

Abstract
A gain-of-function mutation in the myeloproliferative leukemia virus (MPL) gene, which encodes the thrombopoietin receptor, has been identified in patients with essential thrombocythemia and primary myelofibrosis, subgroups of classic myeloproliferative neoplasms (MPNs). The presence of MPL gene mutations is a critical diagnostic criterion for these diseases. Here, we developed a rapid, simple, and cost-effective method of detecting two major MPL mutations, MPLW515L/K, in a single PCR assay; we termed this method DARMS (dual amplification refractory mutation system)-PCR. DARMS-PCR is designed to produce three different PCR products corresponding to MPLW515L, MPLW515K, and all MPL alleles. The amplicons are later detected and quantified using a capillary sequencer to determine the relative frequencies of the mutant and wild-type alleles. Applying DARMS-PCR to human specimens, we successfully identified MPL mutations in MPN patients, with the exception of patients bearing mutant allele frequencies below the detection limit (5%) of this method. The MPL mutant allele frequencies determined using DARMS-PCR correlated strongly with the values determined using deep sequencing. Thus, we demonstrated the potential of DARMS-PCR to detect MPL mutations and determine the allele frequencies in a timely and cost-effective manner.
Introduction
Detecting clonal mutations and measuring mutant allele frequencies in malignant tissues are important tasks in clinical laboratories. In patients with essential thrombocytosis (ET) and primary myelofibrosis (PMF), acquired mutations in genes such as JAK2 tyrosine kinase, calreticulin (CALR), and myeloproliferative leukemia virus (MPL) have been described [1]–[3]. JAK2 and MPL mutations have been demonstrated to play causal roles in MPN development in animal models [4], [5]. Based on these findings, the identification of clonal mutations was deemed a diagnostic criterion for MPN by the World Health Organization (WHO) in 2008.
The major JAK2 mutation found in MPN patients is JAK2V617F (G1849T); therefore, epidemiological studies of MPN examining JAK2V617F have been greatly facilitated by PCR-based assays, such as ARMS (amplification refractory mutation system)-PCR [6], ABC (alternately binding probe competitive)-PCR [7], and allele specific (AS)-qPCR [8]–[11]. In contrast, a gain-of-function mutation in MPL, which encodes the thrombopoietin (TPO) receptor, has been identified in multiple sites within exon 10, making its detection with a simple PCR-based assay difficult. MPL mutations in MPN patients have traditionally been characterized using melting curve assays combined with Sanger sequencing [12], [13], quantitative-PCR [14], [15], or, more recently, deep sequencing [16], [17]. However, these assays generally require expensive machines and reagents and greater workloads. Partly because of the absence of a practical method for detecting MPL mutations, there have been many more epidemiological studies of MPN patients carrying the JAK2 V617F mutation compared with MPL mutations.
Thus far, 10 types of MPL mutations have been identified in or around the transmembrane domain of MPL [17]–[19], and most of these result in the activation of MPL, even in the absence of TPO, and the subsequent activation of downstream targets [5], [20], [21]. These mutations include G1544T and TG1543_1544AA, which result in the substitution of tryptophan with leucine (W515L) or lysine (W515K), respectively, and are found in nearly three-quarters of patients bearing MPL mutations [17]. MPLW515L/K has been shown to promote tumorigenesis in vivo [5], and a higher mutant allele frequency is associated with progression to myelofibrosis [17]. Thus, detecting MPLW515L/K mutations and determining the relative ratios of W515L or W515K to the wild-type allele is critical for improving our understanding of MPN pathogenesis. In this work, we developed a cost-effective and practical method for simultaneously detecting both the MPLW515L and W515K mutations with high sensitivity and for determining the MPLW515L/K allele frequencies using a single PCR assay.
