

Abstract
Ultraviolet matrix-assisted laser desorption/ionization mass spectrometric (UV-MALDI-MS) analysis of highly acidic, thermally labile species such as glycosaminoglycan-derived oligosaccharides is complicated by their poor ionization efficiency and tendency to fragment through the loss of sulfo groups. We have utilized a systematic approach to evaluate the effect of alkali metal counterions on the degree of fragmentation through SO3 loss from a highly sulfated model compound, sucrose octasulfate (SOS). The lithium, sodium, potassium, rubidium, and cesium salts of SOS were analyzed by UV-MALDI-time-of-flight (TOF)MS using an ionic liquid matrix, bis-1,1,3,3-tetramethylguanidinium α-cyano-4-hydroxycinnamate. The positive-ion and negative-ion MALDI mass spectra of five alkali metal salts of SOS were compared in terms of the degree of analyte fragmentation through the SO3 loss and the absolute intensity of a molecular ion signal. Experimental results demonstrate that the lithium, sodium, and potassium salts of SOS undergo some degree of fragmentation through the loss of SO3, whereas the fragmentation through the loss of SO3 in the rubidium and cesium salts of SOS is suppressed. A high detection sensitivity associated with the stability of sulfate half-esters was achieved for the cesium salt of SOS using positive-ion detection. Finally, the cesium salt of chondroitin sulfate A disaccharide was successfully analyzed using UV-MALDI-TOFMS.
Glycosaminoglycans (GAGs) are linear, sulfated polysaccharides that play important roles in angiogenesis, inflammation, as well as cell signaling, proliferation, differentiation, morphogenesis, adhesion, and migration.1 GAGs consist of alternating uronic acid and hexosamine residues linked through (1 →4) glycosidic bonds as in heparin and heparan sulfate, or alternating (1 →3, 1 →4) glycosidic bonds as in chondroitin sulfate. The hexosamine residue can be sulfated at the C-2, C-3, C-4, and C-6 positions, and the uronic acid residue can be sulfated at the C-2 position.1,2 The uronic acid/hexosamine disaccharide building blocks constitute the sequence of a GAG chain and define its biological function through the number and positions of sulfo groups, stereochemistry of uronic acid residues, and type of glycosidic linkages.1–3 Understanding the role of GAGs in biological processes requires the knowledge of their primary structure, which is determined by the disaccharide composition and sequence.1–4
A well-established method for identification of GAGs in biological samples involves the use of polysaccharide lyases.5 These enzymes cleave specific glycosidic linkages through an eliminase mechanism, which results in the formation of a double bond at the non-reducing end of the product detectable by UV absorbance at 232 nm.5 Characterization of the disaccharide mixtures obtained by complete depolymerization using a specific lyase or a combination of lyases allows distinction between different GAGs. A number of techniques can be used for characterization of GAG-derived disaccharide mixtures, including high-performance liquid chromatography (HPLC),6 capillary electrophoresis (CE),7 and liquid chromatography followed by mass spectrometry (LC/MS).8,9 MS-based approaches for characterization of GAG-derived disaccharides afford sensitive and specific analysis, require small sample amounts, and do not rely on the UV-absorbing properties of the analyte. While the number of reports describing applications of LC/MS in the analysis of GAG-derived oligosaccharides is growing, there are very few matrix-assisted laser desorption/ionization mass spectrometric (MALDI-MS) methods for the analysis of sulfated oligosaccharides.
Previous reports of successful UV-MALDI time-of-flight (TOF) and electrospray ionization (ESI) MS analyses of heparin and heparin-like oligosaccharides demonstrated that the fragmentation of the polysulfated oligosaccharides through a loss of sulfate can be suppressed by the formation of a complex with an organic cation such as the guanidinium group of arginine10,11 or tetraethylammonium.12 In each case, the cations were so-called soft Lewis acids.13 In addition, Cs+, a soft Lewis acid,13 was shown to suppress metastable fragmentation through a glycosidic bond dissociation in neutral oligosaccharides.14 We hypothesized that the stability of sulfate half-esters toward cleavage of O–SO3 bonds should increase with the increasing soft character of an alkali metal cation, i.e. in going from lithium to cesium.
