

Abstract
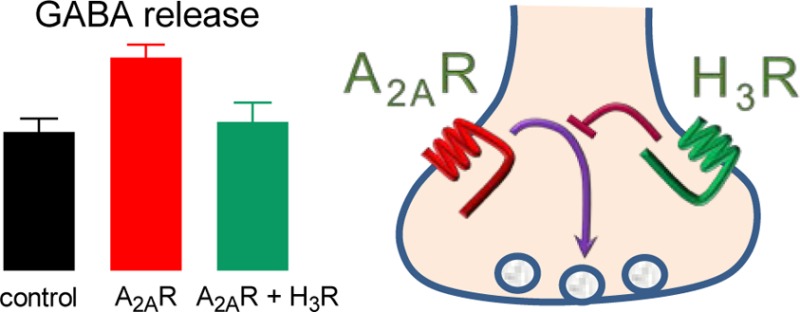
High levels of histamine H3 receptors (H3Rs) are found in the globus pallidus (GP), a neuronal nucleus in the basal ganglia involved in the control of motor behavior. By using rat GP isolated nerve terminals (synaptosomes), we studied whether H3R activation modified the previously reported enhancing action of adenosine A2A receptor (A2AR) stimulation on depolarization-evoked [3H]-GABA release. At 3 and 10 nM, the A2AR agonist CGS-21680 enhanced [3H]-GABA release induced by high K+ (20 mM) and the effect of 3 nM CGS-21680 was prevented by the A2AR antagonist ZM-241385 (100 nM). The presence of presynaptic H3Rs was confirmed by the specific binding of N-α-[methyl-3H]-histamine to membranes from GP synaptosomes (maximum binding, Bmax, 1327 ± 79 fmol/mg protein; dissociation constant, Kd, 0.74 nM), which was inhibited by the H3R ligands immepip, clobenpropit, and A-331440 (inhibition constants, Ki, 0.28, 8.53, and 316 nM, respectively). Perfusion of synaptosomes with the H3R agonist immepip (100 nM) had no effect on K+-evoked [3H]-GABA release, but inhibited the stimulatory action of A2AR activation. In turn, the effect of immepip was blocked by the H3R antagonist clobenpropit, which had no significant effect of its own on K+-induced [3H]-GABA release. These data indicate that H3R activation selectively counteracts the facilitatory action of A2AR stimulation on GABA release from striato-pallidal projections.
Keywords: Adenosine A2A receptor, histamine, histamine H3 receptor, globus pallidus, basal ganglia, GABA release
Histamine regulates a variety of mammalian brain functions including sleep-wake rhythmicity, motor activity, attention and learning as well as different aspects of body homeostasis.1 Upon release from axon terminals of neurons located in the hypothalamic tuberomammillary nucleus the amine acts at three G protein-coupled receptors (H1, H2, and H3) widely expressed in the brain.1,2 Histamine H3 receptors (H3Rs) are mainly located on nerve terminals and control the release and synthesis of histamine and the release of several other neurotransmitters or neuromodulators namely acetylcholine, glutamate, noradrenaline, γ-aminobutyric acid (GABA), dopamine, serotonin (5-hydroxytryptamine, 5-HT), and substance P.2,3
The globus pallidus (GP) forms part of the basal ganglia, a group of subcortical neuronal nuclei involved in the control of motor behavior among other functions.4 There exists strong evidence that the GP plays a critical role in basal ganglia physiology, and thus in the pathophysiology of motor disorders, including Parkinson’s disease, in which alterations in the pattern and synchrony of discharge of pallidal neurons have been reported.5 The neuronal population of the GP is mainly conformed by GABAergic neurons, divided into two groups according to the target of their axons, the subthalamic nucleus (STN) or the striatum.4,5 In turn, the main synaptic afferents to the GP are striato-pallidal GABAergic axons,4 with additional innervation by glutamatergic fibers originated primarily in the STN6,7 and dopaminergic afferents from substantia nigra pars compacta.8
The rat GP is also innervated by histaminergic fibers9 and possesses a high density of H3Rs, mostly located on the axons of neurons projecting to the nucleus, as indicated by the very low levels of the corresponding mRNA found in the GP. Further, H3R mRNA is expressed at high levels by the GABAergic striatal neurons whose axons conform the main synaptic input to the GP.10
Although little is still known about the function of pallidal H3Rs, it is conceivable that these receptors regulate GP synaptic flow at the presynaptic level, as shown for other brain regions.2 In a previous work, we showed that H3R activation reduced the depolarization-evoked release of [3H]-d-aspartate from rat GP slices and inhibited glutamatergic transmission in vivo,11 but failed to detect any significant action on [3H]-GABA release from slices. However, for the terminals of striato-nigral neurons the inhibitory action of H3Rs on depolarization-evoked [3H]-GABA release depends on the concomitant activation of dopamine D1 receptors, which stimulate neurotransmitter release through a signaling pathway involving cAMP and protein kinase A (PKA).12−14 Striatal neurons projecting to the GP do not express D1 receptors but possess adenosine A2A receptors (A2ARs), also coupled to the cAMP/PKA pathway,15 and shown to modulate GABA release in rat GP.16−19 Collaterals of intrinsic GP neurons also release GABA, but these cells do not express A2AR mRNA.20
In the indirect pathway of the basal ganglia, striato-pallidal neurons synapse with GABAergic GP neurons that project to the STN. In turn, glutamatergic STN neurons innervate GABAergic neurons in the substantia nigra pars reticulata (SNr) which control the activity of thalamo-cortical neurons. Under resting conditions, the activity of striatal projection neurons is very low and the spontaneous firing of GP neurons exerts a tonic inhibition on the intrinsic activity of STN neurons.4,21 Stimulation by cortical afferents activates striato-pallidal neurons and thus the indirect pathway making the regulation of GABA release by striato-pallidal terminals critical for basal ganglia function.
In this work, we therefore studied the effect of coactivating A2A and H3 receptors on the depolarization-evoked [3H]-GABA release from rat GP isolated nerve terminals (synaptosomes), and our results showed that H3R activation counteracted the stimulatory action of A2ARs.
Results and Discussion
1. Morphological Characteristics of GP Synaptosomes
Figure 1A illustrates the dissection of the GP from coronal brain slices, and Figure 1B and C shows representative electron micrographs of the GP synaptosomal preparation. There were numerous rounded structures bearing the characteristics of nerve terminals, namely, small and rounded vesicles, mitochondria, and electro-dense regions most likely corresponding to active zones. Figure 1B and C indicates that our preparation was enriched in isolated nerve terminals.
Figure 1.
