

Abstract

Modulation of the neuropeptide S (NPS) system has been linked to a variety of CNS disorders such as panic disorder, anxiety, sleeping disorders, asthma, obesity, PTSD, and substance abuse. In this study, a series of diphenyltetrahydro-1H-oxazolo[3,4-α]pyrazin-3(5H)-ones were synthesized and evaluated for antagonist activity at the neuropeptide S receptor. The absolute configuration was determined by chiral resolution of the key synthetic intermediate, followed by analysis of one of the individual enantiomers by X-ray crystallography. The R isomer was then converted to a biologically active compound (34) that had a Ke of 36 nM. The most potent compound displayed enhanced aqueous solubility compared with the prototypical antagonist SHA-68 and demonstrated favorable pharmacokinetic properties for behavioral assessment. In vivo analysis in mice indicated a significant blockade of NPS induced locomotor activity at an ip dose of 50 mg/kg. This suggests that analogs having improved drug-like properties will facilitate more detailed studies of the neuropeptide S receptor system.
Keywords: Neuropeptide S, structure−activity relationship, NPSR
The discovery and evaluation of numerous neuropeptides is beginning to broaden our understanding of how endogenous peptides regulate a variety of important biological processes. One neuropeptide that is demonstrating a number of important modulatory roles in the central nervous system (CNS) is neuropeptide S (NPS). NPS and its receptor (NPSR) was originally identified in 2002 by Sato et al.,1 but Xu and colleagues initiated the research to delineate the in vivo role(s) of NPS in 2004.2 NPS is a 20 amino acid peptide that functions as an agonist through activation of Gq or Gs protein coupled pathways.2 Defining both receptor and peptide localization in the CNS provided the first indications of NPS function. In situ hybridization showed that NPSR mRNA was expressed widely throughout the CNS.3 However, peptide precursor mRNA was limited to the locus ceruleus (LC) area and the parabrachial nucleus of the brainstem.4 Based on these observations, NPS was hypothesized to be involved in a number of CNS functions.
The human NPSR has two splice variants with a number of polymorphisms. One that has been well characterized occurs at position 107 and generates two variants of the receptor that differ significantly in their pharmacology5 and have been linked to panic disorder6,7 and fear processing.8 Since the discovery of NPS in 2002, modulation of the NPS system has also been linked to anxiety,9,2,10,11 sleeping disorders,2,12 asthma,13 obesity,14,15 PTSD,16,17 and importantly, substance abuse.18−21 NPSR activation has been shown to modulate reward circuits and is expressed in brain areas associated with reward processing such as the ventral tegmental area (VTA), amygdala and substantia nigra.4 Microdialysis studies have demonstrated that central NPS administration stimulates dopamine release in the medial prefrontal cortex,22 and local intra-VTA microinjections of NPS enhance dopamine release in the nucleus accumbens.23 The latter study specifically links NPS to the mesolimbic dopamine system, which is presumed to be a major neuronal pathway modulating reward. NPS has also been shown to increase cocaine self-administration in a corticotrophin releasing factor (CRF) dependent manner.24
Blockade of the NPSR using small molecule antagonists attenuates both drug-seeking and drug-taking behaviors.20,25 The prototypical NPS antagonist SHA-68 (1) shows selective antagonism at the human NPSR at an in vitro potency of 13.7 nM and has been reported to prevent the arousal promoting and anxiolytic-like effects elicited by NPS.26,27 More recently it was found that in rats, peripheral administration of 1 reduced conditioned reinstatement of cocaine seeking in two studies,25,20 yet it was found in one study to reduce cocaine self-administration but only with a concomitant reduction in food self-administration at doses up to 30 mg/kg, ip.20
Several other small molecule NPSR antagonists have recently been reported, primarily by several large pharmaceutical companies, most of which are yet to be evaluated in vivo. For instance, Merck Research Laboratories identified two scaffolds through high-throughput screening (HTS) that have high potency at the NPSR using a calcium mobilization assay. Quinolinone scaffold 3 had a potency of 18 nM and was found to have acceptable drug-like properties,28 and the second series contained a tricyclic imidazole that displayed potencies in the 40–80 nM range.29 In addition, GlaxoSmithKline identified a highly potent naphthopyranopyrimidine scaffold 4.30 Finally, another small molecule scaffold identified to date is NCGC00185684 (5), which resulted from an NIH Molecular Libraries HT screen.31 Compound 5 has good potency in both calcium mobilization and cAMP assays at 1 and 45 nM, respectively, and has recently been shown to reduce operant self-administration of alcohol at 1 mg/kg ip.
Our laboratory has developed a series of NPS antagonists based on the structure of SHA-68 and has recently demonstrated that one of these compounds, RTI-118 (2, Figure 1) blocks cocaine self-administration, cue, stress, and drug induced reinstatement.20 Interestingly, compounds 1 and 2 both reduced cue-induced reinstatement of extinguished cocaine self-administration in rats at similar doses (30 mg/kg), yet only 2 selectively blocked cocaine self-administration. Although at a higher dose of 50 mg/kg 1 was able to reduce cocaine self-administration, it also decreased food-maintained responding, suggesting a nonselective impairment of operant behavior. However, 2 was able to selectively reduce cocaine self-administration at lower doses (10–20 mg/kg, ip) without affecting food self-administration. These results indicate that 2 has better in vivo efficacy for reducing cocaine self-administration compared with 1. Interestingly, 2 (Ke = 109 nM) has roughly 8-fold reduction in antagonist potency versus 1 (13.9 nM) at the NPSR 107I variant when tested in the calcium mobilization assay.27 One distinct structural difference between these compounds is the ability of 2 to be partially ionized at physiological pH and thus more water-soluble. Therefore, the observed difference in efficacy between the two antagonists could be due to differences in drug-like properties.
Figure 1.

Representative classes of NPS antagonists.
We herein report the further evaluation of RTI-118, including the in vitro and in vivo pharmacokinetic properties, as well as additional SAR studies in an effort to shed light on the possible mechanisms for the behavioral differences between the two series. Although five structural classes of small molecule NPS antagonists are currently known, a comprehensive knowledge of the structure–activity relationships is still somewhat limited and a more detailed understanding of the molecular requirements for antagonist activity at the NPSR is required. The continued development of small molecules having improved potency and better drug-like properties than 1 may enable a better understanding of the NPS receptor system. In this study, we describe steps toward the development of a comprehensive pharmacophore with a bias toward increasing aqueous solubility and drug-like properties. Continued development of molecules that have improved drug-like properties will enable more detailed studies of the NPSR as an emerging target for a variety of CNS related disorders.
Chemistry
The synthesis of all target compounds in this study was based on modifications to a central core scaffold. The synthesis of core scaffold 37 was initiated through the condensation of 2 equiv of phenyl lithium with commercially available ethyl 1,4-dibenzylpiperazine-2-carboxylate (Scheme 1). The resulting alcohol 35 was debenzylated using 5% palladium on barium sulfate in ethanol to afford piperazine 36. Cyclization of 36 was accomplished using tert-butyl dicarbonate, triethylamine, and dimethylaminopyridine in tetrahydrofuran.32 This reaction required at least 1 equiv of dimethylaminopyridine to provide clean product. The resulting cyclic tert-butyl carbamate intermediate was deprotected with trifluoroacetic acid in dichloromethane to provide core scaffold 37 in good overall yield.
Scheme 1. Synthesis of Key Intermediate 37.

Reagents and conditions: (a) PhLi, THF, −78 °C; (b) H2 (46 psi), 10% Pd/C, 5% Pd/BaSO4, EtOH; (c) Boc2O, DMAP, NEt3, THF; (d) TFA, DCM.
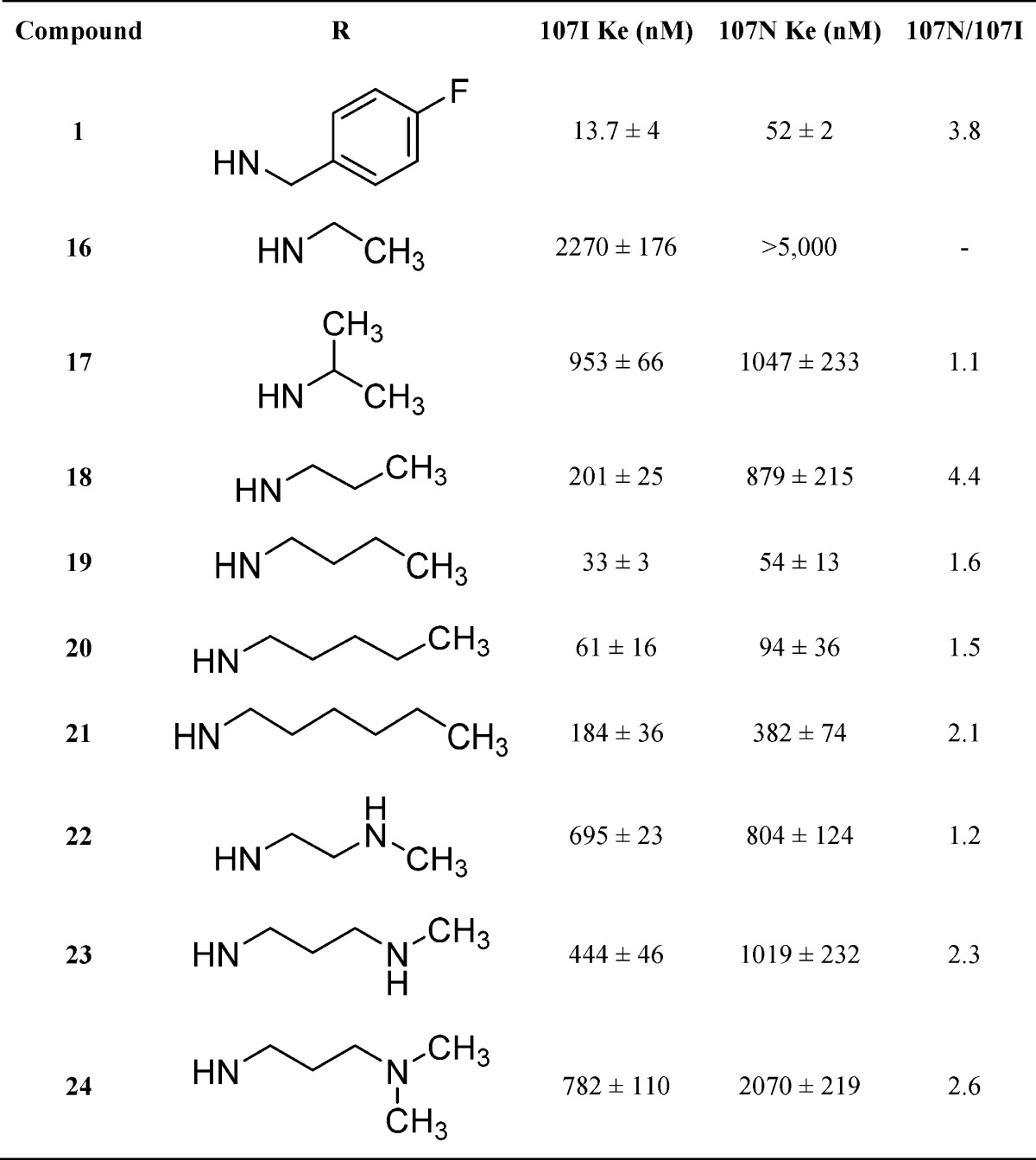
One of the primary goals of this research was to identify substituents that would confer improved aqueous solubility. Rapid condensation of a number of commercially available isocyanates with core scaffold 37 in dichloromethane provided compounds 1, 6, 16–21, 40, and 41 in good yield (Scheme 2). Displacement of the alkyl chlorides 40 and 41 with potassium carbonate, methyl amine, or dimethyl amine in dimethylformamide provided target molecules 22–24, respectively, in moderate yield. Condensation of 3-(piperidin-1-yl)propanoic acid chloride with core scaffold 37 provided amide analog 25 in excellent yield.
Scheme 2. Synthesis of Target Molecules Derived from Isocyanate or Acid Chloride Addition.

Reagents and Conditions: (a) R-isocyanate, CH2Cl2, rt; (b) acid chloride, TEA, 0 °C; (c) CH3NH2, DMF, KHCO3, rt; (d) (CH3)2NH, DMF, KHCO3, rt.
