

Abstract

Parkinson’s disease is a neurodegenerative disease characterized by the motor symptoms of bradykinesia, tremor, and rigidity. Current therapies are based mainly on dopaminergic replacement strategies by administration of either dopamine agonists or dopamine precursor levodopa (L-Dopa). These treatments provide symptomatic relief without slowing or stopping the disease progression, and long-term usage of these drugs is associated with diminished efficacy, motor fluctuation, and dyskinisia. Unfortunately, there had been few novel treatments developed in the past decades. Among nondopaminergic strategies for the treatment of Parkinson’s disease, antagonism of the adenosine A2A receptor has emerged to show great potential. Here we report the optimization of a new chemical scaffold, which achieved exceptional receptor binding affinity and ligand efficiency against adenosine A2A receptor. The leading compounds demonstrated excellent efficacy in the haloperidol induced catalepsy model for Parkinson’s disease.
Keywords: Adenosine receptor, antagonist, GPCR, Parkinson’s disease, lead optimization
Parkinson’s disease is a neurodegenerative disease that affects about 1.5% of the population over the age of 60.1 As the world population ages and life expectancy increases, the number of individuals afflicted by Parkinson’s disease is expected to double by 2030.2 Unfortunately, there had been few novel treatments developed in the past decades. Current therapies are based mainly on dopaminergic replacement strategies by administration of either dopamine agonists or dopamine precursor levodopa (L-Dopa). These treatments provide symptomatic relief without slowing or stopping the disease progression, and long-term usage of these drugs is associated with diminished efficacy, motor fluctuation, and dyskinisia.3,4 Furthermore, the current available dopaminergic drugs have very limited effects for the treatment of nonmotor symptoms of Parkinson’s disease such as depression, dementia, sleep disorder, and other autonomic dysfunctions.5 There is an urgent need to develop effective and durable symptomatic and disease-modifying therapies for the long-term management of Parkinson’s disease.
Among nondopaminergic strategies for the treatment of Parkinson’s disease, antagonism of the adenosine A2A receptor has emerged to show great potential. Adenosine is an important neuromodulator that exerts its functions through interacting with adenosine A1, A2A, A2B, and A3 7-transmembrane G-protein coupled receptor (GPCR) subtypes.6−8 Adenosine A2A receptor is highly expressed in the striatum in central nervous system, where it is colocalized with dopamine D2 receptors on striatopallisal output neuron.9 This discrete location limits the risk of unwanted side effects associated with receptors that have broad central nervous system distribution. A number of A2A receptor antagonists have been shown to improve motor disabilities in animal models of Parkinson’s disease without inducing dyskinesia.10 Examples of A2A receptor antagonists that progressed into clinical development are shown in Figure 1.11,12
Figure 1.

Examples of A2A receptor antagonists in clinical development.
Istradefylline (KW-6002) represents the most advanced A2A receptor antagonist in clinical development.13,14 It was denied approval in the United States due to questionable balance of risk and benefit.15 However, on March 25, 2013, Kyowa Hakko received marketing approval of istradefylline under the name of NOURIAST (20 mg orally administrated once daily) in Japan.16 It represents the world’s first antiparkinsonian agent based on antagonizing adenosine A2A receptor. Preladenant was the most advanced nonxanthine A2A antagonist. It is the most potent and selective compound (A2A 1.1 nM, A1/A2A > 1000) in clinical development.17 Unfortunately, on May 23, 2013, Merck announced the discontinuation of all development activities of preladenant based on the results of three separate phase III trials, which showed no evidence of efficacy when compared with placebo.18 The mixed clinical results demonstrate the challenges in pharmaceutical research and development for a complex disease such as Parkinson’s disease.
We have focused on 2,4,6-trisubstituted pyrimidines in our pursuit of novel A2A receptor antagonists for the treatment of Parkinson’s disease.19−24 Most recently, we reported a bioisosteric replacement of a potentially labile acetamide functional group. This effort led us to a new series of A2A antagonists, shown in Figure 2, with improved receptor binding affinity and chemical stability.25 However, when we subjected compound 3 to a preliminary CYP inhibition test, it demonstrated 80% inhibition of CYP3A4 at 10 μM; it also showed inhibition of other CYP isoforms (Table 2). Although these numbers were not particularly worrisome considering the superb receptor binding affinity of compound 3 (1.1 nM), we decided to address this potential problem in our structure–activity relationship (SAR) campaign. Here we report the optimization campaign based on the initial lead compounds aimed at obtaining better receptor binding affinity, ligand efficiency, CYP inhibition profiles, and in vivo efficacy.
Figure 2.

Replacement of acetamide functional group with bioisosteres.
Table 2. CYP Inhibition of Designed Compounds.
| inhibition
(%) |
||||||
|---|---|---|---|---|---|---|
| compda | clogPb | CYP3A4 | CYP2D6 | CYP1A2 | CYP2C9 | CYP2C19 |
| 3 | 3.29 | 80 | 24 | 64 | 84 | 81 |
| 4 | 2.27 | 52 | 7 | 72 | 69 | 29 |
| 10 | 2.51 | 16 | 18 | 93 | 32 | 36 |
| 12 | 1.83 | 24 | 14 | 95 | 54 | 42 |
| 15 | 3.53 | 64 | 25 | 87 | 65 | 55 |
| 19 | 1.87 | 6 | 16 | 95 | 9 | 8 |
All compounds were tested at 10 μM concentration.
Calculated by Molinspiration.
Results and Discussion
Chemistry
The general synthesis of compounds 9–23 is outlined in Scheme 1. Commercially available 4,6-dichloro-2-(methylthio)pyrimidine 5 was treated with 2,2,2-trifluoroethylamine in the presence of N,N-diisopropylethylamine (DIPEA) in N-methyl-2-pyrrolidone (NMP) at 100 °C to give 6a, or reacted with the desired amine in the presence of sodium hexamethyldisilazide (NaHMDS) to give 6b–e. Treatment of 6a–e with oxone produced the corresponding sulfonyl compounds 7a–e, which were reacted with pyrazole in the presence of NaH in tetrahydrofuran (THF) to give intermediates 8a–e. The chlorine atom of the pyrimidine intermediates 8a–e was displaced with (R)-2-(methoxymethyl)pyrrolidine hydrochloride in the presence of DIPEA in THF at 50 °C or with 1,2,3,4-tetrahydroisoquinoline in THF at 50 °C, to give the final products 9–13 and 14–18, respectively. Treatment of 8a–e with pyrazole in the presence of cesium carbonate in NMP at 95 °C afforded the final compounds 19–23.
Scheme 1. Synthesis of 2,4,6-Trisubstituted Pyrimidines.

Reagents and conditions: (a) For 6a: 2,2,2-trifluoroethylamine, DIPEA, NMP, 100 °C, 8 h; for 6b–e: amine, NaHMDS, THF, rt, 8 h. (b) Oxone, THF, H2O, rt, 10 h. (c) Pyrazole, NaH, THF, rt, 10 h. (d) For 9–13: (R)-2-(methoxymethyl)pyrrolidine hydrochloride, DIPEA, THF, 50 °C, 8 h; for 14–18: 1,2,3,4-tetrahydroisoquinoline, THF, 50 °C, 8 h; for 19–23: pyrazole, Cs2CO3, NMP, 95 °C, 12 h.
Pharmacology and Structure–Activity Relationship
As mentioned in the introduction, compound 3 demonstrated 80% inhibition of CYP3A4 at 10 μM; it also showed inhibition for other CYP isoforms (Table 2). Although these numbers were not particularly worrisome considering the superb receptor binding affinity of compound 3 (1.1 nM), we decided to address this potential problem in our SAR campaign. We hypothesized that the high lipophilicity might play a role in CYP inhibition. The clogP of compound 3 is 3.29. We set to make the new compounds less lipophilic, and therefore, we chose pyrazole as the substituent of the 2 position of the aminopyrimidine core structure. In addition to the benefit of trimming down one unit of clogP when compared with dimethylpyrazole, pyrazole also demonstrated good potency and metabolic stability in our previous experience.19 We kept the same set of R1 and R2 as in our last exercise25 for a clear comparison of the SAR.