Recently, we reported a successful UV-MALDI-TOFMS analysis of underivatized, uncomplexed polysulfated disaccharide sucrose octasulfate (SOS), sodium salt, using ionic liquid matrices (ILMs).15 In the present work, we continue to use this model compound to understand analytical parameters that are important in UV-MALDI-TOFMS analysis of sulfated oligosaccharides, in particular GAG-derived oligosaccharides. In the current study, SOS is used to evaluate the effect of five alkali metal counterions on the extent of the SO3-loss fragmentation. We prepare the lithium, sodium, potassium, rubidium, and cesium salts of SOS and analyze these using UV-MALDI-TOFMS and a new ILM, bis-1,1,3,3-tetramethylguanidinium α-cyano-4-hydroxycinnamate (G2CHCA).16 The experimental results indicate that such parameters as the detection mode, pH of the sample, and the identity of the counterion affect the quality of MALDI mass spectra of SOS.
EXPERIMENTAL
Materials
Sucrose octasulfate (SOS), sodium salt, was a gift from Bukh Meditec (Farum, Denmark). Bovine tracheal chondroitin sulfate A, 70% (balance chondroitin sulfate C), α-cyano-4-hydroxycinnamic acid (CHCA), Dowex 50WX8-100 strongly acidic cation-exchange resin, and the lithium, potassium, rubidium and cesium hydroxides and chlorides were from Sigma (St. Louis, MO, USA). Chondroitinase ABC (chondroitin ABC lyase, EC 4.2.2.4) from Proteus vulgaris was purchased from Seikagaku (Japan). 1,1,3,3-Tetra-methylguanidine was from Acros (NJ, USA). Vivapure S strongly acidic cation-exchange spin-columns were from Vivascience AG (Hannover, Germany). All other reagents were analytical grade; all solvents used were HPLC grade.
Ionic liquid matrix
ILM was prepared by combining 1 equivalent of CHCA dissolved in methanol and 2 equivalents of 1,1,3,3-tetramethylguanidine. Methanol was evaporated under vacuum overnight; the solid was washed several times with acetone and dried at ambient temperature. The matrix solution for MALDI-MS was prepared by dissolving 50–70 mg G2CHCA in 1 mL 100% methanol.
Alkali metal salts of SOS
For milligram-scale preparations of lithium, potassium, rubidium, and cesium salts of SOS, a 5 mg/mL aqueous solution of SOS, sodium salt, was passed through a Dowex 50WX8-100 strong cation-exchange (SCX) column. Acidic fractions (pH <3) were pooled and carefully neutralized with a 0.1 M LiOH, KOH, RbOH, or CsOH solution to pH 7 using pH paper. The neutralized solutions were freeze-dried, and the resulting alkali metal salts of SOS were reconstituted with water to a 1 mM concentration (stock solutions).
Sodium and cesium salts of chondroitin sulfate A disaccharides
Chondroitin sulfate A (CSA) disaccharides were prepared by enzymatic depolymerization of bovine tracheal chondroitin sulfate A with chondroitin sulfate ABC lyase as described previously.5 Briefly, a 50-μL aliquot of 20 mg/mL chon-droitin sulfate solution in a 50 mM Tris/60 mM sodium acetate buffer, pH 8, was incubated with 1 μL of a 1 mU/μL ABC lyase solution at 37°C for 24 h. Microgram-scale preparation of the acidic form of the CSA digest was carried out using Vivapure S mini spin-columns (SCX) pre-equilibrated with 1 N HCl followed by water. The digest mixture was diluted with 4.950 μL of water, and two 50-μL aliquots of the resulting dilution (1% digest) were passed through the SCX membranes. The eluant from one spin-column was mixed with 5 μL of 0.1 M NaOH solution, and the eluant from another spin-column was mixed with 5 μL of 0.1 M CsOH solution. After the addition of base, both samples had pH >8 as determined by pH paper. The resulting solutions of sodium and cesium salts of CSA oligosaccharides were mixed with the matrix solution and applied to a stainless steel target plate for MALDI-MS analyses.
MALDI-TOFMS
Preliminary mass spectra of five alkali metal salts of SOS were acquired on a TofSpec 2E MALDI-TOF mass spectrometer (Micromass, Manchester, UK) in reflectron mode using default instrument parameters: source 20 kV, reflectron 26 kV, extraction lens 19.95 kV, focus lens 16 kV. Ionization was achieved using nitrogen laser at a wavelength of 337 nm. The detection sensitivity experiments with the cesium salt of SOS and analysis of the CSA digest were carried out on an Autoflex II MALDI-TOF mass spectrometer (Bruker Daltonik, Bremen, Germany) equipped with a 337 nm nitrogen laser. For MALDI-MS analyses, 1 vol. part of a sample solution was mixed with 1 vol. part of the matrix solution, and 0.25 μL of this mixture was applied to a stainless steel target. All mass spectra were processed with M over Z software (m/z - freeware edition, Proteometrics, LLC).
