.30 (TFA), 105.81, 65.99, 54.43, 54.38, 39.00, 36.16, 31.34, 28.77, 28.64, 25.31, 23.03. MS: calcd. for C38H55N10O10S– [(M-H)−]: 843.4; LC-MS found: m/z 843.8.
Synthesis of Intermediate 10
A solution of 9 (0.11 mmol) in MeOH was added dropwise to a solution of 6 (0.1 mmol), tris(2-carboxyethyl) phosphine hydrochloride (TCEP, 0.1 mmol), and DIPEA (0.6 mmol) in MeOH under Ar. After reaction at r.t. for additional 30 min, the solution was directly purified by HPLC to afford 10 in 60% yield. 1H NMR (400 MHz, CD3OD) δ 8.51 (s, 1H), 8.33 (d, J = 8.5 Hz, 1H), 7.95 (d, J = 9.4 Hz, 2H), 7.76 (d, J = 8.9 Hz, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.52 (dd, J = 9.3, 2.7 Hz, 1H), 7.47–7.16 (m, 16H), 5.34 (t, J = 9.5 Hz, 1H), 4.49 (m, 1H), 4.38 (m, 1H), 4.28–2.89 (m, 59H), 2.52 (m, 2H), 2.08 (m, 3H), 1.95–1.28 (m, 47H). 13C NMR (101 MHz, CD3OD) δ 173.07, 172.56, 171.30, 171.18, 171.06, 166.46, 161.57 (TFA), 160.85, 160.47, 159.74, 159.51, 156.42, 149.55, 144.69, 144.17, 137.88, 136.75, 136.62, 130.87, 130.49, 130.21, 129.54, 127.99, 126.92, 125.01, 124.09, 123.82, 120.45, 118.17, 117.75, 115.28 (TFA), 112.33, 105.84, 81.72, 79.89, 79.24, 66.99, 66.06, 54.64, 54.11, 51.93, 39.29, 38.97, 36.25, 35.01, 33.46, 31.67, 31.33, 28.94, 28.73, 28.54, 27.63, 27.44, 23.45, 22.93. MS: calcd. for C107H146N19O21S3– [(M-H)−]: 2129.0; MALDI-MS found: m/z 2129.2.
Synthesis of Intermediate 11 (11-I and 11-II)
Compound 10 was treated with a solution of TFA/DCM/TIPSH (95/2.5/2.5, 5 mL) to remove all of the protected groups. After stirring at r.t. for 3 h, the solvent was removed under vacuum, and the residue was dissolved in MeOH (5 mL). 2-(Ethyldisulfanyl)pyridine ((EtS)S-Py) (1.5 equiv) was then added, and the mixture was stirred at r.t. for another 1 h. After removal of MeOH under vacuum, cold Et2O was added. The precipitate was collected by centrifugation, followed by HPLC purification to give 11-II in 79% yield. In order to obtain 11-I for NMR analysis, we further incubated 11-II in PBS buffer (pH 7.4) under Ar at r.t. for additional 24 h to isomerize 11-II to 11-I. 11-I was obtained in 43% yield after HPLC purification.
11-I: 1H NMR (400 MHz, CD3OD) δ 8.41 (d, J = 1.9 Hz, 1H), 8.26 (d, J = 8.5 Hz, 1H), 7.92 (d, J = 8.8 Hz, 1H), 7.86–7.78 (m, 1H), 7.73–7.61 (m, 2H), 7.40 (dd, J = 9.3, 2.6 Hz, 1H), 7.29–7.22 (m, 1H), 5.36 (t, J = 9.7 Hz, 1H), 4.58–4.48 (m, 1H), 4.45–4.35 (m, 1H), 4.35–4.27 (m, 1H), 4.13 (m, 2H), 4.08–3.35 (m, 39H), 3.29–2.94 (m, 19H), 2.82 (q, J = 7.3 Hz, 2H), 2.06 (m, 2H), 1.92 (m, 3H), 1.75 (m, 1H), 1.52–1.40 (m, 8H), 1.37 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 173.00, 171.53, 171.09, 167.70, 166.46, 161.56 (TFA), 160.87, 159.55, 159.41, 149.45, 144.01, 137.92, 136.72, 136.54, 130.79, 130.29, 130.10, 124.86, 124.11, 123.76, 121.18, 120.22, 118.28, 117.65, 115.37 (TFA), 112.18, 105.70, 79.21, 66.03, 55.05, 54.78, 54.09, 53.89, 53.72, 53.58, 53.34, 53.22, 52.28, 50.81, 50.00, 39.05, 39.00, 38.51, 36.23, 34.55, 31.80, 31.68, 31.47, 28.63, 23.22, 22.94, 13.47. MS: calcd. for C73H106N19O19S4+ [(M+H)+]: 1680.7; MALDI-MS found: m/z 1681.1.