The SAR of the synthesized compounds is presented in Table 1. With the combination of 2-pyrazole and 4-(R)-2-(methoxymethyl) pyrrolidine, the best compound was obtained with pyridine. Compound 10 (0.22 nM) was 5 times more potent than preladenant and, to the best of our knowledge, represented one of the highest receptor binding affinity of A2A antagonists. Pyrazine was also a reliable bioisostere as compound 12 demonstrated decent activity (55 nM). Pyrimidine, thiazole, and trifluoroethyl all showed diminished activity compared with pyridine and pyrazine (compounds 11, 13, and 9; 181, 577, and 2884 nM, respectively). The trend continued with the combination of 2-pyrazole and 4-tetrahydroisoquinoline, as the best compound also came from pyridine (compound 15, 0.66 nM). When both 2 and 4 positions were occupied by an aromatic substituent, the best compound was from trifluoroethyl, as compound 19 demonstrated excellent affinity (2.2 nM). Pyrazine, once again, demonstrated decent activity (compound 22, 23 nM). With the data we obtained so far, among the five bioisosteres evaluated, pyridine and pyrazine proved to be excellent surrogates for acetamide, followed by trifluoroethyl and thiazole, pyrimidine proved to be the least favorable biosiostere for the acetamide. The leading compounds with the highest receptor binding affinity 10, 15, and 19 were further profiled in a cell based cAMP assay to confirm them as functional antagonists for the A2A receptor. Compounds 10, 15, and 19 demonstrated inhibition of cAMP production (IC50 2.1 ± 0.5, 1.4 ± 0.4, and 0.5 ± 0.3 nM, respectively). Preladenant was tested in the same assay, and its IC50 was determined to be 70 ± 7.0 nM. Compounds obtained from this new campaign demonstrated exceptional receptor binding affinity (e.g., compounds 10, 15, and 19, 0.22, 0.66, and 2.2 nM, respectively) and ligand efficiency (e.g., compounds 10, 15, and 19, LE 0.51, 0.45, and 0.54, respectively), with desirable physical-chemical properties (e.g., clogP range from 1.87 for compound 19 to 2.51 for compound 10). We are currently using modeling tools to understand the structural basis of the observed SAR results with the aid of crystal structures of A2A receptors bound to antagonists and agonists.26−29
Table 1. SAR of Designed Compounds.
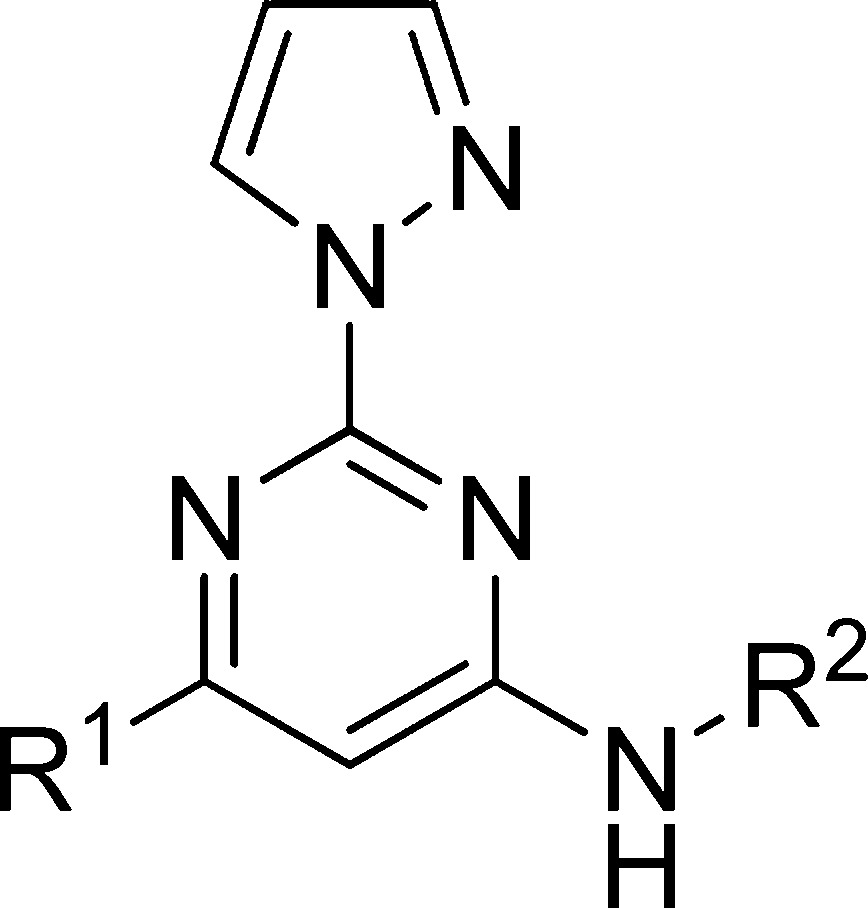
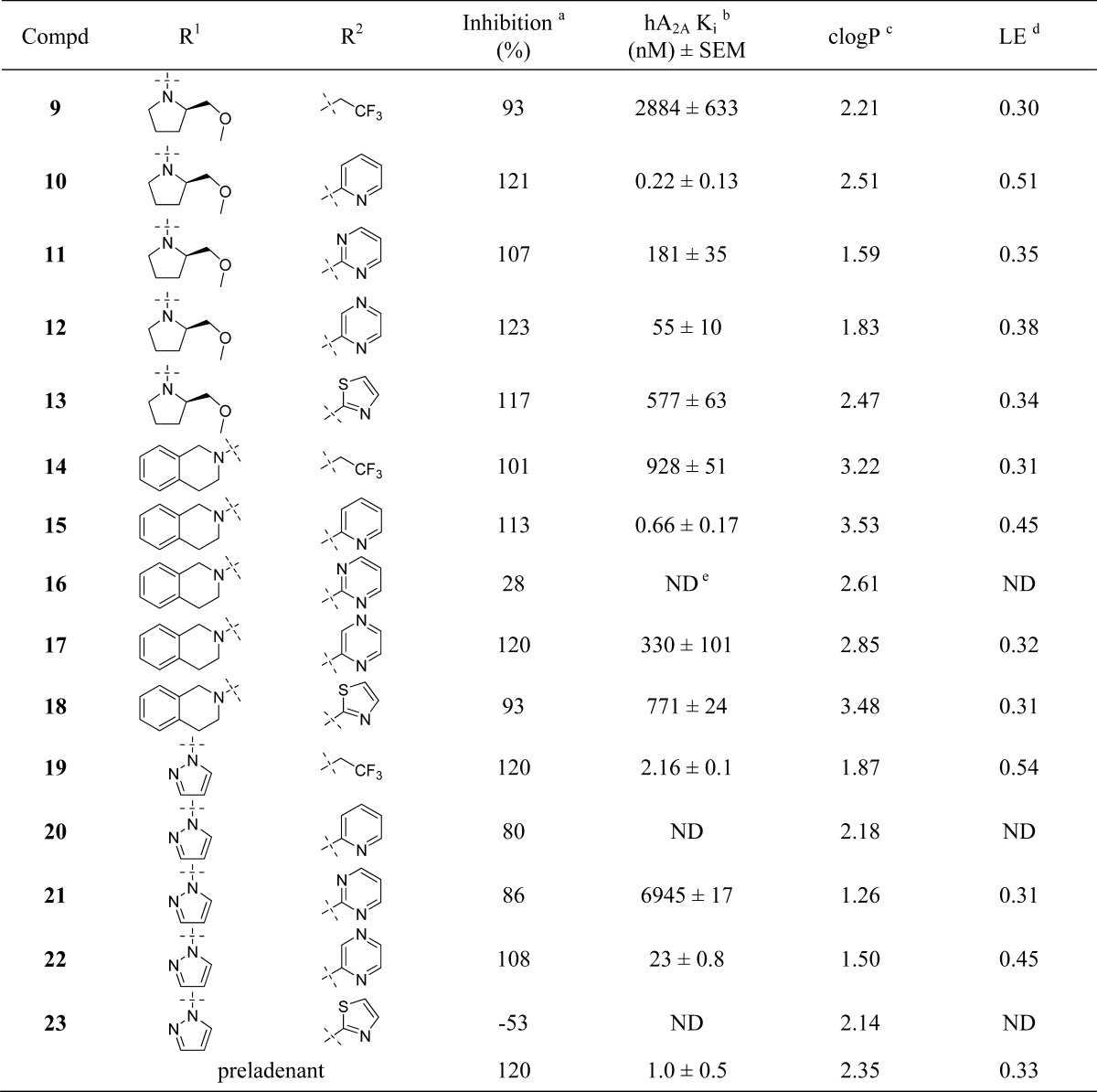
The inhibition of [3H]-ZM 241385 binding at human A2A receptors expressed in HEK293 cells at 100 μmol/L concentration.