Materials and Methods
Standard DNA preparation
A 1,331-base-pair (bp) fragment containing the human MPL sequence (43348414 to 43349744 of NC_000001.11) was PCR-amplified from human genomic DNA (#G3048 Promega, Dane Country, USA) with a set of primers (forward primer: AAATCTGGCATCCTCTGCAGCATGAGTATTATTTG; reverse primer: CAAGAGGTTCTGTTTCAGTGAGTCAGGTCGTGT). The PCR product was subcloned into the pSP73 cloning vector (Promega) to generate pSP73/MPL-WT. An MPLW515L (G1544T) or W515K (TG1543_1544AA) mutation was introduced into pSP73/MPL-WT using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, USA) according to the manufacturer's instructions. The sequences of all of the plasmids were confirmed via Sanger sequencing. A 3,339-bp plasmid carrying the wild-type MPL, W515L, or W515K DNA sequence was linearized by ScaI restriction enzyme digestion, subjected to agarose gel electrophoresis, and purified with the QIAquick gel extraction kit (Qiagen, Hilden, Germany). The purified DNA was mixed at the indicated ratio (see Results and Discussion) to create a standard template for dual amplification refractory mutation system (DARMS)-PCR.
DARMS-PCR
MPL sequences, including TGG1543_1545 (encoding wild-type W515), were amplified via ARMS-PCR [6] with the following modifications. The reaction mixture (20 µL) was composed of 1X AmpliTaq Gold Master Mix (Life Technologies), 120 nM of two outer primers (F_outer and R_outer), 1 µM of F_inner primer, 300 nM of R_inner primer, and template DNA (genomic DNA ranging from 19 to 253 ng). Note that with DARMS-PCR, the frequency of a given MPL mutant allele is determined by comparing the relative signal peak values of that mutant and all MPL alleles; thus, in theory, the template copy number can be variable. Indeed, we confirmed that using the abovementioned range of standard DNA template, the resulting mutant frequencies did not differ significantly from each other (data not shown). The primer sequences and positions are presented in Figure 1A and B. The PCR conditions were as follows: an initial denaturation at 95°C for 10 min; 28 cycles of denaturation at 95°C for 30 sec, annealing at 62°C for 30 sec, and extension at 72°C for 30 sec; and a final extension at 72°C for 7 min.
Figure 1. Detection of MPLW515L/K mutations using DARMS-PCR.

(A) The primers used in the DARMS-PCR assay. The two inner primers harbored sequences (underlined) that matched MPLW515L or W515K, but not the wild-type allele. Other mismatches (capital letters) were introduced into the inner primers to reduce the annealing of the mutant-specific primers to the wild-type sequence. The reverse primers were labeled with FAM (5-carboxyfluorescein hydrate) at the 5′ terminus. (B) A schematic representation of DARMS-PCR products. The two outer primers were designed to generate a 169-base-pair (bp) PCR product from all MPL alleles. The F_inner and R_inner primers annealed specifically to the MPLW515L and W515K alleles, respectively; in combination with the outer primers, they generated 84- and 128-bp PCR products, respectively. From a mutant allele, both 169-bp and 84- or 128-bp fragments were amplified, while, only the 169-bp fragment was generated from the wild-type allele. (C) Demonstration of DARMS-PCR. A capillary electropherogram of DARMS-PCR products showing three peaks derived from wild-type MPL, W515L, and W515K. This result was obtained when PCR was performed with a standard DNA mixture containing equal ratios of MPL wild-type, W515L, and W515K alleles with a total copy number of 105. The horizontal axis represents the fragment length, and the vertical axis represents the fluorescence intensity.
Detection of MPL mutant alleles and determination of allele frequencies
DARMS-PCR products were analyzed via capillary electrophoresis. A capillary electrophoresis mixture was created that contained 1.0 µL of six-fold diluted DARMS-PCR products, 0.2 µL of GeneScan 500 LIZ Size Standard (Life Technologies, Carlsbad, USA), and 15.8 µL of Hi-Di formaldehyde (Life Technologies). A 17-µL aliquot of the mixture was heated at 86°C for 3 min, cooled on ice, and loaded onto an ABI 3130xl Genetic Analyzer (Life Technologies). The sizes of the PCR products were determined based on the size standard. The height of each fluorescence peak corresponding to wild-type MPL, W515K, or W515L was measured. The fluorescence peak values of a standard DNA mixture containing a known wild-type-to-mutant MPL ratio were used to generate the following formulas for calculating the MPL mutant allele frequencies (see Results and Discussion).
