

Abstract
Background
P-glycoprotein (P-gp) mediated drug efflux across the blood–brain barrier (BBB) is an important mechanism underlying poor brain penetration of certain antiepileptic drugs (AEDs). Nanomaterials, as drug carriers, can overcome P-gp activity and improve the targeted delivery of AEDs. However, their applications in the delivery of AEDs have not been adequately investigated. The objective of this study was to develop a nano-scale delivery system to improve the solubility and brain penetration of the antiepileptic drug lamotrigine (LTG).
Methods
LTG-loaded Pluronic® P123 (P123) polymeric micelles (P123/LTG) were prepared by thin-film hydration, and brain penetration capability of the nanocarrier was evaluated.
Results
The mean encapsulating efficiency for the optimized formulation was 98.07%; drug-loading was 5.63%, and particle size was 18.73 nm. The solubility of LTG in P123/LTG can increase to 2.17 mg/mL, making it available as a solution. The in vitro release of LTG from P123LTG presented a sustained-release property. Compared with free LTG, the LTG-incorporated micelles accumulated more in the brain at 0.5, 1, and 4 hours after intravenous administration in rats. Pretreatment with systemic verapamil increased the rapid brain penetration of free LTG but not P123/LTG. Incorporating another P-gp substrate (Rhodamine 123) into P123 micelles also showed higher efficiency in penetrating the BBB in vitro and in vivo.
Conclusion
These results indicated that P123 micelles have the potential to overcome the activity of P-gp expressed on the BBB and therefore show potential for the targeted delivery of AEDs. Future studies are necessary to further evaluate the appropriateness of the nanocarrier to enhance the efficacy of AEDs.
Keywords: antiepileptic drug, lamotrigine, polymeric micelles, P-glycoprotein, pluronic, nanocarrier
Introduction
The drug efflux transporters on the blood–brain barrier (BBB) restrict the access of various central nervous system (CNS) drugs to the target tissue.1 Pathophysiology-associated activation of efflux transporters further strengthens the barrier function in different CNS diseases; in epilepsy, seizures strongly induce expression of the efflux transporter P-glycoprotein (P-gp).2,3 This overexpression has been shown to correlate with reduced brain penetration of several antiepileptic drugs (AEDs) and a reduction or even complete loss of pharmacosensitivity.4–6 Furthermore, a variety of AEDs with different modes of action have been shown to be substrates of P-gp.7,8 Together, these findings point to a role of seizure-induced P-gp overexpression as a limiting factor in epilepsy pharmacotherapy with a lack of satisfactory seizure control in up to 20%–30% of patients.9 Overcoming P-gp overactivity to increase the targeted delivery of AEDs is therefore proposed to be a potential treatment strategy for refractory epilepsy.
An obvious strategy to overcome P-gp activity is direct interference with efflux transport by competitive or noncompetitive P-gp inhibitors. Previous studies have substantiated that neither first- or second-generation P-gp inhibitors are appropriate for clinical application.10,11 On an experimental level, add-on treatment with a third-generation P-gp inhibitor (tariquidar) resulted in almost complete seizure control in rats with pharmacoresistance to phenobarbital and phenytoin.12,13 However, inhibition of P-gp with a more specific third-generation inhibitor at the BBB requires relatively high doses, which might approach the maximum tolerated doses in patients.14 Other methods such as peptide and gene therapy have been explored to counteract P-gp overactivity.15–17 But, direct transporter inhibition by all the abovementioned means also affects basal transporter function throughout the body, thereby limiting the protective function of efflux transport and increasing exposure of sensitive tissues to harmful xenobiotics.18 Taking these concerns into account, overcoming P-gp activity without disruption of barrier function may therefore require a more elegant strategy to promote the delivery of AEDs as well as other P-gp substrates.
The use of drug-loaded nanocarriers, which already show great advantages in treating P-gp mediated multidrug resistant tumors, is a promising alternative to bypassing and/or modulating P-gp and thus promoting the delivery of AEDs without collateral damage.19 Some preliminary studies using nanoparticles like liposomes and polymers to encapsulate antiepileptic agents have shown anticonvulsive effects in epilepsy models.20–22 However, these studies still use inert nanocarriers without biological activity, which may affect their efficacy in P-gp mediated drug-resistant epilepsy. In addition, some of these studies still used delivery of drug-loaded nanoparticles via invasive routes, eg, local intracerebral implantation. Therefore, it is still necessary to develop novel AEDs-incorporated nanoscale delivery systems that not only can be administered non-invasively, but also possess the ability to modulate P-gp activity.
Polymer-based nanotechnology has become one of the most attractive and rapidly-growing areas of nanomedicine-based technology. Examples of such materials are Pluronic® block copolymers (also known as poloxamers) that consist of hydrophilic polyethylene oxide (PEO) and hydrophobic polypropylene oxide (PPO) blocks arranged in an A-B-A tri-block structure: PEO-PPO-PEO.23 It is a prominent feature for Pluronic copolymers that can self-assemble into a spherical micelle structure constructed by ethylene oxide (EO) as a hydrophilic outer shell and propylene oxide (PO) as a hydrophobic inner core. The PO core can encapsulate the hydrophobic drug while the hydrophilic corona maintains the dispersion stability of micelles. Thus, these kinds of block copolymers can result in increased solubility, metabolic stability, and improved circulation time. In addition to their inert carrier functions, recent findings indicate that some Pluronics can be inhibitors of P-gp, which can sensitize multidrug resistant (MDR) tumors to anticancer agents in vitro and in vivo.24 In addition, this polymer was shown to have the ability of modulating the drug efflux transporter (P-gp) activity on the BBB, consequently enhancing the BBB penetration of CNS therapeutics.25–27 Two main mechanisms are suggested to be involved in the P-gp modulating function of the Pluronics. First, the Pluronics can influence mitochondria function and energy conservation in cells expressing P-gp, leading to inhibition of its function.28 Second, interaction of the block copolymer with the P-gp containing membrane also contributes to the inhibition of P-gp activity.29 Pluronic P123 (P123), composed of PEO20-PPO65-PEO20, is one of the most common types of Pluronic copolymer. It has also been demonstrated that P123 has a significant cytotoxicity in MDR cell lines to paclitaxel due to inhibition of the P-gp that was over-expressed in these cells.30 One previous study has utilized this series of polymers to solubilize the AED carbamazepine.31 However, it only treated Pluronic as a solubilizer rather than a nanocarrier.
Lamotrigine (LTG) is a widely used oral AED approved for use in adults with partial seizures, and in pediatrics and adults for the treatment of generalized seizures, either alone or in combination with other anticonvulsants. However, it only shows marginal or no effects in refractory epilepsy,32 partly because it cannot reach therapeutic concentration in epileptogenic focus as a consequence of P-gp mediated drug efflux.7,8,33 In addition, due to its poor water solubility (approximately 170 μg/mL), no parenteral formulation is available. Therefore, LTG therapy cannot be continued when a patient is vomiting, undergoing surgery, or experiencing status epilepticus. Interruption of LTG administration increases the risk of increased seizure activity. Thus, alternative routes of administration are needed to minimize seizure risk when oral administration is not feasible.
The aim of this study was to develop a novel P123 micelles-based delivery system for the antiepileptic drug LTG, intended to be intravenously administered. To achieve this purpose, LTG-loaded P123 micelles were prepared by thin-film hydration, and a central composite design (CCD) was employed to evaluate the effects of selected variables and to optimize the formulation parameters. The optimal preparation was further characterized in terms of particle size distribution, morphological observation and in vitro release. Finally, the in vivo brain targeting capability of LTG-loaded Pluronic micelles was compared with the same drug in Cremophor EL® solution.