11-II: 1H NMR (400 MHz, CD3OD) δ 8.50 (s, 1H), 8.33 (d, J = 8.6 Hz, 1H), 7.97 (dd, J = 11.6, 9.3 Hz, 2H), 7.67 (d, J = 8.5 Hz, 2H), 7.53 (dd, J = 9.3, 2.7 Hz, 1H), 7.36 (d, J = 2.6 Hz, 1H), 5.34 (t, J = 9.4 Hz, 1H), 4.52 (m, 1H), 4.43–4.34 (m, 1H), 4.30 (m, 1H), 4.21 (t, J = 5.9 Hz, 2H), 4.14–3.30 (m, 39H), 3.29–2.88 (m, 19H), 2.82 (q, J = 7.3 Hz, 2H), 2.27–2.03 (m, 2H), 2.01–1.79 (m, 3H), 1.71 (m, 1H), 1.67–1.38 (m, 8H), 1.36 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 173.09, 171.60, 171.26, 167.75, 166.60, 161.67 (TFA), 160.97, 160.61, 159.80, 159.56, 149.58, 144.18, 137.97, 136.81, 136.63, 130.91, 130.44, 130.21, 125.03, 124.17, 123.81, 121.03, 120.30, 118.13, 117.65, 115.23 (TFA), 112.32, 105.83, 79.16, 66.02, 55.15, 54.79, 54.04, 53.26, 52.27, 50.73, 39.11, 38.96, 38.50, 36.19, 35.02, 31.80, 31.64, 31.47, 28.69, 28.52, 23.29, 22.89, 13.46. MS: calcd. for C73H106N19O19S4+ [(M+H)+]: 1680.7; MALDI-MS found: m/z 1681.2.
Synthesis of 1 (1-I and 1-II)
To a solution of 11 in water was carefully added a solution of 0.5 M NaHCO3 to adjust pH value to 7. Then, a solution of GdCl3 (5 equiv. to 11) in water was added. After stirring at r.t. for 10 min, the pH value of the reaction solution was further adjusted to 7 using 0.5 M NaHCO3, and the reaction mixture was stirred at r.t. overnight. After reaction, the reaction mixture was centrifuged (2500 rpm, 3 min), and the supernatant was purified by HPLC to afford two isomers 1-I and 1-II in 93% yield at a ratio of 1:2.
1-I: >99% HPLC purity. MS: MALDI-MS found: m/z 1990.1. HRMS: calculated for C73H100Gd2N19O19S4+ [(M+H)+]: 1990.4808, HR-ESI/MS found: m/z 1990.4784; calculated for C73H99Gd2N19O19S4Na+ [(M+Na)+]: 2012.4627, HR-ESI/MS found: m/z 2012.4692.
1-II: >99% HPLC purity. MS: MALDI-MS found: m/z 1989.4. HRMS: calculated for C73H100Gd2N19O19S4+ [(M+H)+]: 1990.4808, HR-ESI/MS found: m/z 1990.4830; calculated for C73H99Gd2N19O19S4Na+ [(M+Na)+]: 2012.4627, HR-ESI/MS found: m/z 2012.4639.
Synthesis of 1-O
To a solution of 1-I or 1-II in DCM/MeOH (1/1) was added DIPEA (2 equiv) and MnO2 (10 equiv). The reaction mixture was stirred at r.t. overnight. After removal of MnO2 by filtration, the solvent was removed under vacuum, and the residue was purified by HPLC to afford 1-O in 67% yield (>99% HPLC purity). MS: calculated for C73H98Gd2N19O19S4+ [(M+H)+]: 1988.5, MALDI-MS found: m/z 1988.6. HRMS: calculated for C73H99Gd2N19O19S42+ [(M+2H)/2]+: 994.7359, HR-ESI/MS found: m/z 994.7378.