Human specimens
Genomic DNA was purified from peripheral blood collected from 20 patients previously diagnosed with MPN at Juntendo University Hospital (Hongo, Tokyo, Japan) or other participating institutions [22]. This study was conducted in accordance with the Declaration of Helsinki and was approved by the ethics committee of Juntendo University School of Medicine (IRB#21076). Written informed consent for the use of samples and clinical records was obtained from all the patients prior to sample collection. Genomic DNA was purified from 200 µL of each blood sample using the QIAamp DNA Mini Kit (Qiagen). The genomic DNA concentrations were measured with the NanoDrop Lite spectrophotometer (Thermo Scientific, Waltham, USA), and the DNA was stored at −80°C until use.
Targeted deep sequencing
A 169-bp fragment containing the human MPL sequence (43349278 to 43349446 of NC_000001.11) was PCR-amplified from genomic or standard plasmid DNA using forward (5′-AGTAGGGGCTGGCTGGAT-3′) and reverse (5′-GGTCACAGAGCGAACCAAGA-3′) primers (see above). The PCR products were subjected to agarose gel electrophoresis and were purified with the QIAquick gel extraction kit (Qiagen). Sample libraries were prepared with the TruSeq DNA LT Sample Prep Kit (Illumina, San Diego, USA) according to the manufacturer's instructions, and the libraries were deep sequenced with a MiSeq benchtop sequencer (Illumina). The data analysis was performed using CLC Genomics Workbench software version 6.5 (CLC Bio, Aarhus, Denmark) with a minimum coverage of 150,000 and a minimum variant frequency of 1%. The mutant allele frequency was calculated by dividing the number of mutant sequence reads by the sum of the mutant and wild-type reads.
Results and Discussion
Establishment of DARMS-PCR
To develop a single PCR assay for detecting the two major mutant alleles, MPLW515L (G1544T) and W515K (TG1543_1544AA), we modified an ARMS-PCR method [6] by adding a total of four primers to each reaction; we renamed the method DARMS-PCR. Two mutant-specific primers (F_inner and R_inner) and two common primers (F_outer and R_outer) were designed (Figure 1A and B). MPLG1544T, TG1543_1544AA, and a control fragment for all MPL alleles generated unique PCR products that were 128, 84, and 169 bp in size, respectively (Figure 1B). The two reverse primers (R_inner and R_outer) were labeled with the fluorescent dye FAM (5-carboxyfluorescein hydrate), which allowed us to determine the lengths and quantity of the PCR products using a capillary sequencer. According to the design of the assay, when a template containing a mixture of purified DNA fragments representing MPLG1544T, TG1543_1544AA, and wild-type alleles was amplified under optimized conditions (see Materials and Methods), three PCR products corresponding to the different alleles were detected (Figure 1C). All three PCR products produced dual peaks, presumably because of an indefinite adenine addition at the end of each amplicon by Taq polymerase (Figure 1C). The sum of the peak height values from each dual peak was used in the following analyses.
Determination of MPL mutant allele frequency using DARMS-PCR
To examine whether the MPL mutant allele frequency could be quantitatively determined using DARMS-PCR, we created a series of standard DNA templates containing different amounts of wild-type and mutant alleles. Templates containing 10, 20, 40, 60, 80, 90, or 100% mutant (W515L or W515K) allele DNA were used for DARMS-PCR, and the fluorescent intensities of the products were measured with the capillary sequencer (see Materials and Methods). As shown in Figure 2A and B, the fluorescence intensity of the mutant alleles over the intensity of all MPL alleles demonstrated a nearly linear correlation, indicating the accuracy of DARMS-PCR for quantitatively determining MPL mutant allele frequencies. Based on this result, we generated the following formulas to calculate the MPL mutant allele frequencies: MPLW515L allele frequency (%) = 270×[fluorescence value (arbitrary unit) for MPLW515L PCR product]/[fluorescence value (arbitrary unit) for all MPL allele PCR products]; and MPLW515K allele frequency (%) = 200×[fluorescence value (arbitrary unit) for MPLW515K PCR product]/[fluorescence value (arbitrary unit) for all MPL allele PCR products].
Figure 2. Demonstration of DARMS-PCR to determine MPL mutant allele frequencies.