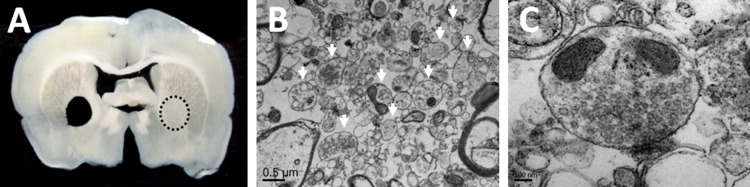
Rat globus pallidus synaptosomes. (A) Brain slice illustrating the dissection of the globus pallidus (dotted circle). (B) Micrograph of the synaptosomal preparation at ×20 000. There are numerous intact rounded structures (arrows) of a range of size bearing the characteristics of nerve terminals, namely, small and rounded vesicles, mitochondria, and electro-dense regions most likely corresponding to active zones. Synaptosomes constitute a significant proportion of the particulate matter present in the preparation. Myelin was identified by its multilamellar structure and mitochondria were identified by the presence of a double membrane and cristae. (C) Micrograph of the synaptosomal preparation at ×50 000 showing a single synaptosome.
2. [3H]-GABA Uptake by Rat GP Synaptosomes
In the rat GP, the GABA transporters GAT-1 and GAT-3 are mainly expressed on axonal terminals and glial cells, respectively.22 Figure 2A shows that SKF-89976A, a high-affinity inhibitor of the GAT-1 transporter, fully inhibited the specific [3H]-GABA uptake by rat GP synaptosomes. The effect was concentration-dependent (Figure 2B) with IC50 0.24 μM (pIC50 6.62 ± 0.06, mean ± standard error, SEM, three experiments). A comparison with the IC50 values reported for cloned rat GABA transporters (GAT-1 0.53 μM, GAT-3 4 390 μM)23 indicated that in our synaptosomal preparation GABA was taken up mostly, if not exclusively by the isolated nerve terminals.
Figure 2.

Inhibition by SKF-89976A of [3H]-GABA uptake by rat GP synaptosomes. Synaptosomes were incubated for 4 min with [3H]-GABA/GABA (50 nM/3 μM) in the absence and presence of the indicated concentrations of the inhibitor SKF-89976A, added 10 min before. (A) Representative experiment. Values are means ± SEM from three replicates. Nonspecific uptake was determined in parallel samples incubated at 4 °C. (B) Analysis of the concentration–response curve for SKF-89976A. [3H]-GABA uptake is expressed as percentage of control values after subtraction of nonspecific uptake. Values are means ± SEM of three replicates from the experiment shown in panel (A). The curve drawn is the best-fit adjust to a logistic equation. The IC50 value is given in the text.
3. [3H]-NMHA Binding to Rat GP Membranes
Saturation [3H]-NMHA binding to GP membranes yielded maximum specific binding (Bmax) 1327 ± 79 fmol/mg protein (three experiments, Figure 3A) and equilibrium dissociation constant (Kd) 0.74 nM (pKd 9.13 ± 0.05). Although the Kd value is similar to that found for membranes obtained from the whole rat GP (Kd 0.58 nM, Bmax 162 ± 29 fmol/mg protein),11 the density is 8-fold higher, indicating that H3Rs were highly concentrated in the GP nerve terminals.
Figure 3.

Binding of N-α-[methyl-3H]-histamine ([3H]-NMHA) to membranes from rat globus pallidus synaptosomes. (A) Saturation binding. Membranes were prepared as described in Methods and then incubated with the indicated concentrations of [3H]-NMHA. Specific receptor binding was determined by subtracting the binding in the presence of 10 μM histamine from total binding. Points are means ± SEM from triplicate determinations from a single experiment, which was repeated a further twice. The line drawn is the best fit to a hyperbola. Best-fit values for the equilibrium dissociation constant (Kd) and maximum binding (Bmax) are given in the text. (B) Inhibition by the H3R agonist immepip and the antagonists/inverse agonists clobenpropit and A-331440. Membranes were incubated with ∼1.5 nM [3H]-NMHA and the indicated drug concentrations. Values are expressed as the percentage of control specific binding and are means ± range from duplicate determinations from a representative experiment. The line drawn is the best fit to a logistic equation for a one-site model. pKi values calculated from the best-fit IC50 estimates are given in the text.
Specific [3H]-NMHA binding was inhibited in a concentration-dependent manner by the H3R agonist immepip and the antagonists/inverse agonists clobenpropit and A-331440 (Figure 3B). Values for the inhibition constants (−log Ki, pKi) from three experiments were 9.55 ± 0.20 (Ki 0.28 nM) for immepip, 8.07 ± 0.21 (Ki 8.53 nM) for clobenpropit, and 6.50 ± 0.04 (Ki 316 nM) for A-331440. These values allowed for the calculation of receptor occupancy by H3R ligands in functional experiments.
4. Characteristics of Depolarization-Evoked [3H]-GABA Release from GP Synaptosomes
Changing the K+ concentration in the superfusion medium from 4 to 20 mM increased [3H]-GABA efflux from rat GP synaptosomes to 341 ± 15% and 278 ± 12% of basal at the peak of release in the first and second depolarizing stimuli, respectively (Figure 4A), with an overall S2/S1 ratio of 0.602 ± 0.021 (45 experiments).
Figure 4.

Effect of the adenosine A2A receptor agonist CGS-21680 on depolarization-evoked [3H]-GABA release from rat globus pallidus synaptosomes. (A) Representative experiments for 3 and 300 nM. Labeled synaptosomes were perfused with KRH solution (4 mM K+), and neurotransmitter release was evoked by raising the K+ concentration in the perfusion medium to 20 mM for the periods indicated by the vertical gray bars. Where required, drugs under test were present for the period indicated by the black bar. Values are expressed as a percentage of [3H]-GABA in fraction 3 and represent the means ± SEM of 4–6 replicates. (B) Statistical analysis for 3 and 10 nM CGS-21680. Values are means ± SEM from three experiments. P values are for the comparison with control release (no drugs added). For panels (B) and (C), the statistical analysis was performed with ANOVA and Dunnett’s post hoc test. (C) Blockade by the A2A receptor antagonist ZM-241385 of the effect of 3 nM CGS-21680. Values are means ± SEM from three experiments. NS, not different from control release.
In synaptosomes perfused with KRH solution without CaCl2, K+-evoked [3H]-GABA release was reduced by 73.6 ± 10.1% (n = 3, P < 0.001 when compared with the release in the presence of 1.8 mM CaCl2, Student’s t test; data not illustrated), indicating that most of the depolarization-induced [3H]-GABA release was due to exocytosis. Basal [3H]-GABA efflux was modestly but significantly reduced when CaCl2 was omitted from the perfusion medium (81.0 ± 1.5% of control values, P = 0.001, Student’s t test).
Depolarization-evoked [3H]-GABA release was not modified by the presence in the perfusion medium of the GAT-1 inhibitor SKF-89976A (3 μM, 103.1 ± 8.0% of control values, P = 0.717, Student’s t test; data not illustrated), indicating that under our experimental conditions the perfusion prevented that the released mixture of [3H]-GABA/GABA yielded the concentration required to significantly activate GABA transport by GP synaptosomes.