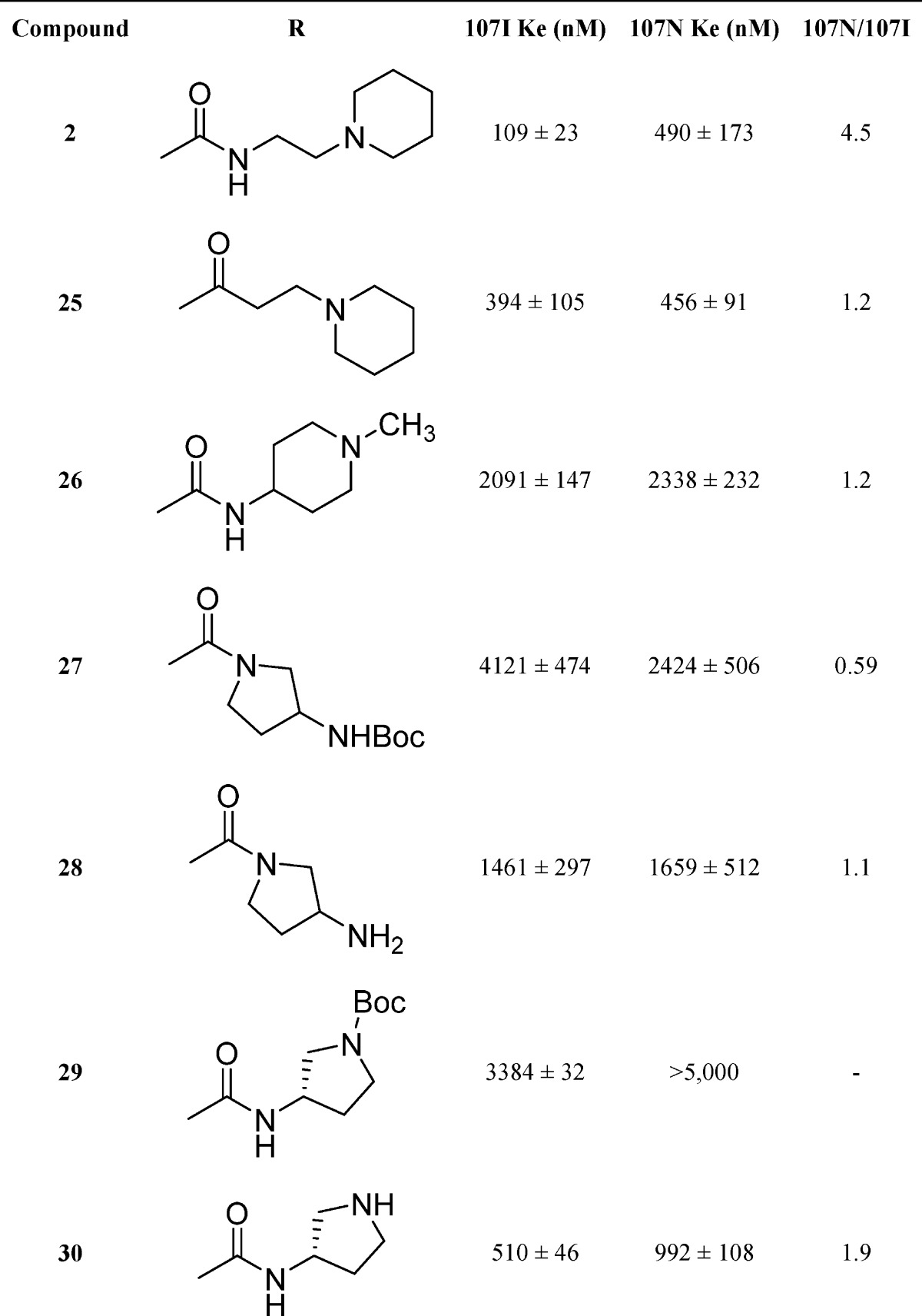
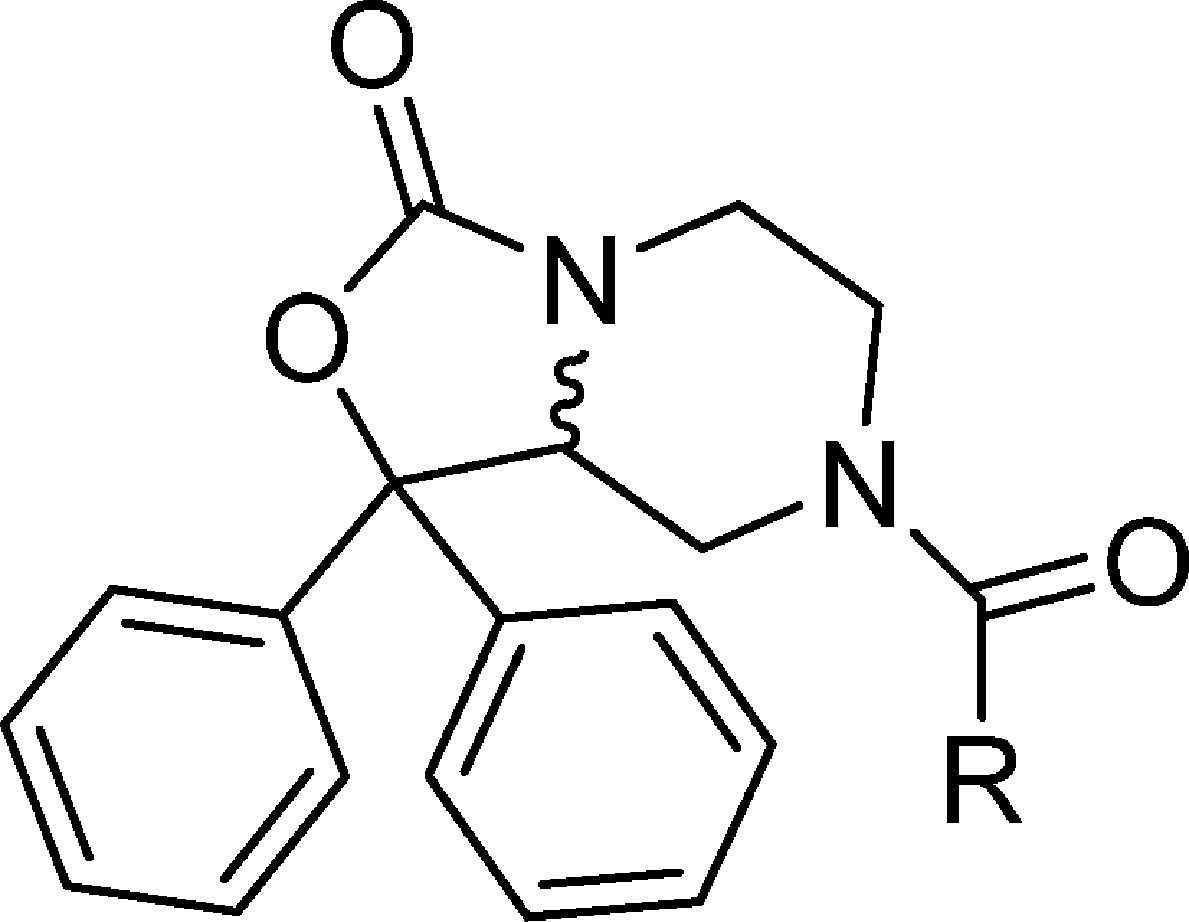
Additional analogs containing basic amines were synthesized using a general method that incorporated the appropriate amine, triphosgene, and triethylamine in tetrahydrofuran. Compounds 2, 7–13, and 26 were prepared from 37 using triphosgene, triethylamine, and piperidine ethyl amine, alternately substituted pyridine methyl amines, alternately substituted pyridine ethyl amines, tert-butyl 2-(aminomethyl)phenylcarbamate, and 1-methylpiperidin-4-amine, respectively (Scheme 3). Deprotection of 13 using trifluoroacetic acid in dichloromethane provided 14. Reductive amination using sodium triacetoxyborohydride in 1,2-dichloroethane with 1 equiv of formaldehyde afforded a mixture of mono and dialkylated products from which 15 was isolated. Target compounds 27–30 were obtained using analogous methods to that described for compound 14.
Scheme 3. Preparation of Target Molecules Using Triphosgene Conditions.

Reagents and conditions: (a) R-amine, (CCl3O)2CO, TEA, THF, 0 °C to rt; (b) TFA, DCM; (c) HCOH, NaH(OAc)3, 1,2-DCE.
The identification of the active enantiomer of core scaffold 37 was performed using a combination of chiral HPLC and X-ray crystallography. Racemic 37 was subjected to chiral HPLC to afford enantiomers 38 and 39 (Scheme 4). The R-camphorsulfonic acid salt of 38 was prepared from R-camphorsulfonic acid in methanol. The resulting salt 38a was subjected to X-ray analysis, and the chirality was determined to be S (Figure 2). Compounds 31 and 32 were then prepared from 38 and 39, respectively, using 4-fluorobenzyl isocyanate in tetrahydrofuran. Compounds 33 and 34 were isolated via separation of 2 by chiral HPLC.
Scheme 4. Identification of the Active Enantiomer of Key Intermediate 37.

Reagents and Conditions: (a) chiral HPLC; b) R-camphorsulfonic acid, MeOH; (c) 4-fluorobenzyl isocyanate.
Figure 2.
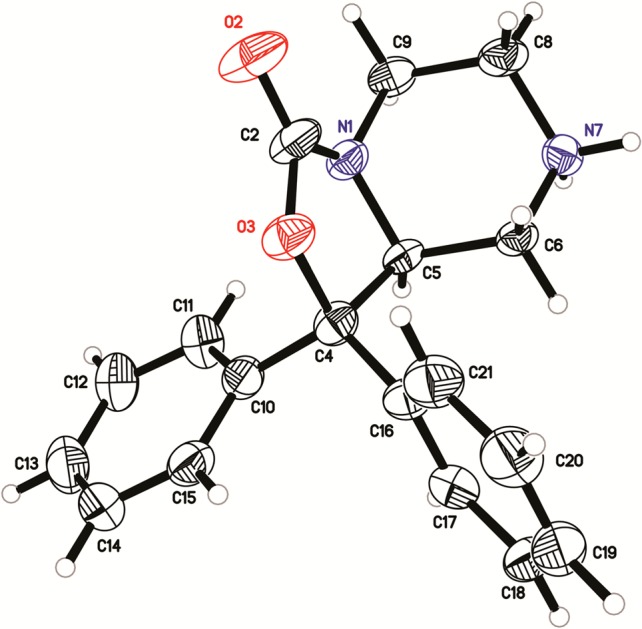
X-ray structure of the inactive enantiomer 38a.
Results and Discussion
One of the prototypical NPSR antagonists, SHA-68 (1), has been studied in a number of in vivo behavioral and in vitro biological assay systems. Pharmacokinetic studies have been performed on 1 and indicate that a significant component of the ip dose enters the CNS,26 yet it behaves differently from 2 in vivo.20 In order to provide a potential basis for this discrepancy, the aqueous solubility of 1 and 2 was evaluated. Aqueous solubility for 2 improved by a factor of 4200 at pH 3 and 3600 at pH 7 compared with 1 as determined by filtration through a 0.4 μm polycarbonate membrane in aqueous buffer (Millipore MultiScreen PCF 96 well filter plate).33 Solubility at pH 3 and 7 for compound 1 is 0.01 and 0.01 μM and that for compound 2 is 42 and 36 μM, respectively. The enhanced solubility of 2 compared with 1 is quite striking and suggests the in vivo differences between 1 and 2 may be related to drug-like properties. Analogs that retain this property and have improved potency will provide additional tools to study the NPSR.
Considering the challenges of formulation and drug delivery for analogs like 1, our laboratory set out to interrogate the tolerances to scaffold modification with the goal of improving aqueous solubility and receptor potency. Compounds were analyzed for agonist activity, and when they displayed no intrinsic activity, antagonist activity at the NPSR was measured using a functional calcium mobilization assay in stably transfected CHO-K1 cells. Each clone individually expressed one of the two predominant polymorphisms, 107I or 107N. The apparent constant Ke was determined by running EC50 curves of the agonist, NPS, with and without the test compounds to measure the rightward shift of the agonist curve. In keeping with literature reports, most compounds were more potent at the 107I variant of the NPSR ranging from 5.5-fold more potent to 0.59-fold. On average compounds were 1–2-fold more potent at the 107I variant. Ke values throughout the results section refer to results obtained using the 107I construct unless specifically noted.
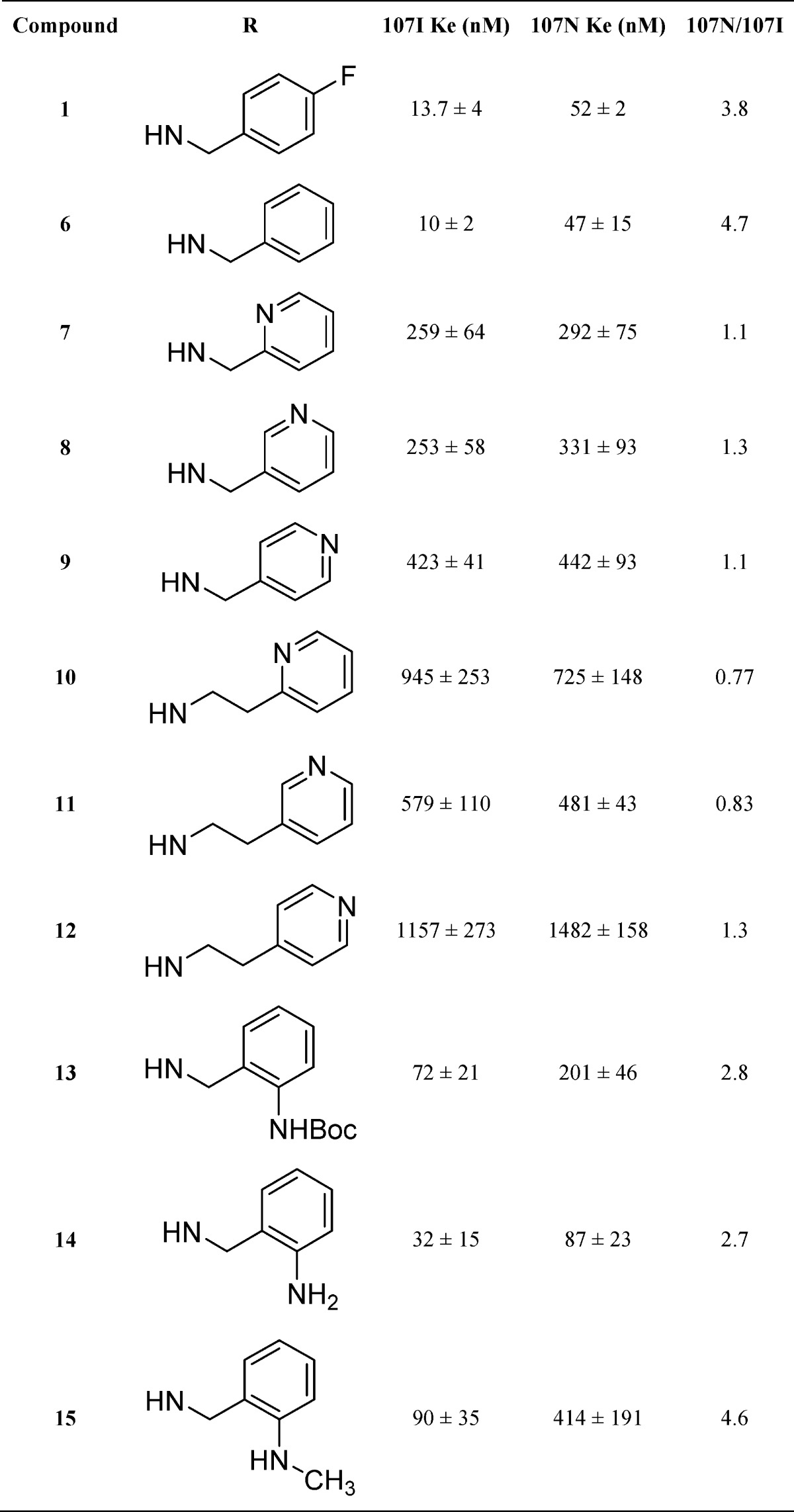
The benzyl urea functions of 1 and 6 have previously been shown as preferred substituents for antagonist potency.27 Based on this information, we introduced a series of benzylpyridines and pyridinylethyl ureas to enhance solubility. Substitution of 1 with a 2-pyridyl (7) reduced potency by 19-fold, while both the 3-pyridyl (8) and 4-pyridyl (9) also displayed a 19- and 30-fold reduction in potency, respectively, versus 1 (Table 1). This suggests that either increased polarity or electron density is not tolerated at this position. Elongation of the linker to ethyl further reduced antagonist potency. Compounds 10–12 had respective potencies of 945, 579, and 1157 nM as antagonists. In another attempt to provide compounds that could remain protonated in a suitable in vivo formulation, 2-aminobenzyl analogs were evaluated. N-Boc protected 13 had a limited effect on potency suggesting that this position could be modified with a variety of substituents and retain activity. Deprotection of 13 afforded free amine 14 (32 nM) that was only 2-fold reduced in activity from 1. Monomethylation provided 15 having a Ke of 90 nM. Based on the data from Table 1, it appears that the benzylic aryl ring is sensitive to alteration and that compounds with pyridyl substitution are not well tolerated.
