

Background: Secondary structure modeling predicts 5′-editing of mitochondrial tRNAs in amoebozoans Dictyostelium discoideum and Polysphondylium pallidum.
Results: Experimental verification reveals unexpected instances of editing and species-specific variations.
Conclusion: Mitochondrial tRNA 5′-editing is a complex process in which 3′–5′ RNA polymerases play a key role.
Significance: Further biochemical characterization of mitochondrial tRNA 5′-editing reactions and enzymes is indicated.
Keywords: Dictyostelium, Enzyme, Mitochondria, RNA Editing, Transfer RNA (tRNA), 3′–5′ Polymerase, tRNA Repair
Abstract
Mitochondrial tRNA (mt-tRNA) 5′-editing was first described more than 20 years ago; however, the first candidates for 5′-editing enzymes were only recently identified in a eukaryotic microbe (protist), the slime mold Dictyostelium discoideum. In this organism, eight of 18 mt-tRNAs are predicted to be edited based on the presence of genomically encoded mismatched nucleotides in their aminoacyl-acceptor stem sequences. Here, we demonstrate that mt-tRNA 5′-editing occurs at all predicted sites in D. discoideum as evidenced by changes in the sequences of isolated mt-tRNAs compared with the expected sequences encoded by the mitochondrial genome. We also identify two previously unpredicted editing events in which G-U base pairs are edited in the absence of any other genomically encoded mismatches. A comparison of 5′-editing in D. discoideum with 5′-editing in another slime mold, Polysphondylium pallidum, suggests organism-specific idiosyncrasies in the treatment of U-G/G-U pairs. In vitro activities of putative D. discoideum editing enzymes are consistent with the observed editing reactions and suggest an overall lack of tRNA substrate specificity exhibited by the repair component of the editing enzyme. Although the presence of terminal mismatches in mt-tRNA sequences is highly predictive of the occurrence of mt-tRNA 5′-editing, the variability in treatment of U-G/G-U base pairs observed here indicates that direct experimental evidence of 5′-editing must be obtained to understand the complete spectrum of mt-tRNA editing events in any species.
Introduction
Transfer RNA species undergo a variety of post-transcriptional processing events to produce the mature tRNA molecules that function in translation (1). These processing steps may include tRNA editing reactions, which result in changes in the tRNA sequence from the sequence encoded by the tRNA gene. Multiple types of editing reactions have been observed to occur at locations throughout a tRNA, including at the 5′ and 3′ ends and within the tRNA body (2–11). These reactions include base deamination wherein C or A nucleotides are edited to become U or I, respectively, and insertion/deletion editing wherein unencoded nucleotides are added to specific locations or encoded nucleotides are removed and replaced. 5′-tRNA editing, first described in the mitochondria of Acanthamoeba castellanii, is an example of the latter type (12). In 5′-editing, pre-tRNA transcripts contain one to three mismatched nucleotides at the 5′-end of the aminoacyl-acceptor stem, and these mismatches are predicted to result in suboptimal tRNA function (see Fig. 1). The mismatched nucleotides are absent in the sequenced mature mt-tRNA,2 which is suggestive of post-transcriptional editing (12–14). The editing reaction minimally comprises a two-step process in which first the mismatched nucleotides are removed from the pre-tRNA transcript and second the correct Watson-Crick base-pairing nucleotides are added back to the 5′-truncated tRNA (Fig. 1). This 5′-end repair step apparently requires a 3′–5′ RNA polymerase capable of using the 3′-aminoacyl-acceptor stem nucleotides as a template for 5′-nucleotide addition (15–17). The requirement for this apparently non-standard enzyme chemistry in the form of a polymerase that acts in the 3′–5′ direction, opposite to all known DNA/RNA polymerases, impeded identification of the 5′-editing enzyme(s) for nearly two decades.
FIGURE 1.
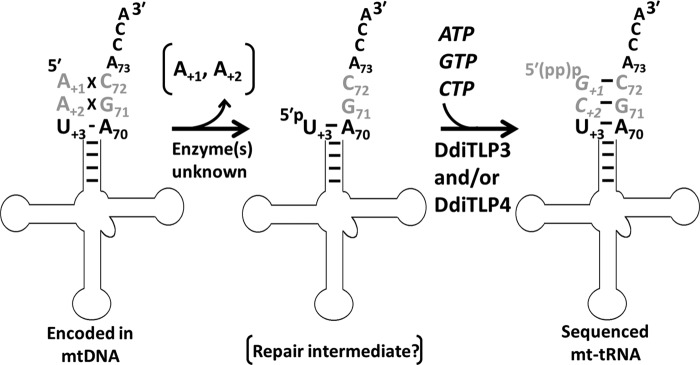
Schematic of mt-tRNA editing in D. discoideum. 5′-Editing of mt-tRNA occurs in at least two steps in which 5′-mismatched nucleotides are removed from the 5′-end of the mt-tRNA, producing a presumed 5′-truncated mt-tRNA intermediate. Biochemical data suggest that the 5′-truncated mt-tRNA is then acted on by one of the TLP enzymes encoded by the D. discoideum nuclear genome, thus repairing the 5′-truncated end via 3′–5′ polymerase activity using the 3′-side of the aminoacyl-acceptor stem as the template for polymerase activity. The presence of an intact 3′-CCA end on the tRNA prior to editing is shown for clarity (and is supported by the sequences obtained in this work), but the precise order of 3′-processing and 5′-editing events has not been determined.
Recently, however, the first two candidate enzymes for participation in mitochondrial 5′-editing, members of the tRNAHis guanylyltransferase (Thg1) superfamily, were identified in the eukaryotic slime mold Dictyostelium discoideum (18, 19). Thg1-like proteins (TLPs) from Bacteria and Archaea exhibit a proficient ability to use the template-dependent 3′–5′ polymerase function to repair 5′-truncated tRNA species; the latter species mirror the truncated tRNA repair intermediates inferred to occur during mt-tRNA 5′-editing (20, 21). Using in vitro enzyme assays, two different D. discoideum TLPs (DdiTLP3 and DdiTLP4) showed equivalent abilities to repair each of two different tRNAs that were predicted to undergo tRNA 5′-editing in D. discoideum mitochondria (15). Whether these two enzymes participate collaboratively or separately in tRNA 5′-editing in vivo remains unknown.
Experimental evidence confirming the existence of tRNA 5′-editing (by comparison of actual mt-tRNA sequences with the encoded mt-tDNA genes and identification of changes in the 5′-nucleotides) has so far only been obtained for a small number of eukaryotes, including A. castellanii, Spizellomyces punctatus, Physarum polycephalum, Monoblepharella sp., and Harpochytrium sp., but not in D. discoideum (6, 12–14, 22, 23). In D. discoideum, the existence of 5′-editing has been inferred based on the presence of mismatched nucleotides in the appropriate positions of the aminoacyl-acceptor stem in the encoded mt-tDNA sequences, but 5′-editing per se has not yet been demonstrated experimentally in this organism. Thus, to fully elucidate the composition and function of the enzymes that carry out the 5′-editing reaction, it is necessary to know the editing status of the mitochondrial tRNA species in this amoebozoan.
In this work, we carried out a comprehensive characterization of the mitochondrial 5′-editing system in D. discoideum by sequencing all eight mt-tRNAs initially predicted to undergo editing in vivo. Additionally, we sequenced several mt-tRNAs that are not predicted to be edited in D. discoideum, revealing the presence of two previously unexpected editing events whereby G-U base pairs are efficiently edited to Watson-Crick base-paired nucleotides. This first example of editing of G-U base pairs in the absence of bona fide non-Watson-Crick mismatches in the aminoacyl-acceptor stem prompted us to undertake a closer investigation of editing activity with respect to G-U base pairs. For this, we compared 5′-editing in D. discoideum with editing in another slime mold, Polysphondylium pallidum, where this process has also been predicted but not experimentally verified. In doing so, we identified different patterns of G-U base pair editing that suggest mechanistic differences between 5′-editing reactions in divergent organisms. In vitro characterization of repair activities catalyzed by DdiTLP3 and DdiTLP4 confirms that both enzymes are able to repair mt-tRNAs that have been shown by direct sequencing to undergo 5′-editing (both the previously proposed as well as newly revealed edited tRNAs) as well as a control tRNA that is not known to be edited in vivo. The general lack of tRNA substrate specificity exhibited by either enzyme during repair of 5′-truncated tRNAs suggests that tRNA substrate selection during 5′-editing is not likely to be exerted at the 3′–5′ polymerase step of the reaction.