5. Effect of A2AR Activation on Depolarization-Evoked [3H]-GABA Release from GP Synaptosomes
The activation of A2ARs has been shown to modulate GABA release from rat GP slices. However, whereas Floran et al.19 reported that the selective agonist CGS-2168024 increased K+-evoked [3H]-GABA release in a wide concentration range (10 nM to 10 μM, EC50 65 nM), Zahniser and co-workers16,17 showed a dual action of the agonist on the release of endogenous GABA induced by electrical stimulation with a facilitatory effect at 1 and 10 nM and inhibition at higher concentrations (100 and 1000 nM).
We therefore tested four concentrations (3, 10, 30, and 300 nM) of the A2AR agonist on K+-evoked [3H]-GABA release from rat GP synaptosomes. Figure 4A illustrates the experimental protocol and the effect of 3 and 300 nM CGS-21680. At 3 and 10 nM, CGS-21680 significantly enhanced K+-evoked [3H]-GABA release to 159.9 ± 20.3 and 143.3 ± 9.4% of control values (S2/S1 ratios: control 0.426 ± 0.029, 3 nM CGS-21680 0.699 ± 0.099, 10 nM CGS-21680 0.670 ± 0.051, three experiments, P < 0.05 for both concentrations when compared with control values, Figure 4B). These concentrations (3 and 10 nM) are below the Ki value reported for CGS-21680 (15 nM),25 indicating an amplification effect in the signaling pathway underlying the receptor action.
The selective A2AR antagonist ZM-24138524 prevented the enhancing action of 3 nM CGS-21680 on K+-evoked [3H]-GABA release (Figure 4C; CGS-21680 135.2 ± 5.9% of controls, CGS-21680 and 100 nM ZM-241385 104.9 ± 10.0% of controls; S2/S1 ratios: control 0.535 ± 0.041; CGS-21680 0.750 ± 0.022, P < 0.05; CGS-21680 and ZM-241385 0.578 ± 0.047, P > 0.05, three experiments). This result confirmed that the stimulatory action of CGS-21680 was receptor-mediated.
In contrast to the effect at 3 and 10 nM, in a different series of experiments (not illustrated), 30 nM CGS-21680 had no significant effect on K+-evoked [3H]-GABA release (105.8 ± 7.7% of controls, S2/S1 ratios: control 0.656 ± 0.040, CGS-21680 0.662 ± 0.039; P > 0.05), whereas at 300 nM in six out of seven experiments there was a reduction in the release when compared with control values (mean 90.2 ± 7.1% of controls), but the analysis of the S2/S1 ratios did not yield statistical significance (S2/S1 ratio 0.601 ± 0.036; P > 0.05). Adenosine A1 receptors are expressed at high density in the rat GP (475 ± 37 fmol/mg protein versus 270 ± 20 fmol/mg protein for A2ARs),16 and electrophysiological evidence indicates that they are also present in striato-pallidal neurons.26 Therefore, and as suggested previously for GP slices,16 one possibility is that at high concentrations CGS-21680 binds and activates a fraction of A1 receptors (Ki for the cloned rat receptor 2600 nM)25 which through their coupling to Gαi/o proteins inhibit both cAMP formation and voltage-activated Ca2+ channels27 and can thus oppose the A2AR-mediated stimulation of GABA release. However, a dual effect of CGS-21680 through different signaling pathways,15 as shown for the A2AR-mediated modulation of acetylcholine release,28 cannot be excluded.
6. Effect of H3R Activation on Depolarization-Evoked [3H]-GABA Release from GP Synaptosomes
When added alone to the perfusion medium, neither the H3R agonist immepip (100 nM) nor the antagonist/inverse agonist clobenpropit (3 μM) had significant effect on depolarization-evoked [3H]-GABA release (111.3 ± 7.0% and 98.4 ± 5.6% of control values, respectively; S2/S1 ratios: control 0.565 ± 0.030, immepip 0.626 ± 0.029, clobenpropit 0.558 ± 0.058, n = 3, P > 0.05 for both drugs when compared with control values; Figure 5A).
Figure 5.

Effect of histamine H3 receptor activation on the stimulatory action of the A2A receptor agonist CGS-21680 on depolarization-evoked [3H]-GABA release from rat globus pallidus synaptosomes. The experimental protocol was as described in the legend to Figure 4. Where required, drugs under test were present for 5 min (CGS-21680 and immepip) or 10 min (clobenpropit) before and during the second depolarizing stimulus. (A) Effect of the H3 receptor agonist immepip (100 nM) or the antagonist/inverse agonist clobenpropit (3 μM) alone on depolarization-evoked [3H]-GABA release. Values are means ± SEM from three experiments. NS, no statistically different from control release (no drugs added), ANOVA and Dunnett’s post hoc test. (B) Inhibition by immepip (100 nM) of the effect of the A2A receptor agonist CGS-21680 (3 nM). Values are means ± SEM from three experiments. The statistical analysis was performed with ANOVA and Dunnett’s test. (C) Effect of different concentrations of immepip. To allow for the variability between experiments, values were expressed as percentage of control release. Values are means ± SEM from the combined data from five experiments with three to five determinations for each concentration of immepip. (D) Blockade by clobenpropit (3 μM) of the effect of immepip (100 nM). Values are means ± SEM from four experiments. The statistical analysis was performed with ANOVA and Tukey’s post hoc test.
However, and as shown in Figure 5B, immepip inhibited the stimulatory effect of A2AR activation on K+-evoked [3H]-GABA release (3 nM CGS-21680, 150.1 ± 13.7% of control values; CGS-21680 and immepip, 102.5 ± 6.3% of controls; S2/S1 ratios: control 0.630 ± 0.036; 3 nM CGS-21680 0.832 ± 0.044; CGS-21680 and immepip 0.581 ± 0.065; P < 0.05 and P > 0.05 for CGS-21680, and CGS-21680 and immepip, respectively, when compared with controls, three experiments). The inhibitory effect of immepip on the A2AR-mediated enhancement of K+-evoked [3H]-GABA release was concentration-dependent, with IC50 5.2 nM (pIC50 8.28 ± 0.27; Figure 5C).