Materials and methods
Materials
Pluronic P123® (P123) and Cremophor EL® (polyoxyl 35 castor oil) were kindly supplied by BASF Corporation (Ludwigshafen, Germany) without further purification. Rhodamine 123 (Rho 123), (±)-verapamil hydrochloride, lamotrigine (LTG) and oxcarbazepine (internal standard [IS]) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). High-performance liquid chromatography (HPLC) grade methanol and acetonitrile were purchased from Merck KgaA (Darmstadt, Germany); 4′,6′-diamidino-2-phenylindole (DAPI) was purchased from Molecular Probes (Eugene, OR, USA); ammonium acetate of analytical grade and dimethyl sulfoxide (DMSO) were obtained from Sinopharm (Shanghai, People’s Republic of China). Water was purified through a Milli-Q® ultraviolet (UV) plus system procured from EMD Millipore (Billerica, MA, USA). All other chemicals used were of analytical grade.
Animals
Sprague Dawley (SD) rats (male, aged 6–8 weeks, 220–300 g) were purchased from Sino-British SIPPR/BK Lab Animal Ltd (Shanghai, People’s Republic of China). Rats were housed under controlled conditions (ambient temperature 23°C–27°C, humidity 65%–75%, 12 hour dark/light cycle). Food and water were freely available. Before being used in the experiments, rats were allowed to adapt to the new conditions for at least 1 week. All experiments were carried out in accordance with guidelines evaluated and approved by the ethics committee of Fudan University. All efforts were made to minimize both the suffering and the number of animals used in this study.
Cell lines
Rat brain capillary endothelial cells (BCECs) were kindly provided by Xin Guo Jiang (Department of Pharmaceutics, School of Pharmacy, Fudan University).34 Primary BCECs were cultured as described previously.35 Briefly, BCECs were expanded and maintained in special Dulbecco’s Modified Eagle Medium (Sigma-Aldrich Co., St Louis, MO, USA) supplemented with 15% heat-inactivated fetal calf serum, 100 μg/mL epidermal cell growth factor, 2 mmol/l L-glutamine, 20 μg/mL heparin, 100 μg/mL penicillin, and 100 μg/mL streptomycin and cultured at 37°C in a humidified atmosphere containing 5% CO2. All cells used in this study were between passage 10 and passage 30.
Preparation of LTG-incorporated Pluronic P123 micelles (P123/LTG micelles)
The LTG loaded P123 micelle was prepared by the thin-film hydration method.36 Briefly, 100 mg of P123 was dissolved in 5 ml methanol and then mixed with different amounts of LTG in a round-bottom flask to obtain the required LTG:P123 ratio. The organic solvent mixture was evaporated under high vacuum to produce a thin film of coprecipitated drug and polymer under 50°C. This film was further dried under vacuum overnight to remove any traces of remaining solvents. After that, the obtained film was hydrated in different amounts of deionized water under 50°C with stirring at 200 rpm for 1 hour to form a micellar suspension. Non-incorporated LTG was removed by filtration of the micellar suspension through a 0.22 μm filter membrane to obtain a clear solution of LTG loaded micelle.
Optimization of P123/LTG micelles through CCD/response surface methodology
A CCD with α=1.414 was employed as per the standard protocol to assure design rotatability. The α value is defined as the distance of the axial runs from the center of the design and equals the fourth root of the number of factorial runs ((22)1/4 = 1.414).37 The amounts of LTG and deionized water (pH 7.4) were selected as the factors, and studied at five levels each. The central point (0, 0) was studied five times. All other formulation and processing variables (amount of P123; temperature of equilibration, etc) were kept invariant throughout the study. Entrapping efficiency (EE%), drug loading efficiency (DL%), and drug concentration in the micelles were taken as the response variables. EE% and DL% were calculated as described below. The optimization study was performed using Statistica 7.0 software (StatSoft, Tulsa, OK, USA). The optimized formulation was repeated in triplicate to evaluate the accuracy of the anticipated responses.
Characterization of the Pluronic P123-based micelles
Micelle size determination
The mean size and distributions of the polymeric micelles were measured by a dynamic light scattering spectrometer (Malvern Zetasizer Nano ZS; Malvern Instruments, Worcestershire, UK). The micellar suspension was analyzed after filtering through a 0.22 μm filter membrane. The intensity autocorrelation was measured at a scattering angle of 90° at 25°C. Measurements were repeated three times each for blank micelles and optimized LTG-loaded micelles.
Surface morphology
The morphological examination of micelles was studied by transmission electron microscopy (TEM; Philips CM 120, Amsterdam, the Netherlands). Briefly, a drop of the micellar solution was placed on a clean copper grid, air-dried and observed under TEM.
Drug-loading and entrapment efficiency
The concentration of LTG in the micelles was determined with a UV-spectrophotometer (UV-2401PC, Wanma Pharmaceutical Co., Ltd, Zhejiang, People’s Republic of China) at 230 nm. The micellar solution was disrupted and suitably diluted with methanol prior to determination. The drug-loading efficiency (DL%) and entrapment efficiency (EE%) of LTG in the polymeric micelles were calculated by the following equations:
| (1) |
| (2) |
In vitro release of LTG from micelles
The release of LTG from the micelles was investigated by a dialysis method with 0.1% Tween-80 volume percent concentration (volume/volume [v/v]) phosphate buffered saline (pH=7.4) solution as the release medium. LTG-incorporated micellar solution (1 mL) was introduced into a pre-swollen dialysis bag with a molecular weight cut-off (MWCO) of 3.5 kDa (Green Bird Science and Technology Development, Shanghai, People’s Republic of China), and the end-sealed dialysis bag was immersed into 50 mL release medium at 37°C±0.5°C with stirring at 100 rpm. While the solubility of LTG in the release medium was 243.27 μg/mL, the maximum concentration of LTG in the medium was 20 μg/mL. Therefore the sink condition was assured. At predetermined time intervals (15, 30, and 45 minutes, and 1, 2, 4, 6, 8, 12, and 24 hours), 0.5 mL of the dissolution medium was withdrawn and the same volume of fresh medium was added. Then, the amount of released LTG was measured by UV-spectrophotometer at 230 nm, and the cumulative release percentage was calculated. For comparison, the release of LTG from stock (methanol) solution was conducted under the same conditions. The drug release of micelles and free LTG formulations were both run in triplicate.
Brain uptake studies
To compare the brain uptake capability of different formulations of LTG, the drug concentrations in serum and brain tissues were measured. All preparations of LTG were injected via tail vein at a dose of 10 mg/kg body weight in SD rats. This dosage was chosen according to previous studies.33,38 Animals were pretreated with intraperitoneal (IP) saline in the presence or absence of the P-gp inhibitor verapamil (20 mg/kg) 90 minutes before LTG administration. The doses, routes of administration, and timing of verapamil administration used were based on earlier studies of P-gp inhibition on the BBB.39,40 Animals were anesthetized with chloroform inhalation at 0.5, 1, or 4 hours after drug administration. Blood samples of 1 mL were collected into heparinized Eppendorf tubes by retro-orbital puncture and the brains were taken out and placed in iced Eppendorf tubes. The blood samples were centrifuged at 3,000 g for 10 minutes and the supernatant (plasma) was collected. Plasma and brain tissue were kept at −80°C until they were further processed. To assess the brain uptake of different formulations, percentages of the injected dose per gram (ID%/g) of brain tissue and ratios of brain–to–plasma were calculated.35,41 Five to seven rats were used for each group.