EXPERIMENTAL PROCEDURES
Sequencing of D. discoideum mt-tRNAs
Total RNA was isolated from D. discoideum cells (strain AX-2) using TRI Reagent (Sigma) according to the manufacturer's instructions. Purified RNA (10 μg; diluted to 1 μg/μl in water) was heated to 95 °C for 5 min, chilled on ice for 10 min, and then circularized using T4 RNA ligase (USB). Circularization reactions (20 μl) contained RNA, 80 μm ATP, 15 mm MgCl2, 3.3 mm dithiothreitol (DTT), 10 μg/ml bovine serum albumin (BSA), 10% dimethyl sulfoxide (DMSO), and 15 units of T4 RNA ligase in 50 mm HEPES, pH 7.5. Reaction mixtures were incubated at 37 °C overnight and then purified by phenol:chloroform extraction and ethanol precipitation.
The circularized RNA was resuspended in 10 μl of double distilled H2O, and 2 μl was used as template for reverse transcription by Superscript III reverse transcriptase (Invitrogen). RNA (∼2 μg) and an appropriate cDNA-RT oligonucleotide (Table 1) for each tRNA (0.2 μm) were annealed in a 10-μl reaction by heating to 95 °C for 5 min followed by slow cooling to room temperature for 30 min; extension reactions were assembled using the entire volume from the annealing reaction and contained 1× first strand buffer (Invitrogen), 5 mm DTT, a 0.5 mm concentration of each dNTP, and 200 units of Superscript III reverse transcriptase in a final volume of 20 μl. Reactions were incubated at 50 °C for 1 h, and then enzyme was heat-inactivated by incubation at 75 °C for 10 min. A portion of each reaction mixture (3 μl) was used as the template for PCR using reverse transcriptase and 3′-oligonucleotides (Table 1) to amplify the ligated 5′- and 3′-end junction with iProof polymerase (Bio-Rad) in 50-μl reactions. PCRs were carried out with 2-min incubation at 98 °C followed by five cycles as follows: 30 s at 98 °C, 30 s at 55 °C, and 20 s at 72 °C and then 25 cycles of 10 s at 98 °C, 30 s at 65 °C, and 20 s at 72 °C. Cycling was followed by 10 min at 72 °C. RT-PCR products of the correct size were extracted from 10% acrylamide, 4 m urea gels by soaking the crushed gel slice in elution buffer containing 0.5 m ammonium acetate, 0.1% SDS, and 5 mm EDTA followed by phenol:chloroform extraction and ethanol precipitation of the extracted DNA.
TABLE 1.
Primers for mt-tRNA amplification and sequencing
| mt-tRNA | 5′ (RT) primer | 3′ primer |
|---|---|---|
| D. discoideum | ||
| AlaUGC | CAACACCCATATTATACC | TATGGGTTCGAGGCCCATTG |
| CysGCA | GCAGAGCACGGTATTAACC | CAGTTCAAGCCTGGTTATC |
| GlnUUG | CAAAAACCATCGCCTTACC | GCGTAGGTTCGAGTCCTAC |
| GluUUC | GAAAAGCCAATGTCCTAACC | CATGAGTTCGATTCTCATAAG |
| IleCAU | GAGTGTGGTGTTCTAACCTAT | ATCGGTTCGAGTCCGATC |
| IleGAU(1) and -(2) | CAAACGAATACTCTAACCAAC | GTCAGAGGTTCGAATCCTC |
| LeuUAA | CTATCGCGTCTACCATTTTC | GTATCAGTTCAAGTCTGATAG |
| LeuUAG | CTAAGACCCTCGCGTCTAC | GTGGGAGTTCAAGTCTCCC |
| MetCAU | GCATGAACCCAATGAACTAAC | GGTGTAGGTTCGAGTCCTAC |
| PheGAA | CAGTCTTTCACTCTAACCAAC | CGGTGGTTCAAATCCACTC |
| ProUGG | CCCAAAACTAACACATTAGCC | GATTATAGGTTCGACTCCTATC |
| TrpCCA | GGAGACATTTGTTCTAACTATTTG | AGATATCAGTGCAAGTCTGG |
| TyrGUA | CAGTCTTCCGCTTTAACCAC | GACGCATAGGTTCGAATCC |
| P. pallidum | ||
| AlaUGC | GCAACACCTCTATTATACCAAT | TAAGGGTTCAAGTCCCTTC |
| AsnGUU | AGCGAATTGCTCTACCTA | GTTAACGAAAAGTTGTTGGTTC |
| GluUUC | GCTAACGTCCTGAACCG | CATGGGTTCAAATCCCATAA |
| IleGAU | AACGATCACTCTACCACTG | CAGTGGTTCAAATCCACTTT |
| LeuUAA | AGTCTATCGCGTCTACCTA | ATCAGTTCAAGTCTGATAG |
| PheGAA | AGTCTTCACTCTACCTACTG | CGGTGGTTCAAATCCACTC |
The entire volume of resulting purified DNA was 5′-phosphorylated in reactions with T4 polynucleotide kinase (New England Biolabs) in 1× T4 polynucleotide kinase buffer and 1 mm ATP, incubated at 37 °C for 30 min, and heat-inactivated at 75 °C for 10 min. 5′-Phosphorylated PCR products were purified by phenol:chloroform extraction and ethanol precipitation and ligated into a pUC19 vector that was blunt end-linearized by treatment with SmaI (New England Biolabs) and dephosphorylated with calf intestinal phosphatase (Invitrogen). After transformation, individual clones were used to isolate plasmid DNA, which was then sequenced. The majority of obtained sequences contained mature 3′-CCA end sequences; only sequences of these 3′-end matured mt-tRNAs are reported here.
Sequencing of P. pallidum mt-tRNAs
P. pallidum strain PPHU8 (ATCC 44421) was cultured in a modified ATCC 712 PYG formulation, and a fraction enriched in mitochondria was isolated by cell disruption followed by differential centrifugation. Mitochondria were sedimented from the postnuclear supernatant by a 20-min centrifugation at 13,000 × g. RNA was isolated from mitochondrial pellets by a phenol:cresol extraction protocol (14), precipitated from the aqueous phase with ethanol, and treated with RNase-free DNase I before being reisolated and stored at −20 °C.
Circularization of tRNAs followed published protocols (14, 24): 1 μl of the resulting circularized tRNA was diluted with 1 μl of 5′ RT primer (1 pmol/μl; Table 1) in a 21.5-μl final volume and incubated at 90 °C for 2 min. The tRNA-primer mixture was allowed to cool to room temperature over a 20-min period and transferred to ice for 15 min. A final reaction volume of 30 μl contained 21.5 μl of cooled tRNA-primer mixture, 0.1 volume of avian myeloblastosis virus buffer (Amersham Biosciences), 1.8 μl of 1 mm dNTPs (60 μm final concentration of each dNTP), and 10 units of avian myeloblastosis virus reverse transcriptase (Amersham Biosciences) and was incubated at 45 °C for 45 min. For PCR of RT products, 20 pmol of the applicable 5′ and 3′ primers (Table 1) and 1 μl of cDNA were used for each reaction. Reaction mixtures (50 μl) consisted of Thermo Buffer (New England Biolabs; 5 μl), 0.2 mm dNTPs (250 μm final concentration of each dNTP), and 2.5 units of Taq DNA polymerase (5 units/μl; New England Biolabs). PCRs (carried out in a PerkinElmer Life Sciences GeneAmp PCR System 2400 thermocycler) were initiated with a 3-min incubation at 94 °C followed by 35 cycles as follows: 30 s at 94 °C, 30 s at 50 or 55 °C (depending on primer melting temperature), and 40 s at 72 °C. Cycling was followed by 12 min at 72 °C. PCR products were electrophoresed in 2% agarose gels. For TOPO TA cloning with pCR 2.1 (Invitrogen), PCR products were ligated and subsequently cloned according to the manufacturer's protocol.