Figure 5D illustrates a different series of experiments in which the effect of immepip (100 nM) was partially blocked by the H3R antagonist/inverse agonist clobenpropit (3 nM CGS-21680, 148.7 ± 9.8% of controls; immepip 104.9 ± 7.5% of controls; CGS 21680 and immepip and 3 μM clobenpropit 125.1 ± 5.0% of controls; S2/S1 ratios: control 0.582 ± 0.054, 3 nM CGS-21680 0.832 ± 0.067, P < 0.001 when compared with control values; immepip 0.558 ± 0.040, P > 0.05 versus controls; CGS-21680 and immepip and clobenpropit 0.706 ± 0.043, P < 0.01 when compared with CGS-21680 and immepip and P < 0.05 when compared with CGS-21680 alone; four experiments). The extent of blockade by clobenpropit of the effect of immepip was 47.2 ± 4.1%, a value within the range calculated (46–54%) on the basis of the affinities of the H3R for the ligands determined from the inhibition of [3H]-NMHA binding to GP synaptosomal membranes (Ki values: immepip 0.28 nM and clobenpropit 8.53 nM). We did not attempt to test larger concentrations of clobenpropit because at micromolar concentrations imidazole-containing compounds can directly inhibit P- and N-type voltage-activated Ca2+ channels (IC50 values for antazoline 10.5 ± 1.1 and 109.5 ± 9.8 μM, respectively).29 Further, the use of the nonimidazole antagonist/inverse agonist A-331440 was prevented by the low affinity of pallidal H3Rs for this drug (Ki 316 nM; Results, section 3).
The partial inhibition by clobenpropit of the agonist effect could also be explained by the antagonist not yielding equilibrium with H3Rs present in GP synaptosomes. However, the binding of 0.2 nM [3H]-clobenpropit (21 °C) to guinea pig cerebro-cortical membranes yields its maximum at 10–13 min,30 and pallidal synaptosomes were perfused with a 15 000-fold higher concentration (3 μM) for 10 min at 37 °C. Therefore, under our experimental conditions, drug affinities, and not an equilibrium issue, appear to account for the partial blockade by clobenpropit of the effect of immepip.
In a previous work, we failed to observe any significant effect of H3R activation on depolarization-stimulated [3H]-GABA release from rat GP slices,11 in contrast to the action reported for slices from rat SNr and striatum.12−14 The nerve terminals of GABAergic striato-nigral neurons are endowed with both dopamine D1 and histamine H3 receptors,31 and in both nigral and striatal slices H3R agonists had no effect when tested alone but markedly inhibited the enhancing effect of D1 receptor stimulation on K+-evoked [3H]-GABA release.12,13
H3Rs couple to Gαi/o proteins and thus their inhibitory effect on neurotransmitter release most likely involves the reduction in depolarization-induced calcium entry,32,33 via the action of Gβγ complexes at the pore-forming α1-subunit of N- and P/Q-type voltage-operated calcium channels.34 In this regard, D1 receptor-mediated facilitation and H3R-mediated inhibition of GABA release from striatal terminals appear to converge at P/Q-type Ca2+ channels, with the facilitatory effect involving the cAMP/PKA pathway.14 In line with an action of H3Rs at P/Q-type channels, in dissociated hypothalamic neurons, the H3R-mediated reduction in the frequency of spontaneous GABAergic inhibitory postsynaptic currents (sIPSCs) was occluded by blocking P/Q-type Ca2+ channels but not by N- or L-type Ca2+ channel blockers.35 Further, A2AR-mediated enhancement of acetylcholine release from striatal synaptosomes relies on two signaling pathways, one involving the cAMP/PKA pathway and P-type Ca2+ channels, and the other mediated by a cholera toxin-insensitive G protein, protein kinase C (PKC), and N-type calcium channels.28 Because both D1 and A2A receptors stimulate adenylyl cyclase activity, one plausible explanation for the selective action of H3R activation on A2AR-mediated facilitation of [3H]-GABA release reported herein is that the cAMP/PKA pathway facilitates the opening of P/Q-type Ca2+ channels present in the striato-pallidal axons and that are also controlled by H3Rs.
Upon transfection, H3Rs formed heteromers with dopamine D1 and D2 receptors,36,37 and in striatal membranes H3R activation decreased the affinity of D2 receptors for their agonists.36 Further, in SK-N-MC cells expressing H3 and D1 receptors, the coupling of the latter shifted from Gαs to Gαi/o proteins, and therefore, their activation no longer resulted in cAMP formation but in inhibition of forskolin-induced cAMP accumulation.37 Thus, another and interesting possibility is that the functional interaction between H3 and A2A receptors reported herein for GABA release is underlain by the formation of H3R–A2AR complexes. In regard to this point, preliminary experiments with rat GP synaptosomal membranes show that the H3R agonist immepip increases in a modest (2-fold) but significant manner the Ki value for the A2AR agonist CGS-21680, suggesting that dimerization between H3 and A2A receptors does take place and reduces A2AR affinity for its agonists.
H3Rs may have spontaneous or constitutive activity, which for cloned receptors depends on the expression level and for native receptors has been reported mainly for H3 autoreceptors.38 For H3Rs expressed in CHO cells, constitutive activity was evident at levels greater than 300 fmol/mg protein39 and the very high density (1327 ± 79 fmol/mg protein) of H3Rs present in GP synaptosomal membranes raised thus the possibility that these receptors possessed constitutive activity. However, the lack of effect of the H3R antagonist/inverse agonist clobenpropit on K+-evoked [3H]-GABA release from rat GP synaptosomes indicated that H3Rs located on GABAergic afferents did not have constitutive activity nor were they significantly occupied by endogenous histamine, most likely removed by the perfusion procedure.
7. Possible Trans-Synaptic Effects of H3R Activation on Depolarization-Evoked [3H]-GABA Release
Given the complexity of the basal ganglia synaptic circuitry, our experimental approach bears intrinsic limitations that include, but are not restricted to, possible trans-synaptic effects of H3R activation. In this regard, in the rat GP, the release of GABA is inhibited by dopamine D2, serotonin 5-HT1B, and group III metabotropic or kainate glutamate receptors,17,40−42 and H3Rs inhibit the release of dopamine, serotonin, and glutamate.11,43,44 However, H3R activation does not inhibit K+-evoked [3H]-dopamine release from GP slices,11 and for glutamate and serotonin the predicted outcome of the activation of H3Rs located on the corresponding nerve terminals would be the disinhibition of GABA release, opposite to the results presented herein. Because the activation of muscarinic receptors stimulate spontaneous [3H]-GABA release from rat GP slices45 and H3R activation reduces acetylcholine release,46 the H3R-mediated reduction in [3H]-GABA release reported herein could also involve an indirect action due to the inhibition of depolarization-induced acetylcholine release from GP synaptosomes.
8. Functional Implications
Out of the three types of GP neurons characterized on the basis of their electrophysiological and morphological properties, type A and B (GABAergic) neurons fire spontaneous action potentials and project mainly to the subthalamic nucleus (STN) although they also directly contact SNr neurons.4,47 STN neurons also possess autonomous generation of action potentials, and their rate and pattern of activity is precisely regulated by GABAergic afferents from the GP and by glutamatergic afferents from the cerebral cortex.48,49
The axons of STN neurons release glutamate upon GABAergic neurons located in both SNr and GP, and according to the disinhibition model of the basal ganglia function4 the H3R-mediated inhibition of GABA release reported herein would translate to increased neuronal firing of GP neurons, leading to reduced firing of SNr neurons by two mechanisms. First, the increased GABAergic input would diminish the activity of STN neurons and hence their excitatory input onto SNr neurons. Second, the disinhibition of GP neurons will increase their direct inhibitory effect on SNr neurons. The reduced activity of SNr neurons will then lead to the disinhibition of thalamo-cortical neurons and thus in facilitation of motor behavior.