Processing of blood and brain tissue for HPLC analysis
Plasma sample extraction for the separation of LTG was based on the method described previously.42,43 Briefly, an aliquot of 100 μL of plasma, previously treated with 10 μL of IS working solution (100 μg/mL), was mixed with 250 μL of 2 mol/L NaOH and then 750 μL of ethyl acetate. The mixtures were vortexed for 2 minutes followed by centrifugation (3,000 g at 15°C for 10 minutes). The upper organic layer was transferred to a clean 10 mL conical glass tube and evaporated to dryness under a stream of nitrogen with gentle heating (at 35°C±0.5°C). The residues were dissolved in 110 μL of methanol, and 20 μL was injected into the HPLC system.
For brain tissue preparation, tissue samples (100 mg) were homogenized with 100 μL water in a high-speed tissue homogenizer in an ice bath (Scientz, JY92, Ningbo, People’s Republic of China). Then 5 μL of the 100 μg/mL IS solution was added into the homogenized tissues. After vortexing, three parts of methanol (300 μL) were added into one part of the homogenate. Standards and control samples of the tissues for drug analysis were prepared in the same manner. Normal tissues were homogenized in water and mixed with known amounts of LTG stock solutions. The resulting mixtures were vortexed for 2 minutes followed by centrifugation (12,000 g at 4°C for 15 minutes). The supernatant (50 μL) was used for HPLC analysis.
HPLC analysis of plasma and brain tissue samples was performed using an RP-HPLC system (Agilent 1260 Infinity series, Agilent Technologies, Santa Clara, CA, USA) by previously-described methods,43,44 with some modifications. A C18 reversed-phase analytical column (250 mm ×4.6 mm, ID 5 μm; Agilent, USA) with a C18 guard column (SecurityGuard C18, 4 mm ×3 mm inner diameter (ID); Phenomenex, Torrance, CA, USA) was used for the separation. The mobile phase consisted of 20 mmol/L ammonium acetate buffer (pH 6.5): acetonitrile (70:30, v/v) at a flow rate of 0.8 mL/min, the temperature of the column was set at 35°C, and a UV detector was set at 230 nm. Retention times for LTG and the internal standard were 6.2 and 10.3 minutes, respectively. The detection limit (LOD) was approximately 6.59 ng/mL at a signal:noise ratio of 3:1 and the lower limit of quantitation (LOQ) was 25 ng/mL. The range of linear response for the plasma sample was 0.5–20 μg/mL (r2>0.9998), and the range of linear response for the brain tissue sample was 0.25–10 μg/g or 0.025–1 μg/100 mg tissue (r2>0.9999). The recoveries of LTG at three different concentrations were similar in both plasma and brain homogenates. Recoveries of LTG in plasma at concentrations of 2.5, 10, and 20 μg/mL were 93.1±7.7, 82.2±2.3, and 88.0%±1.1% (n=5), respectively. Recoveries of LTG in the brain homogenate at concentrations of 0.05, 0.5, and 1 μg/100 mg tissue were 81.3±6.48, 89.5±4.12, and 88.4%±2.3% (n=4), respectively, consistent with a previous report.43
Fluorescent and confocal microscopy
Internalization by BCECs
Rat BCECs were seeded at a density of 104 cells in cell culture dishes (35 mm ×10 mm; Corning-Coaster, Tokyo, Japan), incubated for 24 hours, and checked under the microscope for morphology. For intracellular uptake studies, BCECs were treated with free Rho 123 (dissolved in DMSO and diluted with PBS) or 1% P123 polymeric micelles solutions containing 5 μM Rho 123 at 37°C for 30 minutes.45 After washing with PBS three times, the fluorescence was immediately examined with a confocal laser scanning microscope (Carl Zeiss, LSM 710, Oberkochen, Germany).
Distribution of polymeric micelles in rat brain
Free Rho 123 and Rho 123-incorporated polymeric micelles (5% weight of total polymers) were injected into the tail vein of rats at a dose of 1 mg Rho 123/kg body weight. Pretreatment with verapamil was also applied as described above. Thirty minutes after Rho 123 administration, animals were anesthetized with IP 10% chloral hydrate and their hearts perfused with 100 mL saline and 4% paraformaldehyde solution. The brains were removed, fixed in 4% paraformaldehyde for 48 hours, and placed in 15% sucrose PBS solution for 24 hours until subsidence, then in 30% sucrose for 48 hours until subsidence. Excised brains were then frozen at −80°C in optimum cutting temperature (OCT) embedding medium (Sakura, Torrance, CA, USA). Frozen sections of 20 μm thickness were prepared with a cryotome Cryostat (Leica, CM 1900, Wetzlar, Germany) and stained with 300 nM DAPI for 10 minutes at room temperature. After washing twice with PBS (pH 7.4), the sections were immediately examined under the fluorescence Leica, DMI4000B microscope (Leica).
Statistical analysis
The data are expressed as plus or minus the standard error of the mean (± SEM). The statistical differences between the values of two groups were analyzed using the non-paired t-test if the variance was equivalent; if the variance was not equivalent, then the Mann–Whitney test was performed. Multiple comparisons were performed using analysis of variance (ANOVA) followed by the Bonferroni’s test. The Pearson’s correlation coefficient (r) was used to estimate the correlations between two factors. A value of P<0.05 was considered to be statistically significant. The Statistica software (version 7.0) was used for performing statistical analyses.
Results
Optimization of the formulation of Pluronic P123/LTG micelles
The two-dimensional contour diagrams of drug entrapping efficiency (EE%), drug loading efficiency (DL%) and drug concentration superimposed on each other are shown in Figure 1. The amounts of LTG and de-ionized water needed to obtain optimized levels of EE%, DL% and drug concentration were estimated by Statistica 7.0 as the following: the amount of LTG was 5.5 mg and the volume of water was 4.5 mL (P123, 100 mg). The EE% of the optimized formulation was 98.07%±1.39%, the DL% was 5.12%±0.12%, and the drug concentration was 1.09±0.02 mg/mL (n=3). Upon comparison of the observed responses with the anticipated responses of the checkpoint, the percentage errors in prognosis were 3.76%, 9.06%, and 1.45% for EE%, DL%, and drug concentration (n=3), respectively. The solubility (drug concentration) of LTG was 2.17±0.08 mg/mL when the amounts of P123 and LTG were increased by 2.7-fold (n=4).
Figure 1.
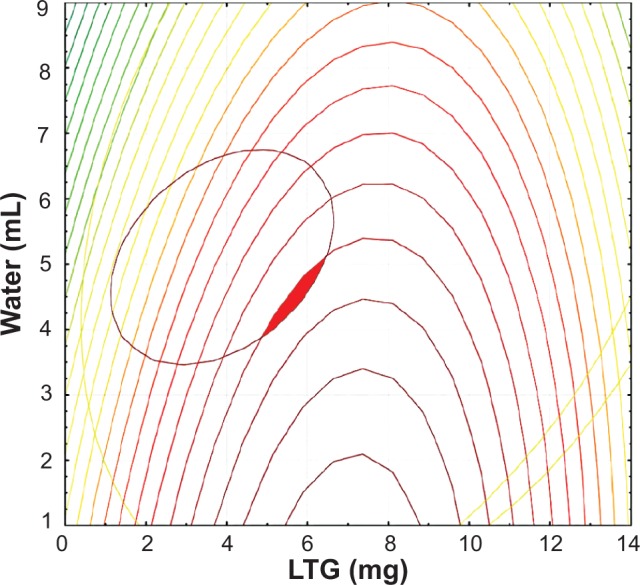
Superimposed two-dimensional contour diagrams of drug loading efficiency (DL%), entrapping efficiency (EE%), and drug concentration in the micelles.