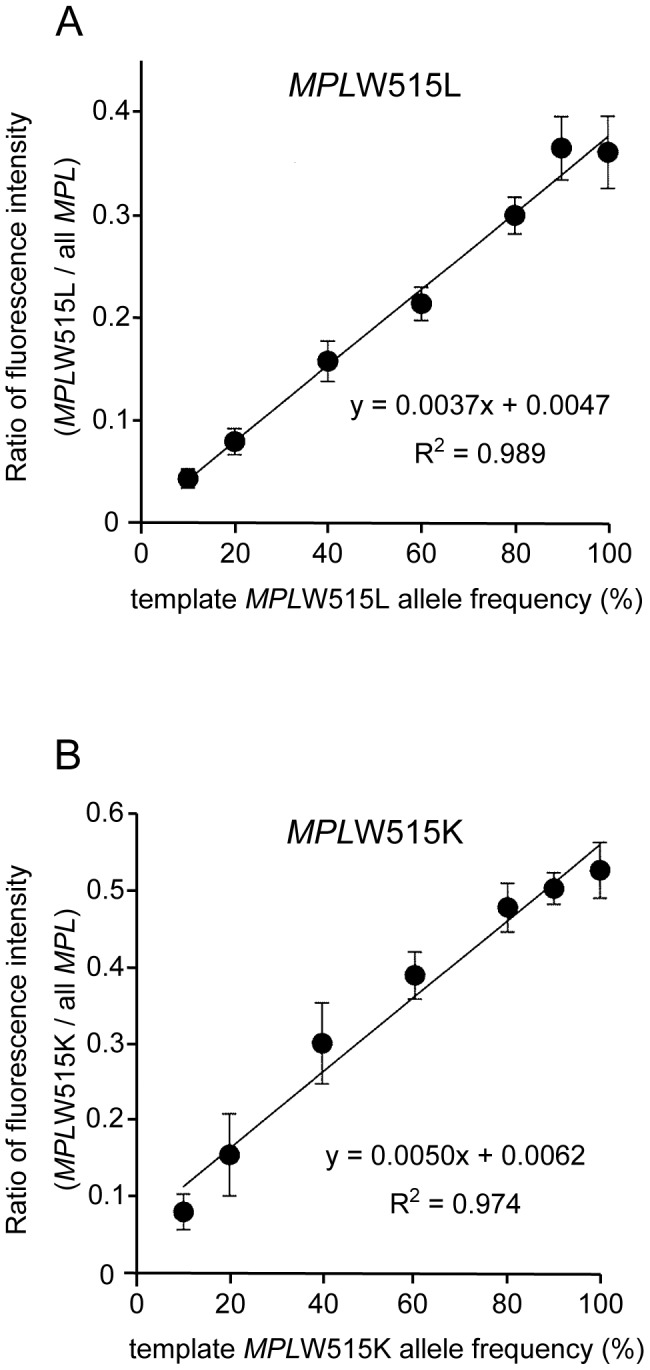
Standard curves for the quantification of MPLW515L (A) and W515K (B) allele frequencies. A series of templates containing different amounts of mutant allele DNA were used for DARMS-PCR. The fluorescence peak value of the mutant allele over the value for all MPL alleles was calculated for each template. The mean values and errors obtained from three independent experiments are shown.
Detection limit of MPL mutations using DARMS-PCR
We then examined the lower detection limit of DARMS-PCR under the conditions used for determining the mutant allele frequencies. We performed DARMS-PCR with DNA containing MPL mutant frequencies of 0, 1, 5, and 10%, and we obtained peak heights of 0, 55, 165, and 221 for MPLW515L and 0, 33, 199, and 731 for W515K (Figure 3). We then determined the average peak heights for nonspecific PCR products, which were defined as noise. The signal-to-noise (S/N) ratio was calculated as the fluorescence value for the peak of the mutant allele divided by the value for noise (Figure 3). When the allele frequency was 5%, the S/N ratios were 1.75 and 1.77 for MPLW515L and W515K, respectively. Thus, we concluded that the detection limit of DARMS-PCR was greater than 5% for both the MPLW515L and 515K mutations.
Figure 3. Lower limit of MPL mutation detection by DARMS-PCR.

Capillary electropherograms for DARMS-PCR assays with low levels of MPLW515L or W515K mutant alleles are presented. The average fluorescence intensities for nonspecific PCR products were defined as noise. S/N ratios, which were calculated as the fluorescence peak value for a mutant PCR product over the value for noise, are shown. An arrow head in the W515L 0% panel indicates a false PCR product (see Results and Discussion).