MR Measurement of r1 Relaxivity
A series of solutions containing 1-I, 1-II, or 1-O at five different concentrations (0.1, 0.2, 0.3, 0.4, and 0.5 mM) in PBS buffer (pH 7.4) were incubated with or without 2 mM TCEP at r.t. overnight. Then, the solutions were placed in 250 μL Eppendorf tubes, bundled together, and MR imaged at 0.5, 1.5, and 3T (Signa HDx, GE Healthcare, Waukesha, Wisconsin) at 37 °C. The scanning procedure began with a localizer and then consisted of a series of inversion-prepared fast spin–echo scans, identical in all aspects (TR 4000 ms, TE minimum, BW 15.6 kHz, field of view 6 cm, slice thickness 3 mm, matrix 128 × 128, NEX 1) except for the inversion time (TI) which was varied as follows: 4000, 2400, 1000, 800, 600, 400, 300, 200, 100, and 50 ms. This scanning procedure produced enough data to fit quantitative T1 relaxation values to each voxel in the image. For quantitative data analysis, the acquired MR images were transferred as DICOM images to an offline workstation, and signal intensities were extracted from each of the 5 samples at each of the 10 TI times by manual region of interest (ROI) placement and voxel averaging within the ROI.
Signal intensity versus TI relationships were fit to the following exponential T1 recovery model by nonlinear least-squares regression: SI (TI) = S0 [1–2 × exp(−TI/T1) + exp(−TR/T1)]. Relaxation rates (R1) were determined as 1/T1. Longitudinal relaxivities (r1, units of mM–1 s–1) were calculated as the slope of R1 vs [Gd] after the determination of true Gd concentration of each sample by the ICP-MS measurement.
MR Detection of GSH Concentration
1-II (200 μM) in PBS buffer (10×) was incubated with various concentration of GSH (0, 1, 10, 50, 100, 250, 500 μM, and 1 and 10 mM) at 37 °C overnight. After incubation, the solutions were transferred into NMR tubes (5 mm O.D.), and the T1 value for each incubation solution was acquired at 0.5 T with Bruker Minispec (mq20 NMR analyzer) at 37 °C, using the standard inversion recovery program.
Cell Culture
HeLa human cervix adenocarcinoma epithelial cells and MDA-MB-468 human breast adenocarcinoma epithelial cells from American Type Culture Collection were cultured in Dulbecco’s modified eagle medium (GIBCO) supplemented with 10% fetal bovine serum (FBS, GIBCO), 100 U mL–1 penicillin, and 100 μg mL–1 streptomycin (GIBCO).
Incubation of 1-I, 1-II, and 1-O in Cell Lysates
The culture medium for adherent MDA-MB-468 (or HeLa) cells was carefully removed and the cells were washed with cold PBS buffer twice. RIPA (RadioImmuno Precipitation Assay) buffer was added to cells using 1 mL of buffer per 5 million cells. Then the cells were kept on ice for 1 h. The cell lysates were gathered, centrifuged at 14 000 × g for 15 min at 4 °C. The supernatant of the cell lysate was collected and incubated with each probe (100 μM) at r.t. overnight, and then injected into an HPLC system for analysis.
MR Detection of Reducing Environment in Cell Lysates with 1-II
Cultured HeLa cells were trypsinized, washed, and centrifuged to form cell pellets. PBS buffer (10×) was added to the cell pellets using 1 mL of buffer per 10 million cells. The cells were lysed with three repeated cycles of freezing and thawing procedure. The cell lysates were then gathered, centrifuged at 14 000 × g for 15 min at 4 °C. The supernatant of the cell lysate was collected and incubated with 1-II (200 μM) at 37 °C overnight. For the inhibition experiment, the collected cell lysates were incubated with 2 mM N-ethylmaleimide at r.t. for 30 min. 1-II was added at a final concentration of 200 μM, and the solution was incubated at 37 °C overnight. After incubation, the solutions were transferred into NMR tubes (5 mm O.D.), and the T1 value for each incubation solution was acquired at 0.5 T with Bruker Minispec (mq20 NMR analyzer) at 37 °C, using the standard inversion recovery program.