Table 1. Potency of Benzyl Urea Analogs of SHA-68 (1) in CHOK1 Cells Expressing Human NPSR Isoforms 107I and 107N.
Previous studies modifying the SHA-68 scaffold (1) indicated that benzylic substituents are preferred but that aliphatic substituents can also lead to analogs with moderate potency.27 We thus set out to evaluate the size requirements for aliphatic substituents in place of the benzyl group on 1 (Table 2). Once the size limitations of the hypothetical binding site were determined, we could then evaluate tolerances to incorporation of solubilizing aliphatic amine groups. Progressive substitution with larger aliphatic moieties starting from ethyl (16, 2270 nM) to isopropyl (17, 953 nM), to propyl (18, 201 nM), and then butyl (19, 33 nM) indicated that a benzylic moiety was not a requirement for optimal potency and alkyl substituted analogs could be prepared with comparable potency to 1. Further elongation of the alkyl chain to pentyl (20, 61 nM) and hexyl (21, 184 nM) started to reduce potency from the optimal activity of butyl (19). This suggested that a lipophilic pocket of limited size accommodated aliphatic and benzylic groups. Once compound 19 was identified as the most potent alkyl analog, we set out to evaluate how incorporating amine groups would be tolerated. Compound 22 is an amino substituted analog of 19 but displays a 21-fold reduction in activity. Increasing the distance of amine from the urea by one carbon (23) slightly improved potency compared with 22, but 23 remained 14-fold less potent than the n-butyl analog 22. Methylation of 23 reduced activity to a Ke of 782 nM for dimethylated analog (24). This data indicates that substituted amino groups can be tolerated as in the case of 2 (Ke = 109 nM) but the disposition of the polar group is critical to activity.
Table 2. Potency of Alkyl Substituted Urea Analogs of SHA-68 in CHOK1 Cells Expressing Human NPSR Isoforms 107I and 107N: Alkyl Substitution of the Urea to Determine Optimal Substituent Size.
Considering that compound 2 displayed moderate potency in earlier studies, we set out to define the receptor tolerances through altering the placement of ionizable amino groups within cyclic substituents (Table 3). Converting the urea functionality to an amide was performed to determine whether a series of commercial amides could be evaluated more rapidly. Replacement of the urea led to reduced potency for 25 having a Ke of 394 nM. This data supports earlier findings where methylation of the urea nitrogen on 1 significantly reduced potency.27 Compound 26 replaced the piperidinylethyl in 2 with an N-methylpiperidine that displayed significantly reduced activity (2091 nM) compared with 2. Both the N-Boc protected and free amino pyrrolidines 27 and 28 also displayed reduced activity whereas pyrrolidine analogs 29 and 30 displayed potencies of 3384 nM and 510 nM, respectively. These data combined with results for the acyclic analogs 16–21 suggest that the receptor site prefers lipophilic substituents of specific size and that amides display reduced activity compared with ureas.
Table 3. Potency of Urea Analogs of SHA-68 in CHOK1 Cells Expressing Human NPSR Isoform 107I: Urea Substituents Designed to Enhance Aqueous Solubility.
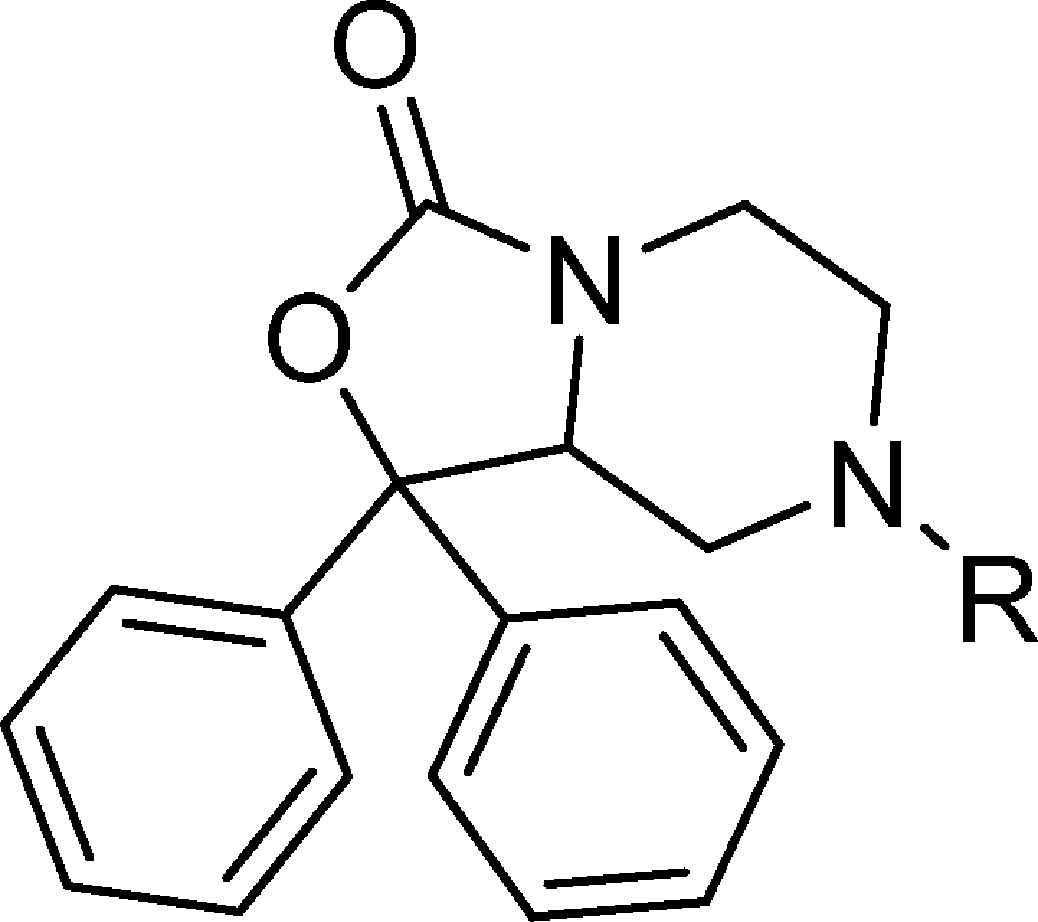
The importance of the chirality in 1 has been demonstrated where only the R isomer was active.34 Similarly, only one bioactive enantiomer was isolated from the racemic mixture of NPS antagonist 3.28 The original chiral study of 1 utilized a multistep synthesis that relied on diastereomeric separation. Considering that one enantiomer is known to retain activity we set out to identify the active enantiomer of the synthetically common intermediate 37 that could be used to rapidly evaluate a number of chiral analogs with this class of compounds. The chiral intermediate described in this study (37) can be readily appended with a large variety of substituents. The ability to further explore SAR using the known biologically active isomer of a synthetic intermediate is of significance. Thus, the intermediate amine 37 was separated into two isomers 38 and 39 using chiral HPLC. The chirality of these compounds was elucidated by preparing the R-camphorsulfonic acid salt of one of the isomers, 38, and performing an X-ray study on the resulting salt 38a. Based on this study 38a had the S configuration. The corresponding 4-fluorobenzyl ureas were then prepared from 38 (S) and 39 (R), respectively, and tested in the calcium mobilization assay. Compound 32 showed a Ke of 5.1 nM, whereas 31 was inactive up to 5 μM, confirming that the R configuration is required for activity at the NPS receptor (Table 4). Similarly, the individual isomers of the piperidylethyl analog 33 and 34 were isolated, and only one isomer, 34, displayed activity (36 nM) at the NPS receptor.
Table 4. Potency of Individual Enantiomers of Lead Compounds 1 and 2 in CHOK1 Cells Expressing Human NPSR Isoform 107I.
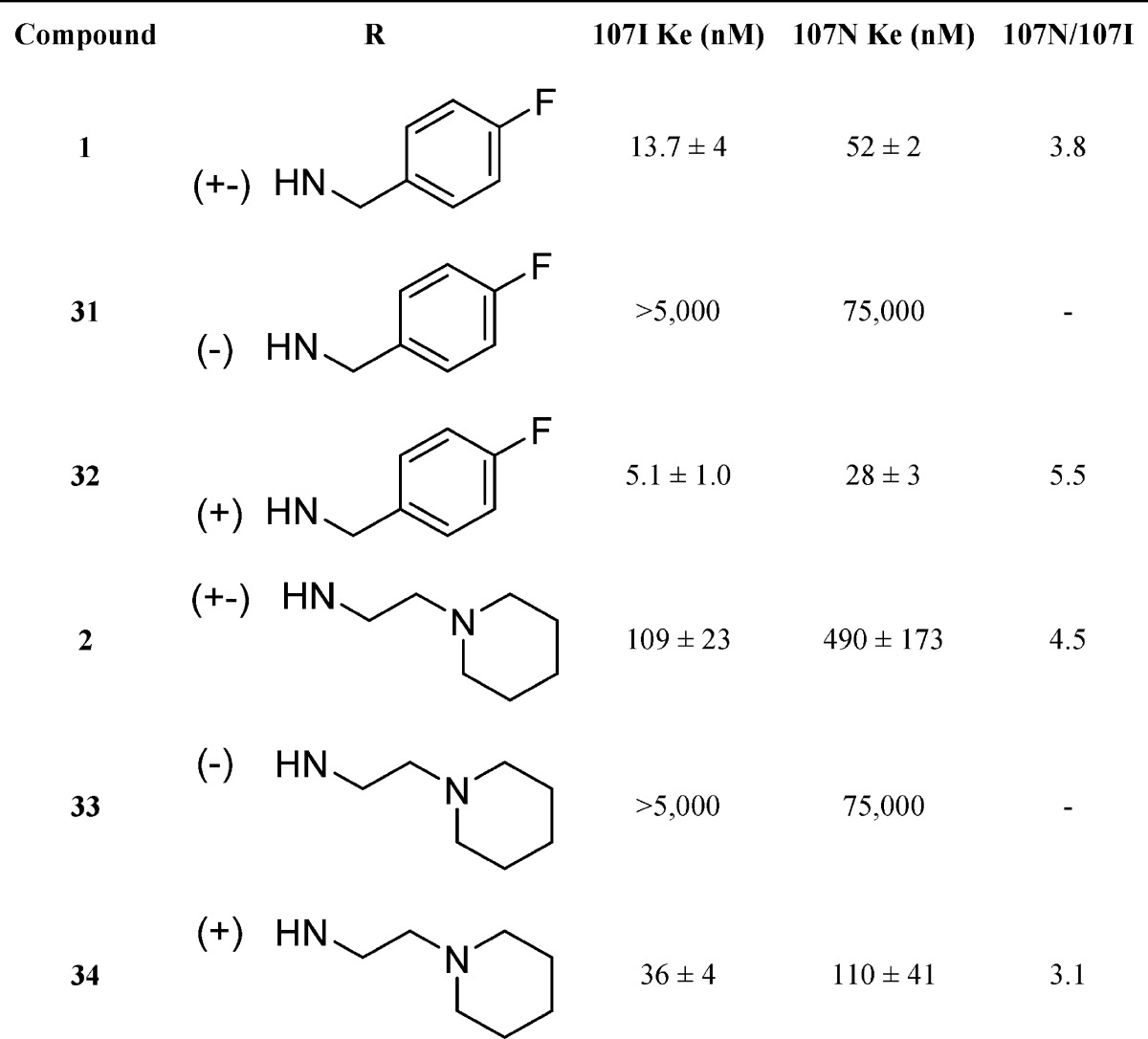
In order to get a more detailed understanding of on-target effects in vivo, compound 34 was further evaluated for its pharmacokinetic properties (Figure 3). Following an ip dose, 34 was rapidly absorbed into systemic circulation, with peak plasma concentration observed at 15 min postdose. After a distribution phase, plasma concentrations declined with an apparent half-life of 33.9 min. Brain concentrations of 34 were measurable as early as 15 min postdose (the first sampling time point), with peak brain concentration observed at 15 min postdose. Compound 34 was eliminated from the brain with an apparent half-life of 143 min. The overall brain to plasma AUC ratio, as determined by AUClast or AUC0-inf ratio, was 0.14 or 0.29, respectively. This study provides a preliminary assessment of the brain penetration of 34 in mice after an ip dose of 5 mg/kg. At 90 min postdose, brain concentrations were approximately 60 nM indicating sufficient brain penetration for NPS receptor modulation considering 34 has a Ke of 36 nM.
Figure 3.
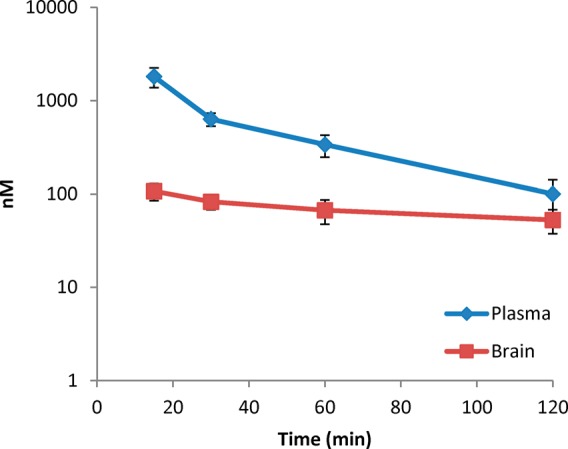
Pharmacokinetic analysis of compound 34 in brain and plasma; 5 mg/kg dose. Sample size, five animals per time point. Data points represent the mean, and error bars represent standard deviation.
In Vivo Activity of 34
NPS is known to induce hyperlocomotion in mice after central administration.2 To test whether this central effect of NPS can be antagonized by 34, we recorded distance traveled at 5 min intervals over 90 min in mice that had received injections of vehicle or 34 (5 and 50 mg/kg ip) and subsequently received central administrations of either vehicle or 1 nmol of NPS icv. As shown in Figure 4, analysis of cumulative activity data revealed that injections of NPS + vehicle increased the cumulative distance traveled by the animals in a statistically significant manner. Compound 34 alone at 50 mg/kg did not modify per se the animal behavior compared with the control group (p = 0.8738, unpaired Student’s t test). When challenged with NPS, 34 at a dose of 5 mg/kg resulted in a nonsignificant reduction (approximately 20%) of the distance traveled as compared with mice receiving NPS + vehicle injections (p = 0.3515, unpaired Student’s t test). At the highest dose (50 mg/kg), 34 significantly attenuated (approximately 60%) the effects of 1 nmol of NPS, demonstrating that 34 (50 mg/kg) was effective at least partially antagonizing NPS-induced activity. Statistically significant effects were obtained with 50 mg/kg, although the antagonist did not fully prevent the effects elicited by the peptide. Together, these data demonstrate that 34 behaves as an NPSR antagonist in vivo and is centrally acting after peripheral administration.