Validation of the DARMS-PCR assay results using deep sequencing
To examine the accuracy of DARMS-PCR for evaluating human specimens, we analyzed genomic DNA samples from 20 MPN patients whose MPL mutation status and allele frequencies had been determined using deep sequencing (see Materials and Methods). The formulas defined in Figure 2 were used to calculate the MPL mutant allele frequencies from the fluorescence peak values, which were determined using DARMS-PCR (Figure 4, Table 1). The results indicated that, using DARMS-PCR, 6 MPLW515L-positive and 3 W515K-positive MPN patients were successfully identified. Patient #120 was not identified due to having an MPL mutant frequency of 1.0% (according to deep sequencing), which is below the DARMS-PCR detection limit (5%) (Table 1). Patient #127, who was initially identified as positive for MPLW515K using DARMS-PCR, was later found to be positive for both MPLW515L and W515K using deep sequencing (Table 1). The mutant allele frequency of MPLW515L in this patient was 2.5%, which was below the detection limit of DARMS-PCR. The remaining 10 patients, who were identified as wild-type for MPL using deep sequencing, had MPL allele frequencies below the positivity threshold of DARMS-PCR and thus were determined to be negative. Overall, although DARMS-PCR had a limitation of detection of mutant allele frequencies lower than 5%, all the patient specimens identified as having MPL mutations using DARMS-PCR were confirmed using deep sequencing. There was a strong correlation between the MPL mutant frequencies determined using DARMS-PCR versus deep sequencing, with errors ranging from −28.0% to 10.4% (Figure 5, Table 1). Further research is required to evaluate the quantitative nature of DARMS-PCR. Nevertheless, these data imply that DARMS-PCR can be used as a diagnostic tool to identify MPL mutations in MPN patients.
Figure 4. Detection of MPL mutations in human specimens.
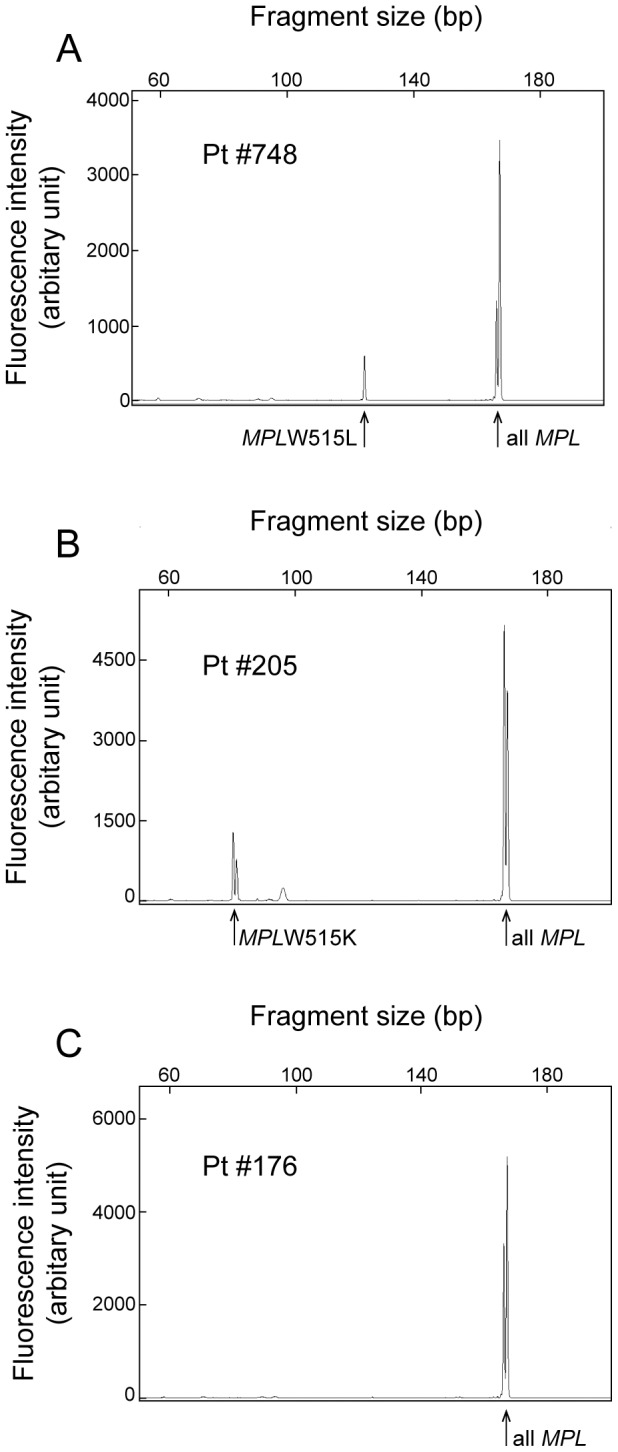
Representative capillary electropherograms from the DARMS-PCR assay with human specimens are presented. Patients #748 (A), #205 (B), and #176 (C) harbored MPLW515L, W515K, and wild-type alleles, respectively. The calculated allele frequencies are presented in Table 1.