Displacement of [3H]-ZM 241385 binding at human A2A receptors expressed in HEK293 cells. Data are expressed as geometric mean values of at least two runs ± the standard error measurement (SEM).
Calculated by Molinspiration.
LE (ligand efficiency) = 1.37pKi/HA (heavy atom).
ND = not determined.
In Vitro Safety and Potential Drug/Drug Interactions
The reference compounds 3, 4 and leading compounds 10, 12 (compound 12 was selected for its decent binding affinity and low clogP), 15, and 19 were evaluated in a preliminary CYP screening assay to assess their potential liability in liver safety and drug/drug interactions. Compound 3 demonstrated 80% inhibition of CYP3A4 at 10 μM; it also showed inhibition for other CYP isoforms (Table 2). We hypothesized that the high lipophilicity might play a role in CYP inhibition. This assumption was validated as compound 4, a close analogue to 3, demonstrated improved CYP profile (except for CYP1A2). Compounds 10 (clogP 2.51), 12 (clogP 1.83), and 19 (clogP 1.87) showed weak CYP3A4 inhibition at 10 μM, while compound 15 (clogP 3.53) demonstrated moderate CYP3A4 inhibition (64%) at 10 μM. Given the exceptional receptor binding affinity of these compounds (e.g., compound 10, 0.22 nM), we project the potential safety and drug/drug interaction liabilities to be marginal.
In Vitro Human and Mouse Liver Microsomal Stability Evaluation
The metabolic stability of compounds 3, 4, 10, 12, 15, and 19 was evaluated in human and mouse liver microsomal tests. Under the current experimental conditions, these compounds demonstrated relatively high clearance and short half-lives in both human and mouse liver microsomes. The high clearance was not directly related to the clogP of the tested compounds, as compound 12 (clogP 1.83, MLM Clint = 132 mL/min/kg, HLM Clint = 24.7 mL/min/kg) demonstrated lower metabolic stability compared with compound 3 (clogP 3.29, MLM Clint = 63 mL/min/kg, HLM Clint = 13.8 mL/min/kg). We had previously obtained metabolic stable compounds using the same substitutions on the aminopyrimidine core,24 with the exception of the pyridine or pyrazine being replaced by acetamide (e.g., compound 1 and 4). It was therefore possible that the new compounds with heterocycles as the bioisosteric replacements for the acetamide posted potential metabolic soft spots which accelerated the clearance. However, the unsubstituted pyrazole might also contribute to the high clearance (e.g., compound 19 with trifluoroethyl as the bioisostere for the acetamide).
Table 3. Metabolic Stability of Designed Compounds.
| Clint (mL/min/kg) |
t1/2 (min) |
|||
|---|---|---|---|---|
| compda | HLMb | MLMc | HLM | MLM |
| 3 | 13.8 | 63 | 51.7 | 38.3 |
| 4 | 29.2 | 152 | 24.4 | 16 |
| 10 | 57 | 684 | 12.6 | 3.6 |
| 12 | 24.7 | 132 | 28.9 | 18.3 |
| 15 | 56 | 195 | 12.6 | 12.4 |
| 19 | 32 | 554 | 22 | 4.4 |
All compounds were tested at 1 μM concentration.
HLM = human liver microsomes.
MLM = mouse liver microsomes.
Haloperidol Induced Catalepsy
We were eager to see if these new compounds could reverse haloperidol induced catalepsy (HIC) in rat to validate the observed in vitro affinity. Because the leading compounds demonstrated fast metabolic clearance, we therefore decided to use intraperitoneal injection (i.p.) route for drug delivery instead of per oral (p.o.) in the HIC to circumvent the first-pass effect in the gastrointestinal tract. KW-6002 5 mg/kg i.p. was used as the standard. Compounds 10, 15, and 19 were tested in HIC, and the results are presented in Figure 3. Compounds 10, 15, and 19 all demonstrated significant reversal of haloperidol induced catalepsy at all three doses (5, 10, and 30 mg/kg). There was no difference between the doses of 10 and 30 mg/kg, indicating a maximum effective dose having been reached at 10 mg/kg (over 90% reversal). The plasma, brain, and striatum exposures of compound 10 were determined using LC–MS/MS (Table 4). Compound 10 demonstrated proportional exposure in plasma, brain, and striatum in the doses of 10 and 30 mg/kg. Compound 10 readily crossed the blood-brain barrier, with brain/plasma (B/P) ratios ranging from 1.58 to 1.67 for doses of 10 and 30 mg/kg, respectively. Even though we did not determine the unbound drug concentration, the good brain/striatum exposure (232 and 121 ng/g, respectively, for the dose of 10 mg/kg) and the exceptional potency of compound 10 (0.22 nM) warrant the observed efficacy in HIC.
Figure 3.

Effect of compounds 10, 15, and 19 in the haloperidol induced catalepsy bar test. Mean descent latency (±standard error) in seconds. The vehicle bar represents the mean and standard error from four experiments. KW-6002 was injected (i.p.) in dose of 5 mg/kg. **p < 0.01, ***p < 0.001 versus Vehicle controls.
Table 4. Plasma and Brain Exposure for Compound 10, at 1 h after i.p. Dosing, in Rats Used in the HIC Model.
| dose (mg/kg) | plasma (ng/mL)a | brain (ng/g)a | striatum (ng/g)a | B/P ratio |
|---|---|---|---|---|
| 10 | 147 ± 68 | 232 ± 50 | 121 ± 88 | 1.58 |
| 30 | 601 ± 447 | 1001 ± 550 | 434b | 1.67 |
Data are expressed as geometric mean values of four runs ± the standard error measurement (SEM).
Pooled data of four rats.
Summary
We optimized a new series of A2A antagonists based on an aminopyrimidine core. The leading compound 10 demonstrated exceptional receptor binding affinity (hA2A 0.22 nM), ligand efficiency (0.51), and desirable physical-chemical properties (clogP 2.51). Compound 10 showed limited liver safety and drug/drug interaction liabilities. Although the metabolic stability in human and mouse liver microsomes was suboptimal, when dosed i.p., compound 10 readily crossed the blood-brain barrier and demonstrated dose proportional exposures in plasma, brain, and striatum. More importantly, compound 10 exhibited excellent reversal of haloperidol induced catalepsy in rat. Further optimization to improve metabolic stability, as well as molecular modeling to understand the structural basis of the observed SAR are ongoing, and results will be presented in due course.
Methods
Chemistry
Reaction progress was monitored by analytical thin layer chromatography (TLC) performed on silica gel HSGF254 precoated plates. Organic solutions were dried over anhydrous Na2SO4, and the solvents were removed under reduced pressure. 1H NMR and 13C NMR spectra were obtained on 400 MHz (Varian) spectrometers. Chemical shifts were given in ppm using tetramethylsilane (TMS) as internal standard. Mass spectra were obtained using an Agilent 1100 LC/MSD Trap SL version mass spectrometer. Final compounds were purified with silica gel 100–200 mesh for column chromatography.