Figure 4.
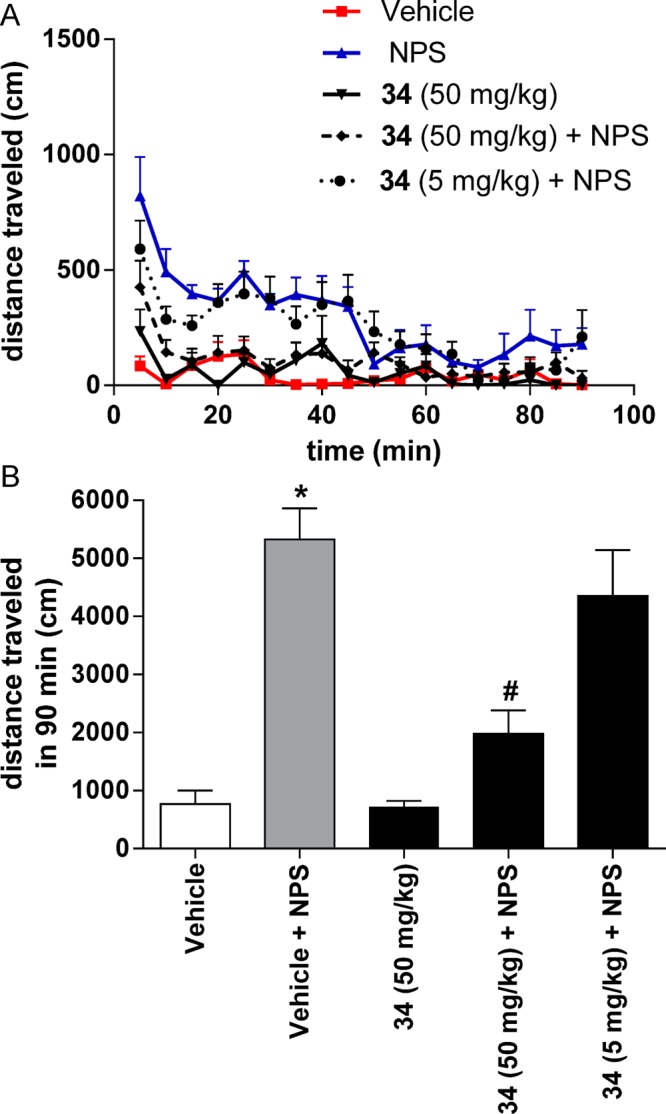
Effects of 34 on motor activity in mice. Mice had been habituated for 60 min before injection. Compound 34 was dissolved in 10% Cremophor EL (vehicle) and injected (ip) 10 min before NPS or vehicle (PBS, 0.1% BSA) were injected centrally (icv). Group sizes: Veh + Veh, n = 8; Veh + NPS, n = 7; 34 + PBS, n = 7; 34 (50 mg/kg) + NPS (1 nmole), n = 8; 34 (5 mg/kg) + NPS (1 nmole), n = 9. (A) Time course of the distance traveled over 90 min. (B) Total distance traveled during the 90 min observation period. *, p < 0.001, Veh + Veh versus Veh + NPS; #, p < 0.001 Veh + NPS versus NPS + 34 (50 mg/kg) (Students t test).
Conclusions
A series of N-substituted diphenyltetrahydro-1H-oxazolo[3,4-α]pyrazin-3(5H)-ones have been synthesized and evaluated for antagonism of the NPS receptor system. The receptor tolerance to both size and polarity of these analogs at several locations were evaluated. The most potent antagonists possessed simple aliphatic ureas such as N-butyl (19) or ureas having basic amine containing substituents that were substituted with hydrophobic groups such as piperidinoethyl (34). Incorporation of protonatable nitrogen improved the aqueous solubility of this series while maintaining NPS receptor potency. Resolution of the key intermediate for the synthesis of these analogs and identification of the absolute configuration of the bioactive isomer now affords the opportunity to rapidly obtain a number of chiral analogs for future study. In vivo behavioral analysis of the most potent compound demonstrated the ability of one such compound (34) to significantly reduce NPS stimulated locomotor activity at a dose of 50 mg/kg. The analogs described in this study provide additional detail as to the requirements for biological activity at the NPSR and represent improved tools to study the NPSR in vivo. The improvement of aqueous solubility opens up possibilities not permitted with less soluble analogs such as iv administration and microinjections into discrete brain regions using an unmodified aqueous buffer system. Identification of additional analogs having improved properties could ultimately lead to pharmacotherapies to treat a variety of CNS disorders.
Methods
General
All standard reagents were commercially available. Compounds were purified by column chromatography on a Teledyne Isco Rf chromoatography unit and by HPLC on an Agilent-Varian HPLC system equipped with Prostar 210 dual pumps, a Prostar 335 diode UV detector and a SEDEX75 (SEDERE, Olivet, France) ELSD detector. The HPLC solvent system was binary, water containing 0.1% trifluoroacetic acid (TFA) and solvent B (acetonitrile containing 0–5% water and 0.1% TFA). A semipreparative Synergy Hydro RP 80A C18 column (4 μm, 250 mm × 21.2 mm column; Phenomenex) or a Gemini-NX RP C18 column (5 μm, 250 mm × 21.2 mm; Phenomenex) was used to purify final compounds at 15 mL/min using a linear gradient from 5% to 50% or 60% B over 30 min. The purity of final compounds was determined using an analytical Synergy Hydro RP80A C18 (4 μm, 250 mm × 4.60 mm column; Phenomenex) or a Gemini-NX C18 (5 μm, 250 mm × 4.6 mm; Phenomenex) with a linear gradient of 5%–95% solvent B over 20 or 30 min at a flow rate of 1 mL/min. Absorbance was monitored at 220 nm. Individual enantiomers were separated, and enantiomeric purity was determined using an analytical Chiralpak-IA column (4.6 mm × 250 mm) with isocratic conditions (hexanes/isopropanol 9/1 or methyl-tert-butylether/ethanol 8/2 or 95/5 containing 0.1% DEA; 1 mL/min). The molecular ion of final compounds was determined using a PE Sciex API 150 EX LC/MS system from PerkinElmer (San Jose, California). Reactions were monitored by thin-layer chromatography (TLC) carried out on precoated 60 Å 250 mm silica gel TLC plates with F-254 indicator visualized under UV light and developed using ceric ammonium molybdate. 1H NMR spectra were recorded at 300 MHz on a Bruker Avance 300 Spectrospin instrument and are reported as follows: chemical shift δ in ppm (multiplicity, coupling constant (Hz), and integration). The following abbreviations were used to explain multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, m = multiplet, br = broad, dd = doublet of doublets, dt = doublet of triplets, ddd = doublet of doublets of doublets, ddt = doublet of doublet of triplets. Optical rotations were determined using a Rudolph Research Autopol III automatic polarimeter.
N-(4-Fluorobenzyl)-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (1)
To a solution of 37 (50 mg, 0.17 mmol) in CH2Cl2 (5 mL) at 0 °C under N2 was added 1-fluoro-4-(isocyanatomethyl)benzene (143 μL, 0.170 mmol). The reaction mixture was stirred at room temperature for 2 h and concentrated, and the residue was purified by medium pressure column chromatography (12 g f SiO2, 100% methylene chloride to 50% chloroform/methanol/ammonium hydroxide 80/20/1) followed by semipreparative HPLC to provide 1 as a white powder (29 mg, 30%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.17 (dd, J = 13.19, 11.30 Hz, 1 H), 2.83–3.00 (m, 1 H), 3.01–3.16 (m, 1 H), 3.63 (dd, J = 13.00, 1.70 Hz, 1 H), 3.86 (dd, J = 13.00, 2.45 Hz, 1 H), 4.03 (dd, J = 13.19, 2.26 Hz, 1 H), 4.26–4.49 (m, 3 H), 4.69–4.81 (m, 1 H), 6.95–7.09 (m, 1 H), 7.21–7.45 (m, 11 H), 7.47–7.56 (m, 2 H). ESI MS m/z: calcd for C26H24FN3O3 445.49, found 446.7 (M + H)+. HPLC (Synergy Hydro, 20 min) tR = 19.47 min (>99%).
3-Oxo-1,1-diphenyl-N-(2-(piperidin-1-yl)ethyl)tetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (2)
To a solution of 37 (0.5 g, 1.70 mmol) in THF (anhydrous, 70 mL) at 0 °C under N2 was added triphosgene (0.17 g, 0.57 mmol), and the reaction mixture was allowed to stir at 0 °C for 5 min. To this mixture was added triethylamine (710 μL, 5.1 mmol), and the reaction was allowed to stir at 0 °C for 5 min. To this mixture was then added piperidine ethylamine (240 μL, 1.67 mmol) at 0 °C, and the reaction mixture was allowed to stir at 0 °C for 2 h and then warmed to room temperature. The resulting suspension was concentrated to dryness and taken up in CH2Cl2 (100 mL). The suspension was made basic with sat. aq. NaHCO3 and extracted with CH2Cl2 (3 × 100 mL). The organic layers were washed with brine (50 mL), dried (MgSO4), and concentrated. The residue was purified by column chromatography (12 g of SiO2, 100% methylene chloride to 40% chloroform/methanol/ammonium hydroxide 80/20/1) provided 2 (470 mg, 62%) as a white solid. 1H NMR (300 MHz, chloroform-d) δ ppm 1.48 (d, J = 4.52 Hz, 2 H), 1.58 (m, J = 4.90 Hz, 4 H), 2.18 (dd, J = 13.19, 11.30 Hz, 1 H), 2.37–2.55 (m, 6 H), 2.84 (td, J = 12.72, 3.58 Hz, 1 H), 3.08 (td, J = 12.62, 3.77 Hz, 1 H), 3.31 (quin, J = 5.18 Hz, 2 H), 3.73–3.97 (m, 3 H), 4.43 (dd, J = 11.11, 3.58 Hz, 1 H), 5.47–5.74 (m, 1 H), 7.28–7.45 (m, 8 H), 7.49–7.58 (m, 2 H). ESI HRMS m/z: calcd for C26H32N4O3 448.57, found 449.25 (M + H)+. HPLC (Synergy Hydro, 20 min) tR = 15.23 min (>99%).
N-Benzyl-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (6)
An analogous procedure to that described for 1 using benzyl isocyanate provided 6 as a white solid (31 mg, 86%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.10–2.22 (m, 1 H), 2.81–2.95 (m, 1 H), 3.00–3.12 (m, 1 H), 3.59–3.70 (m, 1 H), 3.82 (dd, J = 13.00, 3.20 Hz, 1 H), 4.03 (dt, J = 13.28, 1.84 Hz, 1 H), 4.31–4.49 (m, 3 H), 4.89 (br s, 1 H), 7.24–7.42 (m, 13 H), 7.47–7.54 (m, 2 H). ESI HRMS m/z: calcd for C26H25N3O3 427.19, found 450.18 (C26H25N3O3 +Na)+.
3-Oxo-1,1-diphenyl-N-(pyridin-2-ylmethyl)tetrahydro-1H-oxazolo[3,4-α]pyrazine-7(3H)-carboxamide (7)
An analogous procedure to that described for 2 using pyridin-2-ylmethanamine provided 7 as a white powder (42 mg, 58%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.21 (dd, J = 13.19, 11.30 Hz, 1 H), 2.96 (dd, J = 13.56, 3.77 Hz, 1 H), 3.10 (dd, J = 13.19, 3.77 Hz, 1 H), 3.78–3.93 (m, 2 H), 4.04 (dd, J = 13.37, 2.45 Hz, 1 H), 4.44 (dd, J = 11.30, 3.77 Hz, 1 H), 4.48–4.68 (m, 2 H), 5.95–6.09 (m, 1 H), 7.21 (dd, J = 7.16, 5.27 Hz, 1 H), 7.27–7.44 (m, 9 H), 7.49–7.60 (m, 2 H), 7.67 (td, J = 7.63, 1.70 Hz, 1 H), 8.52 (d, J = 4.90 Hz, 1 H). ESI MS m/z: calcd for C25H24N4O3 428.49, found 427.6 (M – H)−, 429.2 (M + H)+, 451.5 (M + Na)+. HPLC (Synergy Hydro, 20 min) tR = 14.61 min (95.0%).
3-Oxo-1,1-diphenyl-N-(pyridin-3-ylmethyl)tetrahydro-1H-oxazolo[3,4-α]pyrazine-7(3H)-carboxamide (8)
An analogous procedure to that described for 2 using pyridin-3-ylmethanamine provided 8 as white powder (52 mg, 71%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.19 (dd, J = 13.56, 11.30 Hz, 1 H), 2.95 (dd, J = 13.00, 3.20 Hz, 1 H), 3.03–3.15 (m, 1 H), 3.61–3.70 (m, 1 H), 3.82–3.93 (m, 1 H), 3.95–4.05 (m, 1 H), 4.34–4.51 (m, 2 H), 4.83 (br s, 1 H), 7.22–7.43 (m, 10 H), 7.48–7.55 (m, 2 H), 7.64 (d, J = 7.16 Hz, 1 H), 8.51–8.57 (m, 1 H). ESI MS m/z: calcd for C25H24N4O3 428.49, found 427.4 (M – H)−, 429.0 (M + H)+, 451.3 (M + Na)+. HPLC (Synergy Hydro, 20 min) tR = 14.20 min (93.4%).
3-Oxo-1,1-diphenyl-N-(pyridin-4-ylmethyl)tetrahydro-1H-oxazolo[3,4-α]pyrazine-7(3H)-carboxamide (9)
An analogous procedure to that described for 2 using pyridin-4-ylmethanamine provided 9 as a white powder (47 mg, 65%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.20 (dd, J = 13.56, 11.30 Hz, 1 H), 2.89–3.02 (m, 1 H), 3.03–3.18 (m, 1 H), 3.62–3.75 (m, 1 H), 3.82–3.94 (m, 1 H), 4.02 (ddd, J = 13.38, 3.58, 1.51 Hz, 1 H), 4.31–4.51 (m, 3 H), 5.01 (br s, 1 H), 7.13–7.23 (m, 2 H), 7.24–7.44 (m, 8 H), 7.47–7.58 (m, 2 H), 8.49–8.64 (m, 2 H). ESI MS m/z: calcd for C25H24N4O3 428.49, found 429.3 (M + H)+; 451.4 (M + Na)+, 427.5 (M – H)−, HPLC (Synergy Hydro, 20 min) tR = 14.04 min (94.1%).