Acknowledgments
This work has been supported by the Stanford University National Cancer Institute (NCI) Centers of Cancer Nanotechnology Excellence (1U54CA151459) and the NCI ICMIC@Stanford (1P50CA114747). P.P. was supported by the Stanford Molecular Imaging Scholars (SMIS) fellowship. We thank Dr. Caroline Harris from Stanford ICP-MS/TIMS facility for assistance with the ICP-MS experiment, Dr. Stephen Lynch from Stanford NMR facility for assistance with the 2-D NMR experiment, and Dr. Eddy Lee for the assistance with collecting the T1-weighted image.
Supporting Information Available
Supporting figures and tables, and NMR, HRMS, and MALDI-MS spectra of compounds are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Terreno E.; Castelli D. D.; Viale A.; Aime S. (2010) Challenges for molecular magnetic resonance imaging. Chem. Rev. 110, 3019–3042. [DOI] [PubMed] [Google Scholar]
- Weissleder R.; Pittet M. J. (2008) Imaging in the era of molecular oncology. Nature 452, 580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C.; Osborne E. A.; Louie A. Y. (2011) Activatable T1 and T2 magnetic resonance imaging contrast agents. Ann. Biomed. Eng. 39, 1335–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffern M. C.; Matosziuk L. M.; Mead T. J. (2014) Lanthanide probes for bioresponsive imaging. Chem. Rev. 114, 4496–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leon-Rodriguez L. M.; Lubag A. J.; Malloy C. R.; Martinez G. V.; Gillies R. J.; Sherry A. D. (2009) Responsive MRI agents for sensing metabolism in vivo. Acc. Chem. Res. 42, 948–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravan P. (2009) Protein-targeted gadolinium-based magnetic resonance imaging (MRI) contrast agents: design and mechanism of action. Acc. Chem. Res. 42, 851–862. [DOI] [PubMed] [Google Scholar]
- Ye F.; Wu X.; Jeong E. K.; Jia Z.; Yang T.; Parker D.; Lu Z.-R. (2008) A peptide targeted contrast agent specific to fibrin-fibronectin complexes for cancer molecular imaging with MRI. Bioconjugate Chem. 19, 2300–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Kolodziej A. F.; Greenfield M. T.; Caravan P. (2011) Heteroditopic binding of magnetic resonance contrast agents for increased relaxivity. Angew. Chem., Int. Ed. 50, 2621–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que E. L.; Chang C. J. (2010) Responsive magnetic resonance imaging contrast agents as chemical sensors for metals in biology and medicine. Chem. Soc. Rev. 39, 51–60. [DOI] [PubMed] [Google Scholar]
- Louie A. Y.; Huber M. M.; Ahrens E. T.; Rothbacher U.; Moats R.; Jacobs R. E.; Fraser S. E.; Meade T. J. (2000) In vivo visualization of gene expression using magnetic resonance imaging. Nat. Biotechnol. 18, 321–325. [DOI] [PubMed] [Google Scholar]
- Arena F.; Singh J. B.; Gianolio E.; Stefania R.; Aime S. (2011) Beta-Gal gene expression MRI reporter in melanoma tumor cells. Design, synthesis, and in vitro and in vivo testing of a Gd(III) containing probe forming a high relaxivity, melanin-like structure upon beta-Gal enzymatic activation. Bioconjugate Chem. 22, 2625–2635. [DOI] [PubMed] [Google Scholar]
- Bogdanov A.; Matuszewski L.; Bremer C.; Petrovsky A.; Weissleder R. (2002) Oligomerization of paramagnetic substrates result in signal amplification and can be used for MR imaging of molecular targets. Mol. Imaging 1, 16–23. [DOI] [PubMed] [Google Scholar]
- Lowe M. P.; Parker D.; Reany O.; Aime S.; Botta M.; Castellano G.; Gianolio E.; Pagliarin R. (2001) pH-Dependent modulation of relaxivity and luminescence in macrocyclic gadolinium and europium complexes based on reversible intramolecular sulfonamide ligation. J. Am. Chem. Soc. 123, 7601–7609. [DOI] [PubMed] [Google Scholar]