Tobacco Acid Pyrophosphatase (TAP) Treatment prior to Circularization
Where indicated, total RNA was treated with TAP to convert 5′-triphosphorylated ends to 5′-monophosphorylated ends that are appropriate for ligation. Reactions (100 μl) contained 10 μg of total RNA (isolated as above) and 4 units of TAP (Epicenter) in 1× TAP buffer as provided by the manufacturer and were incubated at 37 °C for 1 h. Reaction products were purified by phenol:chloroform extraction and ethanol precipitation, and the RNA was resuspended in 10 μl of double distilled H2O for use in the circularization reactions as described above.
Cloning and Transcription of 5′-Truncated D. discoideum mt-tRNAs for in Vitro Assays
In vitro transcripts for mt-tRNA substrates that do not initiate with a G nucleotide (GluUUC and TyrGUA) were produced as 5′-hammerhead ribozyme constructs; a unique targeting sequence was engineered into the ribozyme so that the indicated 5′-end sequence of each mt-tRNA is obtained after co-transcriptional cleavage (15, 25). The CysGCA and TrpCCA transcripts both initiate with G+2 and, therefore, were constructed for direct transcription without a 5′-hammerhead sequence. Sequence-verified clones were used as templates for in vitro transcription by T7 RNA polymerase (15) and then 5′-phosphorylated with T4 polynucleotide kinase and [γ-32P]ATP.
Phosphatase Protection Assays for 5′-End Repair
5′-32P-Labeled mt-tRNAs GluUUC, TyrGUA, TrpCCA, and CysGCA were used as substrates for phosphatase protection assays to test for 5′-nucleotide addition. Reactions were performed as described (15); NTPs or dNTPs were included at 0.1–1 mm concentration in the assays as indicated. Reactions were initiated by addition of enzyme (at the concentrations indicated in each figure) to labeled tRNA and NTP and were carried out in 25 mm HEPES, pH 7.5, 10 mm MgCl2, 3 mm DTT, 125 mm NaCl, 0.2 mg/ml BSA buffer. Digestion with the indicated nuclease (RNase A or RNase T1) followed by calf intestinal phosphatase treatment was carried out as described (20), and reaction products were visualized by TLC and imaged using a Typhoon imager.
RESULTS
Eight tRNA Substrates Containing Mismatched Base Pairs in the Aminoacyl-acceptor Stem are 5′-Edited in D. discoideum
Eighteen mt-tRNA species are encoded in the mitochondrial genome of D. discoideum, and gene sequences for eight of these tRNAs clearly indicate the presence of up to three mismatched nucleotides in the predicted aminoacyl-acceptor stems (Fig. 2). To determine whether these tRNAs undergo 5′-editing to repair the mismatches, we isolated tRNA from axenically grown D. discoideum and sequenced the 5′-end of each of the eight predicted 5′-editing substrates. To obtain tRNA sequences, we used a previously described method in which the bulk tRNA preparations are treated with T4 RNA ligase to join the tRNA 5′- and 3′-ends, and then the sequence of the ligated region is amplified by RT-PCR using specific primers that target each tRNA (22, 26). The resulting PCR products were cloned into a SmaI-digested plasmid for sequencing of individual clones.
FIGURE 2.

Summary of verified mt-tRNA editing events in D. discoideum. Aminoacyl-acceptor stem sequences of the 18 tRNA species encoded by D. discoideum mtDNA (GenBankTM accession number NC_000895.1) are shown. The top row includes mt-tRNAs initially predicted to be substrates for 5′-editing (based on the presence of identifiable mismatches); the bottom row includes mt-tRNAs that do not contain mismatched nucleotides in the first three positions of their aminoacyl-acceptor stem. Mismatched nucleotides in the originally encoded mt-tRNA genes are indicated in red; alterations resulting in Watson-Crick base-paired nucleotides that were verified through this work are indicated by the red nucleotides shown to the left of each arrow. U-G/G-U base pairs in any of the first three positions of the aminoacyl-acceptor stem are indicated in blue; changes at any of these nucleotides supported by the mt-tRNA sequences obtained are also indicated by red nucleotides shown to the left of each arrow.
In the first set of sequenced clones (Table 2), we clearly observed evidence for 5′-editing of six of the eight mt-tRNA species that encode mismatched nucleotides in the acceptor stem (LeuUAG, LeuUAA, GlnUUG, IleGAU(1), IleGAU(2), and TyrGUA). In each case, sequences of the isolated mt-tRNAs contained Watson-Crick base-paired nucleotides in place of the originally encoded mismatched nucleotides, suggesting a mechanism of 5′-editing in D. discoideum similar to that observed in other protozoa (Fig. 1). In one case (tRNALeuUAA) two of four clones appeared to reflect partial editing in that the 3′–5′ repair component of the editing enzyme had apparently added the correct C+3 and U+2 nucleotides but not the final C+1 nucleotide to complete the aminoacyl-acceptor stem (Table 2). Because the number of clones obtained for each mt-tRNA varied, likely due to differences in efficiency of reverse transcription of each mt-tRNA, it is possible that similar partially edited intermediates exist for other tRNAs but were not detected. Nonetheless, sequences that demonstrate the existence of 5′-editing in D. discoideum were readily obtained for each of these six tRNAs, and the observation of these fully edited sequences suggests that the 5′-edited tRNAs likely comprise a significant fraction of the mt-tRNA pool in vivo.
TABLE 2.