3-Oxo-1,1-diphenyl-N-(2-(pyridin-2-yl)ethyl)tetrahydro-1H-oxazolo[3,4-α]pyrazine-7(3H)-carboxamide (10)
An analogous procedure to that described for 2 using 2-(pyridin-2-yl)ethanamine provided 10 as white powder (50 mg, 66%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.17 (dd, J = 13.19, 11.30 Hz, 1 H), 2.84 (td, J = 12.81, 3.39 Hz, 1 H), 2.93–3.11 (m, 3 H), 3.55–3.66 (m, 2 H), 3.72–3.87 (m, 2 H), 3.89–4.00 (m, 1 H), 4.39 (dd, J = 11.11, 3.58 Hz, 1 H), 6.30 (t, J = 4.90 Hz, 1 H), 7.12–7.21 (m, 2 H), 7.23–7.42 (m, 8 H), 7.46–7.55 (m, 2 H), 7.63 (td, J = 7.72, 1.88 Hz, 1 H), 8.43 (dt, J = 4.62, 1.46 Hz, 1 H). ESI MS m/z: calcd for C26H26N4O3 442.52, found 443.4 (M + H)+, 441.4 (M – H)−. HPLC (Synergy Hydro, 20 min) tR = 14.76 min (97.8%).
3-Oxo-1,1-diphenyl-N-(2-(pyridin-3-yl)ethyl)tetrahydro-1H-oxazolo[3,4-α]pyrazine-7(3H)-carboxamide (11)
An analogous procedure to that described for 2 using 2-(pyridin-3-yl)ethanamine provided 11 as a white powder (28 mg, 37%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.14 (dd, J = 13.19, 11.30 Hz, 1 H), 2.78–2.95 (m, 3 H), 3.02 (td, J = 12.72, 3.58 Hz, 1 H), 3.34–3.53 (m, 2 H), 3.60 (d, J = 12.81 Hz, 1 H), 3.81 (dd, J = 12.81, 2.64 Hz, 1 H), 3.94 (dd, J = 13.19, 3.39 Hz, 1 H), 4.39 (dd, J = 11.30, 3.77 Hz, 1 H), 7.17–7.44 (m, 9 H), 7.46–7.56 (m, 3 H), 8.35–8.50 (m, 2 H). ESI MS m/z: calcd for C26H26N4O3 442.52, found 441.3 (M – H)−, 443.5 (M + H)+, 465.4 (M + Na)+. HPLC (Synergy Hydro, 20 min) tR = 14.59 min (97.9%).
3-Oxo-1,1-diphenyl-N-(2-(pyridin-4-yl)ethyl)tetrahydro-1H-oxazolo[3,4-α]pyrazine-7(3H)-carboxamide (12)
An analogous procedure to that described for 2 using 2-(pyridin-4-yl)ethanamine provided 12 as a white powder (55 mg, 72%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.09–2.20 (m, 1 H), 2.78–2.91 (m, 3 H), 3.01 (td, J = 12.72, 3.20 Hz, 1 H), 3.40–3.57 (m, 2 H), 3.59–3.71 (m, 1 H), 3.78 (d, J = 13.19 Hz, 1 H), 3.96 (dd, J = 13.37, 2.83 Hz, 1 H), 4.39 (dd, J = 11.11, 3.58 Hz, 1 H), 7.04–7.11 (m, 2 H), 7.19–7.44 (m, 8 H), 7.44–7.53 (m, 2 H), 8.40 (dd, J = 4.33, 1.70 Hz, 2 H). ESI MS m/z: calcd for C26H26N4O3 442.52, found 441.5 (M – H)−, 443.5 (M + H)+, 465.3 (M + Na)+. HPLC (Synergy Hydro, 20 min) tR = 14.55 min (90.6%).
tert-Butyl-2-((3-oxo-1,1-diphenylhexahydro-1H-oxazolo[3,4-a]pyrazine-7-carboxamido)methyl)-phenylcarbamate (13)
An analogous procedure to that described for 2 using tert-butyl-2-(aminomethyl)phenylcarbamate provided 13 as a viscous oil (75 mg, 44%). 1H NMR (300 MHz, chloroform-d) δ ppm 1.50 (s, 9 H), 2.14 (dd, J = 13.37, 11.11 Hz, 1 H), 2.73–2.89 (m, 1 H), 2.93–3.09 (m, 1 H), 3.66–3.85 (m, 2 H), 3.91–4.02 (m, 1 H), 4.29 (d, J = 5.65 Hz, 1 H), 4.34–4.48 (m, 2 H), 5.44 (s, 1 H), 7.04 (dd, J = 7.54, 1.13 Hz, 1 H), 7.21–7.42 (m, 10 H), 7.44–7.54 (m, 2 H), 7.70 (d, J = 7.91 Hz, 1 H), 8.15 (s, 1 H). ESI MS m/z: calcd for C31H34N4O5 542.25, found 565.3 (M + 23)+.
N-(2-Aminobenzyl)-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (14)
Compound 13 (73 mg, 0.13 mmol) was dissolved in CH2Cl2 (10 mL) at 0 °C. Anhydrous TFA (5 mL) was then added to the solution and allowed to stir at rt for 2 h. The resulting solution was basified with sat. aq. NaHCO3 to pH 11 and extracted with CH2Cl2 (3 × 20 mL). The organic extracts were combined, dried with MgSO4, and concentrated to provide a viscous oil. The oil was purified using medium pressure column chromatography (12 g of SiO2, 100% methylene chloride to 50% chloroform/methanol/ammonium hydroxide 80/20/1) to provide 14 as a viscous oil (59 mg, 99%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.07–2.21 (m, 1 H), 2.79 (td, J = 12.81, 3.77 Hz, 1 H), 2.92–3.13 (m, 1 H), 3.61–3.81 (m, 3 H), 3.93 (dd, J = 13.19, 3.01 Hz, 3 H), 4.25–4.50 (m, 4 H), 5.06 (br s, 1 H), 6.61–6.72 (m, 2 H), 6.99–7.14 (m, 2 H), 7.23–7.40 (m, 8 H), 7.44–7.52 (m, 2 H). ESI MS m/z: calcd for C26H26N4O3 442.20, found 443.5 (M + H)+.
N-(2-(Methylamino)benzyl)-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (15)
To a solution of 14 (55 mg, 0.124 mmol) in 1,2-dichloroethane (5 mL) was added formaldehyde (37% solution in water, 37 μL, 0.50 mmol) and sodium triacetoxyborohydride (105 mg, 0.50 mmol). The reaction mixture was stirred at room temperature for 1 h and then quenched with NaHCO3 (sat. aq. solution; 10 mL), and the aqueous layer was washed with methylene chloride (2 × 10 mL). The organic layer was separated, washed with brine (5 mL), dried (MgSO4), and concentrated under reduced pressure to provide an oil. The oil was purified by column chromatography (12 g of SiO2, 100% methylene chloride to 50% chloroform/methanol/ammonium hydroxide 80/20/1) to provide 15 as a white solid (6 mg, 5%). 1H NMR (300 MHz, DMSO-d6) δ ppm 2.10–2.25 (m, 1 H), 2.54 (s, 1 H), 2.79 (s, 3 H), 3.19–3.31 (m, 1 H), 3.37–3.47 (m, 1 H), 3.50–3.70 (m, 2 H), 4.32 (d, J = 6.40 Hz, 2 H), 4.43 (s, 2 H), 4.67–4.79 (m, 1 H), 6.66–6.78 (m, 2 H), 6.95–7.14 (m, 2 H), 7.25–7.48 (m, 8 H), 7.54–7.65 (m, 2 H). 1H NMR (300 MHz, chloroform-d) δ ppm 2.24 (dd, J = 13.56, 11.30 Hz, 1 H), 2.88 (s, 3 H), 2.91–3.05 (m, 1 H), 3.05–3.23 (m, 1 H), 3.67 (d, J = 12.43 Hz, 2 H), 3.85 (dd, J = 13.00, 2.83 Hz, 1 H), 4.23–4.32 (m, 1 H), 4.38–4.46 (m, 1 H), 4.47–4.61 (m, 3 H), 6.71–6.86 (m, 2 H), 6.96 (d, J = 6.40 Hz, 1 H), 7.11–7.21 (m, 1 H), 7.27–7.42 (m, 8 H), 7.47–7.56 (m, 2 H). ESI MS m/z: calcd for C27H28N4O3 456.54, found 458.3 (M + H)+.
N-Ethyl-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (16)
An analogous procedure to that described for 1 using ethylisocyanate provided 16 as viscous oil (160 mg, 93%). 1H NMR (300 MHz, chloroform-d) δ ppm 1.13 (t, J = 7.16 Hz, 3 H), 2.07–2.20 (m, 1 H), 2.81–2.96 (m, 1 H), 3.07 (td, J = 12.72, 3.58 Hz, 1 H), 3.19–3.35 (m, 2 H), 3.58–3.69 (m, 1 H), 3.84 (dd, J = 13.00, 2.45 Hz, 1 H), 4.00 (ddd, J = 13.28, 3.49, 1.32 Hz, 1 H), 4.42 (dd, J = 11.30, 3.39 Hz, 1 H), 4.50–4.58 (m, 1 H), 7.25–7.42 (m, 8 H), 7.47–7.54 (m, 2 H). ESI HRMS m/z: calcd for C21H23N3O3 365.17, found 388.16 (M + Na)+.
N-Isopropyl-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (17)
An analogous procedure to that described for 1 using i-propylisocyanate provided 17 as a viscous oil (166 mg, 93%). 1H NMR (300 MHz, chloroform-d) δ ppm 1.10–1.21 (m, 6 H), 2.02–2.18 (m, 1 H), 2.79–2.95 (m, 1 H), 3.07 (td, J = 12.62, 3.39 Hz, 1 H), 3.59 (dt, J = 13.19, 1.70 Hz, 1 H), 3.78–4.11 (m, 3 H), 4.33 (d, J = 7.16 Hz, 1 H), 4.42 (dd, J = 11.30, 3.77 Hz, 1 H), 7.25–7.44 (m, 8 H), 7.46–7.56 (m, 2 H). ESI HRMS m/z: calcd for C22H25N3O3 379.19, found 402.18 (M + Na)+.
3-Oxo-1,1-diphenyl-N-propyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (18)
An analogous procedure to that described for 1 using n-propylisocyanate provided 18 as a viscous oil (166 mg, 93%). 1H NMR (300 MHz, chloroform-d) δ ppm 0.91 (t, J = 7.54 Hz, 3 H), 1.46–1.59 (m, 2 H), 2.07–2.20 (m, 1 H), 2.83–2.95 (m, 1 H), 3.07 (td, J = 12.62, 3.39 Hz, 1 H), 3.18 (tdd, J = 7.25, 7.25, 5.46, 1.88 Hz, 2 H), 3.63 (dt, J = 13.19, 1.88 Hz, 1 H), 3.84 (dd, J = 12.81, 2.64 Hz, 1 H), 4.01 (ddd, J = 13.28, 3.67, 1.51 Hz, 1 H), 4.42 (dd, J = 11.30, 3.39 Hz, 1 H), 4.55 (t, J = 5.46 Hz, 1 H), 7.23–7.44 (m, 8 H), 7.47–7.56 (m, 2 H). ESI HRMS m/z: calcd for C22H25N3O3 379.19, found 402.18 (M + Na)+.
N-Butyl-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (19)
An analogous procedure to that described for 1 using n-butylisocyanate provided 19 as a viscous oil (24 mg, 71%). 1H NMR (300 MHz, chloroform-d) δ ppm 0.87–0.97 (m, 3 H), 1.30–1.40 (m, 2 H), 1.41–1.54 (m, 2 H), 2.07–2.19 (m, 1 H), 2.81–2.94 (m, 1 H), 3.07 (td, J = 12.62, 3.39 Hz, 1 H), 3.16–3.27 (m, 2 H), 3.59–3.69 (m, 1 H), 3.83 (dd, J = 13.00, 2.83 Hz, 1 H), 3.95–4.05 (m, 1 H), 4.41 (dd, J = 11.11, 3.58 Hz, 1 H), 4.61 (t, J = 5.27 Hz, 1 H), 7.23–7.44 (m, 8 H), 7.47–7.57 (m, 2 H). ESI HRMS m/z: calcd for C23H27N3O3 393.21, found 416 0.19 (M + Na)+.
3-Oxo-N-pentyl-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (20)
An analogous procedure to that described for 1 using n-pentylisocyanate provided 20 as viscous oil (39 mg, 64%). 1H NMR (300 MHz, chloroform-d) δ ppm 0.81–0.98 (m, 3 H), 1.18–1.41 (m, 4 H), 1.49 (quin, J = 7.25 Hz, 2 H), 2.13 (dd, J = 13.19, 11.30 Hz, 1 H), 2.79–2.97 (m, 1 H), 3.07 (td, J = 12.62, 3.77 Hz, 1 H), 3.14–3.31 (m, 2 H), 3.56–3.72 (m, 1 H), 3.83 (dd, J = 13.19, 2.64 Hz, 1 H), 4.01 (ddd, J = 13.37, 3.58, 1.13 Hz, 1 H), 4.41 (dd, J = 11.30, 3.77 Hz, 1 H), 4.57 (t, J = 5.46 Hz, 1 H), 7.16–7.46 (m, 8 H), 7.47–7.61 (m, 2 H). ESI HRMS m/z: calcd for C24H29N3O3 407.22, found 430.21 (M + Na)+.
N-Hexyl-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (21)
An analogous procedure to that described for 1 using n-hexylisocyanate provided 21 as a viscous oil (39 mg, 61%). 1H NMR (300 MHz, chloroform-d) δ ppm 0.84–0.92 (m, 3 H), 1.25–1.33 (m, 6 H), 1.43–1.53 (m, 2 H), 2.06–2.15 (m, 1 H), 2.89 (dd, J = 13.00, 3.58 Hz, 1 H), 3.05 (dd, J = 12.62, 3.58 Hz, 1 H), 3.16–3.24 (m, 2 H), 3.60–3.69 (m, 1 H), 3.60–3.68 (m, 1 H), 3.83 (dd, J = 13.00, 2.83 Hz, 1 H), 4.01 (ddd, J = 13.38, 3.58, 1.13 Hz, 1 H), 4.41 (dd, J = 11.30, 3.77 Hz, 1 H), 4.59 (s, 1 H), 7.26–7.42 (m, 8 H), 7.47–7.55 (m, 2 H). ESI HRMS m/z: calcd for C25H31N3O3 421.24, found 444.22 (M + Na)+.