Table 1. MPL allele frequencies determined using DARMS-PCR and deep sequencing.
| MPLW515L allele frequency (%)* | MPLW515K allele frequency (%)* | ||||
| MPL allele | Patientno. | DARMS-PCR | Deep sequencing | DARMS-PCR | Deep sequencing |
| MPLW515L | 146 | 77.8 | 79.0 | N.D. | 0.0 |
| 177 | 84.1 | 88.4 | 1.8 | 0.0 | |
| 526 | 24.7 | 17.1 | N.D. | 0.2 | |
| 748 | 33.4 | 43.7 | N.D. | 0.0 | |
| 782 | 19.4 | 47.4 | 0.9 | 0.0 | |
| 830 | 9.7 | 15.4 | 0.4 | 0.0 | |
| MPLW515L/K | 127 | N.D. | 2.5 | 36.3 | 50.7 |
| MPLW515K | 120 | N.D. | 0.1 | N.D. | 1.0 |
| 193 | N.D. | 0.1 | 22.2 | 33.6 | |
| 205 | N.D. | 0.1 | 45.4 | 35.0 | |
| Wild-type MPL | 6 | N.D. | 0.0 | N.D. | 0.0 |
| 50 | N.D. | 0.0 | N.D. | 0.1 | |
| 158 | 1.8 | 0.2 | N.D. | 0.0 | |
| 160 | N.D. | 0.0 | 0.5 | 0.0 | |
| 176 | N.D. | 0.6 | N.D. | 0.0 | |
| 185 | N.D. | 0.3 | 0.8 | 0.0 | |
| 199 | N.D. | 0.0 | 0.6 | 0.5 | |
| 607 | 0.7 | 0.0 | N.D. | 0.0 | |
| 688 | 1.0 | 0.1 | N.D. | 0.0 | |
| 710 | N.D. | 0.2 | 0.4 | 0.0 | |
*Allele frequencies below 1%, as determined using deep sequencing, were defined as negative for the assessed mutation. Note that allele frequencies below 5%, as determined using DARMS-PCR, were also defined as negative based on the results in Figure 3. N.D.: the corresponding peak was not detected.
Figure 5. Comparison of MPL mutant frequencies determined using DARMS-PCR and deep sequencing.
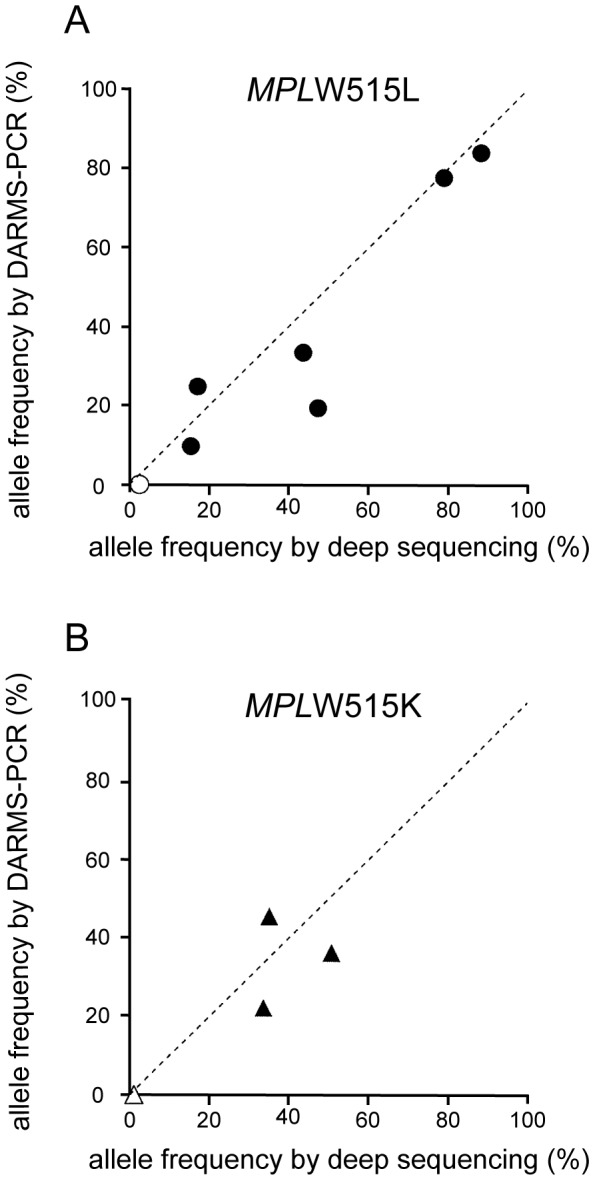
The horizontal axis represents the frequencies of the MPLW515L (A) or W515K (B) mutant alleles determined using deep sequencing, and the vertical axis represents the mutant allele frequencies determined using DARMS-PCR. The specific values are listed in Table 1. Filled symbols represent alleles identified using both DARMS-PCR and deep sequencing. Open symbols represent alleles identified only using deep sequencing.
As observed with the standard template without mutant alleles (shown by the arrowhead at the W515L 0% panel in Figure 3) and with patient specimens that were defined as negative for MPLW515K or W515L with deep sequencing (Table 1), the DARMS-PCR generates false-positive PCR products and thus the subsequent appearance of low but clear allele frequencies. These false-positives presumably originate from faulty priming by inner primers that are capable of annealing to both mutant andwild-type alleles but are designed not to produce PCR products from the wild-type allele with introduced mutations (see Figure 1). Further optimization