Identification of mt-tRNA 5′-editing sites in D. discoideum
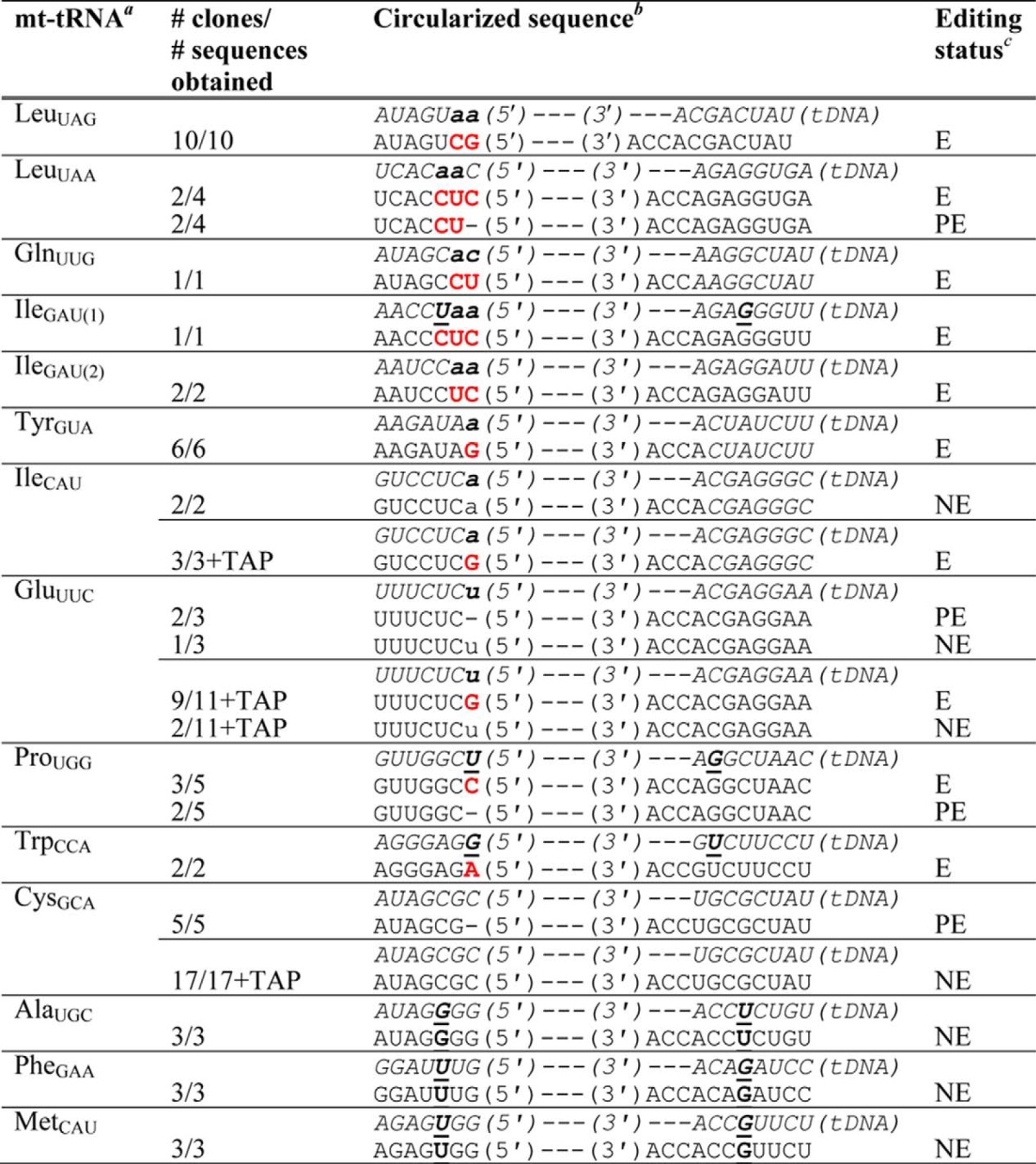
a Fourteen of 18 annotated mt-tRNAs were sequenced for this work; tRNAs LysUUU, AsnGUU, ArgUCU, and HisGUG do not contain annotated mismatches and were not sequenced.
b Aminoacyl-acceptor stem sequences of annotated mt-tRNA genes (shown in italics and labeled as tDNA with 5′- and 3′-ends as indicated) are compared with sequences of clones obtained through this work. Predicted mismatched nucleotides are shown in lowercase bold letters; U-G/G-U base pairs in the annotated or cloned sequences are indicated by underlined nucleotides. Replacement nucleotides are indicated by red letters.
c Editing status is indicated as follows: E, edited, observed changes were all to Watson-Crick base-paired nucleotides; PE, partially edited, evidence obtained for removal of mismatches and/or partial addition of Watson-Crick base-pairing nucleotides; NE, not edited, no evidence for editing was observed in the obtained sequences. All sequences were derived from fully mature 3′-CCA-containing mt-tRNA as indicated.
For the remaining two mt-tRNAs in which 5′-editing was expected to occur (IleCAU and GluUUC), the initially sequenced clones did not reveal sequences of the fully edited mt-tRNAs. For IleCAU, we obtained two clones, both of which were unedited (i.e. retained the +1-72 mismatch), and for GluUUC, we obtained three clones, one of which retained the +1-72 mismatch, whereas the other two showed partial editing in which the N+1 nucleotide had apparently been removed but the correct Watson-Crick-pairing G+1 had not yet been added (Table 2).
To further probe the editing of these mt-tRNAs, we considered a possibility based on the biochemical reaction catalyzed by the presumptive 3′–5′ polymerase component of the 5′-editing enzyme. The 3′–5′ polymerases are members of the Thg1 superfamily of enzymes, which use the 3′-hydroxyl of an incoming NTP to attack an activated 5′-phosphate on the tRNA substrate (27, 28). Thus, the immediate product of the 3′–5′ addition reaction would be a tRNA with a nucleoside triphosphate at its 5′-end (Fig. 1). In Saccharomyces cerevisiae, the Thg1 enzyme removes the 5′-pyrophosphate from the added nucleotide to generate a 5′-monophosphate end, but the ability of TLPs to similarly catalyze the removal of a terminal 5′-triphosphate has not been investigated. We reasoned that if TLPs catalyze 5′-repair but do not efficiently remove the 5′-pyrophosphate from some of the edited mt-tRNAs then the tRNAs would not be circularized efficiently by T4 RNA ligase (because ligase requires a 5′-monophosphate) and thus might not be amplified from the tRNA pool. To test this possibility, we treated the in vivo isolated tRNA with TAP to remove any 5′-triphosphorylated ends prior to ligation and then proceeded with RT-PCR amplification for the two missing tRNAs.
Consistent with our hypothesis, after TAP treatment, we readily obtained fully edited clones of both tRNAs (IleCAU and GluUUC) for which we were previously unable to observe editing (Table 2). The complete shift in the population after TAP treatment (from wholly unedited to substantially edited) suggests that the presence of a 5′-triphosphate was a significant barrier to circularization of at least these two tRNAs. The isolation of the other six circularized tRNAs in edited form even without TAP treatment may mean that the removal of the 5′-triphosphate after repair is more efficient for some substrates than for others or may be due to differences in stability of the 5′-triphosphate in certain mt-tRNAs.
Species-specific Differences in Editing of U-G/G-U Base Pairs during 5′-Editing
For one of the mt-tRNAs sequenced above (IleGAU(1)), we observed that the editing activity went beyond the correction of the two mismatched base pairs at A+1xG72 and A+2xA71 to also “repair” the U+3-G70 base pair in the aminoacyl-acceptor stem (Table 2 and Fig. 2). The ability of the 5′-editing activity to act on U-G/G-U base pairs despite their prevalence in normal tRNA structures has been observed previously as in A. castellanii where two examples of U-G editing were demonstrated, both of which apparently occur during the course of the editing reaction to repair other mismatches in the tRNAs (14).
To further assess the generality of editing of U-G/G-U base pairs, sequences of mt-tRNAs were determined from another eukaryotic slime mold, P. pallidum. In this organism, 5′-editing is also predicted on the basis of mismatches observed in five of 11 mtDNA-encoded tRNAs, and two of these five predicted substrates contain U-G/G-U base pairs in varying contexts within the first three base pairs of the aminoacyl-acceptor stem (Fig. 3). In P. pallidum, as in D. discoideum, isolated mt-tRNA sequences showed evidence of widespread 5′-editing (Table 3). The three mt-tRNAs that contain mismatches but lack U-G/G-U base pairs (IleGAU, LeuUAA, and AsnGUU) were observed to be mostly edited as expected (Table 3). However, in contrast to the observation of substantial editing of U-G/G-U pairs at multiple positions in A. castellanii and D. discoideum, editing of these base pairs in P. pallidum appears to be much rarer. For the two tRNAs that contain mismatches as well as U-G/G-U pairs (AlaUGC and PheGAA), little or no editing of the U-G/G-U base pairs was observed (Table 3). Thus, the ability to repair U-G/G-U base pairs in the course of 5′-editing seems to follow tRNA- or species-specific rules, perhaps depending on organism-specific features of the editing machinery.
FIGURE 3.