Of note, if analyzed separately, the H3R-mediated inhibition of GABA release described herein would have functional actions on the firing of GP neurons opposite to the inhibition of glutamate release described previously.11 In contrast to other neurons of the basal ganglia, striatal projection neurons are mostly silent and require cortical excitation for their activation. It is thus tempting to propose that under resting conditions H3Rs control mainly the feedback circuit GP → STN → GP by reducing glutamate release onto GP neurons,11 leading to increased activity of both STN and SNr neurons and thus to the inhibition of thalamo-cortical neurons. In turn, when motor programs are executed, H3R-mediated inhibition of GABA release may control the circuit striatum → GP → STN → SNr, leading to reduced activity of STN and SNr neurons and disinhibition of thalamo-cortical neurons.
This proposal is highly speculative, and both in vitro and in vivo experiments are required for a better understanding of the function of pallidal H3Rs and its consequences on basal ganglia physiology. One important point to consider is that H3R-mediated inhibition of GABA release from striato-pallidal terminals proposed to result in increased activity of GP neurons and thus in enhanced GABA release onto STN neurons, can paradoxically increase the efficacy of cortical excitatory synaptic inputs to the STN through the activation of GABAA and/or GABAB receptors that inhibit/reset autonomous activity by deactivating postsynaptic voltage-dependent Na+ channels and generate hyperpolarization-induced rebound burst firing through the deinactivation of postsynaptic voltage-dependent Ca2+ and Na+ channels.21
Finally, in rat GP slices, histamine increases neuronal firing (EC50 ∼10 μM) through a postsynaptic, H2 receptor-mediated action.50 This information and our results indicate that histamine controls at the pre- and postsynaptic levels the activity of GP neurons. However, the affinity of H3 and H2 receptors for histamine differs substantially (Ki 5 nM and 2 μM for human receptors, respectively),51,52 and the effect of H3Rs may therefore be exerted tonically, whereas H2 receptor-mediated actions may predominantly take place during the arousal state, in which the activity of histaminergic neurons increases markedly, leading to enhanced transmitter release in the target nuclei.1
9. Conclusion
The GP has emerged as a key point in the control of the basal ganglia motor output,53,54 and in this work we showed that histamine H3 receptor activation counteracts the facilitatory action of adenosine A2A receptors on GABA release from striato-pallidal afferents. Through this presynaptic effect, H3 receptors could contribute to the regulation of basal ganglia function.
Methods
1. Animals
Rats (males, Wistar strain, 250–300 g), bred in the Cinvestav facilities, were used throughout the experiments. All procedures were approved controlled by the Cinvestav Animal Care Committee and in accordance to the guidelines for the care and use of laboratory animals issued by the National Institutes of Health (NIH Publications No. 8023, revised 1978) and the Mexican Council for Animal Care. All efforts were made to minimize animal suffering and to use only as many animals were required for proper statistical analysis.
2. Synaptosome Preparation
After decapitation, the brain was quickly removed from the skull and the forebrain was cut and immersed in ice-cold Krebs-Henseleit solution. Coronal slices (300 μm thick) were then obtained with a vibratome (World Precision Instruments, Sarasota, FL). The pallidal tissue was carefully dissected from the slices, avoiding the adjacent striatum which contains high levels of H3Rs (Figure 1A). The composition of the Krebs-Henseleit solution was as follows (mM): NaCl 116, KCl 3, MgSO4 1, KH2PO4 1.2, NaHCO3 25, d-glucose 11; pH, 7.4 after saturation with O2/CO2, 95:5% v:v. To reduce excitoxicity, CaCl2 was not added to the solution.
Synaptosomes were prepared essentially as described by Cristóvão-Ferreira et al.55 Briefly, GP slices from five rats were placed in 10 mL of 0.32 M sucrose solution containing 10 mM Hepes, 1 mg/mL bovine serum albumin, and 1 mM EDTA (pH 7.4 with NaOH) and then homogenized using 10 strokes of a hand-held homogenizer. The suspension was centrifuged (1500g, 4 °C, 10 min), and the supernatant was collected and centrifuged (14 000g, 12 min, 4 °C). The resulting pellet was resuspended in 5 mL of Percoll (45%) in a modified Krebs-Hepes solution (in mM: NaCl 140, Hepes 10, d-glucose 5, KCl 4.7, EDTA 1, pH 7.3 with NaOH). After centrifugation (2 min, 14 000g, 4 °C), the upper phase was collected and brought up to 20 mL with Krebs-Ringer-Hepes (KRH) solution before further centrifugation (20 000g, 20 min). The supernantant was discarded, and the pellet (synaptosomes) was resuspended in the appropriate solution. The composition of the KRH buffer was as follows (mM): NaCl 113, NaHCO3 25, KCl 4.7, MgCl2 1.2, KH2PO4 1.2, CaCl2 1.8, d-glucose 15, Hepes 20; pH 7.4 with NaOH.
3. Electron Microscopy of the GP Synaptosome Preparation
The synaptosomal preparation was washed twice in 1 mL phosphate-buffered saline solution (PBS) by centrifugation at 15 000g (3 min). The final pellet was fixed (1 h at room temperature) with glutaraldehyde (5% in PBS) followed by mixing by inversion for 10 min. After three washes with 1 mL of PBS, each of 5 min, samples were postfixed in PBS containing 2% osmium tetroxide for 30 min at room temperature. Samples were washed twice in 1 mL of PBS for 5 min and then four times (5 min each) with 1 mL of filtered distilled water before prestaining with uranile (1% in water) for 30 min. After two washes (5 min each) with 1 mL of filtered distilled water, samples were dehydrated in a graded series of alcohol (50, 60, 70 and 80% ethanol, 10 min each, then 90% ethanol for 15 min and finally three times in 100% ethanol for 15 min under stirring). Samples were then infiltrated in Spurr resin under mixing (50% resin/50% ethanol for 3 h, 75% resin/25% ethanol overnight and 100% resin for 6 h with changes of the solution each 2 h for the last step). Finally, samples were incubated for 72 h at 60 °C. Thin sections (∼70 nm thick) were cut on a Reichert Ultracut instrument, stained with lead citrate, and examined in a JEM 1400 transmission electron microscope (Cinvestav Unit of Electron Microscopy). The preparations were analyzed by randomly selecting grid squares at low magnification (×20), at which details of the sample were not visible. Representative fields were then photographed at ×20 000 or ×50 000.