RESULTS AND DISCUSSION
In our previous reports, we demonstrated that ILMs can be successfully used for the MALDI-MS analysis of polysulfated, polycarboxylated oligosaccharides.15,16 The intent of the present work was to improve the quality of MALDI mass spectra acquired with sulfated disaccharides. Using an octasulfated disaccharide, SOS, as a model analyte, we compared MALDI mass spectra of its five alkali metal salts (Fig. 1) in terms of the extent of fragmentation through cleavage of O–SO3 bonds and absolute signal intensity of an intact analyte peak. Polyanionic compounds such as GAG-derived oligosaccharides are usually analyzed by MS in the negative-ion detection mode,8,9,17 but under certain experimental conditions, positive-ion detection permits the acquisition of high-quality mass spectra.12 Thus, for each salt of SOS studied, we compared MALDI mass spectra acquired in the positive-ion and negative-ion modes to establish which detection polarity afforded the lowest extent of SO3 loss and highest detection sensitivity of the molecular ion.
Figure 1.
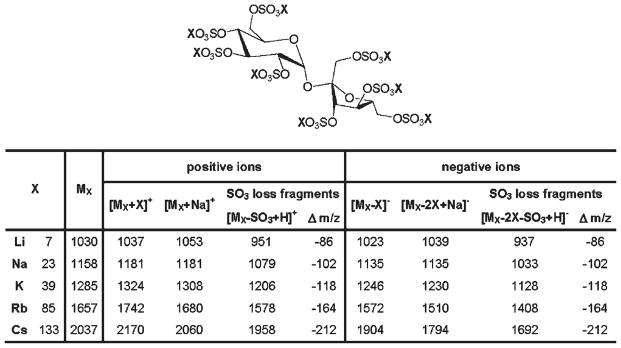
Sucrose octasulfate (SOS): Nominal masses of alkali metal salts of SOS (X8SOS), and m/z values of their expected ions. MX denotes X8SOS, where X = Li, Na, K, Rb, Cs.
Sodium, lithium, and potassium salts of SOS
GAG-derived oligosaccharides are often separated and purified by strong anion-exchange (SAX) chromatography using sodium chloride gradients.6 As the result, these compounds are commonly obtained in the form of sodium salts, making it practical to detect these polyanions by MALDI-MS as sodium salts. This is especially desirable considering the ubiquitous nature of sodium and a high tendency of GAG oligosaccharides to form sodium adducts during mass spectral analysis.8,12,14,15 Using G2CHCA as the matrix, we acquired MALDI mass spectra of several dilutions of the sodium salt of SOS (Na8SOS), in positive-ion and negative-ion reflector mode. For each analyte concentration examined, the instrument parameters were adjusted to yield strong analyte signal in the negative-ion mode and were kept unchanged during the positive-ion acquisition.
Negative-ion MALDI mass spectra of Na8SOS showed higher analyte signal intensity and greater number of SO3-loss fragments than the positive-ion mass spectra acquired from the same target spot under identical instrument parameters (Figs. 2(A) and 2(B)). In the positive-ion mode, the [MNa+Na]+ion dominated the spectrum, and only one fragment due to the loss of SO3 was detected. However, the sensitivity of positive-ion detection was significantly lower than that achieved in the negative-ion mode, and using Na8SOS concentrations below 0.1 mM (TofSpec 2E) did not result in detection of a positive-ion signal. Negative-ion MALDI analysis of Li8SOS resulted in the acquisition of mass spectra with an even greater degree of fragmentation as evident from the signal intensity of the [MLi–Li]−ion relative to the signal intensities of the peaks corresponding to the SO3-loss fragments (Fig. 2(C)). The positive-ion analysis of the Li8SOS sample did not result in detection of the [MLi+Li]+ ion under the experimental conditions used.
Figure 2.

Positive-ion (A) and negative-ion (B) reflector MALDI mass spectra of Na8SOS, and a negative-ion MALDI mass spectrum of Li8SOS (C) acquired with an ILM, G2CHCA. Peaks due to the matrix are marked with triangles; peaks marked with asterisks correspond to the SO3-loss fragments.