- Ronald J. A.; Chen J. W.; Chen Y.; Hamilton A. M.; Rodriguez E.; Reynolds F.; Hegele R. A.; Rogers K. A.; Querol M.; Bogdanov A.; Weissleder R.; Rutt B. K. (2009) Enzyme-sensitive magnetic resonance imaging targeting myeloperoxidase identifies active inflammation in experimental rabbit atherosclerotic plaques. Circulation 120, 592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C.; New E. J. (2013) Promising strategies for Gd-based responsive magnetic resonance imaging contrast agents. Curr. Opin. Chem. Biol. 17, 158–166. [DOI] [PubMed] [Google Scholar]
- Davies G.-L.; Kramberger I.; Davis J. J. (2013) Environmentally responsive MRI contrast agents. Chem. Commun. 49, 9704–9721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsitovich P. B.; Burns P. J.; McKay A. M.; Morrow J. R. (2014) Redox-activated MRI contrast agents based on lanthanide and transition metal ions. J. Inorg. Biochem. 133, 143–154. [DOI] [PubMed] [Google Scholar]
- Loving G. S.; Mukherjee S.; Caravan P. (2013) Redox-activated manganese-based MR contrast agent. J. Am. Chem. Soc. 135, 4620–4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que E. L.; New E. J.; Chang C. J. (2012) A cell-permeable gadolinium contrast agent for magnetic resonance imaging of copper in a Menkes disease model. Chem. Sci. 3, 1829–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres P. J.; MacRenaris K. W.; Vogt S.; Meade T. J. (2008) Cell-permeable MR contrast agents with increased intracellular retention. Bioconjugate Chem. 19, 2049–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Q. N.; Ratnakar J. S.; Kovács Z.; Sherry A. D. (2014) Redox- and hypoxia-responsive MRI contrast agents. ChemMedChem 9, 1116–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarsour E. H.; Kumar M. G.; Chaudhuri L.; Kalen A. L.; Goswami P. C. (2009) Redox control of the cell cycle in health and disease. Antioxid. Redox Signaling 11, 2985–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.; Zhang J.; Wang M.; Mei B.; Guan Y.; Liang G. (2013) Detection of glutathione in vitro and in cells by the controlled self-assembly of nanorings. Anal. Chem. 85, 1280–1284. [DOI] [PubMed] [Google Scholar]
- Lim S. Y.; Hong K. H.; Kim D. I.; Kwon H.; Kim H. J. (2014) Tunable heptamethine-azo dye conjugate as an NIR fluorescent probe for the selective detection of mitochondrial glutathione over cysteine and homocysteine. J. Am. Chem. Soc. 136, 7018–7025. [DOI] [PubMed] [Google Scholar]
- Song Y.; Suntharalingam K.; Yeung J. S.; Royzen M.; Lippard S. J. (2013) Synthesis and characterization of Pt(IV) fluorescein conjugates to investigate Pt(IV) intracellular transformations. Bioconjugate Chem. 24, 1733–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Bao C.; Zhong X.; Zhao C.; Zhu L. (2010) Highly selective detection of glutathione using a quantum-dot-based OFF–ON fluorescent probe. Chem. Commun. 46, 2971–2973. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Song H.; Su Y.; Lv Y. (2013) Turn-on persistent luminescence probe based on graphitic carbon nitride for imaging detection of biothiols in biological fluids. Anal. Chem. 85, 11876–11884. [DOI] [PubMed] [Google Scholar]
- Martinelli J.; Fekete M.; Tei L.; Botta M. (2011) Cleavable beta-cyclodextrin nanocapsules incorporating Gd(III)-chelates as bioresponsive MRI probes. Chem. Commun. 47, 3144–3146. [DOI] [PubMed] [Google Scholar]
- Carrera C.; Digilio G.; Baroni S.; Burgio D.; Consol S.; Fedeli F.; Longo D.; Mortillaro A.; Aime S. (2007) Synthesis and characterization of a Gd(III) based contrast agent responsive to thiol containing compounds. Dalton Trans. 4980–4987. [DOI] [PubMed] [Google Scholar]