4. Binding of N-α-[Methyl-3H]-histamine ([3H]-NMHA) to Synaptosomal Membranes
The synaptosomal pellet was resuspended in 30 mL of 10 mM Tris-HCl solution (4 °C) containing 1 mM EGTA and lysed with a Polytron (3 cycles, 5 s each). The suspension was centrifuged at 32 000g (20 min, 4 °C), and the pellet (synaptosomal membranes) was resuspended in incubation buffer (50 mM Tris-HCl, 5 mM MgCl2, pH 7.4). Protein contents were determined via the bicinchoninic acid assay (BCA; Pierce, Rockford, IL).
Binding experiments (∼10 μg protein aliquots) were carried out and analyzed as described in detail elsewhere.11 Saturation binding data were fitted to a hyperbola and inhibition data to a logistic (Hill) equation (nonlinear regression with Prism 5, Graph Pad Software, San Diego, CA). Values for inhibition constants (Ki) were calculated according to the equation:56Ki = IC50/1 + {[D]/Kd}, where [D] is the concentration of [3H]-NMHA present in the assay and Kd is the mean value for the equilibrium dissociation constant estimated from saturation analysis.
5. Uptake of [3H]-GABA by GP Synaptosomes
Synaptosomes were resuspended in KRH solution supplemented with 10 μM aminooxyacetic acid (to prevent degradation of [3H]-GABA). Aliquots (140 μL) were placed into plastic tubes and incubated for 15 min at 37 °C before the addition of a mixture of [3H]-GABA/GABA in a 50 μL volume to yield the required concentrations (50 nM/3 μM). After 5 min at 37 °C, incubations were filtered through Whatman GF/B glass fiber paper, presoaked in 0.3% polyethylenimine. Filters were washed three times with ice-cold KRH solution and soaked in 4 mL of scintillator, and then the tritium content was determined by scintillation counting. Nonspecific uptake was determined in samples incubated at 4 °C. Where required, the GABA uptake inhibitor SKF-89976A was added in a 10 μL volume 10 min before the labeled neurotransmitter.
6. Depolarization-Evoked [3H]-GABA Release from GP Synaptosomes
Synaptosomes were suspended in KRH solution supplemented with 10 μM aminooxyacetic acid, 2 U/mL adenosine deaminase (to break down endogenous adenosine), and [3H]-GABA/GABA (80 nM/3 μM). After incubation for 30 min at 37 °C, the synaptosomal suspension was apportioned randomly between the chambers of a superfusion apparatus (15 chambers in parallel; 100 μL per chamber) and superfused (1 mL/min) with KRH medium.
Synaptosomes were perfused for 20 min before the collection of 17 fractions of 1 mL (1 min) each. [3H]-GABA release was stimulated by changing to a solution containing high K+ (20 mM, KCl substituted for NaCl) for fractions 4 and 13, returning to normal KRH solution between these fractions and after the second K+ stimulus. Drugs under test were present 5 min (CGS-21680 and immepip), 7 min (ZM-241385), or 10 min (clobenpropit) before and throughout the second K+ stimulus (i.e., fractions 8–13 for CGS-21680 and immepip, fractions 6–13 for ZM-241385 and fractions 8–13 and a 5 min perfusion period between fractions 5 and 6 which was not collected for clobenpropit). The double-pulse protocol allows for the same synaptosomal sample being the control for the effect of drugs under test.
For experiments where the Ca2+-dependence of [3H]-GABA release was tested, synaptosomes were perfused for 15 min before and throughout the collection of fractions with KRH solution without CaCl2 (normal or high K+ as required).
The superfusate fractions were mixed with 4 mL of scintillation liquid, and the tritium content was determined by scintillation counting. The amount of tritium remaining in the synaptosomal tissue was determined by treating each chamber with 0.5 mL of HCl (1 M) for 30 min before addition of scintillator. Tritium efflux into the superfusate was calculated as a fraction of tritium present in the corresponding tissue at the onset of the respective collection period. To allow for variations between chambers, fractional values were transformed to a percentage of the fraction collected immediately before the first change to the high K+ medium (i.e., the release in fraction 3 was set to 100%). To test for statistical differences between treatments, after subtraction of basal release, the area under the release curve for 6 fractions after the change to high K+ (i.e., fractions 3–8 and 12–17) was determined for each individual chamber and the ratio of the second over the first K+ stimuli (S2/S1) was calculated. Statistical comparisons were performed with Student’s t test or one-way ANOVA and post hoc Dunnett’s or Tukey’s test (Graph Pad Prism 5.0) as appropriate.
7. Drugs
The following drugs were purchased from Sigma-Aldrich (Mexico City, Mexico): Adenosine deaminase, aminooxyacetic acid hemihydrochloride, CGS-21680 (2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine hydrochloride hydrate), clobenpropit dihydrobromide, histamine dihydrochloride, immepip dihydrobromide, SKF-89976A (1-(4,4-diphenyl-3-butenyl)-3-piperidinecarboxylic acid hydrochloride), ZM-241385 (4-(2-[7-amino-2-{2-furyl}{1,2,4}triazolo{2,3-a} {1,3,5}triazin-5-yl-amino]ethyl)phenol). [2,3-3H]-γ-Aminobutyric acid ([3H]-GABA, 82 Ci/mmol) and N-α-[methyl-3H]-histamine (85.4 Ci/mmol) were from PerkinElmer (Boston, MA).
Acknowledgments
G.-E.M.-F. and R.M-G. hold Conacyt predoctoral scholarships. We are grateful to Sirenia González-Pozos for her excellent technical assistance in the electron microscopy analysis of the synaptosomal preparation.
Glossary
Abbreviations
- A2AR
adenosine A2A receptor
- GABA
γ-aminobutyric acid
- GP
globus pallidus
- [3H]-NMHA
N-α-[methyl-3H]histamine
- H3R
histamine H3 receptor
- SNr
substantia nigra pars reticulata
- STN
subthalamic nucleus
Author Contributions
G.-E.M.-F. and J.-A.A.-M. designed the study. G.-E.M.-F., J.E.-S., and R.G.-P. conducted experiments. G.-E.M.-F. and J.-A.A.-M. performed data analysis. R.M-G. helped conceive the study. G.-E.M.-F. and J.-A.A.-M. wrote the manuscript.
Supported by Cinvestav and Conacyt (Grant 128205 to J.-A.A.-M.).
The authors declare no competing financial interest.