of the PCR program and primer sequences and positions is required to make the assay more sensitive and accurate. Although the deep-sequencer is apparently more accurate in terms of detecting the MPLW515K/L mutation, we observed potential error calls with less than 1% mutation frequency (see Table 1). This is likely the result of an erratic amplification during the sequencing sample preparation and/or a casual incident during the deep-sequencing. In addition, the current version of DARMS-PCR assesses only two major MPL mutations: MPLW515K/L, which account for approximately 75% of all MPL mutations [17]; thus, 25% of patients bearing rare MPL mutations, such as MPLW515S and MPLS505N [17], cannot be detected with this method. The incorporation of more primers into current DARMS-PCR and/or the establishment of more DARMS-PCR protocols for detecting other rare MPL mutations is required for more comprehensive assessment of MPL mutations.
In summary, we have developed and validated DARMS-PCR, a cost-effective, rapid, and accurate method for identifying two major MPL mutations with a single PCR assay. Although deep sequencing can be the most accurate method for detecting mutant alleles and measuring allele frequencies, it is considerably more expensive and time-consuming than DARMS-PCR. Compared with the specialized thermal cycler equipped with a fluorescence detection system that is needed to perform a melting curve analysis [12], [13], DARMS-PCR requires only a general thermal cycler and a capillary sequencer (or even an agarose gel electrophoresis apparatus). DARMS-PCR is unique in its ability to simultaneously detect two mutations in one single-tube PCR assay; moreover, this method can be applied to detect other mutations as well. By employing this assay, the identification of approximately 75% of MPN patients with MPL mutations should be possible (see Introduction); thus, the use of DARMS-PCR will enhance screening for patients with MPLW515L/K mutations and will deepen our understanding of the pathogenesis of MPN.
Acknowledgments
We thank Kyoko Kubo, Megumi Hasegawa, and Kazuko Kawamura for secretarial assistance, and we thank the other members of the Department of Hematology for encouraging this study. We also acknowledge the Laboratory of Molecular and Biochemical Research, Research Support Center, Juntendo University Graduate School of Medicine for technical help with the ABI 3130xl sequencer.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.