N-(2-(Methylamino)ethyl)-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (22)
Compound 40 (50 mg, 0.14 mmol) was dissolved in THF (15 mL) and methylamine (2 M soln. in THF, 0.70 mL, 1.4 mmol), and NaHCO3 (29 mg, 0.35 mmol), and KI (13 mg, 0.06 mmol) were added. The reaction mixture was heated to 73 °C for 60 h and concentrated to dryness. The residue was purified using medium pressure column chromatography (12 g of SiO2, 100% ethyl acetate to 50% chloroform/methanol/ammonium hydroxide 80/20/1) to provide 22 (15 mg, 30%). 1H NMR (300 MHz, chloroform-d) δ ppm 1.87 (br s, 1 H), 2.14 (dd, J = 13.19, 11.30 Hz, 1 H), 2.41 (s, 3 H), 2.73 (t, J = 5.65 Hz, 2 H), 2.86 (td, J = 12.81, 3.77 Hz, 1 H), 3.08 (td, J = 12.72, 3.58 Hz, 1 H), 3.23–3.38 (m, 2 H), 3.67–3.75 (m, 1 H), 3.84 (dd, J = 13.19, 2.64 Hz, 1 H), 4.02 (ddd, J = 13.37, 3.58, 1.13 Hz, 1 H), 4.41 (dd, J = 11.30, 3.77 Hz, 1 H), 5.26–5.34 (m, 1 H), 7.25–7.42 (m, 8 H), 7.48–7.58 (m, 2 H). ESI HRMS m/z: calcd for C22H26N4O3 394.20, found 395.21 (M + H)+.
N-(3-(Methylamino)propyl)-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (23)
An analogous procedure to that described for 22 using 41 as the starting material provided 23 as a viscous oil (8 mg, 20%). 1H NMR (300 MHz, chloroform-d) δ ppm 1.62–1.67 (m, 3 H), 2.15 (dd, J = 13.37, 11.11 Hz, 1 H), 2.37 (s, 3 H), 2.70–2.78 (m, 2 H), 2.84 (dd, J = 13.19, 3.39 Hz, 1 H), 3.07 (td, J = 12.62, 3.77 Hz, 1 H), 3.28–3.41 (m, 2 H), 3.67–3.77 (m, 1 H), 3.83 (dd, J = 13.37, 2.83 Hz, 1 H), 3.90–3.98 (m, 1 H), 4.41 (dd, J = 11.30, 3.77 Hz, 1 H), 6.92 (d, J = 4.90 Hz, 1 H), 7.26–7.44 (m, 8 H), 7.48–7.56 (m, 2 H). ESI HRMS m/z: calcd for C23H28N4O3 408.22, found 409.22 (M + H)+.
N-(3-(Dimethylamino)propyl)-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (24)
An analogous procedure to that described for 22 using 41 and dimethylamine as starting materials provided 24 as a viscous oil (19 mg, 37%). 1H NMR (300 MHz, chloroform-d) δ ppm 1.61–1.68 (m, 3 H), 2.14–2.21 (m, 6 H), 2.37–2.45 (m, 2 H), 2.79–2.87 (m, 1 H), 3.00–3.08 (m, 1 H), 3.28–3.36 (m, 2 H), 3.62–3.73 (m, 1 H), 3.83 (dd, J = 12.81, 2.64 Hz, 1 H), 3.89 (br s, 1 H), 4.40 (dd, J = 11.11, 3.58 Hz, 1 H), 7.16 (br s, 1 H), 7.28–7.44 (m, 8 H), 7.48–7.55 (m, 2 H). ESI HRMS m/z: calcd for C24H30N4O3 422.23, found 445.22 (M + Na)+.
1,1-Diphenyl-7-(3-(piperidin-1-yl)propanoyl)tetrahydro-1H-oxazolo[3,4-α]pyrazin-3(5H)-one Trifluoroacetate (25)
To a stirred solution of 3-(piperidin-1-yl)propanoic acid was added a 2-fold excess of thionyl chloride, and the reaction mixture was stirred at room temperature for 12 h. The reaction mixture was concentrated to dryness and redissolved in anhydrous CH2Cl2 (10 mL, 0.04 mM). To this solution was added 37 (29 mg, 0.1 mmol) and TEA (56 μL, 0.4 mmol). The mixture was allowed to stir at rt for 6 h and was concentrated to dryness. The residue was purified by column chromatography (12 g of SiO2, 100% methylene chloride to 50% chloroform/methanol/ammonium hydroxide 80/20/1) and then semipreparative HPLC (Synergy Hydro; 15 mL/min 5%–50% B over 30 min) to afford 25 as viscous oil (20 mg, 37%). 1H NMR (300 MHz, chloroform-d) δ ppm 1.32–1.53 (m, 1 H), 2.04 (t, J = 13.00 Hz, 2 H), 2.48–2.96 (m, 5 H), 2.96–3.19 (m, 3 H), 3.19–3.81 (m, 7 H), 3.92 (d, J = 10.17 Hz, 1 H), 4.37 (t, J = 7.72 Hz, 1 H), 4.48–4.69 (m, 1 H), 7.17–7.70 (m, 10 H), 8.77 (br s, 1 H). ESI MS m/z: calcd for C26H31N3O3 433.55, found 434 (M + H)+. HPLC (Synergy Hydro, 20 min) tR = 15.52 min (>99%).
N-(1-Methylpiperidin-4-yl)-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-α]pyrazine-7(3H)-carboxamide trifluoroacetate (26)
An analogous procedure to that described for 2 using 1-methylpiperidin-4-amine provided crude 26, which was purified by semipreparative HPLC (Synergy Hydro; 15 mL/min 5%–60% B over 30 min) to afford 26 (25 mg, 26%). 1H NMR (300 MHz, chloroform-d) δ ppm 1.97–2.20 (m, 5 H), 2.75–2.92 (m, 6 H), 3.05 (td, J = 12.62, 3.39 Hz, 1 H), 3.58 (d, J = 12.06 Hz, 2 H), 3.76–4.05 (m, 4 H), 4.38 (dd, J = 11.11, 3.58 Hz, 1 H), 6.52 (br s, 1 H), 7.20–7.44 (m, 8 H), 7.46–7.58 (m, 2 H), 11.56 (br s, 1 H). ESI MS m/z: calcd for C25H30N4O3 434.54, found 435.2 (M + H)+, 547.7 (M + TFA – H)−. HPLC (Synergy Hydro, 20 min) tR = 14.47 min (>99%).
tert-Butyl-1-(3-oxo-1,1-diphenylhexahydro-1H-oxazolo[3,4-α]pyrazine-7-carbonyl)pyrrolidin-3-ylcarbamate (27)
An analogous procedure to that described for 2 using tert-butyl pyrrolidin-3-ylcarbamate provided 27 as a viscous oil (37 mg, 43%). 1H NMR (300 MHz, chloroform-d) δ ppm 1.42–1.48 (m, 9 H), 1.60–1.92 (m, 1 H), 2.02–2.24 (m, 2 H), 2.87 (br s, 1 H), 3.02–3.25 (m, 2 H), 3.34–3.66 (m, 6 H), 3.71–3.90 (m, 2 H), 4.09–4.21 (m, 1 H), 4.55 (ddd, J = 10.83, 6.88, 3.39 Hz, 1 H), 7.28–7.44 (m, 8 H), 7.50–7.57 (m, 2 H). ESI MS m/z: calcd for C28H34N4O5 506.60, found 507.1 (M + H)+, 505.6 (M – H)−. HPLC (Synergy Hydro, 20 min) tR = 19.45 min (80%).
7-(3-Aminopyrrolidine-1-carbonyl)-1,1-diphenyltetrahydro-1H-oxazolo[3,4-α]pyrazin-3(5H)-one Trifluoroacetate (28)
To a solution of 27 (35 mg, 0.7 mmol) in CH2Cl2 (3 mL) was added trifluoroacetic acid (0.3 mL). The reaction mixture was allowed to stir at rt for 3 h and was concentrated to dryness. Purification by semipreparative HPLC (Synergy Hydro; 15 mL/min 5%–60% B over 30 min) provided 28 as a white solid (28 mg, 77%). 1H NMR (300 MHz, DMSO-d6) δ ppm 1.82–1.95 (m, 1 H), 2.06–2.20 (m, 2 H), 2.65–2.78 (m, 1 H), 3.05–3.21 (m, 1 H), 3.27 (dt, J = 11.59, 4.00 Hz, 1 H), 3.33–3.70 (m, 6 H), 3.75 (d, J = 4.52 Hz, 1 H), 4.64 (ddd, J = 10.83, 6.31, 3.58 Hz, 1 H), 7.29–7.48 (m, 8 H), 7.58 (d, J = 7.54 Hz, 2 H), 8.12 (br s, 3 H). ESI MS m/z: calcd for C28H34N4O5 406.49, found 407.6 (M + H)+. HPLC (Synergy Hydro, 20 min) tR = 13.89 min (>99%).
(3S)-tert-Butyl-3-(3-oxo-1,1-diphenylhexahydro-1H-oxazolo[3,4-α]pyrazine-7-carboxamido)pyrrolidine-1-carboxylate (29)
An analogous procedure to that described for 2 using (S)-tert-butyl 3-aminopyrrolidine-1-carboxylate provided 29 as a viscous oil (46 mg, 53%). 1H NMR (300 MHz, chloroform-d) δ ppm 1.39–1.52 (m, 9 H), 2.08–2.23 (m, 2 H), 2.85–3.01 (m, 1 H), 3.02–3.16 (m, 2 H), 3.41 (br s, 2 H), 3.61 (d, J = 5.65 Hz, 2 H), 3.84 (br s, 1 H), 3.94–4.07 (m, 1 H), 4.27–4.38 (m, 1 H), 4.38–4.49 (m, 2 H), 7.27–7.46 (m, 8 H), 7.47–7.57 (m, 2 H). ESI MS m/z: calcd for C28H34N4O5 506.60, found 407.4 (C28H34N4O5 – Boc + H)+, 507.2 (C28H34N4O5 + H)+. HPLC (Synergy Hydro, 20 min) tR = 19.48 min (83.4%).
3-Oxo-1,1-diphenyl-N-((S)-pyrrolidin-3-yl)tetrahydro-1H-oxazolo[3,4-α]pyrazine-7(3H)-carboxamide Trifluoroacetate (30)
An analogous procedure to that described for 28 provided 30 (35 mg, 75%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.05–2.21 (m, 2 H), 2.35 (d, J = 7.16 Hz, 1 H), 2.71–2.92 (m, 1 H), 3.03 (t, J = 12.24 Hz, 1 H), 3.17–3.62 (m, 4 H), 3.84 (t, J = 13.00 Hz, 2 H), 3.95–4.12 (m, 1 H), 4.40 (ddd, J = 11.02, 7.44, 3.77 Hz, 1 H), 4.60 (br s, 1 H), 7.28–7.43 (m, 8 H), 7.45–7.58 (m, 2 H), 8.78 (br s, 1 H), 9.84 (br s, 1 H). ESI MS m/z: calcd for C28H34N4O5 406.49, found 407.4 (M + H)+, 519.5 (M + TFA – H)−. HPLC (Synergy Hydro, 20 min) tR = 13.97 min (>99%).
(−3aS)-N-(4-Fluorobenzyl)-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (31)
An analogous procedure to that described for 1 using 38 provided 31 as a colorless semisolid (8.0 mg, 30%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.17 (dd, J = 13.19, 11.30 Hz, 1 H), 2.83–3.00 (m, 1 H), 3.01–3.16 (m, 1 H), 3.63 (dd, J = 13.00, 1.70 Hz, 1 H), 3.86 (dd, J = 13.00, 2.45 Hz, 1 H), 4.03 (dd, J = 13.19, 2.26 Hz, 1 H), 4.26–4.49 (m, 3 H), 4.69–4.81 (m, 1 H), 6.95–7.09 (m, 1 H), 7.21–7.45 (m, 11 H), 7.47–7.56 (m, 2 H). tR = 11.47 min, peak 1, Chiralpak-IA 4.6 mm × 250 mm column, in 9:1 hexane/isopropanol at a flow rate of 1 mL/min. ESI MS m/z: calcd for C26H24FN3O3 445.49, found 446.7 (M + H)+. [α]D20 = −151 (c = 0.585, CHCl3).
(+3aR)-N-(4-Fluorobenzyl)-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (32)
An analogous procedure to that described for 1 using 39 provided 32 as a colorless semisolid (8 mg, 30%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.17 (dd, J = 13.19, 11.30 Hz, 1 H), 2.83–3.00 (m, 1 H), 3.01–3.16 (m, 1 H), 3.63 (dd, J = 13.00, 1.70 Hz, 1 H), 3.86 (dd, J = 13.00, 2.45 Hz, 1 H), 4.03 (dd, J = 13.19, 2.26 Hz, 1 H), 4.26–4.49 (m, 3 H), 4.69–4.81 (m, 1 H), 6.95–7.09 (m, 1 H), 7.21–7.45 (m, 11 H), 7.47–7.56 (m, 2 H); tR = 14.77 min, peak 2, Chiralpak-IA 4.6 mm × 250 mm column, in 9:1 hexane/isopropanol at a flow rate of 1 mL/min. ESI MS m/z: calcd for C26H24FN3O3 445.49, found 446.7 (M + H)+.
(−3aS)-3-Oxo-1,1-diphenyl-N-(2-(piperidin-1-yl)ethyl)tetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (33)
Compound 33 was isolated from racemic 2 via semipreparative chiral chromatography on a Chiralpak-IA 20 mm × 250 mm column, utilizing an isocratic method (95:5:0.1 hexane/isopropanol/diethyl amine) at a flow rate of 12 mL/min (100 mg in 400 μL of ethanol/DMSO), to obtain 7.3 mg (tR = 10.45 min, peak 1). [α]D20 = −142 (c = 0.81, methanol).
(+3aR)-3-Oxo-1,1-diphenyl-N-(2-(piperidin-1-yl)ethyl)tetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (34)
Fractions from the isolation of compound 33 that were enriched in compound 34 were pooled (49 mg, 95% enantiomeric purity) and rechromatographed utilizing the same column and isocratic method (40 mg in 400 μL of ethanol/DMSO) to obtain 26.2 mg of pure 34 as a colorless oil (tR = 12.67 min, peak 2); [α]D20 = +138 (c = 0.82, methanol).