While the negative-ion detection of lithium and sodium salts of SOS afforded higher sensitivity than the positive-ion detection, this trend was reversed for the potassium salt of SOS (K8SOS). MALDI mass spectra of K8SOS acquired in the positive-ion mode exhibited an intact analyte peak, [MK+K]+, accompanied by two SO3-loss fragments (Fig. 3(A)). The intensity of the K8SOS molecular peak, [MK–K]−(Fig. 3(B)), was approximately 30% of that of the [MK+K]+ ion acquired with the same instrumental parameters. The extent of fragmentation through the loss of SO3 observed during the MALDI-MS analysis of K8SOS was similar to that of Li8SOS and Na8SOS. The positive-ion and negative-ion MALDI mass spectra acquired with K8SOS exhibited a series of peaks corresponding to the products of K/Na exchange (Δ −16 m/z). However, Li/Na exchange products (Δ 16 m/z) were not detected during the MALDI-MS analysis of Li8SOS.
Figure 3.

Positive-ion (A) and negative-ion (B) reflector MALDI mass spectra of K8SOS, acquired with G2CHCA. Insets show expansion of a series of peaks corresponding to the K/Na exchange products.
Rubidium and cesium salts of SOS
As evident from the MALDI mass spectra of rubidium and cesium salts of SOS (Rb8SOS and Cs8SOS) acquired in the positive-ion and negative-ion reflector modes (Figs. 4 and 5), the use of larger alkali metal counterions for SOS resulted in a greater stability of sulfate half-esters reducing the cleavage of O–SO3 bonds. In contrast to Li, Na, and K salts of SOS, MALDI mass spectra of Rb8SOS and Cs8SOS showed reduced SO3-loss fragmentation not only in the positive-ion mode, but also in the negative-ion mode. A comparison of MALDI mass spectra acquired with five alkali metal salts of SOS demonstrated that the signal intensities of peaks corresponding to molecular species of Rb8SOS and Cs8SOS were greater than that of Li8SOS, Na8SOS, or K8SOS, irrespective of the detection polarity.
Figure 4.

Positive-ion (A) and negative-ion (B) reflector MALDI mass spectra of Rb8SOS, acquired with G2CHCA. Insets show expansion of isotopic clusters of the molecular species. Peaks due to the matrix are marked with triangles.
Figure 5.

Positive-ion (A) and negative-ion (B) reflector MALDI mass spectra of Cs8SOS. The samples for MALDI analysis were prepared by mixing 1 vol. of 1 mM CsOH, 1 vol. of 0.1 mM Cs8SOS in 8 mM CsCl and 1 vol. of G2CHCA solution. Insets show isotopic pattern of the molecular peak.
While MALDI-MS analyses of rubidium and cesium salts of SOS afforded similar sensitivity, the use of the mono-isotopic cesium counterion for SOS was found to be more practical than the use of the rubidium counterion which has two isotopes. The isotopic pattern of the cesium salt of SOS reflected a so-called A +2 nature of sulfur (Fig. 5, insets), which permitted distinction between the analyte peaks from the matrix cluster peaks, CsnCHCAm, in the mass spectrum. Two isotopes of rubidium, 85Rb and 87Rb, occur in a ratio of approximately 100:39, and the presence of rubidium renders the SOS peaks indistinguishable from the peaks corresponding to matrix clusters, RbnCHCAm.
Optimization of sample preparation
Various degrees of X/Na exchange, X =K, Rb, Cs, were observed during MALDI-MS analyses of K8SOS, Rb8SOS, and Cs8SOS. To minimize this problem, 1 mM stock solutions of these salts of SOS were prepared in 8 mM XCl, X =K, Rb, Cs. The optimized concentration of alkali metal chlorides in the solutions of respective alkali metal salts of SOS was determined experimentally. Higher concentrations of alkali metal salts in the MALDI samples negatively affected the analyte signal intensity, whereas lower concentrations of the salt additives were inefficient in suppressing the X/Na exchange. Addition of 1 μL of 1–10 mM XOH, X =K, Rb, Cs, per 2 μL of matrix/analyte mixture was also found to reduce the appearance of X/Na exchange peaks and improve the analyte signal in the positive-ion detection mode.
Sensitivity
We prepared and analyzed a series of Cs8SOS dilutions to determine a detection limit that can be achieved with the present MALDI-MS method. As low as 0.4 pmol Cs8SOS (0.25 μL of 3 μM Cs8SOS solution diluted 50% with the matrix) was detected under the optimized experimental conditions, in the presence of 0.25 μL of 1 mM CsOH (Fig. 6). The detection limit obtained for Cs8SOS was an order of magnitude lower than that for Na8SOS, which was not detected below 4 pmol (Autoflex II). The molecular peak [MCs+Cs]+ was accompanied by a series of peaks corresponding to Cs/Na exchange products (Δ −110 m/z). The appearance of the Cs/Na exchange peaks in MALDI mass spectra acquired with diluted Cs8SOS samples (<0.1 mM) is a limitation of the present method.