- Menchise V.; Digilio G.; Fianolio E.; Cittadino E.; Catanzaro V.; Carrera C.; Aime S. (2011) In vivo labeling of B16 melanoma tumor xenograft with a thiol-reactive gadolinium based MRI contrast agent. Mol. Pharmaceutics 8, 1750–1756. [DOI] [PubMed] [Google Scholar]
- Raghunand N.; Guntle G. P.; Gokhale V.; Nichol G. S.; Mash E. A.; Jagadish B. (2009) Design, synthesis, and evaluation of 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid derived, redox-sensitive contrast agents for magnetic resonance imaging. J. Med. Chem. 53, 6747–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z.-R.; Mohs A. M.; Zong Y.; Feng Y. (2006) Polydisulfide Gd(III) chelates as biodegradable macromolecular magnetic resonance imaging contrast agents. Int. J. Nanomed. 1, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z.; Wu X.-M.; Tan M.-Q.; Jesberger J.; Griswold M.; Lu Z.-R. (2013) Synthesis and evaluation of a polydisulfide with Gd-DOTA monoamide side chains as a biodegradable macromolecular contrast agent for MR blood pool imaging. Contrast Media Mol. Imaging 8, 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.-X.; Lu Z.-R. (2013) Gadolinium-based contrast agents for MR cancer imaging. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 5, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G.; Ronald J.; Chen Y.; Ye D.; Pandit P.; Ma M. L.; Rutt B.; Rao J. (2011) Controlled self-assembling of gadolinium nanoparticles as smart molecular magnetic resonance imaging contrast agents. Angew. Chem., Int. Ed. 50, 6283–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G.; Ren H.; Rao J. (2010) A biocompatible condensation reaction for controlled assembly of nanostructures in living cells. Nat. Chem. 2, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New K.; Bryant L. H.; Brechbiel M. W. (2010) Poly(amidoamine) dendrimer based MRI contrast agents exhibiting enhanced relaxivities derived via metal preligation techniques. Bioconjugate Chem. 21, 1014–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Zong H.; Trivedi E. R.; Vesper B. J.; Waters E. A.; Barrett A. G. M.; Radosevich J. A.; Hoffman B. M.; Meade T. J. (2010) Synthesis and characterization of new prophyrazine-Gd(III) conjugates as multimodal MR contrast agents. Bioconjugate Chem. 21, 2267–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accardo A.; Tesauro D.; Aloj L.; Pedone C.; Morelli G. (2009) Supramolecular aggregates containing lipophilic Gd(III) complexes as contrast agents in MRI. Coord. Chem. Rev. 253, 1293–2213. [Google Scholar]
- Ye D.; Liang G.; Ma M. L.; Rao J. (2011) Controlling intracellular macrocyclization for the imaging of protease activity. Angew. Chem., Int. Ed. 50, 2275–2279. [DOI] [PubMed] [Google Scholar]
- Ye D.; Shuhendler A. J.; Cui L.; Tong L.; Tee S. S.; Tikhomirov G.; Felsher D. W.; Rao J. (2014) Bioorthogonal cyclization-mediated in situ self-assembly of small-molecule probes for imaging caspase activity in vivo. Nat. Chem. 6, 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B.; Jeon J.; Palner M.; Ye D.; Shuhendler A.; Chin F. T.; Rao J. (2013) Positron emission tomography imaging of drug-induced tumor apoptosis with a caspase-triggered nanoaggregation probe. Angew. Chem., Int. Ed. 52, 10511–10514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D.; Shuhendler A. J.; Pandit P.; Brewer K. D.; Tee S. S.; Cui L.; Tikhomirov G.; Rutt B.; Rao J. (2014) Caspase-responsive smart gadolinium-based contrast agent for magnetic resonance imaging of drug-induced apoptosis. Chem. Sci. 10.1039/c4sc01392a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloembergen N.; Morgan L. O. (1961) Proton relaxation times in paramagnetic solutions. Effects of electron spin relaxation. J. Chem. Phys. 34, 842–850. [Google Scholar]