Note: The red area shows the optimized area.
Abbreviation: LTG, lamotrigine.
Particle size distribution and surface morphology
The average micelle size and size distribution for empty micelles and drug-loaded micelles are illustrated in (Figure 2A and B). The mean diameter of empty P123 micelles and drug-incorporated micelles was close to 20 nm with rather narrow size distribution patterns (polydispersity index [PDI] =0.06). Loading micelles with LTG did not visibly affect their size (18.51±0.05 nm for blank micelles versus 18.73±0.23 nm for drug-loaded micelles, n=3, P>0.05) and size distribution. The micelles exhibited spherical shape of moderate uniform particle size under TEM (Figure 2C). The particle size measured from the TEM images was in agreement with that measured by the laser scattering technique. The particle surface was very smooth and no drug crystal was visible.
Figure 2.

Particle size and size distribution of empty Pluronic P123 micelles (A); LTG-loaded micelles (B); TEM image of LTG-loaded P123 micelles (C).
Abbreviations: LTG, lamotrigine; TEM, transmission electron microscopy.
In vitro release of LTG from micelles
The in vitro release of LTG from micellar formulation under sink condition was investigated by the dialysis method with 0.1% Tween-80 solution as the release medium. As shown in Figure 3, about 65% of LTG was released from P123/LTG within the first 4 hours, while almost all LTG was released from the stock solution during the same time period. After 24 hours, 20%–30% of the initially incorporated drug still existed in the micelles and it did not change substantially, even after 48 hours (data not shown).
Figure 3.
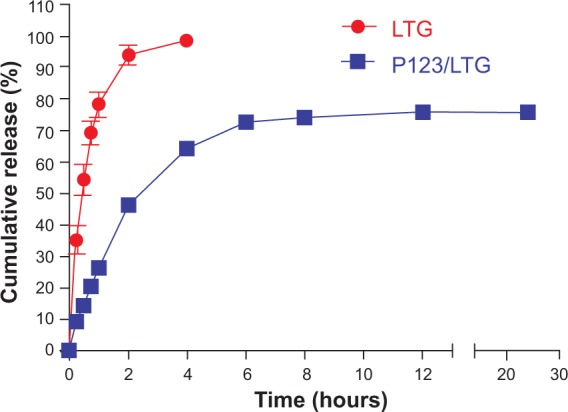
Release profiles of LTG from P123/LTG and the stock solution in 0.1% Tween-80 solution at 37°C.
Note: Each point represents the mean ± SEM (n=3).
Abbreviations: LTG, lamotrigine; SEM, standard error of the mean.
Brain uptake study
The results of the brain uptake of LTG at 0.5, 1, and 4 hours following intravenous (IV) administration at 10 mg/kg free LTG or LTG-incorporated micelles are shown in Figure 4. The serum concentrations of two LTG formulations were comparable at all three time points (LTG: 7.29±0.91 μg/mL, 5.33±0.72 μg/mL, and 5.03±0.17 μg/mL; P123/LTG: 7.76±1.45 μg/mL, 7.12±1.56 μg/mL, and 4.69±0.57 μg/mL). The brain uptake of P123/LTG micelles increased by about two-fold compared with the free LTG treated group (P<0.01). The data from percentage of the injected dose per gram of brain (Figure 4A) were in good agreement with those from the brain/plasma ratio (Figure 4B). The increased brain penetration with micellar formulation still existed 4 hours after drug administration.
Figure 4.
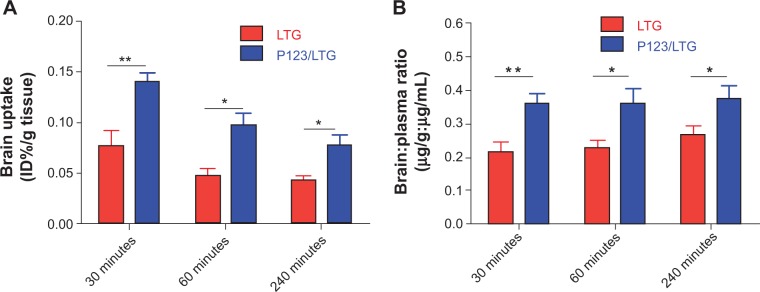
Brain uptake of LTG after IV administration of free LTG and P123/LTG at 30 minutes, 60 minutes, and 240 minutes at a dose of 10 mg LTG/kg body weight.
Notes: (A) Percentages of the injected dose per gram (%ID/g) of brain tissue and (B) Brain:plasma ratios are shown. Data shown are presented as the means ± SEM from 5–6 animals per group. *P<0.05, **P<0.01 against the LTG group.
Abbreviations: LTG, lamotrigine; ID%/g, injected dose per gram; IV, intravenous; SEM, standard error of the mean.
The effect of the P-gp inhibitor verapamil on the 30- minute brain uptake of LTG and P123/LTG is shown in Figure 5. With blockade of P-gp activity by verapamil (20 mg/kg), the efficiency of LTG penetrating into the brain parenchyma was significantly increased, as evident by analysis of the brain/plasma ratio (P<0.01, Figure 5B). The drug concentration (ID%/g) in the brain parenchyma, as represented by ID%/g tissue, showed a similar increment after verapamil inhibition (Figure 5A), but was not statistically different (P=0.061). The plasma concentrations between free LTG alone and free LTG pretreated with verapamil were not significantly different (7.29±0.91 μg/mL versus 8.51±1.40 μg/mL, respectively; P>0.05). The brain penetration efficiency of free LTG solution after systemic verapamil inhibition was comparable to that of LTG micellar formulations (P123/LTG). In contrast, the brain uptake of P123/LTG micelles was not substantially affected by verapamil inhibition (P>0.05).
Figure 5.
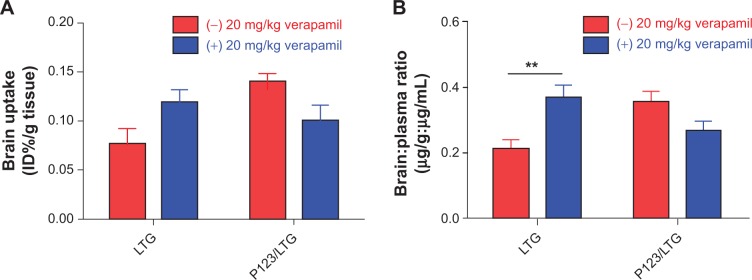
Effect of the P-gp inhibitor verapamil on the brain uptake of LTG in vivo.
Notes: Free LTG or P123/LTG micelles at a dose of 10 mg LTG/kg body weight was administered intravenously with or without pretreatment with verapamil (20 mg/kg IP, −90 minutes [verapamil was administered 90 minutes before the LTG administration]). Brain uptake of LTG (A and B) 30 minutes after drug administration is shown. Data are expressed as the mean ± SEM. **P<0.01 against the control LTG group, n=5–7 per group.