References
- Haas H. L.; Sergeeva O. A.; Selbach O. (2008) Histamine in the nervous system. Physiol. Rev. 88, 1183–1241. [DOI] [PubMed] [Google Scholar]
- Panula P.; Nuutinen S. (2013) The histaminergic network in the brain: basic organization and role in disease. Nat. Rev. Neurosci. 14, 472–487. [DOI] [PubMed] [Google Scholar]
- Feuerstein T. J. (2008) Presynaptic receptors for dopamine, histamine, and serotonin. Handb. Exp. Pharmacol. 184, 289–338. [DOI] [PubMed] [Google Scholar]
- Bolam J. P.; Hanley J. J.; Booth P. A.; Bevan M. D. (2000) Synaptic organisation of the basal ganglia. J. Anat. 196, 527–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C. S.; Surmeier D. J.; Yung W. H. (2005) Striatal information signaling and integration in globus pallidus: timing matters. Neurosignals 14, 281–289. [DOI] [PubMed] [Google Scholar]
- Naito A.; Kita H. (1994) The cortico-pallidal projection in the rat: an anterograde tracing study with biotinylated dextran amine. Brain Res. 653, 251–257. [DOI] [PubMed] [Google Scholar]
- Bevan M. D.; Magill P. J.; Terman D.; Bolam J. P.; Wilson C. J. (2002) Move to the rhythm: oscillations in the subthalamic nucleus-external globus pallidus network. Trends Neurosci. 25, 525–531. [DOI] [PubMed] [Google Scholar]
- Anaya-Martinez V.; Martinez-Marcos A.; Martinez-Fong D.; Aceves J.; Erlij D. (2006) Substantia nigra compacta neurons that innervate the reticular thalamic nucleus in the rat also project to striatum or globus pallidus: implications for abnormal motor behavior. Neuroscience 143, 477–486. [DOI] [PubMed] [Google Scholar]
- Panula P.; Pirvola U.; Auvinen S.; Airaksinen M. S. (1989) Histamine-immunoreactive nerve fibers in the rat brain. Neuroscience 28, 585–610. [DOI] [PubMed] [Google Scholar]
- Pillot C.; Heron A.; Cochois V.; Tardivel-Lacombe J.; Ligneau X.; Schwartz J. C.; Arrang J. M. (2002) A detailed mapping of the histamine H3 receptor and its gene transcripts in rat brain. Neuroscience 114, 173–193. [DOI] [PubMed] [Google Scholar]
- Osorio-Espinoza A.; Alatorre A.; Ramos-Jimenez J.; Garduño-Torres B.; García-Ramírez M.; Querejeta E.; Arias-Montaño J.-A. (2011) Pre-synaptic histamine H3 receptors modulate glutamatergic transmission in rat globus pallidus. Neuroscience 176, 20–31. [DOI] [PubMed] [Google Scholar]
- Garcia M.; Floran B.; Arias-Montano J.-A.; Young J. M.; Aceves J. (1997) Histamine H3 receptor activation selectively inhibits dopamine D1 receptor-dependent [3H]GABA release from depolarization-stimulated slices of rat substantia nigra pars reticulata. Neuroscience 80, 241–249. [DOI] [PubMed] [Google Scholar]
- Arias-Montano J.-A.; Floran B.; Garcia M.; Aceves J.; Young J.-M. (2001) Histamine H3 receptor-mediated inhibition of depolarization-induced, dopamine D1 receptor-dependent release of [3H]-gamma-aminobutryic acid from rat striatal slices. Br. J. Pharmacol. 133, 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Montano J.-A.; Floran B.; Floran L.; Aceves J.; Young J. M. (2007) Dopamine D1 receptor facilitation of depolarization-induced release of gamma-amino-butyric acid in rat striatum is mediated by the cAMP/PKA pathway and involves P/Q-type calcium channels. Synapse 61, 310–319. [DOI] [PubMed] [Google Scholar]
- Fredholm B. B.; IJzerman A. P.; Jacobson K. A.; Klotz K. N.; Linden J. (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 53, 527–552. [PMC free article] [PubMed] [Google Scholar]
- Mayfield R.; Suzuki F.; Zahniser N. R. (1993) Adenosine A2A receptor modulation of electrically evoked endogenous GABA release from slices of rat globus pallidus. J. Neurochem. 60, 2334–2337. [DOI] [PubMed] [Google Scholar]
- Dayne Mayfield R.; Larson G.; Orona R. A.; Zahniser N. R. (1996) Opposing actions of adenosine A2a and dopamine D2 receptor activation on GABA release in the basal ganglia: evidence for an A2a/D2 receptor interaction in globus pallidus. Synapse 22, 132–138. [DOI] [PubMed] [Google Scholar]
- Shindou T.; Nonaka H.; Richardson P. J.; Mori A.; Kase H.; Ichimura M. (2002) Presynaptic adenosine A2A receptors enhance GABAergic synaptic transmission via a cyclic AMP dependent mechanism in the rat globus pallidus. Br. J. Pharmacol. 136, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floran B.; Gonzalez B.; Floran L.; Erlij D.; Aceves J. (2005) Interactions between adenosine A2A and dopamine D2 receptors in the control of [3H]GABA release in the globus pallidus of the rat. Eur. J. Pharmacol. 520, 43–50. [DOI] [PubMed] [Google Scholar]
- Shindou T.; Nonaka H.; Richardson P. J.; Mori A.; Kase H.; Ichimura M. (2003) Adenosine modulates the striatal GABAergic inputs to the globus pallidus via adenosine A2A receptors in rats. Neurosci. Lett. 352, 167–170. [DOI] [PubMed] [Google Scholar]
- Bevan M. D.; Hallworth N. E.; Baufreton J. (2007) GABAergic control of the subthalamic nucleus. Prog. Brain Res. 160, 173–188. [DOI] [PubMed] [Google Scholar]
- Jin X.-T.; Paré J. F.; Smith Y. (2011) Differential localization and function of GABA transporters, GAT-1 and GAT-3, in the rat globus pallidus. Eur. J. Neurosci. 33, 1504–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden L. (1996) GABA transporter heterogeneity: pharmacology and cellular localization. Neurochem. Int. 29, 335–356. [DOI] [PubMed] [Google Scholar]
- Fredholm B. B.; IJzerman A. P.; Jacobson K. A.; Linden J.; Müller C. E. (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors-an update. Pharmacol. Rev. 63, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz K.-N. (2000) Adenosine receptors and their ligands. Naunyn-Schmiedeberg’s Arch. Pharmacol. 362, 382–391. [DOI] [PubMed] [Google Scholar]
- Hernández-González O.; Hernández-Flores T.; Prieto G. A.; Pérez-Burgos A.; Arias-García M. A.; Galarraga E.; Bargas J. (2013) Modulation of Ca2+-currents by sequential and simultaneous activation of adenosine A1 and A2A receptors in striatal projection neurons. Purinergic Signalling 10, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves J.; Queiroz G. (2008) Presynaptic adenosine and P2Y receptors. Handb. Exp. Pharmacol. 184, 339–372. [DOI] [PubMed] [Google Scholar]
- Gubitz A. K.; Widdowson L.; Kurokawa M.; Kirkpatrick K. A.; Richardson P. J. (1996) Dual signalling by the adenosine A2a receptor involves activation of both N- and P-type calcium channels by different G proteins and protein kinases in the same striatal nerve terminals. J. Neurochem. 67, 374–381. [DOI] [PubMed] [Google Scholar]