6-Chloro-2-(methylthio)-N-(2,2,2-trifluoroethyl)pyrimidin-4-amine (6a)
To a sealed tube were added 4,6-dichloro-2-(methylthio)pyrimidine (1.95 g, 10 mmol), 2,2,2-trifluoroethanamine (1.98 g, 20 mmol), N,N-diisopropylethylamine (2.58 g, 20 mmol), and NMP (10 mL). The resulting mixture was heated to 100 °C for 10 h and then poured into H2O (80 mL). The resulting precipitate was filtered, washed with H2O (5 mL) and petro ether (5 mL), and dried in vacuum to give a pale yellow solid (1.96 g, 76%). 1H NMR (400 MHz, CDCl3) δ 6.21 (s, 1H), 5.12 (br s, 1H), 4.22–4.10 (m, 2H), 2.54 (s, 3H).
General Procedure for the Preparation of 6b–e
To a solution of 4,6-dichloro-2-(methylthio)pyrimidine (1.95 g, 10 mmol) and corresponding amine (10 mmol) in THF (15 mL) was added NaHMDS (2 M in THF, 10 mL, 20 mmol) at 0 °C under N2 atmosphere. The resulting mixture was stirred at room temperature for 8 h and then poured into H2O (80 mL). The resulting precipitate was filtered, washed with H2O (5 mL) and petro ether (5 mL), and dried in vacuum to give the title compounds.
6-Chloro-2-(methylthio)-N-(pyridin-2-yl)pyrimidin-4-amine (6b)
Compound 6b as a pale yellow solid (2.3 g, 91%). 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 4.8 Hz, 1H), 7.71 (dd, J = 4.8 Hz, 1H), 7.61 (s, 1H), 7.47 (s, 1H), 7.42 (d, J = 8.0 Hz, 1H), 7.03 (dd, J = 6.8, 5.4 Hz, 1H), 2.59 (s, 3H).
N-(6-Chloro-2-(methylthio)pyrimidin-4-yl)pyrimidin-2-amine (6c)
Compound 6c as a pale yellow solid (2.16 g, 85%). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (br s, 1H), 8.68 (d, J = 4.4 Hz, 2H), 8.08 (s, 1H), 7.16–7.12 (m, 1H), 7.47 (s, 1H), 2.51 (s, 3H).
6-Chloro-2-(methylthio)-N-(pyrazin-2-yl)pyrimidin-4-amine (6d)
Compound 6d as a pale yellow solid (2.03 g, 80%). 1H NMR (400 MHz, CDCl3) δ 8.53 (s, 1H), 8.28 (d, J = 3.6 Hz, 2H), 7.47 (s, 1H), 7.42 (s, 1H), 2.58 (s, 3H).
N-(6-Chloro-2-(methylthio)pyrimidin-4-yl)thiazol-2-amine (6e)
Compound 6e as a yellow solid (1.97 g, 76%). 1H NMR (400 MHz, DMSO-d6) δ 11.95 (br s, 1H), 7.44 (d, J = 2.8 Hz, 1H), 7.22 (d, J = 2.8 Hz, 1H), 6.71 (s, 1H), 2.59 (s, 3H).
General Procedure for the Preparation of 7a–e
To a solution of 6a–e (6.0 mmol) in THF (15 mL) and H2O (3 mL) was added oxone (18.0 mmol) at 0 °C. The resulting mixture was stirred at room temperature for 10 h and then poured into H2O (80 mL). The resulting precipitate was filtered, washed with H2O (5 mL), and dried in vacuum to give the title compounds, which were used directly in the next step without further purification.
6-Chloro-2-(methylsulfonyl)-N-(2,2,2-trifluoroethyl)pyrimidin-4-amine (7a)
Compound 7a as a pale yellow solid (1.3 g, 75%).
6-Chloro-2-(methylsulfonyl)-N-(pyridin-2-yl)pyrimidin-4-amine (7b)
Compound 7b as a pale yellow solid (1.2 g, 70%).
N-(6-Chloro-2-(methylsulfonyl)pyrimidin-4-yl)pyrimidin-2-amine (7c)
Compound 7c as a pale yellow solid (1.3 g, 76%).
6-Chloro-2-(methylsulfonyl)-N-(pyrazin-2-yl)pyrimidin-4-amine (7d)
Compound 7d as a pale yellow solid (1.13 g, 66%).
N-(6-Chloro-2-(methylsulfonyl)pyrimidin-4-yl)thiazol-2-amine (7e)
Compound 7e as an orchid solid (1.64 g, 94%).
General Procedure for the Preparation of 8a–e
To a solution of 7a–e (4.0 mmol) and 1H-pyrazole (8.0 mmol) in THF (15 mL) was added 80% NaH (8.0 mmol) at 0 °C. The resulting mixture was stirred at room temperature for 10 h, and then poured into H2O (80 mL). The resulting precipitate was filtered, washed with H2O (5 mL), and dried in vacuum to give the title compounds.
6-Chloro-2-(1H-pyrazol-1-yl)-N-(2,2,2-trifluoroethyl)pyrimidin-4-amine (8a)
Compound 8a as a white solid (788 mg, 71%). 1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H), 7.80 (s, 1H), 6.47 (s, 1H), 6.40 (s, 1H), 5.58 (br s, 1H), 4.15 (s, 2H).
6-Chloro-2-(1H-pyrazol-1-yl)-N-(pyridin-2-yl)pyrimidin-4-amine (8b)
Compound 8b as a pale yellow solid (982 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 8.56 (s, 1H), 8.40 (d, J = 4.0 Hz, 1H), 8.11 (s, 1H), 7.87 (s, 1H), 7.82 (s, 1H), 7.70 (dd, J = 7.6 Hz, 1H), 7.10–6.99 (m, 2H), 6.49 (s, 1H).
N-(6-Chloro-2-(1H-pyrazol-1-yl)pyrimidin-4-yl)pyrimidin-2-amine (8c)
Compound 8c as a pale yellow solid (927 mg, 85%). 1H NMR (400 MHz, CDCl3) δ 8.63–8.41 (m, 3H), 7.83 (s, 1H), 7.03–6.99 (m, 1H), 6.50 (s, 1H).
6-Chloro-N-(pyrazin-2-yl)-2-(1H-pyrazol-1-yl)pyrimidin-4-amine (8d)
Compound 8d as a pale yellow solid (764 mg, 70%). 1H NMR (400 MHz, CDCl3) δ 8.57 (d, J = 2.8 Hz, 1H), 8.55 (s, 1H), 8.34 (s, 1H), 8.30 (d, J = 2.8 Hz, 1H), 8.06 (s, 1H), 8.00 (s, 1H), 7.84 (s, 1H), 6.51 (s, 1H).
N-(6-Chloro-2-(1H-pyrazol-1-yl)pyrimidin-4-yl)thiazol-2-amine (8e)
Compound 8e as a pale yellow solid (763 mg, 71%). 1H NMR (400 MHz, DMSO-d6) δ 8.71 (s, 1H), 7.92 (s, 1H), 7.53 (d, J = 2.8 Hz, 1H), 7.33 (d, J = 2.8 Hz, 1H), 6.95 (br s, 1H), 6.67 (s, 1H).
General Procedure for the Preparation of 9–13
To a solution of 8a–e (0.3 mmol) and (R)-2-(methoxymethyl)pyrrolidine hydrochloride (0.6 mmol) in THF (1 mL) was added N,N-diisopropylethylamine (1.2 mmol). The resulting mixture was stirred at 50 °C for 8 h.
(R)-6-(2-(Methoxymethyl)pyrrolidin-1-yl)-2-(1H-pyrazol-1-yl)-N-(2,2,2-trifluoroethyl)pyrimidin-4-amine (9)
The reaction mixture was poured into H2O (10 mL), and the resulting precipitate was filtered, washed with H2O (2 mL) and petro ether (2 mL), and dried in vacuum to give the title compound (53 mg, 50%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.49 (s, 1H), 7.74 (s, 1H), 6.41 (s, 1H), 5.45–5.20 (m, 2H), 4.52–4.25 (m, 1H), 4.12–3.88 (m, 2H), 3.75–3.30 (m, 4H), 3.35 (s, 3H), 2.15–1.92 (m, 4H). LCMS (ESI): m/z 356.9.