Funding Statement
This work was funded by Japan Society for the promotion of science (JSPS) (http://www.jsps.go.jp/english/e-grants/) KAKENHI grant #25860416 (SM). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Campbell PJ, Green AR (2006) The myeloproliferative disorders. N Engl J Med 355: 2452–2466. [DOI] [PubMed] [Google Scholar]
- 2. Milosevic JD, Kralovics R (2013) Genetic and epigenetic alterations of myeloproliferative disorders. Int J Hematol 97: 183–197. [DOI] [PubMed] [Google Scholar]
- 3. Guglielmelli P, Nangalia J, Green AR, Vannucchi AM (2014) CALR mutations in myeloproliferative neoplasms: Hidden behind the reticulum. Am J Hematol [DOI] [PubMed] [Google Scholar]
- 4. Skoda RC (2010) JAK2 impairs stem cell function? Blood 116: 1392–1393. [DOI] [PubMed] [Google Scholar]
- 5. Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, et al. (2006) MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 3: e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vannucchi AM, Pancrazzi A, Bogani C, Antonioli E, Guglielmelli P (2006) A quantitative assay for JAK2(V617F) mutation in myeloproliferative disorders by ARMS-PCR and capillary electrophoresis. Leukemia 20: 1055–1060. [DOI] [PubMed] [Google Scholar]
- 7. Morishita S, Komatsu N, Kirito K, Koda AH, Sekiguchi Y, et al. (2011) Alternately binding probe competitive PCR as a simple, cost-effective, and accurate quantification method for JAK2V617F allele burden in myeloproliferative neoplasms. Leuk Res 35: 1632–1636. [DOI] [PubMed] [Google Scholar]
- 8. Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, et al. (2005) Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 366: 1945–1953. [DOI] [PubMed] [Google Scholar]
- 9. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, et al. (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365: 1054–1061. [DOI] [PubMed] [Google Scholar]
- 10. Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, et al. (2005) Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 106: 2162–2168. [DOI] [PubMed] [Google Scholar]
- 11. Antonioli E, Guglielmelli P, Pancrazzi A, Bogani C, Verrucci M, et al. (2005) Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 19: 1847–1849. [DOI] [PubMed] [Google Scholar]
- 12. Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, et al. (2006) MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 108: 3472–3476. [DOI] [PubMed] [Google Scholar]
- 13. Schnittger S, Bacher U, Haferlach C, Beelen D, Bojko P, et al. (2009) Characterization of 35 new cases with four different MPLW515 mutations and essential thrombocytosis or primary myelofibrosis. Haematologica 94: 141–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pancrazzi A, Guglielmelli P, Ponziani V, Bergamaschi G, Bosi A, et al. (2008) A sensitive detection method for MPLW515L or MPLW515K mutation in chronic myeloproliferative disorders with locked nucleic acid-modified probes and real-time polymerase chain reaction. J Mol Diagn 10: 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ruan GR, Jiang B, Li LD, Niu JH, Li JL, et al. (2010) MPL W515L/K mutations in 343 Chinese adults with JAK2V617F mutation-negative chronic myeloproliferative disorders detected by a newly developed RQ-PCR based on TaqMan MGB probes. Hematol Oncol 28: 33–39. [DOI] [PubMed] [Google Scholar]
- 16. Pietra D, Brisci A, Rumi E, Boggi S, Elena C, et al. (2011) Deep sequencing reveals double mutations in cis of MPL exon 10 in myeloproliferative neoplasms. Haematologica 96: 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rumi E, Pietra D, Guglielmelli P, Bordoni R, Casetti I, et al. (2013) Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 121: 4388–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tefferi A (2010) Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 24: 1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Teofili L, Giona F, Torti L, Cenci T, Ricerca BM, et al. (2010) Hereditary thrombocytosis caused by MPLSer505Asn is associated with a high thrombotic risk, splenomegaly and progression to bone marrow fibrosis. Haematologica 95: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chaligne R, Tonetti C, Besancenot R, Roy L, Marty C, et al. (2008) New mutations of MPL in primitive myelofibrosis: only the MPL W515 mutations promote a G1/S-phase transition. Leukemia 22: 1557–1566. [DOI] [PubMed] [Google Scholar]
- 21. Lee TS, Kantarjian H, Ma W, Yeh CH, Giles F, et al. (2011) Effects of clinically relevant MPL mutations in the transmembrane domain revealed at the atomic level through computational modeling. PLoS One 6: e23396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Edahiro Y, Morishita S, Takahashi K, Hironaka Y, Yahata Y, et al. (2014) JAK2V617F mutation status and allele burden in classical Ph-negative myeloproliferative neoplasms in Japan. Int J Hematol 99: 625–634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.