Summary of verified mt-tRNA editing events in P. pallidum. Aminoacyl-acceptor stem sequences of 11 mtDNA-encoded tRNA species in P. pallidum are shown. Clones containing the corresponding mt-tRNA genes were isolated from P. pallidum strain PPHU8 mtDNA and sequenced (36). Sequence comparison indicates that the mitochondrial genome of strain PPHU8 is essentially identical to that of P. pallidum strain PN500 (GenBank accession number EU275726.1). The top row includes mt-tRNAs predicted to be substrates for 5′-editing (based on the presence of identifiable mismatches) prior to this work; the bottom row includes mt-tRNAs that do not contain mismatched nucleotides in the first three positions of their aminoacyl-acceptor stem. Mismatched nucleotides in the originally encoded mt-tRNA genes are indicated in red; alterations that result in Watson-Crick base-paired nucleotides verified here are indicated by red nucleotides shown to the left of each arrow. U-G/G-U base pairs in any of the first three positions of the aminoacyl-acceptor stem are indicated in blue; changes at any of these nucleotides, if observed here, are also indicated by red nucleotides shown to the left of each arrow. Partial editing is indicated by small arrows with the replacement nucleotide that is observed in some but not all clones indicated in parentheses next to the arrowhead.
TABLE 3.
Identification of mt-tRNA 5′-editing sites in P. pallidum
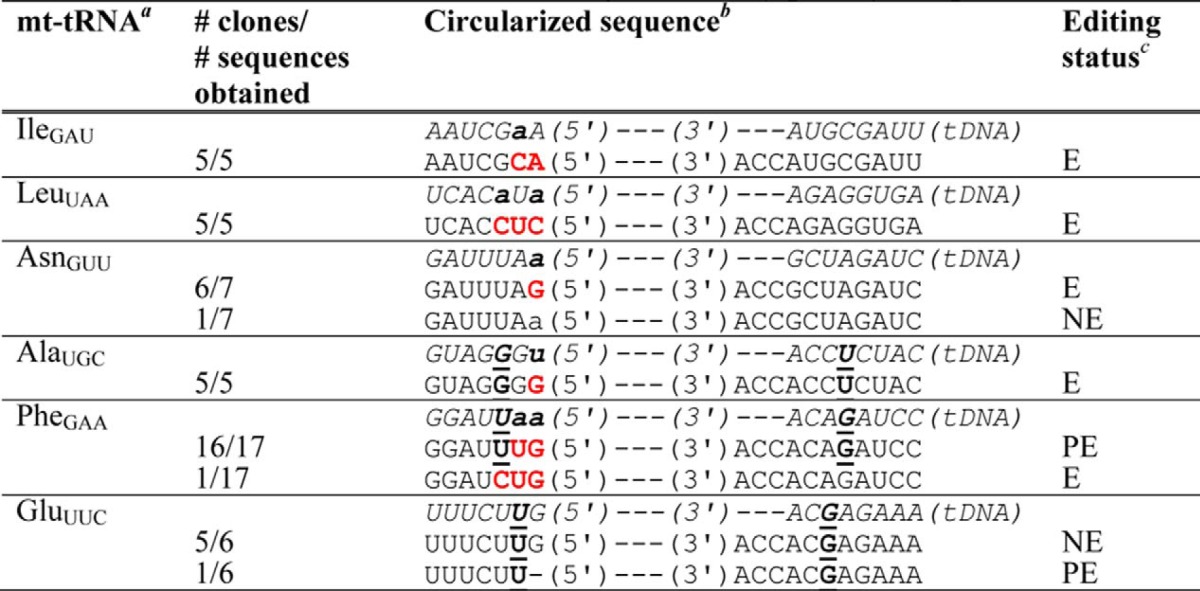
a Six of 11 annotated mt-tRNAs were sequenced for this work; tRNAs ArgUCU, GlnUUG, IleCAU, MetCAU, and HisGUG do not contain annotated mismatches and were not sequenced.
b Aminoacyl-acceptor stem sequences of annotated mt-tRNA genes (shown in italics and labeled as tDNA with 5′- and 3′-ends as indicated) are compared with sequences of clones obtained through this work. Predicted mismatched nucleotides are shown in lowercase bold letters; U-G/G-U base pairs in the annotated or cloned sequences are indicated by underlined nucleotides. Edited nucleotides are indicated by red letters.
c Editing status is indicated as follows: E, edited, observed changes were all to Watson-Crick base-paired nucleotides; PE, partially edited, evidence obtained for removal of mismatches and/or partial addition of Watson-Crick base-pairing nucleotides; NE, not edited, no evidence for editing was observed in the obtained sequences. All sequences were derived from fully mature 3′-CCA-containing mt-tRNA as indicated.
Identification of Two Additional mt-tRNA Editing Substrates in D. discoideum
We chose several of the D. discoideum mt-tRNAs that did not contain mismatches in the encoded sequences, including tRNAs ProUGG, TrpCCA, and CysGCA, to serve as controls for the above sequencing reactions because we expected to obtain the gene-encoded mt-tRNA sequences for these species (Fig. 2). However, ProUGG and TrpCCA, which contain U-G and G-U base pairs, respectively, at the +1-72 position but no other non-Watson-Crick base pairs, unexpectedly also showed evidence of 5′-editing (Table 2). In both cases, the encoded nucleotide at the +1 position was not present; instead the sequenced mt-tRNA contained the correct Watson-Crick base-pairing +1 nucleotide as indicated in Fig. 2. This is the first reported example of 5′-editing occurring in the absence of a bona fide mismatched base pair and implies that the nuclease component of the 5′-editing enzyme (as yet unidentified) must rely on additional features beyond simply the presence of a non-Watson-Crick mismatch.
In light of this observation, sequences of several other mt-tRNAs that similarly contain U-G/G-U base pairs but no identifiable mismatches were examined. Sequences were obtained in the same way for P. pallidum GluUUC, which contains a G+2-U71 base pair, and D. discoideum AlaUGC, PheGAA, and MetCAU, all with G-U/U-G base pairs at the +3-70 position. None of the G-U base pairs were observed to be edited in any of the sequences obtained from either organism (Tables 2 and 3). Thus, the total number of mt-tRNA editing substrates now verified in D. discoideum stands at 10 instead of the eight species that were initially predicted to undergo 5′-editing based on the presence of mismatches in their encoded sequences (Fig. 2).
The sequences obtained for tRNACysGCA suggest some additional complexity in the editing reaction. This tRNA contains no mismatches or U-G/G-U base pairs, so we again expected to isolate sequence identical to that of the mtDNA-encoded tRNA. However, when the standard ligation and amplification procedures were followed, we quantitatively obtained mt-tRNA clones (five of five) that lacked the C+1 nucleotide but instead began with a 5′-terminal G+2 (Table 2). We then treated the same tRNA with TAP prior to the ligation reactions as described above for IleCAU and GluUUC. Interestingly, we readily obtained clones from the TAP-treated RNA, and 15 of 15 of these were full-length, including C+1. This intriguing observation suggests that at least some of the full-length tRNACys may also contain the 5′-triphosphorylated end that is inhibitory to circularization. Further characterization of the processing reactions that generate mt-tRNACys is required to understand whether the 5′-editing machinery plays a role in metabolism of this, or any other, non-mismatch-containing substrates in vivo.
In Vitro Activities of DdiTLP3 and DdiTLP4 Correlate with the Observed in Vivo Editing Pattern
Previous in vitro assays demonstrated that both DdiTLP3 and DdiTLP4 catalyze efficient repair of two D. discoideum mt-tRNAs (IleCAU and LeuUAG) (15), but from this limited number of substrates, it is difficult to discern whether the two candidate enzymes exhibit non-overlapping substrate specificities that could perhaps explain the presence of two putative editing enzymes in D. discoideum. Thus, the in vitro activities of purified DdiTLP3 and DdiTLP4 were tested with four additional substrates, including two mt-tRNAs previously predicted to undergo editing (GluUUC and TyrGUA), one of the newly identified mt-tRNAs (TrpCCA), and a control mt-tRNA (CysGCA) that is not known to undergo editing.