Figure 6.

Positive-ion reflector MALDI mass spectrum of Cs8SOS. The sample was prepared using 1 vol. of 3 μM Cs8SOS solution, 1 vol. of 1 mM CsOH, and 1 vol. of G2CHCA solution, and applying 0.25 μL of the mixture to the target spot. Inset shows isotopic pattern of [MCs+Cs]+ peak.
MALDI-MS analyses of chondroitin sulfate digest
Sodium salts of GAG-derived disaccharides have molecular weights in the range of 500–700 Da and appear in a region of MALDI mass spectra where matrix interference is strong. Using a 50-μL spin-column, the cesium salts of oligosaccharide mixture obtained by enzymatic depolymerization of chondroitin sulfate A (CSA) were prepared and analyzed without any separation or purification steps. Converting sodium salts of CSA disaccharides into the cesium salts shifted their MALDI-MS peaks toward higher m/z values, permitting the use of matrix suppression settings of up to 500 Da without suppressing an analyte signal. A significant improvement in the MALDI-MS signal of the cesium salt of CSA disaccharide as compared to that of the sodium salt of CSA disaccharide (Fig. 7) can be attributed in part to the different matrix suppression settings used in the experiment. Both mass spectra were acquired with the matrix suppression set at 200 Da below the molecular weight of the analyte. However, MALDI mass spectra acquired with the cesium salt of CSA disaccharide showed improved signal intensity regardless of the matrix suppression settings. Therefore, it is likely that much of the MALDI signal enhancement achieved with the cesium salt of CSA disaccharide could result from the increased stability of sulfate half-esters toward thermal fragmentation. The complete absence of SO3-loss fragments in the positive-ion MALDI mass spectra acquired with the sodium and cesium salts of CSA disaccharide makes this method potentially applicable to the analysis of mixtures containing disaccharides having various levels of sulfation.
Figure 7.

Positive-ion MALDI mass spectra of chondroitin sulfate A disaccharide, sodium salt (A) and cesium salt (B) acquired with G2CHCA. Peaks due to the matrix are marked with triangles. Peak marked with closed circle in (B) corresponds to a product of Cs/Na exchange.
CONCLUSIONS
Larger alkali metal cations such as K+, Rb+, and Cs+ are known to suppress metastable fragmentation of glycosidic bonds in neutral oligosaccharides in MALDI Fourier transform ion cyclotron resonance (FTICR)-MS experiments.14,18 Based on the molecular dynamics calculations and empirical observations, this effect was attributed to the inability of the large alkali metal/oligosaccharide complex to undergo charge-induced fragmentation.14 Large alkali metal cations can efficiently coordinate several oxygen atoms,14 which can explain the stability of cesium-coordinated sulfate half-esters toward fragmentation in MALDI-TOFMS described in the present work. The systematic evaluation of the effect of alkali metal counterions on the stability of O–SO3 bonds toward fragmentation in MALDI-MS analysis was carried out using an octasulfated disaccharide, SOS, as a model polyanion. The MALDI mass spectra of five alkali metal salts of SOS, acquired with an ionic liquid matrix, G2CHCA, in positive-ion and negative-ion reflector modes were compared under identical experimental parameters. Two criteria were used to evaluate the experimental results: the extent of the analyte fragmentation through the loss of SO3 and the absolute signal intensity of the analyte molecular species. The cesium salt of SOS afforded MALDI mass spectra with the lowest degree of fragmentation through cleavage of O–SO3 bonds in both positive-ion and negative-ion detection modes. In addition to the suppressed fragmentation, Cs8SOS exhibited a significant MALDI signal enhancement compared to other alkali metal salts of SOS. While the presence of peaks corresponding to Cs/Na exchange products detracted from the quality of MALDI mass spectra of Cs8SOS, a low-picomole detection limit obtained with this counterion compensated for the drawback. The practical utility of the present method is demonstrated by applying it in the analysis of the CSA disaccharides. Strong analyte signal and the complete absence of the SO3-loss fragments make the present method amenable to the direct MALDI-MS analysis of GAG-derived disaccharide mixtures. Future experiments will be directed toward structural characterization of GAG disaccharides by tandem mass spectrometry using the described methodology. A strong MALDI signal corresponding to the protonated cesium salt of CSA disaccharide (Fig. 7(B)) should permit acquisition of structurally informative tandem mass spectra. Protonated oligosaccharides have the greatest metastable decay rate;19 thus, they are expected to fragment efficiently through the glycosidic bond and cross-ring cleavages within the post-source decay timeframe. Zaia and Costello demonstrated that the stabilization of sulfo groups in heparin-like GAGs by metal complexation does not prevent glycosidic bond dissociation in quadrupole orthogonal acceleration TOFMS.20
Acknowledgments
Contract/grant sponsor: NIH; contract/grant number: GM38060.