Abbreviations: LTG, lamotrigine; IP, intraperitoneal; SEM, standard error of the mean; P-gp, P-glycoprotein.
Fluorescent and confocal microscopy study
Another traditional P-gp substrate, Rho 123, was used to confirm the effects of P123 micelles on the BBB. The in vitro internalization experiment was investigated in rat BCECs (Figure 6). The cells treated with free Rho 123 demonstrated very weak fluorescence signals (Figure 6A–C), whereas the signal of those treated with polymeric micelles was strong (Figure 6D–F). The major fraction of the Rho 123-incorporated P123 polymeric micelles was cell-internalized and concentrated in the cytosol (Figure 6F).
Figure 6.

The qualitative evaluation of Rho 123-incorporated polymeric micelles in vitro.
Notes: BCECs were incubated with free Rho 123 (A–C) and Rho 123-incorporated polymeric micelles (D–F) for 30 minutes, and then observed by confocal laser scanning microscopy.
Abbreviation: BCEC, brain capillary endothelial cell.
Rho 123 was also used to assess the brain targeting capability of P123 micelles in vivo. Figure 7 shows the Rho 123 distribution in sections of the cortical layer, hippocampus, substantia nigra, and caudate putamen 0.5 hours after IV injection of free Rho 123 or Rho 123-incorporated micelles at a dose of 1 mg/kg Rho 123. Except for the cortical layer (Figure 7A), the fluorescence of free Rho 123 was almost invisible in the other three regions of the rat brain studied (Figure 7B–D). Pretreatment (IP) with the P-gp inhibitor verapamil 90 minutes before Rho 123 administration increased the Rho 123 accumulation in the cortical layer (Figure 7E) and the red fluorescence signal was also observed in the substantia nigra and caudate putamen (Figure 7G and H). For the micellar formulation, Rho 123 accumulation in the cortical layer (Figure 7I) was comparable to that in verapamil pretreated rats, and much higher than that treated with free Rho 123. Additionally, the Rho 123 fluorescence in the substantia nigra and caudate putamen was even higher than in the verapamil pretreatment group. However, Rho 123 was still obsolete in the hippocampus after systemic P-gp inhibition or delivery with the nanocarrier (Figure 7B, F, and J).
Figure 7.

The qualitative evaluation of Rho 123-incorporated polymeric micelles in vivo.
Notes: Distribution of micelles in the brains of SD rats treated with polymeric micelles (I–L) and free Rho 123 (A–D) 30 minutes after IV administration. The middle row (E–H) shows Rho 123 brain distribution in rats pretreated with verapamil (20 mg/kg, IP) 90 minutes before free Rho 123 administration. Frozen sections (thickness of 20 μm) of the cortical layer (A, E, I), hippocampus (B, F, J), substantia nigra (C, G, K), and caudate putamen (D, H, L) were examined by fluorescence microscopy. The sections were stained with 300 nM DAPI for 10 minutes at room temperature. Red: Rho 123. Blue: cell nuclei. Original magnification: ×200.
Abbreviations: SD, Sprague Dawley; IP, intraperitoneal; DAPI, 4′,6′-diamidino-2-phenylindole; IV, intravenous.
Discussion
In this study, we demonstrated that the Pluronic P123-based polymeric micelles incorporating LTG can be successfully prepared and delivered to the brain. Our major findings are as follows: 1) the LTG can be incorporated into Pluronic P123 micelles and available for intravenous use; 2) P123/LTG micelles exhibit a rapid and enhanced brain penetration efficiency compared with free LTG; and 3) the increased CNS penetration of LTG may partially be attributed to the inhibition of P-gp by the polymeric micellar delivery system. In line with these data, the P123-based nanocarrier is proposed to be a promising drug delivery system to promote the brain delivery of AEDs.
LTG needs a long titration period to achieve an effective serum concentration. It cannot be delivered parenterally, which restrains its clinical use. Cremophor EL and dehydrated ethanol are two common solubilizers for lipophilic drugs. Consistent with previous studies,33,41 10% Cremophor EL used in this study increases the solubility of the LTG to 2.5 mg/mL so that it can be delivered as a solution. Unfortunately, serious side effects attributable to Cremophor EL, including hypersensitivity, nephrotoxicity and neurotoxicity as well as effects on endothelial and vascular muscles, have been reported.46 Amphiphilic block copolymers, which can assemble into nano-scaled core-shell micelles in aqueous media to deliver hydrophobic drugs, have sparked a considerable interest in the last decade.23 However, to our knowledge, LTG has never previously been incorporated into nanomicelles or other nanocarriers. In the current work, we developed novel LTG-loaded nanomicelles made of Pluronic P123. Pluronic copolymers are safe, biocompatible, relatively nontoxic, and have been approved by the US Food and Drug Administration. The critical micelle concentration (CMC) of P123 is extremely low (approximately 0.0068%).47 This property results in some positive functions of formulated micelles, such as more thermodynamic stability upon dilution in a large volume of the blood following intravenous administration. Although the water solubility of LTG could only be increased by about 15-fold in our preparation, it is still available as a solution. Furthermore, the cytotoxicity of P123 micelles is lower than that of Cremophor EL,47 indicating that it may be more suitable to solubilize and deliver drugs.
Besides solubilizing hydrophobic drugs, block copolymer micelles can also target their payloads to specific tissue, eg, brain. Consistent with previous reports,47,48 the particle size of drug-loaded micelles was around 20 nm in our study (Figure 2). Small particles could reduce the uptake and reorganization of the reticuloendothelial system and prolong the systemic circulation time in the blood. Thus, small particle size, ie, diameter below 100 nm, is considered as one of the major prerequisites for the effective brain delivery of nanoparticles.49 In addition, it was found that >90% LTG in the stock solution was released within the first 2 hours, while only 46.3% of LTG was released from the P123 micelles during the same time period (Figure 3), indicating a sustained release behavior, which is similar to previous studies.47,48 It should be noted that about 20% of LTG remained in micelles even after 24 hours, and further release was negligible thereafter (data not shown). This kind of release behavior may be attributed to the crystalline formation of the abundant loaded drugs as a separate phase inside the micellar core, subsequently leading to a slower or even retarded drug release.50,51 This property allows the loaded drug to reach and/or interact with its target with minimal drug loss.