Repair activities were tested using substrates that mimic 5′-truncated intermediates predicted to occur after the nuclease step of the 5′-editing reaction (i.e. for GluUUC, the mismatched U+1 is removed, and the substrate transcript initiates with C+2) (Fig. 4A). The tested substrates all contained an intact 3′-CCA end because the observation of partially edited and unedited intermediates among in vivo tRNA sequences, which all contain mature 3′-CCA ends, indicates that there is no obligatory order between 5′-editing and 3′-CCA addition reactions. A phosphatase protection assay used extensively to characterize 3′–5′ addition with other tRNAs was performed with each 5′-end-labeled substrate incubated in the presence of purified enzyme, ATP (required to activate the 5′-monophosphate for addition), and other NTP(s) as necessary to complete the aminoacyl-acceptor stem of each mt-tRNA. In these assays, nucleotide addition products are observed as phosphatase-resistant labeled oligonucleotides that can be separated by TLC from the inorganic phosphate generated by phosphatase treatment of the unreacted substrate. The ability of each of the four Thg1-related enzymes encoded in D. discoideum (DdiThg1, DdiTLP2, DdiTLP3, and DdiTLP4) and S. cerevisiae Thg1 (SceThg1) to add the indicated N+1 nucleotide and thus repair the 5′-truncated mt-tRNAs was determined (Fig. 4). As observed previously, only DdiTLP3 and DdiTLP4 catalyze efficient repair of any of these substrates, consistent with their ability to catalyze the observed editing reactions documented above by direct mt-tRNA sequencing. Moreover, the similar dependence of the observed activities on the concentration of DdiTLP3 and DdiTLP4, now demonstrated for five of the 10 known edited mt-tRNAs in D. discoideum, reveals no substantial differences between the catalytic activities of either enzyme that might indicate tRNA-specific roles for DdiTLP3 or DditTLP4 in editing. Finally, both enzymes catalyzed proficient repair of the 5′-truncated tRNACysGCA, which is not predicted to undergo editing in vivo, suggesting that the tRNA specificity of the editing reactions is not likely to be dictated by the polymerase component of the editing holoenzyme but perhaps is associated with the unidentified nuclease.
FIGURE 4.

5′-End repair of D. discoideum mt-tRNAs assayed by phosphatase protection. Phosphatase protection assays were performed with 5′-32P-labeled mt-tRNAGluUUC, mt-tRNATyrGUA, mt-tRNATrpCCA, and mt-tRNACysGCA (where the * indicates the position of the labeled phosphate on the reaction products observed in each panel) with 5-fold serial dilutions of each of the four purified DdiThg1/TLP enzymes and SceThg1. Reactions with GluUUC, TyrGUA, and CysGCA contained 7–0.01 μm DdiThg1, 44–0.07 μm DdiTLP2, 5–0.008 μm DdiTLP3, 25–0.04 μm DdiTLP4, and 75–0.12 μm SceThg1; reactions with TrpCCA contained 8–0.01 μm concentrations of each of the enzymes. No-enzyme control lanes are indicated by −. A vertical line marks a break between samples that were resolved on different TLC plates. Identities of oligonucleotides corresponding to the observed 3′–5′ addition products are indicated to the right of each panel; sites of RNase A- or RNase T1-catalyzed digestion that yield these labeled oligonucleotides are indicated by the arrows on the schematic diagram of each mt-tRNA aminoacyl-acceptor stem to the left of each panel.
DISCUSSION
Here, we demonstrate that extensive 5′-editing of mitochondrial tRNA occurs in D. discoideum and P. pallidum (Tables 2 and 3 and Figs. 2 and 3). In D. discoideum, mt-tRNA sequences reveal that 10 of the 18 mtDNA-encoded tRNAs undergo 5′-editing, including one instance of editing of a G-U base pair and two cases of U-G editing (Fig. 2 and Table 2). However, in the slime mold P. pallidum, although 5′-editing of mismatched nucleotides is also observed (for five of 11 annotated mt-tRNAs), U-G/G-U base pairs are observed to be edited much more rarely if at all (Fig. 3 and Table 3). Therefore, there are significant differences in the treatment of U-G/G-U base pairs by the 5′-editing machinery in different species or in the context of different mt-tRNAs, differences that remain to be investigated. We also demonstrate that in vitro 5′-end repair reactions catalyzed by the candidate D. discoideum editing enzymes (DdiTLP3 and DdiTLP4) are consistent with the observed editing events, including the newly identified repair of single U-G/G-U base pairs (Fig. 4).
To date, 5′-editing of mismatches encoded in mt-tRNA has been experimentally verified in seven eukaryotic species, including multiple members of Amoebozoa (D. discoideum, P. pallidum, P. polycephalum, and A. castellanii) and Fungi (S. punctatus, Monoblepharella sp., and Harpochytrium sp.) (6, 12–14, 22, 23). In each case, the presence of mismatched nucleotides encoded in any of the first three positions of the aminoacyl-acceptor stem is highly predictive of the occurrence of 5′-editing to repair these mt-tRNAs, and the majority of mature mt-tRNA sequences reveal that removal and repair of the 5′-mismatched nucleotides has indeed occurred. However, the results presented here demonstrate that the outcome of 5′-editing in any organism cannot be completely predicted on the basis of genome sequencing alone. Among these seven species are examples in which U-G/G-U base pairs are either unedited, partially edited, or fully edited in various contexts, and this variable editing (Fig. 5) does not appear to be a predictable feature for individual mt-tRNAs. Repair of U-G/G-U base pairs in at least two D. discoideum mt-tRNAs in the absence of any other non-Watson-Crick mismatched nucleotides, as documented here, suggests the possibility of an even larger potential pool of mt-tRNA editing substrates beyond just mismatch-containing tRNA. Thus, experimental data, such as direct sequencing of mt-tRNA, are required to rigorously assess the consequences of 5′-editing in any organism.
FIGURE 5.

Differences in 5′-editing of U-G/G-U base pair-containing mt-tRNAs from multiple protozoan species. For each of the six species shown (D. discoideum, A. castellanii, S. punctatus, P. pallidum, Harpochytrium 105, and Monoblepharella 15), sequences are shown for all mt-tRNAs that contain U-G or G-U base pairs and at least one mismatch at any of the first three positions of the aminoacyl-acceptor stem and for which sequences of mt-tRNA are available (this work and Refs. 6, 14, and 23). If evidence for 5′-editing has been observed in any of the sequences obtained, this is indicated by arrows with the replacement nucleotides shown in red to the left of each arrow. Partial editing is indicated by small arrows with the corrected nucleotide that is observed in some but not all clones indicated in parentheses next to the arrowhead. The observed extent of 5′-editing of the indicated U-G/G-U base pairs (based on the number of edited sequences of the total number of sequences obtained for each mt-tRNA) is shown as a percentage below each mt-tRNA. The sequence of A. castellanii MetCAU was obtained from a PCR product instead of individual clones and thus is presumed to reflect 100% editing (14).
The rules that govern how U-G/G-U base pairs are treated in the context of the 5′-editing reaction are apparently not universal. In D. discoideum, we observed robust editing of the U+3-G70 base pair of tRNAIleGAU(1) to a C+3-G70 pair, which occurred concurrently with repair of the mismatched base pairs located at the first two positions of this mt-tRNA. Of the six other species in which 5′-editing has been demonstrated, five of these (A. castellanii, P. pallidum, S. punctatus, Monoblepharella sp., and Harpochytrium sp.) contain at least one tRNA where a G-U pair would be similarly encountered during repair of other mismatched nucleotides; however, there are substantial differences between the observed editing patterns of these tRNAs (Fig. 5). For example, Monoblepharella tRNAMet, like D. discoideum tRNAIleGAU(1), contains mismatches at +1-72 and +2-71 along with a U+3-G70 pair; however, only ∼40% of the isolated Monoblepharella mt-tRNA contained the fully repaired sequences (including C+3-G70), whereas the remainder retained the U-G base pair (23). In addition, examples of mt-tRNA in which G-U base pairs are completely unedited are observed in P. pallidum and Harpochytrium despite the presence of other mismatched nucleotides. The function of 5′-editing of G-U/U-G base pairs is not readily apparent because it is known that these types of base pairs are well tolerated in normal tRNAs. For G-U/U-G base pairs that are edited during the course of editing other bona fide mismatches, it is possible that there is no inherent “benefit” to the tRNA to this editing process. However, for instances of G-U/U-G editing that occur in the absence of other mismatches (see below), there may be some important effect on tRNA structure or function that is not yet apparent due to the limited number of available sequences. This issue remains to be further investigated.