The authors thank Dr. Dmitri Zagorevski for useful discussions. This work was supported by NIH grant # GM38060.
References
- 1.Capila I, Linhardt RJ. Angew Chem Int Ed. 2002;41:390. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 2.Lamari FN, Theocharis AD, Asimakopoulou AP, Malavaki CJ, Karamanos NK. Biomed Chromatogr. 2006;20:539. doi: 10.1002/bmc.669. [DOI] [PubMed] [Google Scholar]
- 3.Mulloy B, Linhardt RJ. Curr Opin Struct Biol. 2001;11:623. doi: 10.1016/s0959-440x(00)00257-8. [DOI] [PubMed] [Google Scholar]
- 4.Linhardt RJ, Loganathan D. In: Biomimetic Polymers. Gebelein CG, editor. Plenum Press; New York: 1990. pp. 135–173. [Google Scholar]
- 5.Linhardt RJ. In: Current Protocols in Molecular Biology. Ausbel FM, editor. Vol. 3. Greene and Wiley; New York: 1999. pp. 17.13B.1–17.13B.16. [Google Scholar]
- 6.Imanari T, Toida T, Koshiishi I, Toyoda H. J Chromatogr A. 1996;720:275. doi: 10.1016/0021-9673(95)00338-x. [DOI] [PubMed] [Google Scholar]
- 7.Volpi N, Maccari F. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;834:1. doi: 10.1016/j.jchromb.2006.02.049. [DOI] [PubMed] [Google Scholar]
- 8.Desaire H, Leary JA. J Am Soc Mass Spectrom. 2000;11:916. doi: 10.1016/S1044-0305(00)00168-9. [DOI] [PubMed] [Google Scholar]
- 9.Saad OM, Leary JA. J Am Soc Mass Spectrom. 2004;15:1274. doi: 10.1016/j.jasms.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 10.Juhasz P, Biemann K. Proc Natl Acad Sci USA. 1994;91:4333. doi: 10.1073/pnas.91.10.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Juhasz P, Biemann K. Carbohydr Res. 1995;270:131. doi: 10.1016/0008-6215(94)00012-5. [DOI] [PubMed] [Google Scholar]
- 12.Gunay NS, Tadano-Aritomi K, Toida T, Ishizuka I, Linhardt RJ. Anal Chem. 2003;75:3226. doi: 10.1021/ac034053I. [DOI] [PubMed] [Google Scholar]
- 13.Pearson RG. J Am Chem Soc. 1963;85:3533. [Google Scholar]
- 14.Cancilla MT, Penn SG, Carroll JA, Lebrilla CB. J Am Chem Soc. 1996;118:6736. [Google Scholar]
- 15.Laremore TN, Murugesan S, Park T-J, Avci FY, Zagorevski DV, Linhardt RJ. Anal Chem. 2006;78:1774. doi: 10.1021/ac051121q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laremore TN, Zhang F, Linhardt RJ. Proc. 54th ASMS Conf. Mass Spectrometry and Allied Topics; Seattle, WA. May 28–June 1, 2006. [Google Scholar]
- 17.Linhardt RJ, Wang HM, Loganathan D, Lamb DJ, Mallis LM. Carbohydr Res. 1992;225:137. doi: 10.1016/0008-6215(92)80045-3. [DOI] [PubMed] [Google Scholar]
- 18.Xie Y, Lebrilla CB. Anal Chem. 2003;75:1590. doi: 10.1021/ac026009w. [DOI] [PubMed] [Google Scholar]
- 19.Ngoka LC, Gal J-F, Lebrilla CB. Anal Chem. 1994;66:692. doi: 10.1021/ac00077a018. [DOI] [PubMed] [Google Scholar]
- 20.Zaia J, Costello CE. Anal Chem. 2003;75:2445. doi: 10.1021/ac0263418. [DOI] [PubMed] [Google Scholar]