(R)-6-(2-(Methoxymethyl)pyrrolidin-1-yl)-2-(1H-pyrazol-1-yl)-N-(pyridin-2-yl)pyrimidin-4-amine (10)
The reaction mixture was poured into H2O (10 mL), and the resulting mixture was extracted with ethyl acetate (10 mL × 3), dried over Na2SO4, and concentrated. The residue was purified by flash chromatography (0.5% MeOH/CH2Cl2) to give the title compound (81 mg, 77%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.56 (d, J = 2.0 Hz, 1H), 8.33 (d, J = 4.4 Hz, 1H), 7.79 (s, 1H), 7.65–7.61 (m, 1H), 7.49 (s, 1H), 7.29 (br s, 1H), 6.99 (d, J = 4.4 Hz, 1H), 6.93–6.89 (m, 1H), 6.45 (s, 1H), 4.51–4.49 (m, 1H), 3.72–3.65 (m, 3H), 3.45 (s, 3H), 3.42 (s, 1H), 2.15–2.06 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 162.3, 158.8, 154.6, 153.7, 147.7, 142.4, 137.8, 128.7, 116.8, 112.3, 107.4, 84.9, 77.2, 72.4, 59.2, 57.1, 47.3. HRMS (ESI) calcd for C18H21N7O (M + H)+, 352.1880; found, 352.1878.
(R)-N-(6-(2-(Methoxymethyl)pyrrolidin-1-yl)-2-(1H-pyrazol-1-yl)pyrimidin-4-yl)pyrimidin-2-amine (11)
The reaction mixture was poured into H2O (10 mL), and the resulting precipitate was filtered, washed with H2O (2 mL) and petro ether (2 mL), and dried in vacuum to give the title compound (26 mg, 25%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.53 (s, 3H), 8.16 (s, 1H), 7.77 (s, 1H), 7.43 (br s, 1H), 6.87–6.85 (m, 1H), 6.43 (s, 1H), 4.60–4.44 (m, 1H), 3.69–3.48 (m, 3H), 3.42 (s, 3H), 2.20–1.96 (m, 4H). LCMS (ESI): m/z 352.9.
(R)-6-(2-(Methoxymethyl)pyrrolidin-1-yl)-N-(pyrazin-2-yl)-2-(1H-pyrazol-1-yl)pyrimidin-4-amine (12)
The reaction mixture was poured into H2O (10 mL), and the resulting mixture was extracted with ethyl acetate (10 mL × 3), dried over Na2SO4, and concentrated. The residue was purified by flash chromatography (0.5% MeOH/CH2Cl2) to give the title compound (50 mg, 47%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.53 (s, 1H), 8.39 (s, 1H), 8.22 (s, 1H), 8.14 (d, J = 2.0 Hz, 1H), 7.78 (s, 1H), 7.10 (br s, 1H), 6.44 (s, 1H), 4.62–4.48 (m, 1H), 3.75–3.53 (m, 3H), 3.50–3.42 (m, 1H), 3.42 (s, 3H), 2.22–2.00 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 162.4, 158.2, 154.7, 150.4, 142.6, 141.3, 136.4, 135.7, 128.8, 107.6, 85.5, 77.2, 72.4, 59.2, 57.3, 47.4. HRMS (ESI) calcd for C17H20N8O (M + H)+, 353.1833; found, 353.1830.
(R)-N-(6-(2-(Methoxymethyl)pyrrolidin-1-yl)-2-(1H-pyrazol-1-yl)pyrimidin-4-yl)thiazol-2-amine (13)
The reaction mixture was poured into H2O (10 mL), and the resulting precipitate was filtered, washed with H2O (2 mL) and petro ether (2 mL), and dried in vacuum to give the title compound (24 mg, 22%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.70 (s, 1H), 7.85 (s, 1H), 7.49 (d, J = 3.2 Hz, 1H), 6.96 (d, J = 3.2 Hz, 1H), 6.49 (s, 1H), 5.96 (br s, 1H), 4.68–4.55 (m, 1H), 3.80–3.43 (m, 4H), 3.42 (s, 3H), 2.25–2.00 (m, 4H). LCMS (ESI): m/z 357.8.
General Procedure for the Preparation of 14–18
To a solution of 8a–e (0.3 mmol) in THF (1 mL) was added 1,2,3,4-tetrahydroisoquinoline (0.9 mmol). The resulting mixture was stirred at 50 °C for 8 h.
6-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-(1H-pyrazol-1-yl)-N-(2,2,2-trifluoroethyl)pyrimidin-4-amine (14)
The reaction mixture was poured into H2O (10 mL), and the resulting mixture was extracted with ethyl acetate (10 mL × 3), dried over Na2SO4, and concentrated. The residue was purified by flash chromatography (0.5% MeOH/CH2Cl2) to give the title compound (57 mg, 51%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H), 7.74 (s, 1H), 7.27–7.23 (m, 4H), 6.43 (s, 1H), 5.49 (s, 1H), 5.25 (br s, 1H), 4.76 (s, 2H), 4.09–4.07 (m, 2H), 3.94–3.92 (m, 2H), 2.98–2.97 (m, 2H). LCMS (ESI): m/z 374.9.
6-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-(1H-pyrazol-1-yl)-N-(pyridin-2-yl)pyrimidin-4-amine (15)
The reaction mixture was poured into H2O (10 mL), and the resulting mixture was extracted with ethyl acetate (10 mL × 3), dried over Na2SO4, and concentrated. The residue was purified by flash chromatography (0.5% MeOH/CH2Cl2) to give the title compound (35 mg, 32%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.60 (dd, J = 2.6, 0.6 Hz, 1H), 8.36 (dd, J = 5.0, 1.2 Hz, 1H), 7.78 (dd, J = 1.5, 0.7 Hz, 1H), 7.66–7.57 (m, 2H), 7.34 (s, 1H), 7.26–7.18 (m, 4H), 7.04 (d, J = 8.3 Hz, 1H), 6.91 (ddd, J = 7.2, 5.0, 0.8 Hz, 1H), 6.45 (dd, J = 2.6, 1.6 Hz, 1H), 4.84 (s, 2H), 3.98 (s, 2H), 3.00 (t, J = 5.9 Hz, 2H). LCMS (ESI): m/z 369.9.
N-(6-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-(1H-pyrazol-1-yl)pyrimidin-4-yl)pyrimidin-2-amine (16)
The reaction mixture was poured into H2O (10 mL), and the resulting precipitate was filtered, washed with H2O (2 mL) and petro ether (2 mL), and dried in vacuum to give the title compound (38 mg, 34%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 8.60 (d, J = 4.7 Hz, 2H), 8.14 (s, 1H), 7.82 (s, 1H), 7.71 (s, 1H), 7.29–7.26 (m, 4H), 6.96–6.91 (m, 1H), 6.49 (s, 1H), 4.91 (s, 2H), 4.04 (s, 2H), 3.05 (t, J = 5.6 Hz, 2H). LCMS (ESI): m/z 370.9.
6-(3,4-Dihydroisoquinolin-2(1H)-yl)-N-(pyrazin-2-yl)-2-(1H-pyrazol-1-yl)pyrimidin-4-amine (17)
The reaction mixture was poured into H2O (10 mL), and the resulting mixture was extracted with ethyl acetate (10 mL × 3), dried over Na2SO4, and concentrated. The residue was purified by flash chromatography (0.5% MeOH/CH2Cl2) to give the title compound (49 mg, 44%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.61 (d, J = 2.0 Hz, 1H), 8.50 (s, 1H), 8.29 (s, 1H), 8.17 (d, J = 2.0 Hz, 1H), 7.81 (s, 1H), 7.75 (s, 1H), 7.30–7.23 (m, 4H), 6.47 (s, 1H), 4.86 (s, 2H), 3.99 (s, 2H), 3.03 (t, J = 5.9 Hz, 2H). LCMS (ESI): m/z 370.9.
N-(6-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-(1H-pyrazol-1-yl)pyrimidin-4-yl)thiazol-2-amine (18)
The reaction mixture was poured into H2O (10 mL), and the resulting precipitate was filtered, washed with H2O (2 mL) and petro ether (2 mL), and dried in vacuum to give the title compound (25 mg, 22%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.86 (s, 1H), 7.86 (s, 1H), 7.48 (s, 1H), 7.25–7.22 (m, 4H), 6.97 (s, 1H), 6.51 (s, 1H), 6.05 (s, 1H), 4.81 (s, 2H), 3.99–3.97 (m, 2H), 3.01–3.00 (m, 2H). LCMS (ESI): m/z 375.8.