Even when the position of the G-U/U-G base pair is considered (i.e. whether it is located immediately next to the mismatch versus removed by one canonical base pair), a generalizable “rule” for editing of G-U base pairs is not evident. Although the G+3-U70 in P. pallidum tRNAAla is separated by one standard Watson-Crick base pair from the mismatch at +1-72 and is not edited, the U+3-G70 pair in A. castellanii tRNALeuUAA is located in a similar sequence context and is fully edited (Fig. 5). Furthermore, some G-U or U-G base pairs are located directly adjacent to a mismatched base pair and are efficiently edited to Watson-Crick pairs (i.e. those from D. discoideum IleGAU(1), A. castellanii MetCAU, and Monoblepharella ProUGG or GlyUCC), whereas other base pairs in a similar context are either partially edited (P. pallidum PheGAA and Monoblepharella MetCAU and GluUUC) or wholly unedited (Harpochytrium MetCAU). The different outcomes in terms of editing may reflect differences in catalytic activities of TLPs or the editing nucleases from different organisms. Thus, biochemical characterization of different editing enzymes from multiple species that carry out 5′-editing will be required to directly address this question.
This work revealed editing of the +1-72 G-U/U-G base pairs of D. discoideum tRNAPro and tRNATrp, which is to our knowledge the only demonstrated example of mt-tRNA 5′-editing that occurs in the absence of any other obvious mismatched nucleotides. This observation is consistent with previous biochemical data obtained with the partially purified mitochondrial 5′-editing enzyme in A. castellani in which the active fraction exhibited nucleotide incorporation activity with commercially obtained Escherichia coli tRNATyr (17). Because the E. coli tRNA lacks any mismatched nucleotides in the aminoacyl-acceptor stem, one possible explanation for this result would be a constitutive removal/repair activity that is not strictly dependent on the presence of mismatched nucleotides in the stem but instead acts more broadly on tRNA in vivo. The existence of editing activities that do not require mismatched nucleotides would not be apparent from simple sequence comparisons because such editing would involve no changes in the identity of the encoded bases in the aminoacyl-acceptor stem.
Relevant to this possibility is the observation reported here that for D. discoideum tRNACys full-length mt-tRNA sequences could not be obtained without first treating the tRNA with TAP (presumably to remove additional ligation-inhibiting 5′-phosphates) (Table 2). A possible explanation for this striking difference is that the mature tRNACysGCA present in D. discoideum contains a 5′-triphosphorylated C+1 nucleotide that is inhibitory to ligation, although the presence of other unknown 5′-phosphate modifications that inhibit ligation cannot be excluded. Because the standard cleavage reaction catalyzed by the 5′-maturation enzyme RNase P would generate a 5′-monophosphorylated C+1 nucleotide, an alternative mechanism may have generated the 5′-end of mt-tRNACys. One intriguing possibility is that C+1 is added by the same enzymes that catalyze 5′-editing in vivo. We note that tRNACys is the only one of the “unedited” mt-tRNAs in D. discoideum that contains a C at the +1 position and that other tRNAs containing C+1 are known to be miscleaved with some frequency by RNase P in bacteria (29). In this scenario, RNase P miscleavage could generate a 5′-truncated substrate (tRNACys initiating at G+2) that would be repaired by DdiTLP3 and/or DdiTLP4 as was observed with the in vitro activity assays (Fig. 4).
The question of the true identity of the 5′-editing enzyme in vivo in D. discoideum remains unresolved by these assays, and based on these in vitro results, it remains possible that either DdiTLP3, DdiTLP4, or both enzymes together are required to participate in editing in the cell. Intriguingly, both DdiTLP3 and DdiTLP4 were observed to catalyze a single major addition product corresponding to the correct full-length mt-tRNA in the in vitro assays, suggesting that the enzymes possess some mechanism to control the 3′–5′ addition reaction and avoid addition of excess nucleotides to the 5′-end of the tRNA during repair. The molecular basis for this activity and whether these enzymes utilize a strategy similar to that of the yeast Thg1 enzyme, which uses removal of a 5′-pyrophosphate residue from the added nucleotides to limit addition (30), remain to be addressed through further experiments. The structures of eukaryotic Thg1 and a bacterial TLP have been solved and indicate that Thg1/TLP family enzymes are tetramers, which has also been confirmed by solution measurements (31, 32). Therefore, it is possible that a heterotetramer containing both DdiTLP3 and DdiTLP4 comprises the active form of the editing enzyme. In vitro mixing experiments in which equal concentrations of both DdiTLP3 and DdiTLP4 were mixed prior to initiating reactions did not reveal any significant stimulation of 5′-repair activity beyond that observed for either enzyme alone (data not shown), but these observations do not rule out the possibility that such a heterotetramer forms co-translationally or after mitochondrial import. DdiTLP3, but not DdiTLP4, is predicted to contain an N-terminal tag for mitochondrial localization; however, localization of either of these enzymes has not been experimentally determined and would be required to confirm unequivocally or rule out a role in mitochondrial tRNA 5′-editing for either enzyme in living cells (33).
In D. discoideum, five of the 10 edited mt-tRNAs and one unedited mt-tRNA have now been shown to undergo efficient repair by both DdiTLP3 and DdiTLP4 (Ref. 15 and Fig. 4). Although substrate-specific activities of either enzyme on one or more of the remaining edited tRNAs could yet be identified that might explain the presence of two possible editing enzymes in D. discoideum, the weight of the evidence obtained to date argues against this possibility. Instead, the 5′-repair component of the editing enzyme seems to act broadly on any 5′-truncated mt-tRNAs that are generated by the nuclease activity, suggesting that any substrate recognition component of the 5′-editing enzyme would appear to be exerted at the 5′-nuclease step. This behavior contrasts sharply with the strong tRNA substrate selectivity exhibited by the eukaryotic Thg1 members of the 3′–5′ polymerase enzyme family that exhibit a >104-fold preference for nucleotide addition to tRNAHis over other tRNA species (34). The apparent lack of inherent tRNA substrate selectivity also opens the door to participation of TLPs in editing-like activities that do not depend on the presence of mismatched nucleotides but instead may involve alternative tRNA processing or quality control pathways. This may be particularly important with respect to the as-yet-unidentified functions of TLP enzymes in Bacteria and Archaea, which do not contain any 5′-mismatches in their encoded tRNAs (20, 21, 35). Studies to characterize the biological function of these enzymes are underway and may reveal pathways for general maintenance of tRNA 5′-end integrity.
Acknowledgments
We thank Jonatha Gott for technical assistance and Bhalchandra Rao for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM087543 (to J. E. J.) and Canadian Institutes of Health Research Grant MOP-4124 (to M. W. G.).
- mt-tRNA
- mitochondrial tRNA
- Thg1
- tRNAHis guanylyltransferase
- TLP
- Thg1-like protein
- Ddi
- D. discoideum
- TAP
- tobacco acid pyrophosphatase
- mtDNA
- mitochondrial DNA
- Sce
- S. cerevisiae.