The presence of P-gp on the BBB influences delivery of various central nervous system drugs.1 The poor penetration of free Rho 123 across the BBB in our study also supports this notion. Similar to the results from previous studies with local verapamil administration,33 systemic administration of verapamil in the present study increased the LTG uptake by 2-fold in normal rodent brain (Figure 5). The enhanced brain penetration of LTG has also been observed in P- glycoprotein (Mdr1a/1b) knockout mice,41 further supporting the role of P-gp on the brain penetration of LTG. Except for LTG, previous reports have indicated that P-gp inhibitors can also promote the brain delivery of phenobarbital, felbamate, phenytoin, and oxcarbamazepine in animals.33,52,53 Unfortunately, findings from clinical trials did not support the application of the currently available P-gp inhibitors in combination with P-gp substrates due to complex drug interactions and other hurdles.10,11
Nanotechnology has been proposed to be an important alternative to P-gp inhibitors in delivering P-gp substrates.14 However, only a limited amount of data are available concerning nano-delivery systems for targeted delivery of AEDs and other anticonvulsive agents.20,54–56 Most of them only use stealth nanomaterials and apply invasive ways to deliver drugs. Pluronic block copolymers are a kind of polymeric nanomaterial that can function not only as inert carriers, but also as biological response modifiers. Here, we reported that the AED LTG can be successfully encapsulated with these polymers at reasonable EE% and DL% (Figure 1). More importantly, LTG-incorporated P123 micelles achieved higher brain accumulation at 0.5, 1, and 4 hours after IV administration, compared with LTG in 10% Cremophor EL solution at the same dose (Figure 4). Since plasma levels between the two formulations did not differ significantly at three time points, we can at least rule out that the increased brain uptake of micelles is merely related to the pharmacokinetic change. The introduction of a brain-to-serum ratio can help to preclude the possibility that the difference was caused by blood contamination in the brain homogenate. Herein, we demonstrated that the brain uptake of P123/LTG micelles increased by about 2-fold compared with the free LTG treated group in vivo (Figure 4). We believe this is meaningful, as the P-gp expression level was just increased by ~2-fold in refractory epilepsy.5,57,58 The relatively low increment of brain uptake with micelles in this study may be attributable to the already high brain penetration of free LTG, which is highly hydrophobic. It should also be noted that the vehicle containing Cremophor EL, which was used to solubilize free LTG, had previously been shown to inhibit P-gp.59 Similar enhancement of brain uptake was also observed in other brain-targeted nanocarriers.35,60 Batrakova et al reported that Pluronic P85 can sensitize multidrug resistant cancer cells61 and enhance the delivery of P-gp substrates to the brain26 through a common mechanism: inhibition of P-gp activity. Zhang et al showed that P123 micelles also have the ability to inhibit P-gp and consequently sensitize MDR cells.30 But the effects of P123 micelles on P-gp highly expressed BBB have not been extensively investigated. Our study found that pretreatment with verapamil can increase the brain penetration of free LTG instead of P123/LTG, suggesting a similar effect of P123 micelles and verapamil on the BBB. Furthermore, incorporating another P-gp substrate Rho 123 into P123 micelles showed similar results (Figure 6D–F and 7I–L). With these in mind, we proposed that the enhanced brain uptake of LTG by the P123 nanocarrier may partially be due to its rapid and transient P-gp modulating effects, although other mechanisms such as bypass of P-gp have not been completely ruled out.
Among Pluronic polymers, Pluronic P85 has the strongest ability to inhibit P-gp and can significantly increase the brain penetration of CNS drugs. But it is not an ideal drug carrier due to its small micelle core and poor solubilization capability.62 In the present study, as a proof-of-concept, we proved that antiepileptic drug LTG can be successfully and feasibly encapsulated by P123 micelles. More importantly, it was found that the drug-loaded micelles can lead to a rapid and enhanced brain uptake of LTG, while they do not greatly increase the serum concentrations. In addition, it has been suggested that the polymeric-based nanocarriers enter brain tissues without the irreversible disruption of the BBB.60 In brain diseases such as epilepsy, the P-gp level is increased to the point that a therapeutic brain concentration cannot be achieved at tolerable drug doses, leading to drug refractoriness.1 Previous studies have shown that the improved antiepileptic effects of some AEDs can be achieved by increasing their concentrations in epileptogenic focus.12,13,53 Therefore, future studies need to address the question of whether the increase in LTG brain concentrations observed in the present study is associated with an improved efficacy and whether it is possible to overcome resistance in chronic epilepsy models.
Conclusion
Pluronic P123 micelles can efficiently solubilize otherwise poorly soluble LTG, enabling it to be delivered as a solution. In fact, the encapsulation of drug by the micelles exhibited a significantly enhanced and rapid delivery of LTG into the brain. The effect of micelles is equivalent, but not synergistic to the P-gp inhibitor verapamil. Further studies are necessary to evaluate the anticonvulsant and reversal effects of the investigated LTG micellar formulations on the P-gp overexpressed refractory epilepsy model. In conclusion, our findings suggest that the P123-based nanocarrier is a promising vehicle for targeted delivery of AEDs and other anticonvulsant agents, which may have implications in the treatment of refractory epilepsy.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Grant Number 81100964/H0913 and Grant Number 81072594). We greatly acknowledge Shuang Zhang and Yan Bao for their technical assistance in experiments.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005;6(8):591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- 2.Bauer B, Hartz AM, Pekcec A, Toellner K, Miller DS, Potschka H. Seizure-induced up-regulation of P-glycoprotein at the blood–brain barrier through glutamate and cyclooxygenase-2 signaling. Mol Pharmacol. 2008;73(5):1444–1453. doi: 10.1124/mol.107.041210. [DOI] [PubMed] [Google Scholar]
- 3.Bankstahl JP, Hoffmann K, Bethmann K, Loscher W. Glutamate is critically involved in seizure-induced overexpression of P-glycoprotein in the brain. Neuropharmacology. 2008;54(6):1006–1016. doi: 10.1016/j.neuropharm.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 4.Wen T, Liu YC, Yang HW, et al. Effect of 21-day exposure of phenobarbital, carbamazepine and phenytoin on P-glycoprotein expression and activity in the rat brain. J Neurol Sci. 2008;270(1–2):99–106. doi: 10.1016/j.jns.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 5.van Vliet EA, van Schaik R, Edelbroek PM, et al. Region-specific overexpression of P-glycoprotein at the blood–brain barrier affects brain uptake of phenytoin in epileptic rats. J Pharmacol Exp Ther. 2007;322(1):141–147. doi: 10.1124/jpet.107.121178. [DOI] [PubMed] [Google Scholar]