General Procedure for the Preparation of 19–23
To a sealed tube was added 8a–e (0.3 mmol), 1H-pyrazole (1.2 mmol), Cs2CO3 (0.9 mmol), and NMP (1 mL). The resulting mixture was stirred at 95 °C for 12 h.
2,6-Di(1H-pyrazol-1-yl)-N-(2,2,2-trifluoroethyl)pyrimidin-4-amine (19)
The reaction mixture was poured into H2O (10 mL), and the resulting precipitate was filtered, washed with H2O (2 mL) and petro ether (2 mL), and dried in vacuum to give the title compound (21 mg, 23%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1H), 8.58 (d, J = 2.0 Hz, 1H), 7.84 (s, 1H), 7.78 (s, 1H), 7.00 (s, 1H), 6.50 (s, 2H), 5.68 (br s, 1H), 4.20–4.18 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 164.9, 158.4, 155.2, 143.6, 143.5, 129.5, 128.3, 124.2 (q, J = 254.3 Hz), 108.5, 108.4, 77.2, 30.8 (q, J = 53.7 Hz). HRMS (ESI) calcd for C12H10F3N7 (M + Na)+, 332.0842; found, 332.0840.
2,6-Di(1H-pyrazol-1-yl)-N-(pyridin-2-yl)pyrimidin-4-amine (20)
The reaction mixture was poured into H2O (10 mL), and the resulting mixture was extracted with ethyl acetate (10 mL × 3), dried over Na2SO4, and concentrated. The residue was purified by flash chromatography (0.5% MeOH/CH2Cl2) to give the title compound (23 mg, 25%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.66 (d, J = 2.0 Hz, 1H), 8.63 (s, 1H), 8.45 (d, J = 4.4 Hz, 1H), 8.36 (s, 1H), 7.91 (br s, 1H), 7.86 (s, 1H), 7.84 (s, 1H), 7.74–7.70 (m, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.04–7.01 (m, 1H), 6.53 (s, 2H). LCMS (ESI): m/z 304.9.
N-(2,6-Di(1H-pyrazol-1-yl)pyrimidin-4-yl)pyrimidin-2-amine (21)
The reaction mixture was poured into H2O (10 mL), and the resulting precipitate was filtered, washed with H2O (2 mL) and petro ether (2 mL), and dried in vacuum to give the title compound (29 mg, 32%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.92 (s, 1H), 8.69 (s, 2H), 8.68 (s, 1H), 8.63 (s, 1H), 8.56 (s, 1H), 7.86 (s, 2H), 6.99 (s, 1H), 6.54 (s, 2H). LCMS (ESI): m/z 305.9.
N-(Pyrazin-2-yl)-2,6-di(1H-pyrazol-1-yl)pyrimidin-4-amine (22)
The reaction mixture was poured into H2O (10 mL), and the resulting precipitate was filtered, washed with H2O (2 mL) and petro ether (2 mL), and dried in vacuum to give the title compound (25 mg, 27%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.75 (s, 1H), 8.67 (d, J = 1.8 Hz, 1H), 8.63 (d, J = 1.8 Hz, 1H), 8.38 (s, 2H), 8.29 (d, J = 1.8 Hz, 1H), 8.08 (s, 1H), 7.88 (s, 1H), 7.85 (s, 1H), 6.54 (s, 2H). LCMS (ESI): m/z 305.9.
N-(2,6-Di(1H-pyrazol-1-yl)pyrimidin-4-yl)thiazol-2-amine (23)
The reaction mixture was poured into H2O (10 mL), and the resulting precipitate was filtered, washed with H2O (2 mL) and petro ether (2 mL), and dried in vacuum to give the title compound (55 mg, 51%) as a pale yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 8.92 (s, 1H), 8.81 (s, 1H), 7.95 (s, 2H), 7.52 (s, 1H), 7.39 (br s, 1H), 7.31 (s, 1H), 6.67 (s, 2H). LCMS (ESI): m/z 310.8.
Cell Culture
HEK293 cells stably expressing human A2A (HEK 293-A2A cells) were obtained from Academy of Military Medical Sciences (Beijing China). The cells were cultured in DF12 supplemented with 10% fetal bovine serum (FBS), 1% penicillin, streptomycin, and 400 μg/mL G418. The cells were maintained at 37 °C in an incubator under a humidified atmosphere of 95% air and 5% CO2.
Membrane Preparation
Cultured cells in 10 × 10 cm dishes were washed by ice-cold phosphate-buffered saline (PBS) once and then scraped into an EP tube. They were then collected by using the 4 °C centrifuge with the lysis buffer containing 5 mM Tris-HCl pH 7.4, 5 mM EDTA·2Na, and 5 mM EGTA in the presence of PMSF (1:1000) and incubated for 30 min on ice with extraction by using a 1 mL needle for 15 times. After centrifugation at 40 000g for 20 min, the supernatant was discarded. Pellets were resuspended in reaction buffer (50 mM Tris-HCl pH 7.4, 2 mM MgCl2) with mechanical extraction by using a 1 mL needle for 15 times and then centrifuged at 40 000g for 20 min. The final pellets were suspended in reaction buffer with mechanical extraction. The aliquot of membranes were frozen at −80 °C.
Radioligand Binding Assay
An aliquot of membranes (50–100 μg of protein) was treated with adenosine deaminase (2 U/mL, type X bovine spleen, Sigma) and incubated at 37 °C for 30 min with 1.0 nM [3H]-ZM 241385 (50.00 Ci/mmol ART 0884) for the A2A human membranes in the presence of varying concentrations of competing ligand. Nonspecific binding was determined in the presence of 10 μM NECA for A2A membranes. Incubation was terminated by rapid filtration over 12-well filter plate (Millipore) equipped with GF/B filter papers (Whatman, U.K.). Filters were washed three times with ice-cold 50 mM Tris-HCl buffer. The filter papers, after being oven-dried, were incubated with a scintillation cocktail (methylbenzene 500 mL, POPOP 50 mg, PPO 2 g), and radioactivity was counted on a Beckman LS-6500 (PerkinElmer) counter. Binding data were fitted in one-site competition binding model for IC50 determination, performed with GraphPad Prism (GraphPad Software, San Diego, CA), and Ki values were calculated based on the Cheng-Prusoff equation (Ki = IC50/(1 + (L/Kd))).
cAMP Functional Assay
HEK-293 cells transfected with A2A receptor were used. All cells were cultured in DMEM-F12 nutrient mixture supplemented with 10% FBS, 0.05% Geneticin, 1% penicillin, and 1% streptomycin at 37 °C in 5% CO2. The cells were detached with 0.25% trypsin solution (0.25% trypsin). The cells were collected at 1100r/min for 3 min at room temperature and resuspended in Hank’s balanced salt solution (138 mM NaCl, 5 mM KCl, 1.3 mM CaCl2, 0.5 mM MgCl2, 0.4 mM MgSO4, 0.3 mM KH2PO4, 0.3 mM Na2HPO4, 5.6 mM glucose, pH 7.4) containing 100 μM 3-isobutyl-1-methylxanthine (Sigma, I7018) as phosphodiesterase inhibitor. The cells were loaded into 384-well plates (∼4000 cells/well) and incubated with test compound (10–4–10–11 M) for 90 min at 37 °C, and then the A2A receptor agonist NECA (1 μM final concentration) was added to the sample and the mixture was incubated again for 10 min at room temperature. The cAMP production was measured by using a Cisbio kit according to the manufacturer’s instructions. Briefly, the fluorescence signals at 665 and 620 nm were measured and the ratio was determined. The cyclic AMP levels were calculated according to the corresponding standard curve. IC50 values were obtained from concentration–response curves after log transformation.