(1,4-Dibenzylpiperazin-2-yl)diphenylmethanol (35)
To a solution of phenyl lithium (74.7 mL, 1.8 M in dibutylether) and THF (anhydrous, 170 mL) at −78 °C was added dropwise a solution of ethyl 1,4-dibenzylpiperazine-2-carboxylate (9 g, 26.59 mmol) in THF (anhydrous, 100 mL). After complete addition, the reaction mixture was warmed to room temperature over 30 min and then stirred at rt for 1 h. The reaction mixture was quenched with H2O (200 mL) and extracted with CH2Cl2 (2 × 250 mL). The organic layer was dried (MgSO4), concentrated, and purified by medium pressure column chromatography (80 g of SiO2, 100% CH2Cl2) to provide 35 as a yellow solid (11.92 g, quant.). 1H NMR (300 MHz, chloroform-d) δ ppm 1.56 (br s, 1 H), 2.32–2.48 (m, 2 H), 2.48–2.61 (m, 2 H), 2.79 (d, J = 11.68 Hz, 1 H), 3.28–3.46 (m, 2 H), 3.53–3.78 (m, 4 H), 6.96–7.35 (m, 16 H), 7.35–7.45 (m, 2 H), 7.45–7.54 (m, 2 H). ESI MS m/z: calcd for C31H32N2O 448.6, found 449.7 (M + H)+, 448.0 (M – H)−.
Diphenyl(piperazin-2-yl)methanol (36)
To a solution of 35 (2.54 g, 5.76 mmol) in CH2Cl2 (100 mL) was added 1 N HCl in Et2O (8.5 mL, 8.5 mmol). The reaction mixture was allowed to stir at room temperature for 1 h and concentrated to dryness, and the residual white powder was dried under high vacuum for 1 h. The residue was redissolved in hot ethanol (110 mL) and hydrogenated (46 psi) over 5% Pd/BaSO4 (1 g) and 10% Pd/C (1 g) at rt for 20 h. The reaction mixture was filtered through Celite, and the filtrate was concentrated to dryness. The resulting residue was suspended in CH2Cl2 (200 mL) and washed with aq. NaOH (1 N, 50 mL). The aqueous layer was extracted with CH2Cl2 (2 × 200 mL); the combined organic layers were washed with brine (2 × 50 mL), dried (MgSO4), and concentrated to dryness to provide crude 36 (1.16 g) as a yellow solid. 1H NMR (300 MHz, chloroform-d) δ ppm 1.60 (br s, 2 H), 2.56–2.77 (m, 3 H), 2.83–3.05 (m, 3 H), 3.70 (dd, J = 9.42, 3.39 Hz, 1 H), 4.48 (br s, 1 H), 7.11–7.38 (m, 6 H), 7.49 (dd, J = 8.29, 1.13 Hz, 2 H), 7.57–7.67 (m, 2 H). ESI MS m/z: calcd for C17H20N2O 268.36, found 269.4 (M + H)+, 251.2 (M – HO) +. HPLC tR = 10.57 min (>99%).
1,1-Diphenyltetrahydro-1H-oxazolo[3,4-α]pyrazin-3(5H)-one (37)
To a solution of 36 (1g, 3.73 mmol) in THF (anhydrous, 100 mL) was added di-t-butyl dicarbonate (2.03 g, 9.32 mmol), NEt3 (1.3 mL, 9.32 mmol), and DMAP (1.13 g, 9.32 mmol). The reaction mixture was allowed to stir at rt for 2 h, concentrated to dryness, and redissolved in CH2Cl2 (100 mL). The solution was washed with aq. sat. NaHCO3 (50 mL) and brine (50 mL), dried (MgSO4), and concentrated. The resulting semisolid was redissolved in CH2Cl2 (200 mL), and TFA (2.85 mL, 37.3 mmol) was added. The reaction mixture was stirred at room temperature for 8 h, concentrated to dryness, and redissolved in CH2Cl2 (100 mL). The solution was washed with aq. sat. NaHCO3 (2 × 50 mL), dried (MgSO4), and concentrated. The residue was purified by column chromatography (80 g of SiO2, 100% methylene chloride to 40% chloroform/methanol/ammonium hydroxide 80/20/1) to provide 37 as a white solid (900 mg, 82% over three steps). 1H NMR (300 MHz, DMSO-d6) δ ppm 2.38–2.48 (m, 1 H), 2.89–3.09 (m, 1 H), 3.09–3.21 (m, 1 H), 3.21–3.43 (m, 2 H), 3.82 (dd, J = 14.13, 3.96 Hz, 1 H), 4.91 (dd, J = 11.68, 3.39 Hz, 1 H), 7.25–7.61 (m, 10 H), 9.33 (br s, 1 H). ESI MS m/z: calcd for C18H18N2O2 294.35, found 295.2 (M + H)+. HPLC (Synergy Hydro, 20 min) tR = 13.59 min (95.1%).
(−3aS)-1,1-Diphenyltetrahydro-1H-oxazolo[3,4-α]pyrazin-3(5H)-one (38)
Compound 38 was isolated from racemic 37 via semipreparative chiral chromatography on a Chiralpak-IA 20 mm × 250 mm column, utilizing an isocratic method (95:5:0.1 hexane/isopropanol/diethyl amine) at a flow rate of 12 mL/min over 13 identical separations (40 mg in 150 μL of ethanol/DMSO per run), to obtain 104.8 mg as a colorless oil (tR = 17.2 min, peak 1); [α]D20 = −258.5 (c = 0.265, MeOH).
(−3aS)-1,1-Diphenyltetrahydro-1H-oxazolo[3,4-α]pyrazin-3(5H)-one R-Camphor Sulfonate (38a)
Compound 38 (0.088 g, 0.30 mmol) was dissolved in MeOH (15 mL) and (R)-camphor sulfonic acid (0.069 g, 0.30 mmol) was added. The suspension was warmed until the solid dissolved and was allowed to stand at room temperature for 8 h. The resulting crystals were collected and dried under reduced pressure to afford 38a as colorless needles (0.106 g), mp =211–213 °C; [α]D20 = −151.43 (c = 0.175, MeOH). Compound 38a was sent for X-ray analysis.
(+3aR)-1,1-Diphenyltetrahydro-1H-oxazolo[3,4-α]pyrazin-3(5H)-one (39)
Fractions from the isolation of compound 38 that were enriched in compound 39 were pooled (126 mg, 93% enantiomeric purity) and rechromatographed over six separations (16 mg in 100 μL of ethanol/DMSO per run) utilizing the same column and isocratic method to obtain 85.7 mg of pure 39 as a colorless oil (tR = 23.4 min, peak 2). [α]D20 = +252.8 (c = 0.345, MeOH).
N-(2-Chloroethyl)-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (40)
An analogous procedure to that described for 1 using 2-chloroethyl isocyanate provided 40 as a viscous oil (39 mg, 61%). 1H NMR (300 MHz, chloroform-d) δ ppm 2.09–2.26 (m, 1 H), 2.74–2.95 (m, 1 H), 3.08 (td, J = 12.62, 3.77 Hz, 1 H), 3.44–3.68 (m, 4 H), 3.70–3.87 (m, 2 H), 3.98 (dd, J = 13.56, 2.26 Hz, 1 H), 4.42 (dd, J = 11.30, 3.77 Hz, 1 H), 5.33 (t, J = 5.09 Hz, 1 H), 7.18–7.43 (m, 8 H), 7.44–7.60 (m, 2 H).
N-(3-Chloropropyl)-3-oxo-1,1-diphenyltetrahydro-1H-oxazolo[3,4-a]pyrazine-7(3H)-carboxamide (41)
An analogous procedure to that described for 1 using 3-chloropropylisocyanate provided 41 as a viscous oil (195 mg, %). 1H NMR (300 MHz, chloroform-d) δ ppm 1.92–2.02 (m, 2 H), 2.14 (dd, J = 13.56, 11.30 Hz, 1 H), 2.77–2.94 (m, 1 H), 3.06 (td, J = 12.62, 3.77 Hz, 1 H), 3.38 (qd, J = 6.22, 2.83 Hz, 2 H), 3.52–3.64 (m, 2 H), 3.64–3.76 (m, 1 H), 3.83 (dd, J = 13.19, 2.64 Hz, 1 H), 3.97 (ddd, J = 13.37, 3.58, 1.13 Hz, 1 H), 4.41 (dd, J = 11.11, 3.58 Hz, 1 H), 5.00 (t, J = 5.65 Hz, 1 H), 7.24–7.43 (m, 8 H), 7.46–7.55 (m, 2 H).
X-ray Crystallographic Analysis
Single-crystal X-ray diffraction data on compound 38a was collected using Cu Kα radiation and a Bruker Platinum-135 CCD area detector. The crystal was prepared for data collection by coating with high viscosity microscope oil. The oil-coated crystal was mounted on a micromesh mount (Mitergen, Inc.) and transferred to the diffractometer, and a data set was collected at 150 K. The 0.205 × 0.010 × 0.005 mm3 crystal was monoclinic in space group P21, with unit cell dimensions a = 11.5583(6), b = 7.2211(4), c = 16.4478(9) Å and β = 103.333(2)°. Data was 72.7% complete to 66.79°θ (∼0.83 Å) with an average redundancy of 1.67. The structure was solved by direct methods and refined by full-matrix least-squares on F2 values using the programs found in the SHELXTL suite (Bruker, SHELXTL v6.10, 2000, Bruker AXS Inc., Madison, WI). Corrections were applied for Lorentz, polarization, and absorption effects. Parameters refined included atomic coordinates and anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms on carbons were included using a riding model [coordinate shifts of C applied to H atoms] with C–H distance set at 0.96 Å. The final anisotropic full matrix least-squares refinement on F2 with 336 variables and one restraint converged at R1 = 3.85% for the observed data and wR2 = 9.89% for all data. The absolute configuration was determined from the X-ray data using the method of Flack.35 Complete information on data collection and refinement is available in the Supporting Information.
Atomic coordinates for 38a have been deposited with the Cambridge Crystallographic Data Centre (deposition number 978117). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK [fax: +44(0)-1223-336033 or e-mail: deposit@ccdc.cam.ac.uk.
In Vitro Functional Assays
Functional determinations, identification of functional antagonists at the NPSR, utilized RD-HGA16 cells (Molecular Devices), a CHO cell line stably overexpressing the promiscuous Gq-protein Ga16. Two individual cell lines were created that stably express each NPSR variant (NPS Ile107 and Asn107). Cells were loaded with the calcium sensitive dye calcium3 (Molecular Devices) for 1 h, and compounds were assayed in separate experiments for intrinsic activity and for the ability to inhibit NPS activity as measured by calcium mobilization in the FlexStation assay. All test compounds were evaluated for activity at least three times, and each experiment was conducted on a different day. Apparent constant Ke is determined by running the EC50 curves of the agonist, NPS, with and without the test compounds and measuring the rightward shift of the agonist curve. Ke values were calculated using the equation Ke = [L]/((EC50–/EC50+) – 1) where [L] is the test compound concentration, EC50– is the EC50 of NPS alone, and EC50+ is the EC50 of NPS in the presence of test compound. Therefore, the Ke value is independent of the concentration of test compounds. A three-parameter logistic equation was fit to the concentration response data with Prism (v5 for Windows, GraphPad Software; San Diego, CA) to calculate the EC50 and Ke values. NPS EC50 values for all experiments were in the range of 0.25–1.13 nM at the 107I and 5.1–11.2 nM for the 107N variant.
NPS Induced Locomotion
Male C57Bl/6 mice (National Cancer Institute, Bethesda, MD), age 8–12 weeks, were group housed (four animals per cage) under controlled conditions (temperature 21 ± 2 °C; relative humidity 50%–60%; 12 h light–dark cycle, lights on 6:00 AM) with free access to food and water. Prior to drug injections, mice were briefly anesthetized with halothane.
Locomotion of mice was monitored in rectangular plexiglass boxes (60 cm × 40 cm × 50 cm) and cumulative distance traveled (total distance in centimeters that the animal traveled during the test) was measured with an automated activity monitor system (Versamax, Accuscan, Columbus, OH). Mice were allowed to habituate to the recording chamber for 60 min. Vehicle or compound 34 (in PBS, 10% Cremophor EL) were injected ip 10 min before the central administration of either NPS (1 nmol in 2 μL of PBS, 0.1% BSA) or vehicle (PBS, 0.1% BSA), as describe in Xu et al.2 Mice were allowed to recover for 5 min and then placed into the apparatus. Recording of locomotor activity continued for 90 min.
Pharmacokinetic Analysis
Male C57Bl/6 mice were purchased from Charles River Laboratories (Raleigh, NC) at a target weight of 25–30 g corresponding to ages of approximately 8 weeks at receipt. Mice were dosed at 5 mg/kg with a single dose of 34 as a solution in 2.5% dimethylacetamide, 2.5% Cremophor EL, and 95% phosphate buffered saline, at a final concentration of 0.5 mg/mL. Blood samples were collected via cardiac puncture from five mice per time point at target time points of 15 min, 30 min, 1 h, and 2 h postdose. At sacrifice, the brain of each animal was removed, weighed, and immediately flash-frozen using liquid nitrogen within low density polyethylene vials. Whole brain samples were homogenized using a Potter–Elvehjem type homogenizer in ethanol/water (50:50, v/v) solution, which was added in a ratio of 3 mL per gram of brain. A 50 μL aliquot of brain homogenate or plasma was transferred to microcentrifuge tubes followed by 200 μL of deionized/distilled water and 1.2 mL of a 10 ng/mL solution of reserpine internal standard in acetonitrile. The samples were vortexed and centrifuged in a model 5415C microcentrifuge (Eppendorf North America, Westbury NY) at 16000g and transferred to autosampler vials for analysis. Calibration standards were prepared by combining 40 μL blank plasma or blank brain homogenate and 10 μL of 34 spiking solution in microcentrifuge tubes and 1.2 mL internal standard solution in acetonitrile. Sample analysis was conducted using an Applied Biosystems API 5000 triple quadrupole mass spectrometer (Foster City Ca) interfaced to Agilent 1100 binary pump and autosampler. A Valco valve and Agilent 1100 isocratic pump were used to divert the first minute of the run to waste while infusing 50:50 acetonitrile/water makeup solvent to the mass spectrometer. Chromatography was accomplished using a Phenomenex Luna C18 column (50 mm × 2 mm i.d., 5 μm particle size) fitted with a guard cartridge. Injection volumes were 10 μL. Two mobile phase solutions were used: solution A was 0.1% formic acid in water, and solution B was 0.1% formic acid in methanol, and the flow rate was 0.5 mL/min. Initial chromatographic conditions were 90% A then decreasing to 5% A over 4 min, then returning to initial conditions for 2 min for column equilibration. Compound 34 was monitored in positive electrospray mode with an MRM transition of 449.20 → 155.10 with DP = 50, CE = 39, and CXP = 36. Reserpine internals standard was monitored with a transition of 609.20 → 195.00 with DP = 231, CE = 41, and CXP = 14. Mass spectrometer parameters were as follows: CUR = 13, GS1 = 50, GS2 = 50, IS = 4000, TEM = 600 °C, and CAD = 8.