REFERENCES
- 1. Phizicky E. M., Hopper A. K. (2010) tRNA biology charges to the front. Genes Dev. 24, 1832–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alfonzo J. D., Blanc V., Estévez A. M., Rubio M. A., Simpson L. (1999) C to U editing of the anticodon of imported mitochondrial tRNATrp allows decoding of the UGA stop codon in Leishmania tarentolae. EMBO J. 18, 7056–7062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Antes T., Costandy H., Mahendran R., Spottswood M., Miller D. (1998) Insertional editing of mitochondrial tRNAs of Physarum polycephalum and Didymium nigripes. Mol. Cell. Biol. 18, 7521–7527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gott J. M., Emeson R. B. (2000) Functions and mechanisms of RNA editing. Annu. Rev. Genet. 34, 499–531 [DOI] [PubMed] [Google Scholar]
- 5. Grosjean H., Benne R. (eds) (1998) Modification and Editing of RNA, ASM Press, Washington, D. C [Google Scholar]
- 6. Laforest M.-J., Roewer I., Lang B. F. (1997) Mitochondrial tRNAs in the lower fungus Spizellomyces punctatus: tRNA editing and UAG ‘stop’ codons recognized as leucine. Nucleic Acids Res. 25, 626–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leigh J., Lang B. F. (2004) Mitochondrial 3′ tRNA editing in the jakobid Seculamonas ecuadoriensis: a novel mechanism and implications for tRNA processing. RNA 10, 615–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Randau L., Stanley B. J., Kohlway A., Mechta S., Xiong Y., Söll D. (2009) A cytidine deaminase edits C to U in transfer RNAs in Archaea. Science 324, 657–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rubio M. A., Ragone F. L., Gaston K. W., Ibba M., Alfonzo J. D. (2006) C to U editing stimulates A to I editing in the anticodon loop of a cytoplasmic threonyl tRNA in Trypanosoma brucei. J. Biol. Chem. 281, 115–120 [DOI] [PubMed] [Google Scholar]
- 10. Schürer H., Schiffer S., Marchfelder A., Mörl M. (2001) This is the end: processing, editing and repair at the tRNA 3′-terminus. Biol. Chem. 382, 1147–1156 [DOI] [PubMed] [Google Scholar]
- 11. Yokobori S.-I., Pääbo S. (1995) tRNA editing in metazoans. Nature 377, 490. [DOI] [PubMed] [Google Scholar]
- 12. Lonergan K. M., Gray M. W. (1993) Editing of transfer RNAs in Acanthamoeba castellanii mitochondria. Science 259, 812–816 [DOI] [PubMed] [Google Scholar]
- 13. Lonergan K. M., Gray M. W. (1993) Predicted editing of additional transfer RNAs in Acanthamoeba castellanii mitochondria. Nucleic Acids Res. 21, 4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Price D. H., Gray M. W. (1999) Confirmation of predicted edits and demonstration of unpredicted edits in Acanthamoeba castellanii mitochondrial tRNAs. Curr. Genet. 35, 23–29 [DOI] [PubMed] [Google Scholar]
- 15. Abad M. G., Long Y., Willcox A., Gott J. M., Gray M. W., Jackman J. E. (2011) A role for tRNAHis guanylyltransferase (Thg1)-like proteins from Dictyostelium discoideum in mitochondrial 5′-tRNA editing. RNA 17, 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bullerwell C. E., Gray M. W. (2005) In vitro characterization of a tRNA editing activity in the mitochondria of Spizellomyces punctatus, a chytridiomycete fungus. J. Biol. Chem. 280, 2463–2470 [DOI] [PubMed] [Google Scholar]
- 17. Price D. H., Gray M. W. (1999) A novel nucleotide incorporation activity implicated in the editing of mitochondrial transfer RNAs in Acanthamoeba castellanii. RNA 5, 302–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jackman J. E., Gott J. M., Gray M. W. (2012) Doing it in reverse: 3′-to-5′ polymerization by the Thg1 superfamily. RNA 18, 886–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jackman J. E., Phizicky E. M. (2006) tRNAHis guanylyltransferase catalyzes a 3′-5′ polymerization reaction that is distinct from G−1 addition. Proc. Natl. Acad. Sci. U.S.A. 103, 8640–8645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rao B. S., Maris E. L., Jackman J. E. (2011) tRNA 5′-end repair activities of tRNAHis guanylyltransferase (Thg1)-like proteins from Bacteria and Archaea. Nucleic Acids Res. 39, 1833–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abad M. G., Rao B. S., Jackman J. E. (2010) Template-dependent 3′–5′ nucleotide addition is a shared feature of tRNAHis guanylyltransferase enzymes from multiple domains of life. Proc. Natl. Acad. Sci. U.S.A. 107, 674–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gott J. M., Somerlot B. H., Gray M. W. (2010) Two forms of RNA editing are required for tRNA maturation in Physarum mitochondria. RNA 16, 482–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laforest M.-J., Bullerwell C. E., Forget L., Lang B. F. (2004) Origin, evolution, and mechanism of 5′ tRNA editing in chytridiomycete fungi. RNA 10, 1191–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yokobori S., Pääbo S. (1995) Transfer RNA editing in land snail mitochondria. Proc. Natl. Acad. Sci. U.S.A. 92, 10432–10435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mörl M., Lizano E., Willkomm D. K., Hartmann R. K. (2005) in Handbook of RNA Biochemistry (Hartmann R. K., Schon A., Westhof E., eds) pp. 22–35, Wiley-VCH, Weinheim, Germany [Google Scholar]
- 26. Rao B. S., Mohammad F., Gray M. W., Jackman J. E. (2013) Absence of a universal element for tRNAHis identity in Acanthamoeba castellanii. Nucleic Acids Res. 41, 1885–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gu W., Jackman J. E., Lohan A. J., Gray M. W., Phizicky E. M. (2003) tRNAHis maturation: an essential yeast protein catalyzes addition of a guanine nucleotide to the 5′ end of tRNAHis. Genes Dev. 17, 2889–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smith B. A., Jackman J. E. (2012) Kinetic analysis of 3′–5′ nucleotide addition catalyzed by eukaryotic tRNAHis guanylyltransferase. Biochemistry 51, 453–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen W.-Y., Singh D., Lai L. B., Stiffler M. A., Lai H. D., Foster M. P., Gopalan V. (2012) Fidelity of tRNA 5′-maturation: a possible basis for the functional dependence of archaeal and eukaryal RNase P on multiple protein cofactors. Nucleic Acids Res. 40, 4666–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith B. A., Jackman J. E. (2014) Saccharomyces cerevisiae Thg1 uses 5′-pyrophosphate removal to control addition of nucleotides to tRNAHis. Biochemistry 53, 1380–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hyde S. J., Eckenroth B. E., Smith B. A., Eberley W. A., Heintz N. H., Jackman J. E., Doublié S. (2010) tRNAHis guanylyltransferase (THG1), a unique 3′-5′ nucleotidyl transferase, shares unexpected structural homology with canonical 5′-3′ DNA polymerases. Proc. Natl. Acad. Sci. U.S.A. 107, 20305–20310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hyde S. J., Rao B. S., Eckenroth B. E., Jackman J. E., Doublié S. (2013) Structural studies of a bacterial tRNAHIS guanylyltransferase (Thg1)-like protein, with nucleotide in the activation and nucleotidyl transfer sites. PLoS One 8, e67465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Claros M. G., Vincens P. (1996) Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 241, 779–786 [DOI] [PubMed] [Google Scholar]
- 34. Jackman J. E., Phizicky E. M. (2006) tRNAHis guanylyltransferase adds G−1 to the 5′ end of tRNAHis by recognition of the anticodon, one of several features unexpectedly shared with tRNA synthetases. RNA 12, 1007–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heinemann I. U., Randau L., Tomko R. J., Jr., Söll D. (2010) 3′-5′ tRNAHis guanylyltransferase in bacteria. FEBS Lett. 584, 3567–3572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schindel E. T. (2004) Editing of Mitochondrial tRNAs in Polysphondylium pallidum. M.Sc. thesis, Dalhousie University [Google Scholar]