Catalepsy
Haloperidol (1.5 mg/kg) and KW-6002 (5 mg/kg) and compounds (30 mg/kg, 10 mg/kg, 5 mg/kg) were dissolved in 5% DMSO, 5% Tween 80, 5% cremophor, and 85% saline. Male Wistar (about 220 g) rats were habituated to the vivarium for 1 week prior to testing. Animals (n = 4/total N = 20) were randomly assigned to different groups, and behavioral testing was performed blindly to each group. The data about different dosages were collected once a week to allow for complete drug washout during testing sessions. One hour prior to testing, the animals were intraperitoneally (i.p.) injected with KW-6002 or compounds, followed by injection of haloperidol (i.p.) 30 min before the testing. The behavioral test trial (maximum duration of 200 s) began by placing the forepaws of rats on a 10 cm high bar (horizontally placed) and hind quarters on a platform. Time was recorded when the animal fully removed both paws from the bar or climbed onto the bar with all of its limbs. After the haloperidol-induced catalepsy test (samples collected about 1 h), the plasma and brain were collected for the preclinical assays.
Assessment of Metabolic Stability in Liver Microsomes
To evaluate metabolic stability in vitro, test compounds were incubated with mouse and human liver microsomes in the presence of NADPH (in triplicate). The reaction mixture consisted of microsomes (0.2 mg/mL), phosphate buffer (100 mM, pH 7.4), NADPH (1 mM), and test compounds (1 μM). Prior to the start of the incubation, the mixture was warmed up at 37 °C for 5 min. The reaction was initiated by the addition of NADPH to a final concentration of 1 mM. Aliquots of 50 μL samples were taken at six time points (0, 10, 20, 30, 45, and 60 min), and then placed into Eppendorf tubes containing 100 μL of cold acetonitrile with internal standard. Samples were then centrifuged at 13 000 rpm for 10 min, and an aliquot of 10 μL of the supernatants was subject to LC–MS/MS analysis. Substrate depletion parameters were calculated using the following equations:
| 1 |
where k is the slope from the linear regression analysis of the ln[%remaining] of test compound as a function of incubation time.
| 2 |
where a is 20 mg/g for mouse and human, and b is 87.5 g/kg for mouse and 25.7 g/kg for human.30
| 3 |
where Qh is the hepatic blood flow (90 mL/min for mouse and 20.7 mL/min for human).
Evaluation of CYP Inhibitory Potency
Inhibitory potency of test compounds against human P450 enzymes was investigated in human liver microsomes. The five major CYP isoforms and their respective probe substrates were as follows: CYP1A2 (phenacetin, 30 μM), CYP2C9 (tolutamide, 100 μM), CYP2C19 (S-mephenytoin, 40 μM), CYP2D6 (dextromethorphan, 5 μM), and CYP3A4 (midazolam, 1 μM). All probe substrates were used at concentrations near or below their Kms (Michaelis constant). Triplicate incubations were conducted in the 200 μL incubation mixtures at 37 °C. The mixture contained HLM (0.2 mg/mL), phosphate buffer (100 mM, pH 7.4), NADPH (1 mM), tested compounds (10 μM), and individual CYP probe substrate. The mixtures were preincubated for 10 min to allow inhibitor/enzyme interactions before the initiation of the reaction with NADPH. After a specific time period (10 min for CYP1A2, 2D6 and 3A4; 30 min for CYP2C9 and 2C19), reactions were terminated by the addition of 100 μL of cold acetonitrile containing an appropriate internal standard. Samples were then centrifuged and injected into the LC–MS/MS apparatus to quantify the concentrations of specific metabolites formed by individual CYP enzymes.
LC–MS/MS Analysis
All samples were analyzed by using a LC–MS/MS system consisting of an API4000 Qtrap mass spectrometer equipped with a turbo-V ionization source (Applied Biosystems, Ontatrio, Canada), two LC-20AD pumps with a CBM-20A controller, DGU-20A solvent degasser, and a SIL-20A autosampler (Shimadzu, Columbia, MD). An Agela Venusil XBP C18 column (50 × 2.1 mm; 5 μm particle size) was used to achieve HPLC separation. Column temperature was held at 40 °C. Flow rate was 0.3 mL/min with the total run time of 6 min.
For MS/MS quantitation, the API4000 Qtrap mass spectrometer was operated in the ESI positive mode with multiple reaction-monitoring (MRM). All compounds and the internal standard were monitored with dwell time set to 100 ms. Other MS/MS parameters were set as follows: curtain gas, 30 psi; nebulizer gas (GS1), 55 psi; turbo gas (GS2), 55 psi; ion spray voltage, 4500 V; and ion source temperature, 500 °C. For the selected ion transitions, the declustering potential (DP) and the collision energy (CE) were optimized for the best sensitivity for each analyte. Data were collected and processed using AB Sciex Analysist 1.5.2 data collection and integration software.
Glossary
Abbreviations
- B/P
brain/plasma
- CE
collision energy
- DIPEA
N,N-diisopropylethylamine
- DMSO
dimethyl sulfoxide
- L-Dopa
levodopa
- DP
declustering potential
- EDTA·2Na
ethylenediaminetetraacetic acid disodium salt
- EGTA
ethylene glycol-bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- FBS
fetal bovine serum
- GPCR
G protein-coupled receptor
- HA
heavy atom
- HIC
haloperidol induced catalepsy
- HLM
human liver microsomes
- i.p.
intraperitoneal injection
- LE
ligand efficiency
- MLM
mouse liver microsomes
- MRM
multiple reaction-monitoring
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- NaHMDS
sodium hexamethyldisilazide
- ND
not determined
- NECA
5′-(N-ethylcarboxamido)adenosine
- NMP
N-methyl-2-pyrrolidone
- PBS
phosphate-buffered saline
- PMSF
phenylmethanesulfonyl fluoride
- p.o.
per oral
- POPOP
bis(phenyloxazolyl) benzene
- PPO
2,5-diphenyloxazole
- SAR
structure–activity relationship
- SEM
standard error of the mean
- THF
tetrahydrofuran
- Tris-HCl
tris(2-carboxyethyl)phosphine hydrochloride
Supporting Information Available
1H and 13C NMR files of synthetic intermediates and final products as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† J.Z. and Z.Y.contributed equally to this paper. X.H.Z. conceived the project, and oversaw and designed the chemistry. J.Z., Z.Y., and H.M. performed synthetic chemistry work. M.W. and H.Z. performed and interpreted in vitro DMPK experiments. X.L. and L.L. performed the pharmacology and in vivo experiments, and X.C.Z. interpreted the data.
This work was supported by National Natural Science Foundation of China (Grant No. 81273372, 81130023, 31373382); National Basic Research Plan (973) of the Ministry of Science and Technology of China (2011CB5C4403); China Postdoctoral Science Foundation (2013M541729), BM2013003, and PAPD (A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions).
The authors declare no competing financial interest.