- Driggers E. M.; Hale S. P.; Lee J.; Terrett N. K. (2008) The exploration of macrocycles for drug discovery―an underexploited structural class. Nat. Rev. Drug. Discovery 7, 608–624. [DOI] [PubMed] [Google Scholar]
- Feng W.; Yamato K.; Yang L.; Ferguson J. S.; Zhong L.; Zou S.; Yuan L.; Zeng X. C.; Gong B. (2009) Efficient kinetic macrocyclization. J. Am. Chem. Soc. 131, 2629–2637. [DOI] [PubMed] [Google Scholar]
- Marsault E.; Peterson M. L. (2011) Macrocycles are great cycles: application, opportunities, and challenges of synthetic macrocycles in drug discovery. J. Med. Chem. 54, 1961–2004. [DOI] [PubMed] [Google Scholar]
- Seo S. H.; Jones T. V.; Seyler H.; Peters J. O.; Kim T. H.; Chang J. Y.; Tew G. N. (2006) Liquid crystalline order from ortho-phenylene ethynylene macrocycles. J. Am. Chem. Soc. 128, 9264–9265. [DOI] [PubMed] [Google Scholar]
- Yang W.-Y.; Ahn J.-H.; Yoo Y.-S.; Oh N.-E.; Lee M. (2005) Supramolecular barrels from amphiphilic rigid-flexible macrocycles. Nat. Mater. 4, 399–503. [DOI] [PubMed] [Google Scholar]
- Mulder W. J.; Strijkers G. J.; van Tilborg G. A.; Griffioen A. W.; Nicolay K. (2006) Lipid-based nanoparticles for contrast-enhanced MRI and molecular imaging. NMR Biomed. 19, 142–164. [DOI] [PubMed] [Google Scholar]
- Kielar F.; Tei L.; Terreno E.; Botta M. (2010) Large relaxivity enhancement of paramagnetic lipid nanoparticles by restricting the local motions of the Gd(III) chelates. J. Am. Chem. Soc. 132, 7836–7837. [DOI] [PubMed] [Google Scholar]
- Mastarone D. J.; Harrison V. S.; Eckermann A. L.; Parigi G.; Luchinat C.; Meade T. J. (2011) A modular system for the synthesis of multiplexed magnetic resonance probes. J. Am. Chem. Soc. 133, 5329–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garimella P. D.; Datta A.; Romanini D. W.; Raymond K. N.; Francis M. B. (2011) Multivalent, high-relaxivity MRI contrast agents using rigid cysteine-reactive gadolinium complexes. J. Am. Chem. Soc. 133, 14704–14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livramento J. B.; Toth E.; Sour A.; Borel A.; Merbach A. E.; Ruloff R. (2005) High relaxivity confined to a small molecular space: a metallostar-based, potential MRI contrast agent. Angew. Chem., Int. Ed. 44, 1480–1484. [DOI] [PubMed] [Google Scholar]
- Boden C. D. J.; Pattenden G.; Ye T. (1995) The synthesis of optically-active thiazoline and thiazole derived peptides from N-protected alpha-amino-acids. Synlett 417–419. [Google Scholar]
- Niwa K.; Nakamura M.; Ohmiya Y. (2006) Stereoisomeric bio-inversion key to biosynthesis of firefly d-luciferin. FEBS Lett. 580, 5283–5287. [DOI] [PubMed] [Google Scholar]
- Caravan P. (2006) Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem. Soc. Rev. 35, 512–523. [DOI] [PubMed] [Google Scholar]
- Wang S.-Q.; Wu Q.-H.; Wang H.-Y.; Zheng X.-X.; Shen S.-L.; Zhang Y.-R.; Miao J.-Y.; Zhao B.-X. (2014) Novel pyrazoline-based fluorescent probe for detecting glutathione and its application in cells. Biosens. Bioelectron. 55, 386–390. [DOI] [PubMed] [Google Scholar]
- Ortega A. L.; Mena S.; Estrela J. M. (2011) Glutathione in cancer cell death. Cancer 3, 1285–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacciatore I.; Cornacchia C.; Pinnen F.; Mollica A.; Di Stefano A. (2010) Prodrug approach for increasing cellular glutathione levels. Molecules 15, 1242–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.