- Milhaud D.; Fagni L.; Bockaert J.; Lafon-Cazal M. (2002) Inhibition of voltage-gated Ca2+ channels by antazoline. NeuroReport 13, 1711–1714. [DOI] [PubMed] [Google Scholar]
- Harper E. A.; Shankley N. P.; Black J. W. (1999) Characterization of the binding of [3H]-clobenpropit to histamine H3-receptors in guinea-pig cerebral cortex membranes. Br. J. Pharmacol. 128, 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J. H.; Yanai K.; Watanabe T. (1994) Marked increase in histamine H3 receptors in the striatum and substantia nigra after 6-hydroxydopamine-induced denervation of dopaminergic neurons: an autoradiographic study. Neurosci. Lett. 178, 19–22. [DOI] [PubMed] [Google Scholar]
- Takeshita Y.; Watanabe T.; Sakata T.; Munakata M.; Ishibashi H.; Akaike N. (1998) Histamine modulates high-voltage-activated calcium channels in neurons dissociated from the rat tuberomammillary nucleus. Neuroscience 87, 797–805. [DOI] [PubMed] [Google Scholar]
- Molina-Hernandez A.; Nunez A.; Sierra J. J.; Arias-Montano J.-A. (2001) Histamine H3 receptor activation inhibits glutamate release from rat striatal synaptosomes. Neuropharmacology 41, 928–934. [DOI] [PubMed] [Google Scholar]
- De Waard M.; Hering J.; Weiss N.; Feltz A. (2005) How do G proteins directly control neuronal Ca2+ channel function?. Trends Pharmacol. Sci. 26, 427–436. [DOI] [PubMed] [Google Scholar]
- Jang I.-S.; Rhee J.-S.; Watanabe T.; Akaike N.; Akaike N. (2001) Histaminergic modulation of GABAergic transmission in rat ventromedial hypothalamic neurones. J. Physiol. 534, 791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrada C.; Ferré S.; Casadó V.; Cortés A.; Justinova Z.; Barnes C.; Canela E. I.; Goldberg S. R.; Leurs R.; Lluis C.; Franco R. (2008) Interactions between histamine H3 and dopamine D2 receptors and the implications for striatal function. Neuropharmacology 55, 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrada C.; Moreno E.; Casadó V.; Bongers G.; Cortés A.; Mallol J.; Canela E. I.; Leurs R.; Ferré S.; Lluís C.; Franco R. (2009) Marked changes in signal transduction upon heteromerization of dopamine D1 and histamine H3 receptors. Br. J. Pharmacol. 157, 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrang J. M.; Morisset S.; Gbahou F. (2007) Constitutive activity of the histamine H3 receptor. Trends Pharmacol. Sci. 28, 350–357. [DOI] [PubMed] [Google Scholar]
- Rouleau A.; Ligneau X.; Tardivel-Lacombe J.; Morisset S.; Gbahou F.; Schwartz J. C.; Arrang J. M. (2002) Histamine H3-receptor-mediated [35S]GTPg[S] binding: evidence for constitutive activity of the recombinant and native rat and human H3 receptors. Br. J. Pharmacol. 135, 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rav-Acha M.; Bergman H.; Yarom Y. (2008) Pre- and postsynaptic serotoninergic excitation of globus pallidus neurons. J. Neurophysiol. 100, 1053–1066. [DOI] [PubMed] [Google Scholar]
- Macinnes N.; Duty S. (2008) Group III metabotropic glutamate receptors act as hetero-receptors modulating evoked GABA release in the globus pallidus in vivo. Eur. J. Pharmacol. 580, 95–99. [DOI] [PubMed] [Google Scholar]
- Jin X.-T.; Paré J. F.; Raju D. V.; Smith Y. (2006) Localization and function of pre and postsynaptic kainate receptors in the rat globus pallidus. Eur. J. Neurosci. 23, 374–386. [DOI] [PubMed] [Google Scholar]
- Schlicker E.; Fink K.; Detzner M.; Gothert M. (1993) Histamine inhibits dopamine release in the mouse striatum via presynaptic H3 receptors. J. Neural Transm.: Gen. Sect. 93, 1–10. [DOI] [PubMed] [Google Scholar]
- Threlfell S.; Cragg S. J.; Kalló I.; Turi G. F.; Coen C. W.; Greenfield S. A. (2004) Histamine H3 receptors inhibit serotonin release in substantia nigra pars reticulata. J. Neurosci. 24, 8704–8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayadjanian N.; Menétrey A.; Besson M. J. (1997) Activation of muscarinic receptors stimulates GABA release in the rat globus pallidus. Synapse 26, 131–139. [DOI] [PubMed] [Google Scholar]
- Prast H.; Tran M. H.; Fischer H.; Kraus M.; Lamberti C.; Grass K.; Philippu A. (1999) Histaminergic neurons modulate acetylcholine release in the ventral striatum: role of H3 histamine receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 360, 558–564. [DOI] [PubMed] [Google Scholar]
- Cooper A. J.; Stanford I. M. (2000) Electrophysiological and morphological characteristics of three subtypes of rat globus pallidus neurone in vitro. J. Physiol. 527, 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenz D.; Kital S. T. (1999) A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus. Nature 400, 677–682. [DOI] [PubMed] [Google Scholar]
- Magill P. J.; Bolam J. P.; Bevan M. D. (2000) Relationship of activity in the subthalamic nucleus-globus pallidus network to cortical electroencephalogram. J. Neurosci. 20, 820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.; Wang J. J.; Yung W. H.; Chan Y. S.; Chow B. K. (2005) Excitatory effect of histamine on neuronal activity of rat globus pallidus by activation of H2 receptors in vitro. Neurosci. Res. 53, 288–297. [DOI] [PubMed] [Google Scholar]
- Flores-Clemente C.; Osorio-Espinoza A.; Escamilla-Sánchez J.; Leurs R.; Arias J. M.; Arias-Montaño J. A. (2013) A single-point mutation (Ala280Val) in the third intracellular loop alters the signalling properties of the human histamine H3 receptor stably expressed in CHO-K1 cells. Br. J. Pharmacol. 170, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leurs R.; Smit M. J.; Menge W. M.; Timmerman H. (1994) Pharmacological characterization of the human histamine H2 receptor stably expressed in Chinese hamster ovary cells. Br. J. Pharmacol. 112, 847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeso J. A.; Rodriguez-Oroz M. C.; Benitez-Temino B.; Blesa F. J.; Guridi J.; Marin C.; Rodriguez M. (2008) Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov. Disord. 23, 548–559. [DOI] [PubMed] [Google Scholar]
- Hikosaka O.; Isoda M. (2010) Switching from automatic to controlled behavior: cortico-basal ganglia mechanisms. Trends Cognit. Sci. 14, 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristóvão-Ferreira S.; Vaz S. H.; Ribeiro J. A.; Sebastião A. M. (2009) Adenosine A2A receptors enhance GABA transport into nerve terminals by restraining PKC inhibition of GAT-1. J. Neurochem. 109, 336–347. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. (1973) Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108. [DOI] [PubMed] [Google Scholar]