Supplementary Material
References
- Lees A. J.; Hardy J.; Revesz T. (2009) Parkinson’s disease. Lancet 373, 2055–2066. [DOI] [PubMed] [Google Scholar]
- Dorsey E.; Constantinescu R.; Thompson J.; Biglan K.; Holloway R.; Kieburtz K.; Marshall F.; Ravina B.; Schifitto G.; Siderowf A. (2007) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68, 384–386. [DOI] [PubMed] [Google Scholar]
- Ahlskog J. E.; Muenter M. D. (2001) Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov. Disord. 16, 448–458. [DOI] [PubMed] [Google Scholar]
- Rascol O.; Brooks D. J.; Korczyn A. D.; De Deyn P. P.; Clarke C. E.; Lang A. E. (2000) A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N. Engl. J. Med. 342, 1484–1491. [DOI] [PubMed] [Google Scholar]
- Chaudhuri K.; Schapira A. H. (2009) Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 8, 464–474. [DOI] [PubMed] [Google Scholar]
- Ralevic V.; Burnstock G. (1998) Receptors for purines and pyrimidines. Pharmacol. Rev. 50, 413–492. [PubMed] [Google Scholar]
- Fredholm B. B.; AP I. J.; Jacobson K. A.; Klotz K. N.; Linden J. (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 53, 527–552. [PMC free article] [PubMed] [Google Scholar]
- Fredholm B. B.; Abbracchio M. P.; Burnstock G.; Daly J. W.; Harden T. K.; Jacobson K. A.; Leff P.; Williams M. (1994) Nomenclature and classification of purinoceptors. Pharmacol. Rev. 46, 143–156. [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P.; Le Moine C.; Fisone G.; Fredholm B. B. (1999) Distribution, biochemistry and function of striatal adenosine A2A receptors. Prog. Neurobiol. 59, 355–396. [DOI] [PubMed] [Google Scholar]
- Fredduzzi S.; Moratalla R.; Monopoli A.; Cuellar B.; Xu K.; Ongini E.; Impagnatiello F.; Schwarzschild M. A.; Chen J. F. (2002) Persistent behavioral sensitization to chronic L-DOPA requires A2A adenosine receptors. J. Neurosci. 22, 1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki F. S., and Shimada J. (2000) Adenosine Antagonist and Parkinson’s Disease (Kase H., Richardson P. J., and Jenner P., Eds.); Academic Press: New York; pp 31−43. [Google Scholar]
- Shah U.; Hodgson R. (2010) Recent progress in the discovery of adenosine A2A receptor antagonists for the treatment of Parkinson’s disease. Curr. Opin. Drug Discovery Dev. 13, 466–480. [PubMed] [Google Scholar]
- Jenner P. (2005) Istradefylline, a novel adenosine A2A receptor antagonist, for the treatment of Parkinson’s disease. Expert Opin. Invest. Drugs 14, 729–738. [DOI] [PubMed] [Google Scholar]
- Jenner P.; Mori A.; Hauser R.; Morelli M.; Fredholm B. B.; Chen J. F. (2009) Adenosine, adenosine A2A antagonists, and Parkinson’s disease. Parkinsonism Relat. Disord. 15, 406–413. [DOI] [PubMed] [Google Scholar]
- Kyowa Hakko receives not approvable letter from FDA for Istradefylline (KW-6002), Kyowa Hakko Kirin Co Ltd Press Release, February28, 2008. [Google Scholar]
- Approval for manufacturing and marketing of NOURIAST® tablets 20 mg, a novel antiparkinsonian agent, Kyowa Hakko Kirin Co Ltd Press Release, March25, 2013. [Google Scholar]
- Neustadt B. R.; Hao J.; Lindo N.; Greenlee W. J.; Stamford A. W.; Tulshian D.; Ongini E.; Hunter J.; Monopoli A.; Bertorelli R.; Foster C.; Arik L.; Lachowicz J.; Ng K.; Feng K. I. (2007) Potent, selective, and orally active adenosine A2A receptor antagonists: arylpiperazine derivatives of pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines. Bioorg. Med. Chem. Lett. 17, 1376–1380. [DOI] [PubMed] [Google Scholar]
- Merck provides update on phase III clinical program for Preladenant, the company’s investigational Parkinson’s disease medicine, Merck Press Release, May23, 2013. [Google Scholar]
- Slee D. H.; Zhang X.; Moorjani M.; Lin E.; Lanier M. C.; Chen Y.; Rueter J. K.; Lechner S. M.; Markison S.; Malany S.; Joswig T.; Santos M.; Gross R. S.; Williams J. P.; Castro-Palomino J. C.; Crespo M. I.; Prat M.; Gual S.; Diaz J. L.; Wen J.; O’Brien Z.; Saunders J. (2008) Identification of novel, water-soluble, 2-amino-N-pyrimidin-4-yl acetamides as A2A receptor antagonists with in vivo efficacy. J. Med. Chem. 51, 400–406. [DOI] [PubMed] [Google Scholar]
- Slee D. H.; Chen Y.; Zhang X.; Moorjani M.; Lanier M. C.; Lin E.; Rueter J. K.; Williams J. P.; Lechner S. M.; Markison S.; Malany S.; Santos M.; Gross R. S.; Jalali K.; Sai Y.; Zuo Z.; Yang C.; Castro-Palomino J. C.; Crespo M. I.; Prat M.; Gual S.; Diaz J. L.; Saunders J. (2008) 2-Amino-N-pyrimidin-4-ylacetamides as A2A receptor antagonists: 1. Structure-activity relationships and optimization of heterocyclic substituents. J. Med. Chem. 51, 1719–1729. [DOI] [PubMed] [Google Scholar]
- Slee D. H.; Moorjani M.; Zhang X.; Lin E.; Lanier M. C.; Chen Y.; Rueter J. K.; Lechner S. M.; Markison S.; Malany S.; Joswig T.; Santos M.; Gross R. S.; Williams J. P.; Castro-Palomino J. C.; Crespo M. I.; Prat M.; Gual S.; Diaz J. L.; Jalali K.; Sai Y.; Zuo Z.; Yang C.; Wen J.; O’Brien Z.; Petroski R.; Saunders J. (2008) 2-Amino-N-pyrimidin-4-ylacetamides as A2A receptor antagonists: 2. Reduction of hERG activity, observed species selectivity, and structure-activity relationships. J. Med. Chem. 51, 1730–1739. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Rueter J. K.; Chen Y.; Moorjani M.; Lanier M. C.; Lin E.; Gross R. S.; Tellew J. E.; Williams J. P.; Lechner S. M.; Markison S.; Joswig T.; Malany S.; Santos M.; Castro-Palomino J. C.; Crespo M. I.; Prat M.; Gual S.; Diaz J. L.; Saunders J.; Slee D. H. (2008) Synthesis of N-pyrimidinyl-2-phenoxyacetamides as adenosine A2A receptor antagonists. Bioorg. Med. Chem. Lett. 18, 1778–1783. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Tellew J. E.; Luo Z.; Moorjani M.; Lin E.; Lanier M. C.; Chen Y.; Williams J. P.; Saunders J.; Lechner S. M.; Markison S.; Joswig T.; Petroski R.; Piercey J.; Kargo W.; Malany S.; Santos M.; Gross R. S.; Wen J.; Jalali K.; O’Brien Z.; Stotz C. E.; Crespo M. I.; Diaz J. L.; Slee D. H. (2008) Lead optimization of 4-acetylamino-2-(3,5-dimethylpyrazol-1-yl)-6-pyridylpyrimidines as A2A adenosine receptor antagonists for the treatment of Parkinson’s disease. J. Med. Chem. 51, 7099–7110. [DOI] [PubMed] [Google Scholar]
- Lanier M. C.; Moorjani M.; Luo Z.; Chen Y.; Lin E.; Tellew J. E.; Zhang X.; Williams J. P.; Gross R. S.; Lechner S. M.; Markison S.; Joswig T.; Kargo W.; Piercey J.; Santos M.; Malany S.; Zhao M.; Petroski R.; Crespo M. I.; Diaz J. L.; Saunders J.; Wen J.; O’Brien Z.; Jalali K.; Madan A.; Slee D. H. (2009) N-[6-Amino-2-(heteroaryl)pyrimidin-4-yl]acetamides as A2A receptor antagonists with improved drug like properties and in vivo efficacy. J. Med. Chem. 52, 709–717. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Li X.; Ma H.; Zheng J.; Zhen X.; Zhang X. (2014) Replacement of amide with bioisosteres led to a new series of potent adenosine A2A receptor antagonists. Bioorg. Med. Chem. Lett. 24, 152–155. [DOI] [PubMed] [Google Scholar]
- Jaakola V. P.; Griffith M. T.; Hanson M. A.; Cherezov V.; Chien E. Y.; Lane J. R.; Ijzerman A. P.; Stevens R. C. (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F.; Wu H.; Katritch V.; Han G. W.; Jacobson K.; Gao Z.-G.; Cherezov V.; Stevens R. C. (2011) Structure of an agonist-bound human A2A adenosine receptor. Science 332, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G.; Warne T.; Edwards P. C.; Bennett K.; Langmead C. J.; Leslie A. G.; Tate C. G. (2011) Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore A. S.; Robertson N.; Errey J. C.; Ng I.; Hollenstein K.; Tehan B.; Hurrell E.; Bennett K.; Congreve M.; Magnani F.; Tate C. G.; Weir M.; Marshall F. H. (2011) Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 19, 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston J. B.; Carlile D. J. (1997) Prediction of hepatic clearance from microsomes, hepatocytes, and liver slices. Drug Metab. Rev. 29, 891–922. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.