- 6.Rizzi M, Caccia S, Guiso G, et al. Limbic seizures induce P-glycoprotein in rodent brain: functional implications for pharmacoresistance. J Neurosci. 2002;22(14):5833–5839. doi: 10.1523/JNEUROSCI.22-14-05833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang C, Kwan P, Zuo Z, Baum L. The transport of antiepileptic drugs by P-glycoprotein. Adv Drug Deliv Rev. 2012;64(10):930–942. doi: 10.1016/j.addr.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Luna-Tortos C, Fedrowitz M, Loscher W. Several major antiepileptic drugs are substrates for human P-glycoprotein. Neuropharmacology. 2008;55(8):1364–1375. doi: 10.1016/j.neuropharm.2008.08.032. [DOI] [PubMed] [Google Scholar]
- 9.Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51(6):1069–1077. doi: 10.1111/j.1528-1167.2009.02397.x. [DOI] [PubMed] [Google Scholar]
- 10.Saltz L, Murphy B, Kemeny N, et al. A phase I trial of intrahepatic verapamil and doxorubicin. Regional therapy to overcome multidrug resistance. Cancer. 1994;74(10):2757–2764. doi: 10.1002/1097-0142(19941115)74:10<2757::aid-cncr2820741004>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 11.Lhomme C, Joly F, Walker JL, et al. Phase III study of valspodar (PSC 833) combined with paclitaxel and carboplatin compared with paclitaxel and carboplatin alone in patients with stage IV or suboptimally debulked stage III epithelial ovarian cancer or primary peritoneal cancer. J Clin Oncol. 2008;26(16):2674–2682. doi: 10.1200/JCO.2007.14.9807. [DOI] [PubMed] [Google Scholar]
- 12.van Vliet EA, van Schaik R, Edelbroek PM, et al. Inhibition of the multidrug transporter P-glycoprotein improves seizure control in phenytoin-treated chronic epileptic rats. Epilepsia. 2006;47(4):672–680. doi: 10.1111/j.1528-1167.2006.00496.x. [DOI] [PubMed] [Google Scholar]
- 13.Brandt C, Bethmann K, Gastens AM, Loscher W. The multidrug transporter hypothesis of drug resistance in epilepsy: Proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol Dis. 2006;24(1):202–211. doi: 10.1016/j.nbd.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 14.Nieto Montesinos R, Beduneau A, Pellequer Y, Lamprecht A. Delivery of P-glycoprotein substrates using chemosensitizers and nanotechnology for selective and efficient therapeutic outcomes. J Control Release. 2012;161(1):50–61. doi: 10.1016/j.jconrel.2012.04.034. [DOI] [PubMed] [Google Scholar]
- 15.Ao L, Wu Y, Kim D, et al. Development of peptide-based reversing agents for P-glycoprotein-mediated resistance to carfilzomib. Mol Pharm. 2012;9:2197–2205. doi: 10.1021/mp300044b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu N, Liu H, Zhang YF, et al. Effects of brain ikkbeta gene silencing by small interfering rna on P-glycoprotein expression and brain damage in the rat kainic acid-induced seizure model. CNS Neurol Disord Drug Targets. 2013 Aug 27; doi: 10.2174/18715273113129990106. Epub. [DOI] [PubMed] [Google Scholar]
- 17.Chen L, Cheng X, Tian L, Yang T, Hermann S, Zhou D. Inhibition of P-glycoprotein over-expression by shRNA-mdr1b in rat astrocytes. Neurochem Res. 2009;34(3):411–417. doi: 10.1007/s11064-008-9797-3. [DOI] [PubMed] [Google Scholar]
- 18.Fromm MF. Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol Sci. 2004;25(8):423–429. doi: 10.1016/j.tips.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 19.Rossi MA. Targeting anti-epileptic drug therapy without collateral damage: nanocarrier-based drug delivery. Epilepsy Curr. 2012;12(5):199–200. doi: 10.5698/1535-7511-12.5.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori N, Kurokouchi A, Osonoe K, et al. Liposome-entrapped phenytoin locally suppresses amygdaloid epileptogenic focus created by db-cAMP/EDTA in rats. Brain Res. 1995;703(1–2):184–190. doi: 10.1016/0006-8993(95)01095-5. [DOI] [PubMed] [Google Scholar]
- 21.Huang WC, Hu SH, Liu KH, Chen SY, Liu DM. A flexible drug delivery chip for the magnetically-controlled release of anti-epileptic drugs. J Control Release. 2009;139(3):221–228. doi: 10.1016/j.jconrel.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Veronesi MC, Aldouby Y, Domb AJ, Kubek MJ. Thyrotropin-releasing hormone d,l polylactide nanoparticles (TRH-NPs) protect against glutamate toxicity in vitro and kindling development in vivo. Brain Res. 2009;1303:151–160. doi: 10.1016/j.brainres.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 23.Batrakova EV, Kabanov AV. Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J Control Release. 2008;130(2):98–106. doi: 10.1016/j.jconrel.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabanov AV, Batrakova EV, Alakhov VY. Pluronic block copolymers for overcoming drug resistance in cancer. Adv Drug Deliv Rev. 2002;54(5):759–779. doi: 10.1016/s0169-409x(02)00047-9. [DOI] [PubMed] [Google Scholar]
- 25.Kim JY, Choi WI, Kim YH, Tae G. Brain-targeted delivery of protein using chitosan- and RVG peptide-conjugated, pluronic-based nanocarrier. Biomaterials. 2013;34(4):1170–1178. doi: 10.1016/j.biomaterials.2012.09.047. [DOI] [PubMed] [Google Scholar]
- 26.Batrakova EV, Miller DW, Li S, Alakhov VY, Kabanov AV, Elmquist WF. Pluronic P85 enhances the delivery of digoxin to the brain: in vitro and in vivo studies. J Pharmacol Exp Ther. 2001;296(2):551–557. [PubMed] [Google Scholar]
- 27.Saxena V, Hussain MD. Poloxamer 407/TPGS mixed micelles for delivery of gambogic acid to breast and multidrug-resistant cancer. Int J Nanomedicine. 2012;7:713–721. doi: 10.2147/IJN.S28745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirillova GP, Mokhova EN, Dedukhova VI, et al. The influence of pluronics and their conjugates with proteins on the rate of oxygen consumption by liver mitochondria and thymus lymphocytes. Biotechnol Appl Biochem. 1993;18(Pt 3):329–339. [PubMed] [Google Scholar]
- 29.Batrakova EV, Li S, Vinogradov SV, Alakhov VY, Miller DW, Kabanov AV. Mechanism of pluronic effect on P-glycoprotein efflux system in blood–brain barrier: contributions of energy depletion and membrane fluidization. J Pharmacol Exp Ther. 2001;299(2):483–493. [PubMed] [Google Scholar]
- 30.Zhang W, Shi Y, Hao JG, Fang XL. Mechanism of inhibition of P-glycoprotein mediated efflux by Pluronic P123/F127 block copolymers: Relationship between copolymer concentration and inhibitory activity. Eur J Pharm Biopharm. 2012;83(2):266–274. doi: 10.1016/j.ejpb.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 31.Kadam Y, Yerramilli U, Bahadur A. Solubilization of poorly water-soluble drug carbamezapine in pluronic micelles: effect of molecular characteristics, temperature and added salt on the solubilizing capacity. Colloids Surf B Biointerfaces. 2009;72(1):141–147. doi: 10.1016/j.colsurfb.2009.03.027. [DOI] [PubMed] [Google Scholar]
- 32.Shorvon SD. Drug treatment of epilepsy in the century of the ILAE: the second 50 years, 1959–2009. Epilepsia. 2009;50(Suppl 3):93–130. doi: 10.1111/j.1528-1167.2009.02042.x. [DOI] [PubMed] [Google Scholar]
- 33.Potschka H, Fedrowitz M, Loscher W. P-Glycoprotein-mediated efflux of phenobarbital, lamotrigine, and felbamate at the blood–brain barrier: evidence from microdialysis experiments in rats. Neurosci Lett. 2002;327(3):173–176. doi: 10.1016/s0304-3940(02)00423-8. [DOI] [PubMed] [Google Scholar]