Pharmacokinetic analyses were conducted using WinNonlin (version 5.2; Pharsight Corporation, Mountainview CA). Mean plasma concentration vs time data for 34 was analyzed using noncompartmental methods (models 200, WinNonlin) to determine pharmacokinetic parameter estimates. The following parameters were estimated: maximum observed plasma concentration and time at which the maximum concentration was observed (Cmax and Tmax, respectively); terminal elimination rate constant (λz) and terminal half-life (t1/2); area under the plasma concentration vs time curve (AUC) to last time point with measurable 34 concentration (AUClast) and AUC extrapolated to infinity (AUC0-inf). The concentration–time data of 34 in the brain were also analyzed similarly. Given the limited amount of data collected in this study, statistical analyses were not conducted.
Acknowledgments
We also thank Dr. Stewart Clark, Mr. Keith Warner, and Ms. Tiffany Langston, for their valuable technical assistance.
Glossary
Abbreviations
- ANOVA
Analysis of variance
- API
atmospheric pressure ionization
- cAMP
cyclic adenosine monophosphate
- CNS
central nervous system
- CRF
corticotrophin releasing factor
- CSA
camphor sulfonic acid
- DEA
diethylamine
- DMSO
dimethyl sulfoxide
- GPCR
G-protein coupled receptor
- HTS
high throughput screen
- LC/MS
liquid chromatography–mass spectrometry
- LC
locus ceruleus
- NPS
neuropeptide S
- NPSR
neuropeptide S receptor
- PTSD
posttraumatic stress disorder
- TFA
trifluoroacetic acid
- TLC
thin-layer chromatography
- VTA
ventral tegmental area
Supporting Information Available
HPLC analysis and MS of target compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by a United States National Institute of Mental Health Research Grant (MH081247-01). The X-ray crystallographic work was supported by NIDA through Interagency Agreement No. Y1-DA1101 with the Naval Research Laboratory (NRL).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sato S. Shintani Y.; Miyajima N.; Yoshimura K., Novel G-protein coupled receptor protein and DNA thereof. World Patent Application WO 2002031145 A1, 2002.
- Xu Y. L.; Reinscheid R. K.; Huitron-Resendiz S.; Clark S. D.; Wang Z.; Lin S. H.; Brucher F. A.; Zeng J.; Ly N. K.; Henriksen S. J.; de Lecea L.; Civelli O.; Neuropeptide S. (2004) A neuropeptide promoting arousal and anxiolytic-like effects. Neuron 43(4), 487–497. [DOI] [PubMed] [Google Scholar]
- Xu Y. L.; Gall C. M.; Jackson V. R.; Civelli O.; Reinscheid R. K. (2007) Distribution of neuropeptide S receptor mRNA and neurochemical characteristics of neuropeptide S-expressing neurons in the rat brain. J. Comp. Neurol. 500(1), 84–102. [DOI] [PubMed] [Google Scholar]
- Clark S. D.; Duangdao D. M.; Schulz S.; Zhang L.; Liu X.; Xu Y. L.; Reinscheid R. K. (2011) Anatomical characterization of the neuropeptide S system in the mouse brain by in situ hybridization and immunohistochemistry. J. Comp. Neurol. 519(10), 1867–1893. [DOI] [PubMed] [Google Scholar]
- Reinscheid R. K.; Xu Y. L. (2005) Neuropeptide S and its receptor: A newly deorphanized G protein-coupled receptor system. Neuroscientist 11(6), 532–538. [DOI] [PubMed] [Google Scholar]
- Domschke K.; Reif A.; Weber H.; Richter J.; Hohoff C.; Ohrmann P.; Pedersen A.; Bauer J.; Suslow T.; Kugel H.; Heindel W.; Baumann C.; Klauke B.; Jacob C.; Maier W.; Fritze J.; Bandelow B.; Krakowitzky P.; Rothermundt M.; Erhardt A.; Binder E. B.; Holsboer F.; Gerlach A. L.; Kircher T.; Lang T.; Alpers G. W.; Strohle A.; Fehm L.; Gloster A. T.; Wittchen H. U.; Arolt V.; Pauli P.; Hamm A.; Deckert J. (2010) Neuropeptide S receptor gene — converging evidence for a role in panic disorder. Mol. Psychiatry 16, 938–948. [DOI] [PubMed] [Google Scholar]
- Okamura N.; Hashimoto K.; Iyo M.; Shimizu E.; Dempfle A.; Friedel S.; Reinscheid R. K. (2007) Gender-specific association of a functional coding polymorphism in the Neuropeptide S receptor gene with panic disorder but not with schizophrenia or attention-deficit/hyperactivity disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 31(7), 1444–1448. [DOI] [PubMed] [Google Scholar]
- Raczka K. A.; Gartmann N.; Mechias M. L.; Reif A.; Buchel C.; Deckert J.; Kalisch R. (2010) A neuropeptide S receptor variant associated with overinterpretation of fear reactions: a potential neurogenetic basis for catastrophizing. Mol. Psychiatry 15, 1067–1074. [DOI] [PubMed] [Google Scholar]
- Leonard S. K.; Dwyer J. M.; Sukoff Rizzo S. J.; Platt B.; Logue S. F.; Neal S. J.; Malberg J. E.; Beyer C. E.; Schechter L. E.; Rosenzweig-Lipson S.; Ring R. H. (2008) Pharmacology of neuropeptide S in mice: therapeutic relevance to anxiety disorders. Psychopharmacology (Berlin) 197(4), 601–611. [DOI] [PubMed] [Google Scholar]
- Rizzi A.; Vergura R.; Marzola G.; Ruzza C.; Guerrini R.; Salvadori S.; Regoli D.; Calo G. (2008) Neuropeptide S is a stimulatory anxiolytic agent: a behavioural study in mice. Br. J. Pharmacol. 154, 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale G.; Filaferro M.; Ruggieri V.; Pennella S.; Frigeri C.; Rizzi A.; Guerrini R.; Calo G. (2008) Anxiolytic-like effect of neuropeptide S in the rat defensive burying. Peptides 29(12), 2286–2291. [DOI] [PubMed] [Google Scholar]
- Gottlieb D. J.; O’Connor G. T.; Wilk J. B. (2007) Genome-wide association of sleep and circadian phenotypes. BMC Med. Genet. 8(Suppl 1), S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitinen T.; Polvi A.; Rydman P.; Vendelin J.; Pulkkinen V.; Salmikangas P.; Makela S.; Rehn M.; Pirskanen A.; Rautanen A.; Zucchelli M.; Gullsten H.; Leino M.; Alenius H.; Petays T.; Haahtela T.; Laitinen A.; Laprise C.; Hudson T. J.; Laitinen L. A.; Kere J. (2004) Characterization of a common susceptibility locus for asthma-related traits. Science 304(5668), 300–304. [DOI] [PubMed] [Google Scholar]
- Cline M. A.; Godlove D. C.; Nandar W.; Bowden C. N.; Prall B. C. (2007) Anorexigenic effects of central neuropeptide S involve the hypothalamus in chicks (Gallus gallus). Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol. 148(3), 657–663. [DOI] [PubMed] [Google Scholar]
- Cline M. A.; Prall B. C.; Smith M. L.; Calchary W. A.; Siegel P. B. (2008) Differential appetite-related responses to central neuropeptide S in lines of chickens divergently selected for low or high body weight. J. Neuroendocrinol 20(7), 904–908. [DOI] [PubMed] [Google Scholar]
- Meis S.; Bergado-Acosta J. R.; Yanagawa Y.; Obata K.; Stork O.; Munsch T. (2008) Identification of a neuropeptide S responsive circuitry shaping amygdala activity via the endopiriform nucleus. PLoS One 3(7), e2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungling K.; Seidenbecher T.; Sosulina L.; Lesting J.; Sangha S.; Clark S. D.; Okamura N.; Duangdao D. M.; Xu Y. L.; Reinscheid R. K.; Pape H. C. (2008) Neuropeptide S-mediated control of fear expression and extinction: Role of intercalated GABAergic neurons in the amygdala. Neuron 59(2), 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioccioppo R., Economidou D., Cannella N., Braconi S., Stopponi S. (2007) Neuropeptide s system activation facilitates conditioned reinstatement of cocaine-seeking in the rat. Presented at the Society for Neuroscience 2007, San Diego, 271.18/Z1.
- Badia-Elder N. E.; Henderson A. N.; Bertholomey M. L.; Dodge N. C.; Stewart R. B. (2008) The effects of neuropeptide S on ethanol drinking and other related behaviors in alcohol-preferring and -nonpreferring rats. Alcohol.: Clin. Exp. Res. 32(8), 1380–1387. [DOI] [PubMed] [Google Scholar]
- Schmoutz C. D.; Zhang Y.; Runyon S. P.; Goeders N. E. (2012) Antagonism of the neuropeptide S receptor with RTI-118 decreases cocaine self-administration and cocaine-seeking behavior in rats. Pharmacol., Biochem. Behav. 103(2), 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas A.; Gines P.; Runyon B. A. (2009) Is albumin infusion necessary after large volume paracentesis?. Liver Int. 29(5), 636–640. [DOI] [PubMed] [Google Scholar]; 640 - 641.discussion
- Si W.; Aluisio L.; Okamura N.; Clark S. D.; Fraser I.; Sutton S. W.; Bonaventure P.; Reinscheid R. K. (2010) Neuropeptide S stimulates dopaminergic neurotransmission in the medial prefrontal cortex. J. Neurochem 115, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki T.; Kim J.; Sasaki K. (2010) Microinjection of neuropeptide S into the rat ventral tegmental area induces hyperactivity and increases extracellular levels of dopamine metabolites in the nucleus accumbens shell. Peptides 31(5), 926–931. [DOI] [PubMed] [Google Scholar]
- Paneda C.; Huitron-Resendiz S.; Frago L. M.; Chowen J. A.; Picetti R.; de Lecea L.; Roberts A. J. (2009) Neuropeptide S reinstates cocaine-seeking behavior and increases locomotor activity through corticotropin-releasing factor receptor 1 in mice. J. Neurosci. 29(13), 4155–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallupi M.; Cannella N.; Economidou D.; Ubaldi M.; Ruggeri B.; Weiss F.; Massi M.; Marugan J.; Heilig M.; Bonnavion P.; de Lecea L.; Ciccocioppo R. (2010) Neuropeptide S facilitates cue-induced relapse to cocaine seeking through activation of the hypothalamic hypocretin system. Proc. Natl. Acad. Sci. U. S. A. 107(45), 19567–19572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura N.; Habay S. A.; Zeng J.; Chamberlin A. R.; Reinscheid R. K. (2008) Synthesis and pharmacological in vitro and in vivo profile of 3-oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (SHA 68), a selective antagonist of the neuropeptide S receptor. J. Pharmacol. Exp. Ther. 325(3), 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Gilmour B. P.; Navarro H. A.; Runyon S. P. (2008) Identifying structural features on 1,1-diphenyl-hexahydro-oxazolo[3,4-a]pyrazin-3-ones critical for Neuropeptide S antagonist activity. Bioorg. Med. Chem. Lett. 18(14), 4064–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed J. Y.; Zartman A. E.; Kett N. R.; Gotter A. L.; Uebele V. N.; Reiss D. R.; Condra C. L.; Fandozzi C.; Lubbers L. S.; Rowe B. A.; McGaughey G. B.; Henault M.; Stocco R.; Renger J. J.; Hartman G. D.; Bilodeau M. T.; Trotter B. W. (2010) Synthesis and evaluation of a new series of Neuropeptide S receptor antagonists. Bioorg. Med. Chem. Lett. 20(15), 4700–4703. [DOI] [PubMed] [Google Scholar]
- Trotter B. W.; Nanda K. K.; Manley P. J.; Uebele V. N.; Condra C. L.; Gotter A. L.; Menzel K.; Henault M.; Stocco R.; Renger J. J.; Hartman G. D.; Bilodeau M. T. (2010) Tricyclic imidazole antagonists of the Neuropeptide S Receptor. Bioorg. Med. Chem. Lett. 20(15), 4704–4708. [DOI] [PubMed] [Google Scholar]
- Micheli F.; Di Fabio R.; Giacometti A.; Roth A.; Moro E.; Merlo G.; Paio A.; Merlo-Pich E.; Tomelleri S.; Tonelli F.; Zarantonello P.; Zonzini L.; Capelli A. M. (2010) Synthesis and pharmacological characterization of 5-phenyl-2-[2-(1-piperidinylcarbonyl)phenyl]-2,3-dihydro-1H-pyrrolo[1,2-c]imidazo l-1-ones: a new class of Neuropeptide S antagonists. Bioorg. Med. Chem. Lett. 20(24), 7308–7311. [DOI] [PubMed] [Google Scholar]
- Thorsell A.; Tapocik J. D.; Liu K.; Zook M.; Bell L.; Flanigan M.; Patnaik S.; Marugan J.; Damadzic R.; Dehdashti S. J.; Schwandt M. L.; Southall N.; Austin C. P.; Eskay R.; Ciccocioppo R.; Zheng W.; Heilig M. (2013) A novel brain penetrant NPS receptor antagonist, NCGC00185684, blocks alcohol-induced ERK-phosphorylation in the central amygdala and decreases operant alcohol self-administration in rats. J. Neurosci. 33(24), 10132–10142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knölker H. J.; Braxmeier T. (1998) Isocyanates. Part 5. Synthesis of chiral oxazolidin-2-ones and imidazolidin-2-ones via DMAP-catalyzed isocyanation of amines with di-tert-butyl dicarbonate. Tetrahedron Lett. 39(51), 9407–9410. [Google Scholar]
- Roy D.; Ducher F.; Laumain A.; Legendre J. Y. (2001) Determination of the aqueous solubility of drugs using a convenient 96-well plate-based assay. Drug Dev. Ind. Pharm. 27(1), 107–109. [DOI] [PubMed] [Google Scholar]
- Trapella C.; Pela M.; Del Zoppo L.; Calo G.; Camarda V.; Ruzza C.; Cavazzini A.; Costa V.; Bertolasi V.; Reinscheid R. K.; Salvadori S.; Guerrini R. (2011) Synthesis and separation of the enantiomers of the neuropeptide S receptor antagonist (9R/S)-3-oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (SHA 68). J. Med. Chem. 54(8), 2738–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flack H. D. (1983) On Enantiomorph-Polarity Estimation. Acta Crystallogr. A39, 876–881. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.