- 34.Lu W, Tan YZ, Hu KL, Jiang XG. Cationic albumin conjugated pegylated nanoparticle with its transcytosis ability and little toxicity against blood–brain barrier. Int J Pharm. 2005;295(1–2):247–260. doi: 10.1016/j.ijpharm.2005.01.043. [DOI] [PubMed] [Google Scholar]
- 35.Shao K, Huang R, Li J, et al. Angiopep-2 modified PE-PEG based polymeric micelles for amphotericin B delivery targeted to the brain. J Control Release. 2010;147(1):118–126. doi: 10.1016/j.jconrel.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Jackson JK, Burt HM. Development of amphiphilic diblock copolymers as micellar carriers of taxol. Int J Pharm. 1996;132(1–2):195–206. [Google Scholar]
- 37.Hamed E, Sakr A. Application of multiple response optimization technique to extended release formulations design. J Control Release. 2001;73(2–3):329–338. doi: 10.1016/s0168-3659(01)00356-x. [DOI] [PubMed] [Google Scholar]
- 38.Ebert U, Reissmuller E, Loscher W. The new antiepileptic drugs lamotrigine and felbamate are effective in phenytoin-resistant kindled rats. Neuropharmacology. 2000;39(10):1893–1903. doi: 10.1016/s0028-3908(00)00039-3. [DOI] [PubMed] [Google Scholar]
- 39.O’Brien FE, Clarke G, Fitzgerald P, Dinan TG, Griffin BT, Cryan JF. Inhibition of P-glycoprotein enhances transport of imipramine across the blood–brain barrier: microdialysis studies in conscious freely moving rats. Br J Pharmacol. 2012;166(4):1333–1343. doi: 10.1111/j.1476-5381.2012.01858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clarke G, O’Mahony SM, Cryan JF, Dinan TG. Verapamil in treatment resistant depression: a role for the P-glycoprotein transporter? Hum Psychopharmacol. 2009;24(3):217–223. doi: 10.1002/hup.1008. [DOI] [PubMed] [Google Scholar]
- 41.Nakanishi H, Yonezawa A, Matsubara K, Yano I. Impact of P-glycoprotein and breast cancer resistance protein on the brain distribution of antiepileptic drugs in knockout mouse models. Eur J Pharmacol. 2013;710(1–3):20–28. doi: 10.1016/j.ejphar.2013.03.049. [DOI] [PubMed] [Google Scholar]
- 42.Forssblad E, Eriksson AS, Beck O. Liquid chromatographic determination of plasma lamotrigine in pediatric samples. J Pharm Biomed Anal. 1996;14(6):755–758. doi: 10.1016/0731-7085(95)01669-4. [DOI] [PubMed] [Google Scholar]
- 43.Castel-Branco MM, Almeida AM, Falcao AC, Macedo TA, Caramona MM, Lopez FG. Lamotrigine analysis in blood and brain by high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 2001;755(1–2):119–127. doi: 10.1016/s0378-4347(01)00044-5. [DOI] [PubMed] [Google Scholar]
- 44.Mallayasamy SR, Arumugamn K, Jain T, et al. A sensitive and selective HPLC method for estimation of lamotrigine in human plasma and saliva: application to plasma-saliva correlation in epileptic patients. Arzneimittelforschung. 2010;60(10):599–606. doi: 10.1055/s-0031-1296332. [DOI] [PubMed] [Google Scholar]
- 45.Shao K, Wu J, Chen Z, et al. A brain-vectored angiopep-2 based polymeric micelles for the treatment of intracranial fungal infection. Biomaterials. 2012;33(28):6898–6907. doi: 10.1016/j.biomaterials.2012.06.050. [DOI] [PubMed] [Google Scholar]
- 46.Weiss RB, Donehower RC, Wiernik PH, et al. Hypersensitivity reactions from taxol. J Clin Oncol. 1990;8(7):1263–1268. doi: 10.1200/JCO.1990.8.7.1263. [DOI] [PubMed] [Google Scholar]
- 47.Wei Z, Hao J, Yuan S, et al. Paclitaxel-loaded Pluronic P123/F127 mixed polymeric micelles: formulation, optimization and in vitro characterization. Int J Pharm. 2009;376(1–2):176–185. doi: 10.1016/j.ijpharm.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 48.Zhao L, Shi Y, Zou S, Sun M, Lil L, Zhail G. Formulation and in vitro evaluation of quercetin loaded polymeric micelles composed of pluronic P123 and D-a-tocopheryl polyethylene glycol succinate. J Biomed Nanotechnol. 2011;7(3):358–365. doi: 10.1166/jbn.2011.1298. [DOI] [PubMed] [Google Scholar]
- 49.Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv Drug Deliv Rev. 2001;47(1):65–81. doi: 10.1016/s0169-409x(00)00122-8. [DOI] [PubMed] [Google Scholar]
- 50.Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Ther. 2006;112(3):630–648. doi: 10.1016/j.pharmthera.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 51.Djordjevic J, Barch M, Uhrich KE. Polymeric micelles based on amphiphilic scorpion-like macromolecules: novel carriers for water-insoluble drugs. Pharm Res. 2005;22(1):24–32. doi: 10.1007/s11095-004-9005-3. [DOI] [PubMed] [Google Scholar]
- 52.Potschka H, Loscher W. In vivo evidence for P-glycoprotein-mediated transport of phenytoin at the blood–brain barrier of rats. Epilepsia. 2001;42(10):1231–1240. doi: 10.1046/j.1528-1157.2001.01901.x. [DOI] [PubMed] [Google Scholar]
- 53.Clinckers R, Smolders I, Meurs A, Ebinger G, Michotte Y. Quantitative in vivo microdialysis study on the influence of multidrug transporters on the blood–brain barrier passage of oxcarbazepine: concomitant use of hippocampal monoamines as pharmacodynamic markers for the anticonvulsant activity. J Pharmacol Exp Ther. 2005;314(2):725–731. doi: 10.1124/jpet.105.085514. [DOI] [PubMed] [Google Scholar]
- 54.Kubek MJ, Domb AJ, Veronesi MC. Attenuation of kindled seizures by intranasal delivery of neuropeptide-loaded nanoparticles. Neurotherapeutics. 2009;6(2):359–371. doi: 10.1016/j.nurt.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bennewitz MF, Saltzman WM. Nanotechnology for delivery of drugs to the brain for epilepsy. Neurotherapeutics. 2009;6(2):323–336. doi: 10.1016/j.nurt.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boison D, Scheurer L, Tseng JL, Aebischer P, Mohler H. Seizure suppression in kindled rats by intraventricular grafting of an adenosine releasing synthetic polymer. Exp Neurol. 1999;160(1):164–174. doi: 10.1006/exnr.1999.7209. [DOI] [PubMed] [Google Scholar]
- 57.Bankstahl JP, Loscher W. Resistance to antiepileptic drugs and expression of P-glycoprotein in two rat models of status epilepticus. Epilepsy Res. 2008;82(1):70–85. doi: 10.1016/j.eplepsyres.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 58.Sisodiya SM, Lin WR, Harding BN, Squier MV, Thom M. Drug resistance in epilepsy: expression of drug resistance proteins in common causes of refractory epilepsy. Brain. 2002;125(Pt 1):22–31. doi: 10.1093/brain/awf002. [DOI] [PubMed] [Google Scholar]
- 59.Woodcock DM, Jefferson S, Linsenmeyer ME, et al. Reversal of the multidrug resistance phenotype with cremophor EL, a common vehicle for water-insoluble vitamins and drugs. Cancer Res. 1990;50(14):4199–4203. [PubMed] [Google Scholar]
- 60.Haque S, Md S, Alam MI, Sahni JK, Ali J, Baboota S. Nanostructure-based drug delivery systems for brain targeting. Drug Dev Ind Pharm. 2012;38(4):387–411. doi: 10.3109/03639045.2011.608191. [DOI] [PubMed] [Google Scholar]
- 61.Batrakova EV, Li S, Elmquist WF, Miller DW, Alakhov VY, Kabanov AV. Mechanism of sensitization of MDR cancer cells by Pluronic block copolymers: Selective energy depletion. Br J Cancer. 2001;85(12):1987–1997. doi: 10.1054/bjoc.2001.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kabanov AV, Batrakova EV, Miller DW. Pluronic block copolymers as modulators of drug efflux transporter activity in the blood–brain barrier. Adv Drug Deliv Rev. 2003;55(1):151–164. doi: 10.1016/s0169-409x(02)00176-x. [DOI] [PubMed] [Google Scholar]